
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Génotypage de l'ADN microsatellite et cytométrie des flux Analyses des tissus Hydatidiform Hydradins intégrés paraffins fixés à la formaline
Dans cet article
Résumé
Les grains de beauté hydatidiformes sont des grossesses humaines anormales avec des étiologies hétérogènes qui peuvent être classées en fonction de leurs caractéristiques morphologiques et de leur contribution parentale aux génomes des molaires. Ici, les protocoles du génotypage et de la cytométrie d'écoulement microsatellites de la paraffine-intégrée des tissus paraffines formalines sont décrits en détail, ainsi que l'interprétation et l'intégration des résultats.
Résumé
La taupe hydatidiform (HM) est une grossesse humaine anormale caractérisée par la prolifération trophoblastique excessive et le développement embryonnaire anormal. Il existe deux types de HM basés sur l'évaluation morphologique microscopique, hm complet (CHM) et HM partiel (PHM). Ceux-ci peuvent être subdivisés en fonction de la contribution parentale aux génomes molaires. Une telle caractérisation de HM, par des analyses de morphologie et de génotype, est cruciale pour la gestion patiente et pour la compréhension fondamentale de cette pathologie intrigante. Il est bien documenté que l'analyse morphologique de HM est sujette à une grande variabilité interobservateur et ne suffit pas à elle seule à classer avec précision HM en CHM et PHM et les distinguer des avortements hydropiques non-molaires. L'analyse du génotypage est principalement effectuée sur l'ADN et les tissus à partir de produits de conception intégrés à la paraffine (FFPE) qui ont une qualité inférieure à l'optimal et peuvent donc conduire à de mauvaises conclusions. Dans cet article, des protocoles détaillés pour les analyses de génotypage multiplex et de cytométrie du flux des tissus molaires FFPE sont fournis, ainsi que l'interprétation des résultats de ces méthodes, leur dépannage, et l'intégration avec l'évaluation morphologique , l'immunohistochimiede KIP2 p57, et l'hybridation in situ de fluorescence (FISH) pour atteindre un diagnostic correct et robuste. Ici, les auteurs partagent les méthodes et les leçons apprises au cours des 10 dernières années de l'analyse d'environ 400 produits de conception.
Introduction
Une taupe hydatidiform (HM) est une grossesse humaine anormale caractérisée par le développement embryonnaire anormal, l'hyperprolifération du trophoblaste, et la dégénérescence hydropic des villosités chorioniques (CV). Historiquement, HM était divisé en deux types, HM complet (CHM) et HM partiel (PHM) basé seulement sur l'évaluation morphologique1. Cependant, il a été démontré que l'évaluation morphologique seule ne suffit pas à classer HM dans les deux sous-types (CHM et PHM) et les distinguer des fausses couches non molaires2,3,4.
Puisque CHM et PHM ont des propensions différentes aux malignités, il est donc important de déterminer avec précision le type génotypique de HM pour fournir le suivi et la gestion appropriés aux patients. Par conséquent, au cours des dernières décennies, plusieurs méthodologies ont été développées et développées dans le but d'identifier la contribution parentale aux tissus molaires et d'atteindre une classification correcte de HM. Il s'agit notamment de l'analyse karyotype, du polymorphisme de baguage chromosomique, de la typage sérologique de l'antigène leucocyte humain (HLA), du polymorphisme de longueur de fragment de restriction, du nombre variable de répétitions de tandem, du génotypage microsatellite, de la cytométrie du débit et du p57 immunohistochimie KIP2. Cela a permis une subdivision précise des conceptions de HM basées sur la contribution parentale à leurs génomes, comme suit: CHM, qui sont diploïdes androgènes monospermiques ou diploïdes dispermiques androgénétiques androgènes, et PHM, qui sont triploïdes, dispermiques dans 99% et monospermic dans 1% des cas5,6,7,8. En outre, il existe un autre type de HM génotypique qui a émergé au cours des deux dernières décennies, qui est biparental diploïde. Ce dernier est principalement récurrent et peut affecter un seul membre de la famille (cas simplex) ou au moins deux membres de la famille (cas familiaux). Ces taupes biparentales diploïdes sont principalement provoquées par des mutations récessives dans NLRP7 ou KHDC3L dans les patients9,10,11,12. Diploid biparental HM chez les patients présentant des mutations récessives dans NLRP7 peut être diagnostiqué comme CHM ou PHM par analyse morphologique et ceci semble être associé à la sévérité des mutations dans les patients13,14. En plus de la classification de HM en fonction de leurs génotypes, l'introduction et l'utilisation de plusieurs méthodes de génotypage ont permis la distinction des différentes entités molaires des fausses couches non molaires, telles que les conceptions biparentales diploïdes anéuploides et d'autres types de conceptions5,15. De telles conceptions peuvent avoir une certaine prolifération de trophoblastet et la morphologie vilaine anormale qui imitent, dans une certaine mesure, quelques dispositifs morphologiques de HM.
Le but de cet article est de fournir des protocoles détaillés pour le génotypage multiplexe et la cytométrie de flux des tissus paraffinés (FFPE) de formaline fixe, et des analyses complètes des résultats de ces méthodes et de leur intégration avec d'autres méthodes pour diagnostic correct et concluant des tissus molaires.
Access restricted. Please log in or start a trial to view this content.
Protocole
Cette étude a été approuvée par la Commission d'examen institutionnel de McGill. Tous les patients ont donné leur consentement écrit pour participer à l'étude et faire récupérer leurs produits de conception FFPE (POC) dans divers départements de pathologie.
REMARQUE: Bien qu'il existe plusieurs méthodes de génotypage et de détermination ploidy par cytométrie de flux, les protocoles fournis ici décrivent une méthode d'analyse à l'aide d'une plate-forme pour chacun.
1. Génotypage
-
Sélection du meilleur bloc FFPE
- Pour chaque produit de conception FFPE (POC), préparez des sections tachées d'hématoxylin et d'éosine (H et E) d'une épaisseur de 4 m d'épaisseur, telles que décrites dans les sections 1.2 et 1.3, une pour chaque bloc disponible, pour l'évaluation morphologique par microscopie.
- À l'aide des diapositives h et E et d'un microscope léger, sélectionnez le bloc FFPE qui a la plus grande quantité de villosités chorioniques (CV), et si possible, le bloc qui a CV séparé des tissus maternels et non mêlés.
-
Sectionnement
- Placer le bloc choisi sur la glace pendant 15 min pour faciliter le sectionnement.
