
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Microsatellite DNA genotipagem e fluxo citometria de análise Ploidy de formalina-fixa parafina-Embedded Hydatidiform molar tecidos
Neste Artigo
Resumo
As toupeiras Hydatidiform são gravidezes humanas anormais com etiologias heterogêneas que podem ser classificadas de acordo com suas características morfológicas e contribuição parental aos genomas do molar. Aqui, os protocolos de genotipagem multiplex do ADN do microssatélites e de fluxo citometria de tecidos parafina-encaixados formalin-fixados do molar são descritos em detalhe, junto com a interpretação e a integração dos resultados.
Resumo
A toupeira Hydatidiform (HM) é uma gravidez humana anormal caracterizada pela proliferação trophoblastic excessiva e pelo desenvolvimento embrionário anormal. Existem dois tipos de HM com base na avaliação morfológica microscópica, HM completo (CHM) e HM parcial (PHM). Estes podem ser mais subdivididos com base na contribuição parental para os genomas molar. Essa caracterização da HM, por meio de análises morfológicas e genotípicas, é crucial para a gestão do paciente e para o entendimento fundamental dessa intrigante patologia. É poço-documentado que a análise morfológica do HM está sujeita à variabilidade interobservador larga e não é suficiente no seus próprios para classificar exatamente HM no chm e no PHM e distingui-los dos Abortions hidrópica do não-molar. A análise de Genotyping é executada na maior parte no ADN e nos tecidos dos produtos parafina-encaixados formalin-fixos (FFPE) da concepção, que têm menos do que a qualidade óptima e podem conseqüentemente conduzir às conclusões erradas. Neste artigo, são fornecidos protocolos detalhados para a análise de genotipagem multiplex e citometria de fluxo de tecidos molar de FFPE, juntamente com a interpretação dos resultados desses métodos, sua solução de problemas e a integração com a avaliação morfológica , P57KIP2 Immunohistochemistry, e a hibridação in situ da fluorescência (peixes) para alcangar um diagnóstico correto e robusto. Aqui, os autores compartilham os métodos e lições aprendidas nos últimos 10 anos a partir da análise de aproximadamente 400 produtos de concepção.
Introdução
Uma toupeira Hydatidiform (HM) é uma gravidez humana anormal caracterizada pelo desenvolvimento embrionário anormal, pela hiperproliferação do trophoblast, e pela degeneração hidrópica de Villi coriónico (CV). Historicamente, HM costumava ser dividido em dois tipos, HM completo (CHM) e HM parcial (PHM) com base apenas na avaliação morfológica1. No entanto, tem sido demonstrado que a avaliação morfológica isoladamente não é suficiente para classificar HM nos dois subtipos (chm e PHM) e distingui-los de abortos não-molar2,3,4.
Porque o chm e o PHM têm propensões diferentes às malignidades, é conseqüentemente importante determinar exatamente o tipo genotípica de HM para fornecer a continuação e a gerência apropriadas aos pacientes. Conseqüentemente, nas últimas décadas, diversas metodologias têm sido desenvolvidas e evoluídas com o objetivo de identificar a contribuição parental para os tecidos molares e alcançar uma correta classificação da HM. Estes incluem a análise do karyotype, o polimorfismo da borda cromossomática, a tipagem sorológicos do antígeno humano da leucócito (HLA), o polimorfismo do comprimento do fragmento da limitação, número variável de repetições em tandem, genotyping do microssatélites, citometria do fluxo, e P57 KIP2 immunohistochemistry. Isto permitiu a subdivisão exata de concepções do HM baseadas na contribuição parental a seus genomes, como segue: chm, que são chimerism androgenética monospérmicos ou diploid androgenética diploid, e PHM, que são triploid, chimerism em 99% e monospérmicos em 1% dos casos5,6,7,8. Além disso, há um outro tipo genotípica de HM que emergiu nas duas décadas passadas, que é biparental diploid. O último é na maior parte periódico e pode afetar um único membro da família (casos simples) ou pelo menos dois membros da família (casos familial). Estas toupeiras biparental diploid são causadas na maior parte por mutações recessive em NLRP7 ou em KHDC3L nos pacientes9,10,11,12. O HM biparental diploid nos pacientes com mutações recessive em NLRP7 pode ser diagnosticado como o chm ou o PHM pela análise morfológica e este parece ser associado com a severidade das mutações nos pacientes13,14. Além da classificação de HM de acordo com seus genótipos, a introdução e o uso de vários métodos de genotipagem permitiram distinguir as diferentes entidades molares de abortos não-molar, tais como concepções biparentais diploides aneuploides e outros tipos de concepções5,15. Tais concepções podem ter alguma proliferação do trofoblasto e morfologia villous anormal que imitam, em certa medida, algumas características morfológicas de hm.
O objetivo deste artigo é fornecer protocolos detalhados para genotipagem multiplex e citometria de fluxo de tecidos de parafina (FFPE) fixos em formalina, e análises abrangentes dos resultados desses métodos e sua integração com outros métodos para diagnóstico correto e conclusivo de tecidos molares.
Access restricted. Please log in or start a trial to view this content.
Protocolo
Este estudo de pesquisa foi aprovado pelo Comitê de revisão institucional da McGill. Todos os pacientes forneceram consentimento por escrito para participar do estudo e ter seus produtos de concepção de FFPE (POCs) recuperados de vários departamentos de patologia.
Nota: Embora existam vários métodos para a determinação de genotipagem e ploidia por citometria de fluxo, os protocolos aqui fornecidos descrevem um método de análise usando uma plataforma para cada um.
1. genotipagem
-
Seleção do melhor bloco de FFPE
- Para cada produto de concepção de FFPE (POC), prepare seções coradas de 4 μm de espessura de hematoxilina e eosina (H & E), conforme descrito nas seções 1,2 e 1,3, uma para cada bloco disponível, para avaliação morfológica por microscopia.
- Usando os slides H & e e um microscópio de luz, selecione o bloco FFPE que tem a maior quantidade de vilosidades coriônicas (CV), e se possível, o bloco que tem CV separado de, e não misturado com, tecidos maternos.
-
Corte
- Coloque o bloco escolhido no gelo por 15 min para facilitar o corte.
- Ajuste o micrótomo para cortar seções com 4 μm de espessura para avaliação morfológica microscópica e 10 μm de espessura para extração de DNA.
- Coloc o bloco frio no micrótomo e corte uma seção de cada bloco para a mancha de H & e e as seções 10 − 30 do bloco escolhido, dependendo da quantidade de CV no bloco, para a extração do ADN.
Nota: Para os blocos que estão cheios de CV, 10 seções são suficientes para a extração de DNA. Se apenas cerca de 10% do bloco contém CV, enquanto o resto são tecidos maternos, então 20 − 30 seções são necessários para garantir quantidades suficientes de DNA. - Usando fórceps, transfira cada seção a um banho de água de 45 ° c. Pegue a seção do banho de água com uma corrediça carregada positivamente (tabela dos materiais) que seja etiquetada previamente com o número de identificação da amostra usando um lápis.
- Coloque os slides contendo as seções em um forno a 65 ° c para permitir que as seções sigam os slides. Mantenha os slides para H & E no forno por 25 min. Mantenha os slides para extração de DNA no forno por 20 min.
Nota: O menor tempo de incubação torna os tecidos ligeiramente menos aderentes às lâminas e, consequentemente, facilita a remoção dos tecidos maternos.
-
Coloração de H & E
- Permita que os slides esfriem até a temperatura ambiente (10 min).
- Preparação do reagente
- Prepare a solução de trabalho eosina Y (0,25%) como por a tabela 1. Misture bem e armazene à temperatura ambiente.
- Prepare a solução de hematoxilina de trabalho diluindo a solução de estoque de hematoxilina 5x em água (ou seja, misture 80 mL de água com 20 mL de hematoxilina).
Nota: Envolva a solução do estoque do hematoxilina na folha para o armazenamento.
- Prepare frascos de coloração com os reagentes corretos uma capa de fumaça de acordo com a tabela 2.
- Realize a coloração H & E submergindo os slides nos frascos de coloração apropriados para o período de tempo correto de acordo com a tabela 2.
- Monte as secções de 4 μm para análise morfológica com suporte de montagem e lamínula com lamelas de vidro (tabela de materiais).
Nota: As secções de 10 μm para genotipagem não devem ser coverescorregas. - Deixe as seções de 10 μm a capa de fumaça por um mínimo de 3 h para que os odores tóxicos de xileno se dissipar.
Atenção: Todas as etapas de coloração precisam de ser executadas uma capa das emanações. Os produtos do xileno precisam de ser mantidos a capa em todas as vezes porque os odores do xileno são tóxicos. Além disso, xileno e hematoxilina precisam ser descartados em recipientes especiais. Uma vez que estes recipientes estão cheios, precisam de ser descartados como recomendado pela organização de segurança do laboratório.
| Reagente | Quantidade |
| Solução de estoque de eosina Y (1%) | 250 mL |
| 80% etanol | 750 mL |
| Ácido acético glacial (concentrado) | 5 mL de |
Tabela 1: solução de trabalho eosina Y (0,25%) Preparação.
| Reagente usado (100 mL por o escaninho) | Duração |
| 1) xileno | 5 minutos |
| 2) xileno | 5 minutos |
| 3) 100% etanol | 2 minutos |
| 4) 95% etanol | 2 minutos |
| 5) 70% etanol | 2 minutos |
| 6) 50% etanol | 2 minutos |
| 7) água destilada | 5 minutos |
| 8) hematoxilina | 4 minutos |
| 9) água destilada | 5 minutos |
| 10) eosina | 1 minuto |
| 11) 95% etanol | 5 minutos |
| 12) 100% etanol | 5 minutos |
| 13) xileno | 5 minutos |
| 14) xileno | 5 minutos |
Tabela 2: reagentes e durações para o protocolo de coloração H & E.
-
Isolamento de CV
- um estereomicroscópio leve, use fórceps e pequenos pedaços de lenços de papel umedecidos com água (tabela de materiais) para raspar os tecidos maternos indesejados de H & e-manchado 10 μm seções grossas.
Nota: O objetivo final é manter nada além de CV ou membranas fetais (quando presentes) nas lâminas e, assim, remover todos os outros tecidos. Esta etapa pode precisar de muito tempo e paciência, dependendo do bloco, pois requer atenção meticulosa aos detalhes. - Tenha uma segunda pessoa verific duas vezes as corrediças após a limpeza para assegurar-se de que estejam livres dos tecidos maternos.
- Tire fotos dos slides limpos ou documente o seguinte para ajudar na interpretação dos dados: 1) se o tecido foi difícil de limpar, hemorrágico ou muito limpo, 2) o número de secções utilizadas, e 3) a quantidade aproximada de tecidos limpos.