- Ajustez le microtome pour couper des sections de 4 m d'épaisseur pour l'évaluation morphologique microscopique et de 10 m d'épaisseur pour l'extraction de l'ADN.
- Placez le bloc froid dans le microtome et coupez une section de chaque bloc pour la coloration de H et E et 10-30 sections du bloc choisi, selon la quantité de CV dans le bloc, pour l'extraction de l'ADN.
REMARQUE: Pour les blocs pleins de CV, 10 sections sont suffisantes pour l'extraction de l'ADN. Si seulement environ 10% du bloc contient le CV tandis que le reste sont des tissus maternels, alors 20-30 sections sont nécessaires pour assurer des quantités suffisantes d'ADN. - À l'aide de forceps, transférer chaque section dans un bain d'eau de 45 oC. Ramassez la section du bain d'eau avec une glissière chargée positivement(Tableau des matériaux)qui est préalablement étiquetée avec le numéro d'identification de l'échantillon à l'aide d'un crayon.
- Placez les lames contenant les sections dans un four à 65 oC pour permettre aux sections d'adhérer aux glissières. Gardez les glissières de H et E au four pendant 25 min. Gardez les glissières pour l'extraction de l'ADN dans le four pendant 20 min.
REMARQUE: Le temps d'incubation plus court rend les tissus légèrement moins adhérents aux diapositives et facilite par conséquent l'ablation des tissus maternels.
-
Coloration de H et E
- Laisser refroidir les glissières à température ambiante (10 min).
- Préparation du réactif
- Préparer la solution de travail Eosin Y (0,25%) selon le tableau 1. Bien mélanger et conserver à température ambiante.
- Préparer la solution d'hématoxyline de travail en diluant la solution de stock d'hématoxyline 5x dans l'eau (c.-à-d., mélanger 80 ml d'eau avec 20 ml d'hématoxyline).
REMARQUE: Enveloppez la solution de stock d'hématoxylin dans le papier d'aluminium pour le stockage.
- Préparer les bocaux de coloration avec les réactifs corrects sous une hotte de fumée selon le tableau 2.
- Effectuez la coloration de H et E en submergeant les diapositives dans les bocaux de coloration appropriés pour la période appropriée selon le tableau 2.
- Monter les sections de 4 m pour l'analyse morphologique avec le milieu de montage et le couvercle avec des couvertures en verre (Tableau des matériaux).
REMARQUE: Les sections de 10 m pour le génotypage ne doivent pas être masquées. - Laissez les sections de 10 m sous le capot de fumée pendant un minimum de 3 h afin que les odeurs toxiques de xylène se dissipent.
MISE EN GARDE: Toutes les étapes de coloration doivent être effectuées sous une hotte de fumée. Les produits à xylène doivent être conservés sous le capot en tout temps parce que les odeurs de xylène sont toxiques. De plus, le xylène et l'hématoxylin doivent être jetés dans des contenants spéciaux. Une fois que ces contenants sont pleins, ils doivent être jetés comme recommandé par l'organisme de sécurité du laboratoire.
| réactif | quantité |
| Solution d'actions Eosin Y (1%) | 250 ml |
| 80% d'éthanol | 750 ml |
| Acide acétique glaciaire (concentré) | 5 mL |
Tableau 1 : Solution de travail Eosin Y (0,25 %) préparation.
| Réactif utilisé (100 ml par bac) | durée |
| 1) Xylène | 5 min |
| 2) Xylène | 5 min |
| 3) 100% éthanol | 2 min |
| 4) 95% d'éthanol | 2 min |
| 5) 70% éthanol | 2 min |
| 6) 50% d'éthanol | 2 min |
| 7) Eau distillée | 5 min |
| 8) Hématoxylin | 4 min |
| 9) Eau distillée | 5 min |
| 10) Eosin | 1 min |
| 11) 95% d'éthanol | 5 min |
| 12) 100% éthanol | 5 min |
| 13) Xylène | 5 min |
| 14) Xylène | 5 min |
Tableau 2 : Réactifs et durées du protocole de coloration H et E.
-
Isolement du CV
- Sous un microscope léger, utilisez des forceps et de petits morceaux de lingettes en papier hydratées à l'eau(Tableau des matériaux)pour gratter les tissus maternels indésirables des sections de 10 m d'épaisseur.
REMARQUE: L'objectif final est de ne garder que des membranes CV ou fœtales (lorsqu'elles sont présentes) sur les toboggans et donc d'enlever tous les autres tissus. Cette étape peut avoir besoin de beaucoup de temps et de patience, selon le bloc, car elle nécessite une attention méticuleuse aux détails. - Demandez à une deuxième personne de vérifier les glissières après le nettoyage pour s'assurer qu'elles sont exemptes de tissus maternels.
- Prenez des photos des diapositives nettoyées ou documentez ce qui suit pour aider à l'interprétation des données : 1) si le tissu était difficile à nettoyer, hémorragique ou très propre, 2) le nombre de sections utilisées, et 3) la quantité approximative de tissus nettoyés.
REMARQUE : La figure 1 fournit un exemple de diapositive facile à nettoyer. Pour un bloc contenant à peu près cette quantité de CV, 10 sections sont suffisantes pour l'extraction de l'ADN. La diapositive de la figure 2 a très peu de CV qui sont mélangés avec les tissus maternels, ce qui rend très difficile et long à nettoyer. Pour un bloc contenant à peu près cette quantité de CV, 30 sections sont nécessaires pour l'extraction de l'ADN. - Recueillir le CV à l'aide de petits morceaux de papier humide. À l'aide des forceps, arracher un petit morceau des lingettes de papier humidifiées et l'utiliser pour recueillir le CV.
- Placez les lingettes en papier avec leur CV attaché dans un tube étiqueté de 1,5 ml.
- Minimisez la quantité de lingettes en papier utilisées dans cette étape car trop peut obstruer la colonne d'extraction d'ADN et, par conséquent, réduire la quantité finale d'ADN recueilli. En moyenne, visez à utiliser moins de sept petits morceaux de lingettes en papier par échantillon. Si cela n'est pas possible en raison de la présence de grandes quantités de CV, diviser l'échantillon en deux tubes pour faciliter l'extraction.
- Sous un microscope léger, utilisez des forceps et de petits morceaux de lingettes en papier hydratées à l'eau(Tableau des matériaux)pour gratter les tissus maternels indésirables des sections de 10 m d'épaisseur.