Nota: a Figura 1 fornece um exemplo de um slide que é fácil de limpar. Para um bloco que contenha aproximadamente esta quantidade de CV, 10 seções são suficientes para a extração do ADN. O slide na Figura 2 tem muito poucos CV que se misturam com os tecidos maternos, tornando-se muito difícil e demorado para limpar. Para um bloco que contenha aproximadamente esta quantidade de CV, 30 seções são necessários para a extração do ADN. - Colete o CV usando pequenas peças umedecidas de lenços de papel. Usando o fórceps, rasgue uma parte minúscula fora das limpezas de papel umedecidas e use-a para recolher o CV.
- Coloque os pedaços de lenços de papel com o seu CV anexado num tubo rotulado de 1,5 mL.
- Minimize a quantidade de limpezas de papel usadas nesta etapa como demasiada pode obstruir a coluna da extração do ADN e conseqüentemente reduzir a quantidade final de ADN coletado. Em média, o objetivo de usar menos de sete pequenos pedaços de papel toalhetes por amostra. Se isso não for possível devido à presença de grandes quantidades de CV, divida a amostra entre dois tubos para facilitar a extração.
- um estereomicroscópio leve, use fórceps e pequenos pedaços de lenços de papel umedecidos com água (tabela de materiais) para raspar os tecidos maternos indesejados de H & e-manchado 10 μm seções grossas.

Figura 1: slide representativo para genotipagem. Parte superior: uma corrediça que precise de ser "limpada" para tornar-se livre de tecidos maternos. Inferior: o mesmo slide mostrado depois de ter sido limpo e agora não contém nada, mas CV para extração de DNA. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: slide representativo para genotipagem. Parte superior: uma corrediça que precise de ser "limpada" para tornar-se livre de tecidos maternos. Inferior: o mesmo slide mostrado depois de ter sido limpo e agora não contém nada, mas CV para extração de DNA. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
- Siga o protocolo da extração do ADN do jogo de FFPE (tabela dos materiais) para executar a extração do ADN.
Nota: Alguns kits recomendam o uso de 15 − 20 μL de tampão de eluição para a eluição final. Da experiência, a eluição com 15 μL do amortecedor da eluição trabalha bem para a maioria de amostras. As diluições podem ser preparadas a partir do DNA de estoque, conforme necessário.
-
Quantificação do DNA
- Usando um dispositivo do espectrofotômetro do laboratório, carregue 1 μL do ADN e absorvância da medida em 260 nanômetro para a quantificação.
- Carregue 1 μL de DNA em um gel de agarose a 2% e faça eletroforese em gel em uma voltagem de 80 − 100 V para avaliação qualitativa.
- Com base nos resultados das etapas 1.6.1 e 1.6.2, escolha o volume de DNA a ser usado na amplificação em tandem curto Multiplex (STR) da reacção em cadeia da polimerase (PCR). Apontar para usar um mínimo de 1000 ng de DNA na amplificação de PCR que se segue.
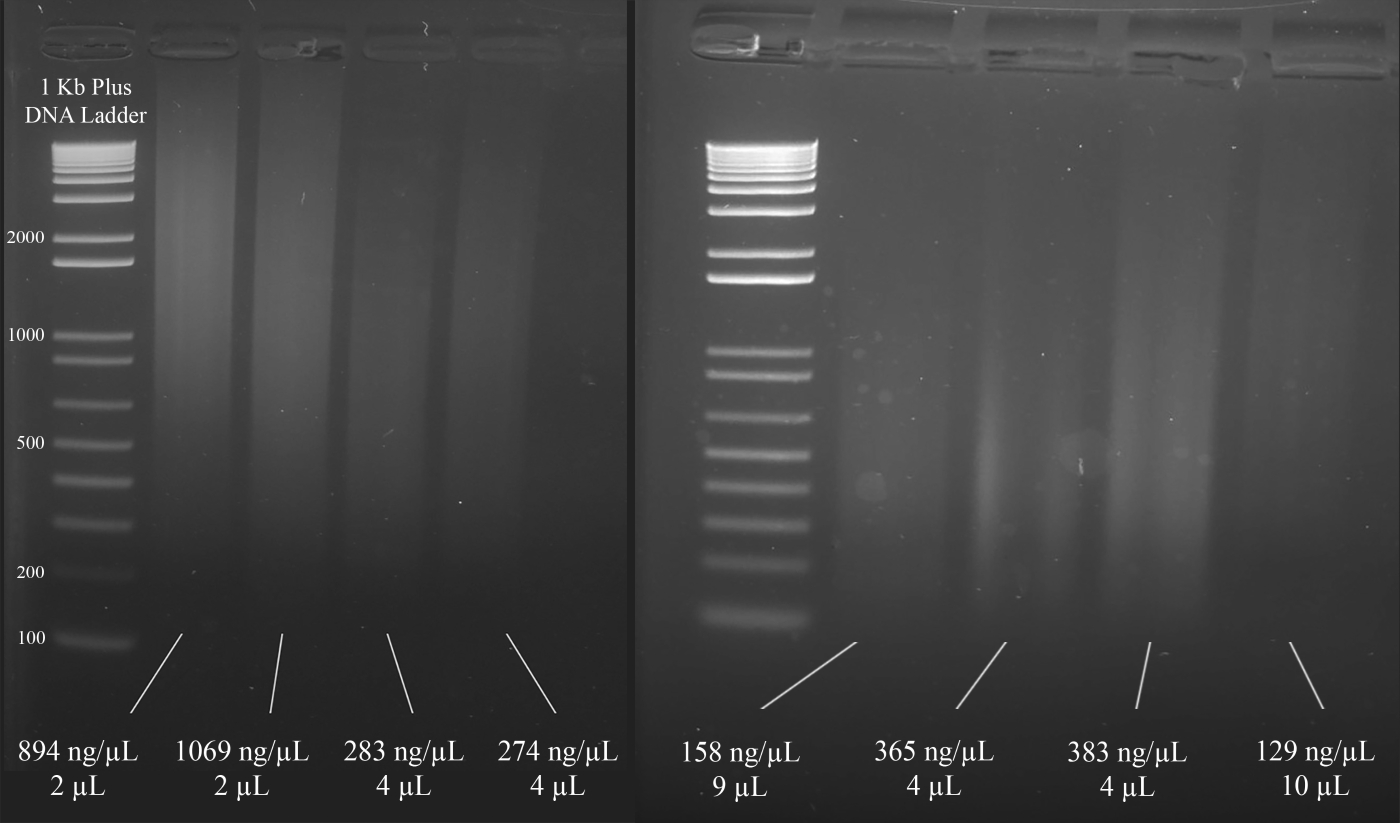
Nota: a Figura 3 demonstra exemplos representativos de géis, juntamente com as concentrações do DNA (com base nos resultados do espectrofotômetro), e o volume da solução de DNA que é recomendado para o multiplex Str PCR que se segue.