Figure 1 : Diapositive représentative pour le génotypage. Haut : Une diapositive qui doit être « nettoyée » pour se libérer des tissus maternels. En bas: La même diapositive montrée après qu'il a été nettoyé et contient maintenant rien d'autre que CV pour l'extraction de l'ADN. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 2 : Diapositive représentative pour le génotypage. Haut : Une diapositive qui doit être « nettoyée » pour se libérer des tissus maternels. En bas: La même diapositive montrée après qu'il a été nettoyé et contient maintenant rien d'autre que CV pour l'extraction de l'ADN. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
- Suivez le protocole de l'extraction d'ADN du kit FFPE (Tableau des Matériaux) pour effectuer l'extraction de l'ADN.
REMARQUE: Certains kits recommandent d'utiliser 15 à 20 l de tampon d'élution pour l'élution finale. D'expérience, l'élution avec 15 l de tampon d'élution fonctionne bien pour la plupart des échantillons. Les dilutions peuvent être préparées à partir de l'ADN du stock au besoin.
-
Quantification de l'ADN
- À l'aide d'un dispositif de spectrophotomètre de laboratoire, chargez 1 L d'ADN et mesurez l'absorption à 260 nm pour la quantification.
- Chargez 1 L d'ADN sur un gel agarose de 2 % et exécutez l'électrophorèse de gel à une tension de 80 à 100 V pour l'évaluation qualitative.
- Sur la base des résultats des étapes 1.6.1 et 1.6.2, choisissez le volume d'ADN à utiliser dans l'amplification de la réaction en chaîne de polymériase (PCR) à répétition en tandem court multiplexe (STR). Visez à utiliser un minimum de 1000 ng d'ADN dans l'amplification PCR qui suit.
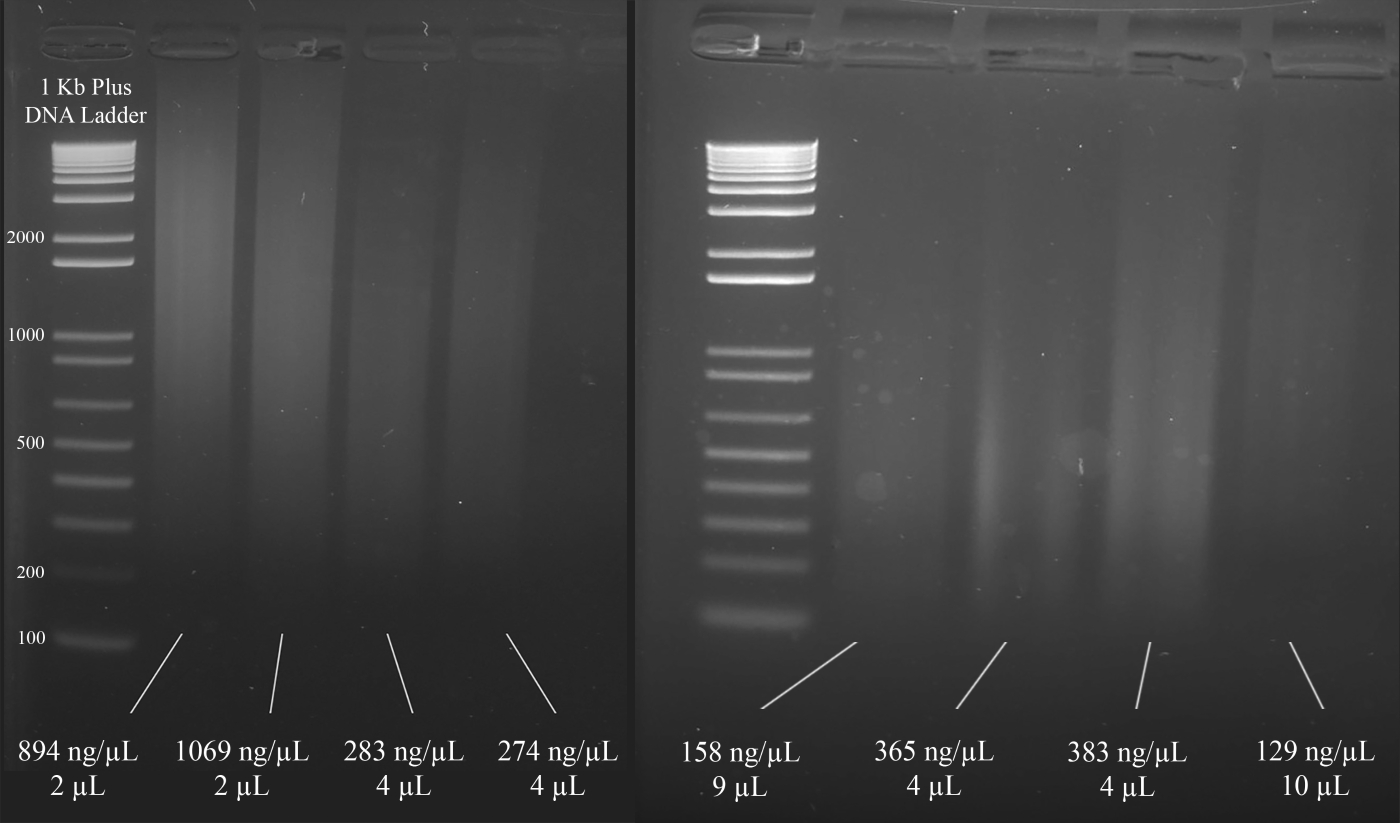
REMARQUE : La figure 3 démontre des exemples représentatifs de gels ainsi que les concentrations de l'ADN (basé sur les résultats du spectrophotomètre) et le volume de la solution d'ADN recommandée pour le multiplex STR PCR qui suit.

Figure 3 : Gel représentatif pour la quantification de l'ADN. Sont inclus les concentrations de chaque ADN, mesurées à l'aide d'un spectrophotomètre, et les quantités utilisées pour le multiplexe PCR. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
-
Amplification PCR
- Effectuer le génotypage microsatellite fluorescent à l'aide d'un système STR multiplex (Tableau des matériaux).
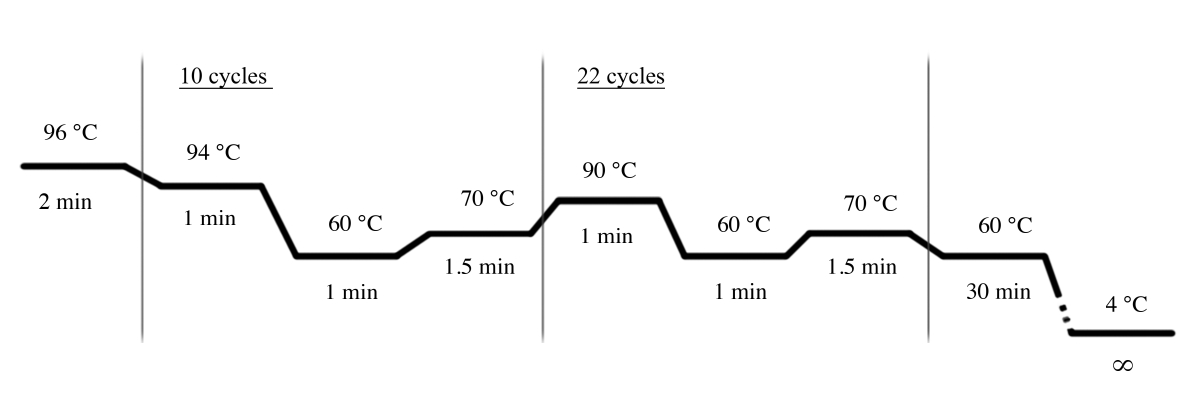
- Utilisez les conditions PCR indiquées à la figure 4 pour l'amplification PCR à l'aide du système STR multiplex (Tableau des matériaux).
REMARQUE: Les amorces suivantes sont utilisées dans ce système STR multiplex : D18S51, D21S11, TH01, D3S1358, Penta E, FGA, TPOX, D8S1179, vWA, Amelogenin, CSF1PO, D16S539, D7S820, D13S317, D5S818, et Penta D.

Figure 4 : Conditions du cycle PCR pour le système MULTIPLEx STR. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
-
Résoudre les produits PCR par électrophoresis capillaire.
- Suspendre 1 l de chaque échantillon amplifié dans 0,5 l de la voie standard interne du système multiplex et 9,5 l de formamide fortement déionisé (Tableau des matériaux).
- Exécuter des échantillons à travers un instrument d'électrophorèse capillaire (Table of Materials) à l'aide d'une matrice de séparation appropriée ( Table ofMaterials) pour l'instrument et l'ensemble de teinture du système multiplex.
- Analyse des données
- Analysez les données à l'aide d'un logiciel d'analyse des fragments d'ADN et comparez les allèles POC aux allèles parentaux pour déterminer leur origine.
- Configurez une norme de taille.
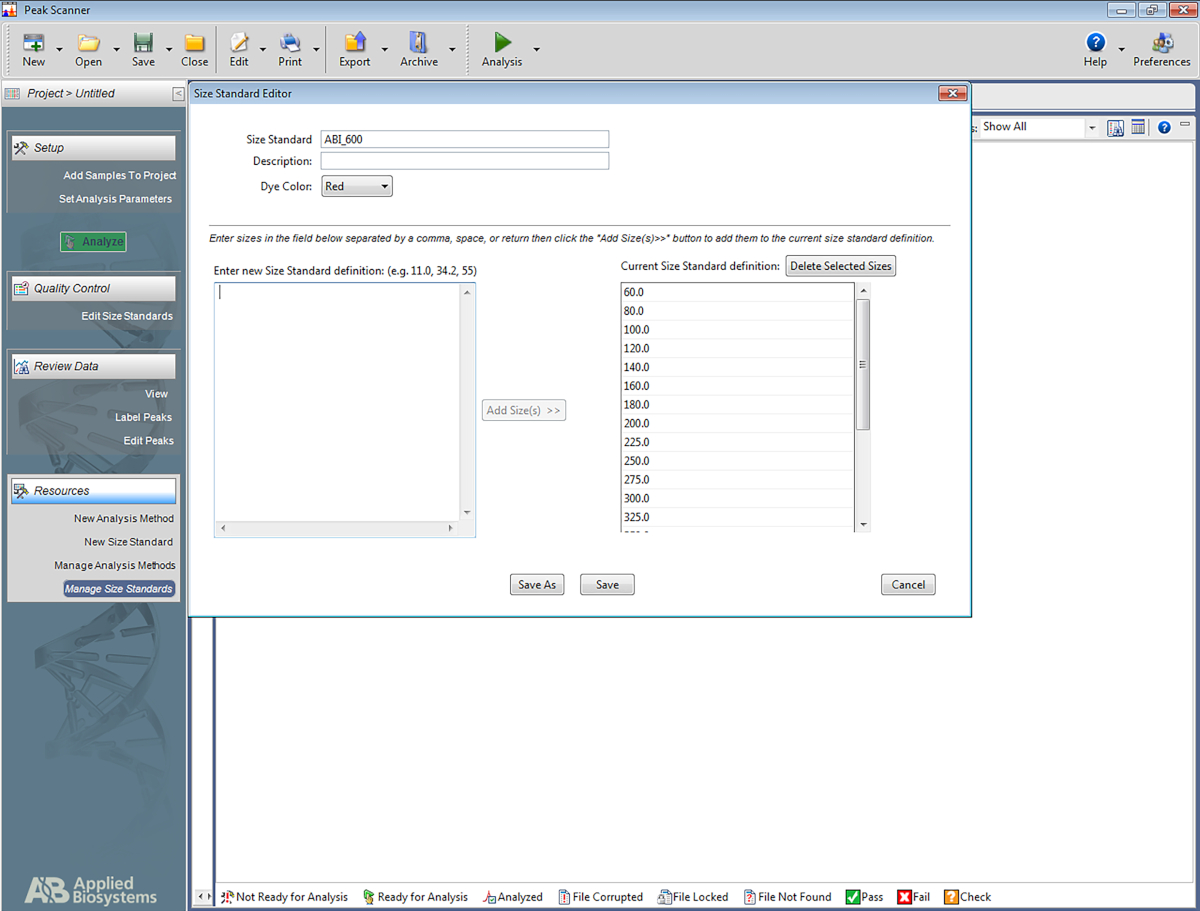
REMARQUE: Cela permet au logiciel de reconnaître l'échelle qui est utilisée dans le système STR multiplex, et d'attribuer des paires de base aux amplicons en fonction de l'échelle. Les étapes suivantes sont pour un logiciel spécifique (Tableau des matériaux) mais peut être utile pour la mise en place d'autres types de logiciels ainsi.- Ouvrez le logiciel. Cliquez sur Démarrer le nouveau projet, puis sur New Size Standard.
- Donnez un nom à la norme de taille (p. ex., ABI-600).
- Dans la boîte nommée Entrez nouvelle définition de la normede taille : entrez ce qui suit : 60, 80, 100, 120, 140, 160, 180, 200, 225, 250, 275, 300, 325, 350, 375, 400, 425, 450, 475, 500, 550, 600. Cliquez ensuite sur Ajouter de la taille.