Figura 3: gel representativo para quantificação do DNA. São incluídas as concentrações de cada DNA, conforme medido com um espectrofotômetro, e as quantidades utilizadas para a PCR multiplex. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
-
Amplificação do PCR
- Realize a genotipagem de microssatélites fluorescentes usando um sistema de STR Multiplex (tabela de materiais).
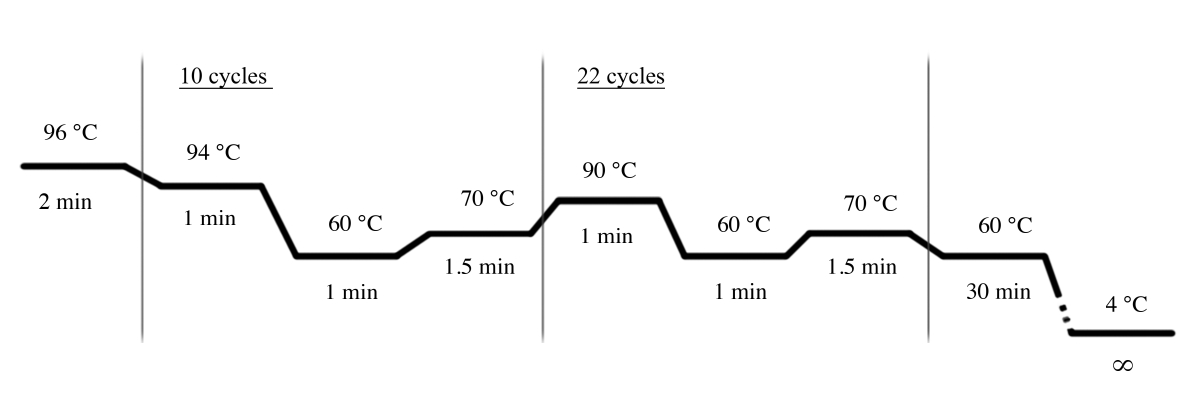
- Use as condições de PCR mostradas na Figura 4 para a amplificação de PCR usando o sistema multiplex Str (tabela de materiais).
Nota: Os seguintes primers são usados neste sistema multiplex STR: D18S51, D21S11, TH01, D3S1358, penta E, FGA, TPOX, D8S1179, vWA, amelogenin, CSF1PO, D16S539, D7S820, D13S317, D5S818, e penta D.

Figura 4: condições do ciclo de PCR para o sistema MULTIPLEX Str. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
-
Resolva os produtos do PCR pela electroforese capilar.
- Suspender 1 μL de cada amostra amplificada em 0,5 μL da faixa padrão interna do sistema multiplex e 9,5 μL de formamida altamente desionizada (tabela de materiais).
- Execute amostras através de um instrumento capilar da electroforese (tabela dos materiais) usando uma matriz apropriada da separação (tabela dos materiais) para o instrumento e o jogo do corante do sistema multiplex.
- Análise de dados
- Analise os dados com um software de análise de fragmentos de DNA e compare os alelos POC com os alelos parentais para determinar sua origem.
- Configure um padrão de tamanho.