REMARQUE : Les numéros saisis apparaîtront sous la case de droite, qui est nommée Définition standard de la taille actuelle (voir figure 5). - Cliquez sur Enregistrer.
- Pour importer et analyser un fichier, cliquez sur Ajouter des fichiers, et choisissez le fichier fsa à analyser. Cliquez sur Ajouter des fichiers sélectionnés, puis sur OK. Ensuite, suivez ces étapes:
- Localisez la colonne Taille Standard et choisissez ABI-600 (ou quel que soit le nom donné à la norme de taille).
- Dans le cadre de la méthode d'analyse,cliquez sur Dimensionnement par défaut - NPP, puis cliquez sur le bouton Analyse verte.
- Le fichier est maintenant prêt à être consulté. Ajuster les options d'affichage pour afficher les données comme vous le souhaitez.
- Dépannage - méthode d'analyse
REMARQUE: Le logiciel peut parfois ne pas identifier les pics et les aligner correctement. Cela se produit lorsque les pics sont trop bas ou trop élevés. Les deux méthodes d'analyse suivantes peuvent être corrigées et doivent être essayées avant qu'un échantillon ne soit retesté.- Méthode d'analyse 1 pour les pics élevés :
- Cliquez sur la nouvelle méthode d'analyse et nommez-la High Peaks (ou un autre nom selon vos préférences personnelles).
- Cliquez sur Gamme, puis sur La plage partielle pour l'analyse et le dimensionnement. Ensuite, tapez 100 pour le point de départ et la taille de départ.
- Pour le point d'arrêt, entrez 10 000. Pour la taille d'arrêt,entrez 1000.
- Ensuite, cliquez sur les hauteurs minimales de pointe et de changer les chiffres de telle sorte que le seuil de pointe pour les couleurs est la suivante: Bleu: 50; Vert: 50; Jaune: 20; Rouge: 100; Orange: 5000.
- Enregistrer la nouvelle méthode d'analyse.
- Méthode d'analyse 2 pour les pics bas :
- Cliquez sur la nouvelle méthode d'analyse et nommez-la Low Peaks (ou un autre nom selon vos préférences personnelles).
- Cliquez sur Gamme, puis sur La plage partielle pour l'analyse et le dimensionnement. Ensuite, tapez 100 pour le point de départ et la taille de départ.
- Pour le point d'arrêt, entrez 10 000. Pour la taille d'arrêt,entrez 1000.
- Ensuite, cliquez sur Les drapeaux de qualité et de changer la plage de laissez-passer de telle sorte qu'il se lit de 0,5 à 1. Modifier la plage de faible qualité de telle sorte qu'il se lit de 0,0 à 0,0. Changement Supposons la linéarité à ce qui suit: de (bp) 100,0 à (bp) 800.0.
- Enregistrer la nouvelle méthode d'analyse.
REMARQUE: Il est maintenant possible de réanalyser un fichier en choisissant Low Peaks ou High Peaks selon la méthode d'analyse, puis en cliquant sur le bouton Analyse verte.
- Méthode d'analyse 1 pour les pics élevés :

Figure 5 : Capture d'écran montrant l'éditeur standard de taille. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
2. Cytométrie de flux
-
Choisir le bloc FFPE idéal
- À l'aide de diapositives H et E et d'un microscope léger, sélectionnez un bloc FFPE qui a environ 50 à 70 % de ses tissus composés de CV.
REMARQUE : La figure 6 est un exemple représentatif d'un bloc approprié pour l'analyse de cytométrie du débit, car elle est composée d'environ 50 % de CV (la moitié droite de la section) et de 50 % de tissus maternels (moitié gauche). La présence de tissus maternels est importante parce qu'ils servent de contrôle interne pour le pic diploïde. - Pour les blocs qui n'ont pas la quantité idéale de CV, enrichissez-vous pour le CV au fur et à mesure que la section est effectuée. Pour ce faire, identifier de quel côté des sections fraîchement coupées contient plus de CV selon sa diapositive correspondante. Sur cette base, utilisez une lame pour couper l'autre moitié qui doit être jeté afin d'enrichir pour CV.
REMARQUE : La figure 7 montre un bloc qui n'a pas suffisamment de CV pour l'analyse de cytométrie du débit. Pour des blocs comme celui-ci, les sections doivent être coupées de telle sorte que la moitié qui contient moins de CV est jetée afin d'augmenter les quantités de CV par rapport aux tissus maternels, comme le montre la figure. Assurez-vous de couper plus de sections pour compenser ce qui est jeté.
- À l'aide de diapositives H et E et d'un microscope léger, sélectionnez un bloc FFPE qui a environ 50 à 70 % de ses tissus composés de CV.

Figure 6 : Section H et E représentant un bloc POC idéal pour la cytométrie du débit. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 7 : Section H et E représentant un bloc plus difficile pour la cytométrie du débit. Cette section représentative de la H-E montre que seule la moitié inférieure de cette section doit être utilisée pour l'analyse de la cytométrie du débit, dans le but d'enrichir pour le CV. La zone décrite, étiquetée « CV », est principalement composée de CV. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
- Sectionnement
- Laisser les blocs sur la glace pendant 15 min pour faciliter le sectionnement.
- À l'aide du meilleur bloc FFPE possible, couper quatre sections de 50 m d'épaisseur (ou deux sections de 100 m d'épaisseur) à l'aide d'un microtome.
REMARQUE: Pour la cytométrie de flux, il est préférable d'avoir des sections plus épaisses. - Dans le cas où un bloc FFPE idéal n'est pas disponible, visez à maintenir le rapport CV/tissu maternel néanmoins. Par exemple, si seulement 30 % du bloc est constitué de CV alors que le reste a des tissus maternels, retirez au moins la moitié de la section qui contient les tissus maternels et utilisez plus de sections pour compenser (voir la figure 7).
- Placer les sections dans des tubes étiquetés de 15 ml.
REMARQUE: Assurez-vous de ruban adhésif sur les étiquettes parce que les réactifs organiques utilisés dans l'étape suivante peuvent dissoudre et enlever l'encre.
- Protocole de cytométrie de flux à partir de tissus FFPE
- Déparaffinisation et réhydratation
- Effectuer les lavages suivants (tableau 3) sous une hotte de fumée.
- Remplir le tube de 15 ml de 6 ml du réactif approprié, suivant l'ordre présenté dans le tableau 3, laisser les sections dans les réactifs pour la durée respective, puis retirer le réactif à l'aide d'aspiration sous vide et d'une pipette Pasteur en verre.
- Entre chaque étape, trempez la pipette Pasteur d'abord dans 70% d'éthanol, puis dans de l'eau distillée, puis passez à l'étape suivante.
- Soyez très prudent de ne pas enlever les morceaux de tissu avec le réactif. Inclinez le tube de 15 ml à un angle de 60 degrés pour faciliter l'aspiration du réactif liquide sans dessiner de tissus.
MISE EN GARDE: Les liquides jetés contiennent du xylène et doivent être éliminés dans des contenants de déchets de xylène.
- Préparation de la solution
- Préparer la solution de citrate en dissolvant 2 g d'acide citrique dans 1 L d'eau double distillée. Amenez le pH à 6. Conserver à 4 oC.
- Préparer la solution de pepsine en dissolvant 0,01 g de pepsine en 2 ml de 9 parties par millier de NaCl, pH 1,64. C'est pour un échantillon.
MISE EN GARDE: La pepsine est toxique et peut facilement se disperser et s'envoler. Portez un masque lors de la manipulation de la pepsine sous sa forme de poudre et essuyez toute la zone de travail après l'avoir utilisé. - Propidium Iodide (PI)-ribonuclease Préparation d'une solution pour un échantillon.
- Mélanger 50 l'IP avec 450 oL de PBS (pour diluer 10x).
- Ajouter 50 ll de ribonuclée A (1 mg/ml) au mélange. Conserver enveloppé dans du papier d'aluminium en tout temps.
- Digestion et coloration
- Ajouter une solution de citrate de 4 oC aux tubes de 15 ml, puis placer dans un bain d'eau de 80 oC pendant 2 h.
- Laisser refroidir la solution à température ambiante (15 min). Retirez la solution de citrate.
- Ajouter 6 ml de 1x PBS, vortex, et attendre 1/2 min pour permettre aux tissus de se déposer au fond. Retirez le 1x PBS à l'aide d'aspiration sous vide et d'une pipette Pasteur en verre.
- Ajouter 1 ml de solution de pepsine (préchauffé e à 37 oC) et placer dans un bain sec de 37 oC pendant 30 min. Vortex toutes les 10 min. Préparer la solution PI-ribonuclease A dans les 10 dernières min de cette incubation.
- Ajouter 6 ml de 1x PBS, vortex, et attendre 1/2 min pour permettre aux tissus de se déposer au fond. Retirez le 1x PBS à l'aide d'aspiration sous vide et d'une pipette Pasteur en verre.
- Ajouter 550 ll de la solution PI-ribonuclease A et placer les échantillons dans un bain sec de 37 oC pendant 30 min.
REMARQUE: À ce stade, les échantillons peuvent être enveloppés dans du papier d'aluminium et laissés pendant la nuit à 4 oC jusqu'au lendemain matin. - Filtrer la solution à l'issue d'un maillage de filtration de 48 m. Recueillir le filtrate dans des tubes à fond rond en polystyrène, qui peuvent être utilisés avec le cytomètre d'écoulement. Utilisez des forceps pour placer un morceau de maille de filtration de 5 cm sur 5 cm dans la partie supérieure du tube, de sorte que le liquide peut être taché à travers le maillage et dans le tube.
REMARQUE: Les échantillons sont maintenant prêts à être exécutés avec le cytomètre de débit. Gardez-les enveloppés dans du papier d'aluminium jusqu'à ce qu'ils soient prêts à être exécutés.
- Exécutez des échantillons avec un cytomètre de débit à l'aide du technicien de la plate-forme de cytométrie de flux de l'organisation.
REMARQUE: Le canal PE est utilisé pour détecter l'ADN taché d'IP et le débit doit être réglé pour ralentir lors de l'acquisition. Assurez-vous que la tension est choisie de telle sorte que le pic diploïde est à peu près à 200 le long de l'axe X PE-A pour faciliter l'analyse et l'interprétation. Visez à enregistrer un minimum de 20 000 événements par échantillon.
- Déparaffinisation et réhydratation
| Réactif utilisé (6 ml chacun) | durée |
| 1) Xylène | 2 x 10 min |
| 2) 100% éthanol | 2 x 10 min |
| 3) 95% d'éthanol | 10 min |
| 4) 70% éthanol | 10 min |
| 5) 50% d'éthanol | 10 min |
| 6) Eau distillée | 2 x 10 min |
Tableau 3 : Réactifs et durées de déparaffinisation et de réhydratation.
-
Analyse des données de cytométrie de flux
- Analyser les données à l''eau à l'eide urétique des flux(Tableau des matériaux).
REMARQUE: Les étapes suivantes sont pour un logiciel spécifique (Tableau des matériaux) mais peut être utile pour la mise en place d'autres types de logiciels ainsi.- Après avoir analysé les échantillons sur un cytomètre de débit, téléchargez les fichiers FCS 2.0 pour analyse.
- Ouvrez le logiciel d'analyse de cytométrie de flux, cliquez sur Fichier ( Nouveau document.
- Cliquez sur l'icône
 Histogram ( ), puis faites glisser le pointeur pour faire un rectangle.
Histogram ( ), puis faites glisser le pointeur pour faire un rectangle. - Parcourez le fichier FCS, puis cliquez sur Open. Le long de l'axe x, cliquez sur FCS-A, puis sélectionnez PE-A.
- Cliquez sur l'icône
 Dot Plot ( ) puis faites glisser le pointeur pour créer un autre rectangle sous l'intrigue de l'histogramme. Ensuite, naviguez pour le même fichier FCS qui a été sélectionné pour l'histogramme.
Dot Plot ( ) puis faites glisser le pointeur pour créer un autre rectangle sous l'intrigue de l'histogramme. Ensuite, naviguez pour le même fichier FCS qui a été sélectionné pour l'histogramme. - Changer l'axe x de la parcelle de point à PE-A et l'axe y à PE-W.