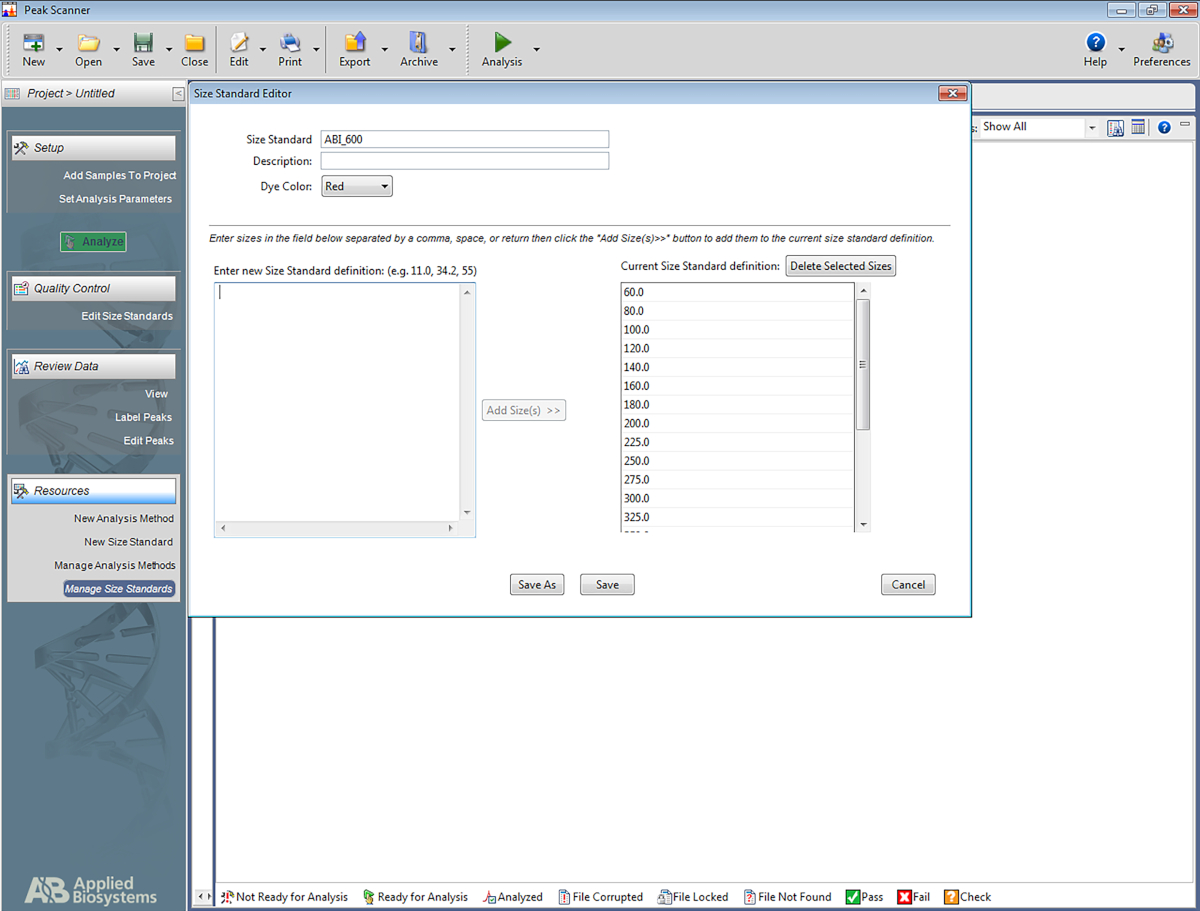
Nota: Isto permite que o software reconheça a escada que é usada no sistema do Str do multiplex, e atribua os emparelhadas aos amplicões baseados na escada. Os seguintes passos são para um software específico (tabela de materiais), mas pode ser de ajuda para a criação de outros tipos de software também.- Abra o software. Clique em Iniciar novo projeto e, em seguida, em novo tamanho padrão.
- Dê ao tamanho padrão um nome (por exemplo, ABI_600).
- Na caixa denominada Inserir novo tamanho padrão definição: digite o seguinte: 60, 80, 100, 120, 140, 160, 180, 200, 225, 250, 275, 300, 325, 350, 375, 400, 425, 450, 475, 500, 550, 600. Em seguida, clique em Adicionar tamanho (s).
Nota: os números inseridos aparecerá a caixa à direita, que é denominado definição padrão de tamanho atual (consulte a Figura 5). - Clique em Save.
- Para importar e analisar um arquivo, clique em Adicionar arquivose escolha o arquivo FSA a ser analisado. Clique em Adicionar arquivos selecionados e, em seguida, em OK. Em seguida, siga estes passos:
- Localize a coluna tamanho padrão e escolha ABI_600 (ou qualquer nome foi dado ao padrão de tamanho).
- Em método de análise, clique em dimensionamento padrão-NPP e, em seguida, clique no botão verde Analyze .
- O arquivo agora está pronto para visualização. Ajuste as opções de visualização para visualizar os dados conforme desejado.
- Solução de problemas-método de análise
Nota: O software pode, por vezes, não conseguir identificar picos e alinhá-los corretamente. Isto acontece quando os picos são demasiado baixos ou demasiado elevados. Os dois métodos de análise a seguir podem corrigir isso e devem ser tentados antes que uma amostra seja retestada.- Método de análise 1 para picos altos:
- Estale sobre o método novo da análise e nomeie-o altos picos (ou um outro nome como por a preferência pessoal).
- Clique no intervalo e, em seguida, em intervalo parcial para a análise e dimensionamento. Em seguida, digite 100 para o ponto inicial e o tamanho inicial.
- Para o ponto de parada, digite 10.000. Para o tamanho de parada, insira 1000.
- Em seguida, clique em alturas mínimas de pico e alterar os números de modo que o limiar de pico para as cores é o seguinte: azul: 50; Verde: 50; Amarelo: 20; Vermelho: 100; Laranja: 5000.
- Salve o novo método de análise.
- Método de análise 2 para picos baixos:
- Estale sobre o método novo da análise e nomeie-o baixo picos (ou um outro nome como por a preferência pessoal).
- Clique no intervalo e, em seguida, em intervalo parcial para a análise e dimensionamento. Em seguida, digite 100 para o ponto inicial e o tamanho inicial.
- Para o ponto de parada, digite 10.000. Para o tamanho de parada, insira 1000.
- Em seguida, clique em bandeiras de qualidade e alterar o intervalo de passagem de tal forma que ele lê de 0,5 para 1. Mude a escala da baixa qualidade tal que lê de 0,0 a 0,0. Alterar assume linearidade para o seguinte: de (BP) 100,0 a (BP) 800,0.
- Salve o novo método de análise.
Nota: Agora é possível reanalisar um arquivo escolhendo picos baixos ou picos altos em método de análise e, em seguida, clicando no botão verde Analyze .
- Método de análise 1 para picos altos:

Figura 5: captura de tela mostrando o editor de tamanho padrão. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
2. citometria de fluxo
-
Escolhendo o bloco FFPE ideal
- Usando corrediças de H & e e um microscópio claro, selecione um bloco de FFPE que tenha aproximadamente 50 − 70% de seus tecidos compor do CV.
Nota: a Figura 6 é um exemplo representativo de um bloco apropriado para a análise de citometria de fluxo, pois é composto de aproximadamente 50% cv (metade direita da seção) e 50% de tecidos maternos (metade esquerda). A presença de tecidos maternos é importante porque servem como controle interno para o pico diploide. - Para os blocos que não têm a quantidade ideal de CV, enriquecer para CV como o corte é realizada. Para fazer isso, identifique qual lado das seções recém-cortadas contém mais CV de acordo com seu slide H & E correspondente. Com base nisso, use uma lâmina para cortar a outra metade que precisa ser descartada, a fim de enriquecer para CV.
Nota: a Figura 7 mostra um bloco que não tem CV suficiente para análise de citometria de fluxo. Para blocos como este, as seções precisam ser cortadas de tal forma que a metade que contém menos CV é descartada, a fim de aumentar as quantidades de CV em relação aos tecidos maternos, como mostrado na figura. Certifique-se de cortar mais seções para compensar o que é Descartado.
- Usando corrediças de H & e e um microscópio claro, selecione um bloco de FFPE que tenha aproximadamente 50 − 70% de seus tecidos compor do CV.

Figura 6: seção H &Amp; E representando um bloco POC que é ideal para citometria de fluxo. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 7: Secção H &Amp; E que representa um bloqueio mais difícil para a citometria de fluxo. Esta seção representativa de H & E mostra que apenas a metade inferior desta seção deve ser utilizada para análise de citometria de fluxo, com o objetivo de enriquecer para o CV. A área esboçada, rotulada "CV", é composta principalmente de CV. por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
- Corte
- Deixe os blocos no gelo por 15 min para facilitar o corte.
- Usando o melhor bloco FFPE possível, corte quatro seções que são 50 μm de espessura (ou 2 100 μm de espessura seções) usando um micrótomo.
Nota: Para a citometria de fluxo é preferível ter seções mais grossas. - No caso em que um bloco de FFPE ideal não está disponível, o objetivo de manter a proporção de CV para o tecido materno, no entanto. Por exemplo, se apenas 30% do bloco é composto de CV, enquanto o resto tem tecidos maternos, em seguida, remover pelo menos metade da seção que contém os tecidos maternos e usar mais seções para compensar (ver Figura 7).
- Coloque as secções em tubos de 15 mL rotulados.
Nota: Seja certo gravar sobre as etiquetas porque os reagentes orgânicos usados na etapa seguinte podem dissolver e remover a tinta.
- Protocolo de citometria de fluxo de tecidos FFPE
- Desparaffinização e reidratação
- Realize as seguintes lavas (tabela 3) uma capa de fumos.
- Encha o tubo de 15 mL com 6 mL do reagente apropriado, seguindo a ordem apresentada na tabela 3, deixe as secções nos reagentes para a respectiva duração e, em seguida, retire o reagente utilizando sucção a vácuo e uma pipeta Pasteur de vidro.
- Entre cada passo, mergulhe a pipeta Pasteur primeiro em 70% etanol, em seguida, em água destilada, e depois proceder para o próximo passo.
- Tenha muito cuidado para não remover partes de tecido juntamente com o reagente. Incline o tubo de 15 mL para um ângulo de 60 graus para facilitar a sucção do Reagente líquido sem desenhar os tecidos.
Atenção: Os líquidos descartados contêm xileno e devem ser eliminados em recipientes de resíduos de xileno.
- Preparação da solução
- Prepare a solução de citrato dissolvendo 2 g de ácido cítrico em 1 L de água destilada dupla. Traga o pH para 6. Conservar a 4 ° c.
- Prepare a solução de pepsina dissolvendo 0, 1 g de pepsina em 2 mL de 9 partes por mil NaCl, pH 1,64. Isto é para uma amostra.
Atenção: Pepsin é tóxico e pode facilmente dispersar e tornar-se transportado por via aérea. Use uma máscara ao segurar a pepsina em sua forma de pó e limpe toda a área de trabalho depois de usá-la. - Iodeto de propiídio (PI)-ribonuclease uma preparação de solução para uma amostra.
- Misturar 50 μL de PI com 450 μL de PBS (para diluir 10x).
- Adicionar 50 μL de ribonuclease a (1 mg/mL) à mistura. Mantenha embrulhado em folha em todos os momentos.
- Digestão e coloração
- Adicionar 4 ° c solução de citrato para os tubos de 15 mL, em seguida, colocar em um banho de água 80 ° c para 2 h.
- Deixe a solução arrefecer até à temperatura ambiente (15 min). Retire a solução de citrato.
- Adicione 6 mL de 1X PBS, vortex, e aguarde 1 − 2 min para permitir que os tecidos se instalem na parte inferior. Retire o 1X PBS usando sucção a vácuo e uma pipeta Pasteur de vidro.
- Adicione 1 mL de solução de pepsina (pré-aquecida a 37 ° c) e coloque em um banho seco de 37 ° c por 30 min. Vortex a cada 10 min. Prepare a PI-ribonuclease uma solução nos últimos 10 min desta incubação.