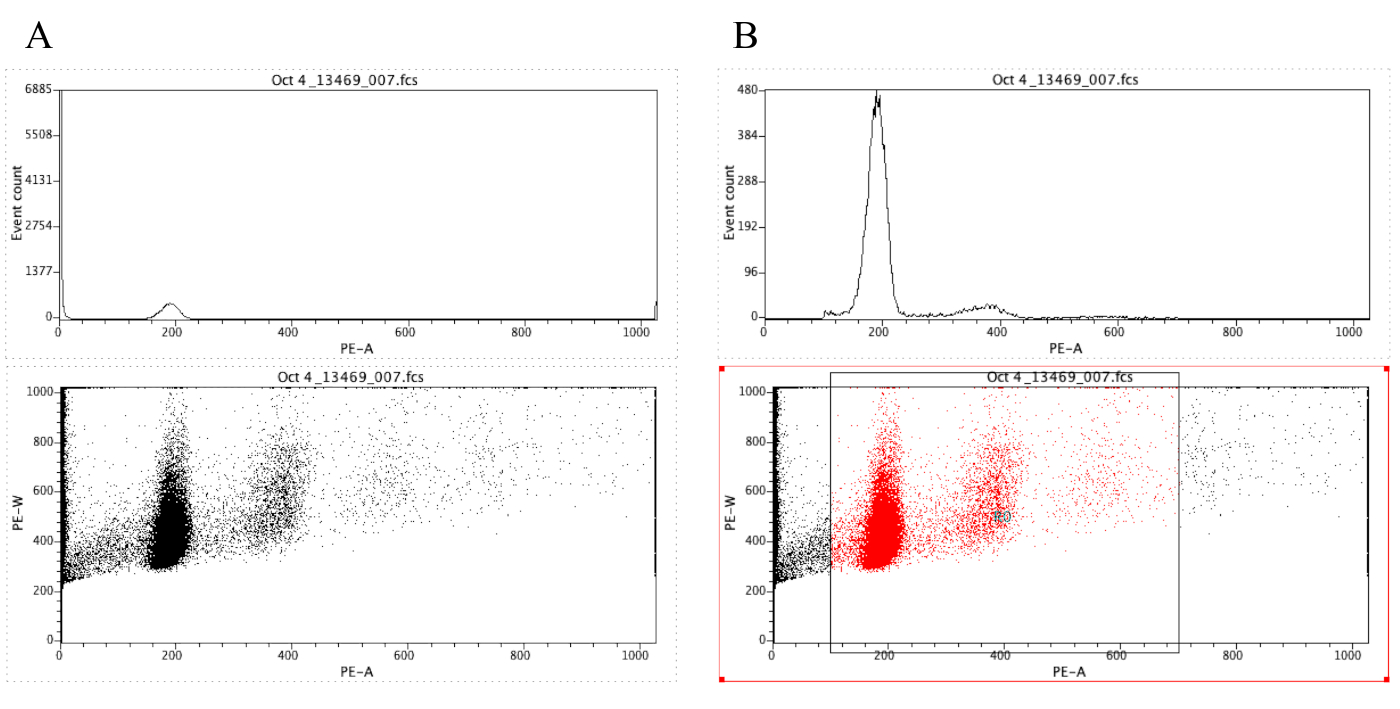
REMARQUE : La figure 8A démontre l'apparence des parcelles à ce stade. - Cliquez sur l'icône Région
 ( ) et dessinez une case sur l'intrigue à points qui commence avant le pic diploïde (environ 100 sur l'axe x de la figure 8B) et qui se termine autour de 700 sur l'axe x, comme le montre l'intrigue à points de la figure 8 B.
( ) et dessinez une case sur l'intrigue à points qui commence avant le pic diploïde (environ 100 sur l'axe x de la figure 8B) et qui se termine autour de 700 sur l'axe x, comme le montre l'intrigue à points de la figure 8 B.
REMARQUE: Le pic diploïde de la figure 8 est à peu près à 200 sur l'axe X. Ceci est choisi arbitrairement car les échantillons sont enregistrés à travers le cytomètre de débit, simplement pour faciliter l'analyse et l'interprétation des résultats. - Cliquez sur Terrain (fr) Modifier les régions / Portes, puis tapez R0 dans la cellule qui est à côté de la cellule G0 sous stratégie. Cliquez ensuite sur Fermer.
- Cliquez n'importe où sur l'histogramme, puis sur terrain ( Format Plot/Overlay. Sous la porte,sélectionnez G0 et R0, puis cliquez sur OK.
REMARQUE: C'est l'étape de gating qui permet de mieux visualiser les pics ploidy. L'histogramme devrait maintenant ressembler à l'histogramme de la figure 8B. Il est possible de jouer avec la porte créée (en déplaçant la boîte tirée à l'étape 2.4.1.7) afin de se concentrer sur des régions spécifiques de la parcelle de point. - Pour étiqueter les parcelles, cliquez
 sur l'icône de la zone de texte ( ), puis faites glisser le pointeur pour créer une boîte en haut du document, puis tapez les informations suivantes : ID patient, ID POC et le bloc utilisé (puisqu'il peut y avoir plusieurs blocs pour un POC) , pourcentage de CV présent sur le bloc, tension utilisée pour faire fonctionner l'échantillon et date.
sur l'icône de la zone de texte ( ), puis faites glisser le pointeur pour créer une boîte en haut du document, puis tapez les informations suivantes : ID patient, ID POC et le bloc utilisé (puisqu'il peut y avoir plusieurs blocs pour un POC) , pourcentage de CV présent sur le bloc, tension utilisée pour faire fonctionner l'échantillon et date.
- Analyser les données à l''eau à l'eide urétique des flux(Tableau des matériaux).

Figure 8 : Capture d'écran affichant un histogramme et une parcelle de point d'un échantillon représentatif qui n'est pas gated (A) et fermé (B). Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
Access restricted. Please log in or start a trial to view this content.
Résultats
La complexité des tissus molaires et le fait qu'ils peuvent avoir divers génotypes nécessite une analyse rigoureuse et l'utilisation de plusieurs méthodes telles que l'évaluation morphologique, l'immunohistochimie p57, le génotypage microsatellite, la cytométrie du débit et le FISH. Par exemple, un patient (1790) a été référé avec deux PHM qui se sont avérés être triploïdes par l'analyse de microarray des POC seulement. Le patient a été donc diagnostiqué avec PHM récu...
Access restricted. Please log in or start a trial to view this content.
Discussion
HM sont des grossesses humaines anormales avec des étiologies hétérogènes et ont différents types histologiques et génotypiques, ce qui rend leur classification précise et le diagnostic difficile. L'évaluation morphologique histopathologique s'est souvent avérée inexacte et n'est donc pas fiable en soi pour classer HM en CHM et PHM et les distinguer des fausses couches non-molaires. Par conséquent, un diagnostic précis de HM exige l'utilisation d'autres méthodes telles que le génotypage d'ADN microsatellite...
Access restricted. Please log in or start a trial to view this content.
Déclarations de divulgation
Les auteurs n'ont rien à révéler.