- Adicione 6 mL de 1X PBS, vortex, e aguarde 1 − 2 min para permitir que os tecidos se instalem na parte inferior. Retire o 1X PBS usando sucção a vácuo e uma pipeta Pasteur de vidro.
- Adicionar 550 μL da solução de PI-ribonuclease e colocar as amostras num banho seco de 37 ° c durante 30 min.
Nota: Neste ponto, as amostras podem ser embrulhadas em folha e deixada durante a noite a 4 ° c até a manhã seguinte. - Filtre a solução através de uma malha de filtração de 48 μm. Colete o filtrado em tubos de poliestireno de fundo redondo, que pode ser usado com o citometro de fluxo. Use fórceps para colocar um pedaço de 5 cm por 5 cm de malha de filtração na parte superior do tubo, de tal forma que o líquido pode ser pipetado através da malha e no tubo.
Nota: As amostras estão agora prontas para serem executadas com o citometro de fluxo. Mantê-los embrulhados em folha até que eles estão prontos para ser executado.
- Execute amostras com um citômetro do fluxo com a ajuda do técnico da plataforma da citometria do fluxo da organização.
Nota: O canal de PE é usado para detectar o DNA manchado de PI e a taxa de fluxo deve ser ajustada para retardar durante a aquisição. Assegure-se de que a tensão esteja escolhida de modo que o pico diploid esteja aproximadamente em 200 ao longo do eixo-x do PE-a para facilitar a análise e a interpretação. Apontar para gravar um mínimo de 20.000 eventos por amostra.
- Desparaffinização e reidratação
| Reagente utilizado (6 mL cada) | Duração |
| 1) xileno | 2 x 10 min |
| 2) 100% etanol | 2 x 10 min |
| 3) 95% etanol | 10 minutos |
| 4) 70% etanol | 10 minutos |
| 5) 50% etanol | 10 minutos |
| 6) água destilada | 2 x 10 min |
Tabela 3: reagentes e durações para desparaffinização e reidratação.
-
Análise de dados de citometria de fluxo
- Analise dados com um software de análise de citometria de fluxo (tabela de materiais).
Nota: Os seguintes passos são para um software específico (tabela de materiais), mas pode ser de ajuda para a criação de outros tipos de software também.- Depois de executar as amostras em um citometro de fluxo, baixar FCS 2,0 arquivos para análise.
- Abra o software de análise de citometria de fluxo, clique em arquivo | Novo documento.
- Clique no ícone de histograma (
 ) e, em seguida, arraste o ponteiro para criar um retângulo.
) e, em seguida, arraste o ponteiro para criar um retângulo. - Procure o arquivo FCS e, em seguida, clique em abrir. Ao longo do eixo x, clique em FCS-a e, em seguida, selecione PE-a.
- Clique no ícone de gráfico de pontos
 () e arraste o ponteiro para criar outro retângulo abaixo do gráfico de histograma. Em seguida, procure o mesmo arquivo FCS que foi selecionado para o histograma.
() e arraste o ponteiro para criar outro retângulo abaixo do gráfico de histograma. Em seguida, procure o mesmo arquivo FCS que foi selecionado para o histograma. - Altere o eixo x do gráfico de pontos para PE- a e o eixo y para PE-W.
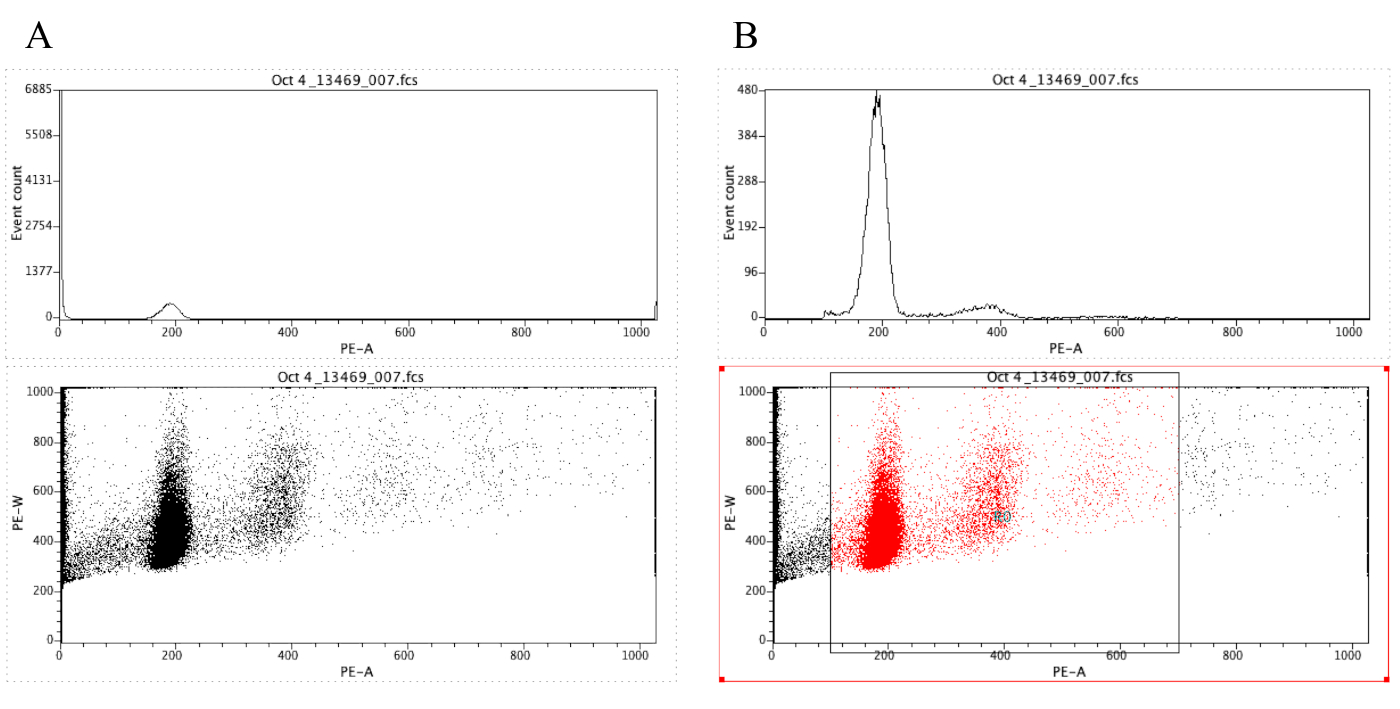
Nota: a Figura 8a demonstra a aparência das plotagens neste momento. - Clique no ícone de região (
 ) e desenhe uma caixa no gráfico de pontos que começa antes do pico diploide (em torno de 100 no eixo x na Figura 8B) e que termina em torno de 700 no eixo x, como mostrado no gráfico de pontos na Figura 8 O B.