Remerciements
Les auteurs remercient Sophie Patrier et Marianne Parésy d'avoir partagé le protocole original de cytométrie du débit, et Promega et Qiagen d'avoir fourni des fournitures et des réactifs. Ce travail a été appuyé par le Réseau québécois en reproduction et l'Institut canadien de recherche en santé (MOP-130364) à R.S.
Access restricted. Please log in or start a trial to view this content.
matériels
| Name | Company | Catalog Number | Comments |
| BD FACS Canto II | BD BioSciences | 338960 | |
| Capillary electrophoresis instrument: Genomes Applied Biosystems 3730xl DNA Analyzer | Applied biosystems | 313001R | Service offered by the Centre for Applied Genomics (http://www.tcag.ca) |
| Citric acid | Sigma | 251275 | |
| Cytoseal 60, histopathology mounting medium | Fisher | 23244257 | |
| Eosin Y stock solution (1%) | Fisher | SE23-500D | |
| FCSalyzer - flow cytometry analysis software | SourceForge | - | https://sourceforge.net/projects/fcsalyzer/ |
| FFPE Qiagen kit | Qiagen | 80234 | |
| Forceps | Fine Science Tools | 11295-51 | For sectioning and for the cleaning process |
| Glacial Acetic Acid (Concentrated) | Sigma | A6283-500mL | |
| Glass coverslips: Cover Glass | Fisher | 12-541a | |
| Hematoxylin | Fisher | CS401-1D | |
| Highly deionized formamide: Hi-Di Formamide | Thermofisher | 4311320 | |
| IHC platform: Benchmark Ultra | Roche | - | |
| Kimwipes | Ultident | 30-34120 | |
| Microtome | Leica | RM2135 | |
| Microtome blades | Fisher | 12-634-1C | |
| Nitex filtering mesh, 48 μm | Filmar | 74011 | http://www.filmar.qc.ca/index.php?filet=produits&id=51&lang=en ; any other filter is suitable, but this is an inexpensive and effective option from a non-research company |
| p57 antibody | Cell Marque | 457M | |
| Pasteur pipette | VWR | 53499-632 | |
| PCR machine | Perkin Elmer, Applied Biosystems | GeneAmp PCR System 9700 | |
| PeakScanner 1.0 | Applied Biosystems | 4381867 | Software for genotyping analysis. |
| Pepsin from porcine gastric mucosa | Sigma | P7012 | |
| Polystyrene round-bottom tubes | BD Falcon | 352058 | |
| Positively charged slides: Superfrost Plus 25x75mm | Fisher | 1255015 | |
| PowerPlex 16 HS System | Promega Corporation | DC2102 | |
| Propidium Iodide | Sigma | P4864 | |
| Ribonuclease A from bovine pancreas | Sigma | R4875 | |
| Separation matrix: POP-7 Polymer | Thermofisher | 4352759 | |
| UltraPure Agarose | Fisher | 16500-500 | |
| Xylene | Fisher | X3P1GAL |
Références
- Szulman, A. E., Surti, U. The syndromes of hydatidiform mole. II. Morphologic evolution of the complete and partial mole. American Journal of Obstetrics & Gynecology. 132 (1), 20-27 (1978).
- Fukunaga, M., et al. Interobserver and intraobserver variability in the diagnosis of hydatidiform mole. The American Journal of Surgical Pathology. 29 (7), 942-947 (2005).
- Gupta, M., et al. Diagnostic reproducibility of hydatidiform moles: ancillary techniques (p57 immunohistochemistry and molecular genotyping) improve morphologic diagnosis for both recently trained and experienced gynecologic pathologists. The American Journal of Surgical Pathology. 36 (12), 1747-1760 (2012).
- Howat, A. J., et al. Can histopathologists reliably diagnose molar pregnancy? Journal of Clinical Pathology. 46 (7), 599-602 (1993).
- Banet, N., et al. Characteristics of hydatidiform moles: analysis of a prospective series with p57 immunohistochemistry and molecular genotyping. Modern Pathology. 27 (2), 238-254 (2014).
- Lipata, F., et al. Precise DNA genotyping diagnosis of hydatidiform mole. Obstetrics & Gynecology. 115 (4), 784-794 (2010).
- Buza, N., Hui, P. Partial hydatidiform mole: histologic parameters in correlation with DNA genotyping. International Journal of Gynecologic Pathology. 32 (3), 307-315 (2013).
- Fisher, R. A., et al. Frequency of heterozygous complete hydatidiform moles, estimated by locus-specific minisatellite and Y chromosome-specific probes. Human Genetics. 82 (3), 259-263 (1989).
- Murdoch, S., et al. Mutations in NALP7 cause recurrent hydatidiform moles and reproductive wastage in humans. Nature Genetics. 38 (3), 300-302 (2006).
- Parry, D. A., et al. Mutations causing familial biparental hydatidiform mole implicate c6orf221 as a possible regulator of genomic imprinting in the human oocyte. American Journal of Human Genetics. 89 (3), 451-458 (2011).
- Nguyen, N. M., Slim, R. Genetics and Epigenetics of Recurrent Hydatidiform Moles: Basic Science and Genetic Counselling. Current Obstetrics and Gynecology Reports. 3, 55-64 (2014).
- Sebire, N. J., Savage, P. M., Seckl, M. J., Fisher, R. A. Histopathological features of biparental complete hydatidiform moles in women with NLRP7 mutations. Placenta. 34 (1), 50-56 (2013).
- Nguyen, N. M., et al. Comprehensive genotype-phenotype correlations between NLRP7 mutations and the balance between embryonic tissue differentiation and trophoblastic proliferation. Journal of Medical Genetics. 51 (9), 623-634 (2014).
- Brown, L., et al. Recurrent pregnancy loss in a woman with NLRP7 mutation: not all molar pregnancies can be easily classified as either "partial" or "complete" hydatidiform moles. International Journal of Gynecologic Pathology. 32 (4), 399-405 (2013).
- Colgan, T. J., Chang, M. C., Nanji, S., Kolomietz, E. A Reappraisal of the Incidence of Placental Hydatidiform Mole Using Selective Molecular Genotyping. The International Journal of Gynecological Cancer. 26 (7), 1345-1350 (2016).
- Murphy, K. M., McConnell, T. G., Hafez, M. J., Vang, R., Ronnett, B. M. Molecular genotyping of hydatidiform moles: analytic validation of a multiplex short tandem repeat assay. The Journal of Molecular Diagnostics. 11 (6), 598-605 (2009).
Access restricted. Please log in or start a trial to view this content.
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.