) e desenhe uma caixa no gráfico de pontos que começa antes do pico diploide (em torno de 100 no eixo x na Figura 8B) e que termina em torno de 700 no eixo x, como mostrado no gráfico de pontos na Figura 8 O B.
Nota: O pico diploide na Figura 8 é aproximadamente em 200 no eixo x. Isto é escolhido arbitrariamente porque as amostras são gravadas através do cytometer do fluxo, para facilitar simplesmente a análise e a interpretação dos resultados. - Clique em Plot | Edite regiões/portõese digite R0 na célula que está ao lado da célula G0 em estratégia. Em seguida, clique em fechar.
- Clique em qualquer lugar no histograma, em seguida, em Plot | Formatar plotagem/sobreposição. portão, selecione G0 = R0 e, em seguida, clique em OK.
Nota: Esta é a etapa de gating que permite que um melhor visualize os picos do ploidia. O histograma agora deve se parecer com o histograma na Figura 8B. É possível jogar ao redor com a porta criada (movendo a caixa desenhada na etapa 2.4.1.7) a fim centrar-se sobre regiões específicas do lote do ponto. - Para rotular os gráficos, clique no ícone de área de
 texto (), em seguida, arraste o ponteiro para criar uma caixa na parte superior do documento e, em seguida, digite as seguintes informações: ID do paciente, POC ID e o bloco usado (uma vez que pode haver vários blocos para um POC) , por cento CV presente no bloco, tensão usada para executar a amostra, e a data.
texto (), em seguida, arraste o ponteiro para criar uma caixa na parte superior do documento e, em seguida, digite as seguintes informações: ID do paciente, POC ID e o bloco usado (uma vez que pode haver vários blocos para um POC) , por cento CV presente no bloco, tensão usada para executar a amostra, e a data.
- Analise dados com um software de análise de citometria de fluxo (tabela de materiais).

Figura 8: captura de tela exibindo um histograma e um gráfico de pontos de uma amostra representativa que é ungated (a) e gated (B). Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
Access restricted. Please log in or start a trial to view this content.
Resultados
A complexidade dos tecidos molares e o fato de que eles podem ter vários genótipos necessitam de uma análise rigorosa e o uso de vários métodos, como avaliação morfológica, imunohistoquímica P57, genotipagem de microssatélites, citometria de fluxo e FISH. Por exemplo, um paciente (1790) foi consultado com os dois PHM que foram encontrados para ser triploid pela análise do microarray dos POCs somente. O paciente foi diagnosticado conseqüentemente com o PHM periódico. O Microsa...
Access restricted. Please log in or start a trial to view this content.
Discussão
Hm são gravidezes humanas anormais com etiologias heterogêneas e têm tipos histológicos e genotípica diferentes, que faz sua classificação exata e o diagnóstico que desafiam. A avaliação morfológica histopatológica foi provada frequentemente imprecisa e é conseqüentemente não confiável no seus próprios classificar HM em CHM e em PHM e distingui-los dos abortos do não-molar. Conseqüentemente, um diagnóstico exato do HM exige o uso de outros métodos tais como o Genotyping multiplex do ADN do microssat?...
Access restricted. Please log in or start a trial to view this content.
Divulgações
Os autores não têm nada a revelar.
Agradecimentos
Os autores agradecem Sophie Patrier e Marianne Parésy por compartilharem o protocolo original de citometria de fluxo, e Promega e Qiagen para fornecimento de suprimentos e reagentes. Este trabalho foi apoiado pelo Réseau Québécois en reprodução e o Instituto canadense de pesquisa em saúde (MOP-130364) para o RS
Access restricted. Please log in or start a trial to view this content.
Materiais
| Name | Company | Catalog Number | Comments |
| BD FACS Canto II | BD BioSciences | 338960 | |
| Capillary electrophoresis instrument: Genomes Applied Biosystems 3730xl DNA Analyzer | Applied biosystems | 313001R | Service offered by the Centre for Applied Genomics (http://www.tcag.ca) |
| Citric acid | Sigma | 251275 | |
| Cytoseal 60, histopathology mounting medium | Fisher | 23244257 | |
| Eosin Y stock solution (1%) | Fisher | SE23-500D | |
| FCSalyzer - flow cytometry analysis software | SourceForge | - | https://sourceforge.net/projects/fcsalyzer/ |
| FFPE Qiagen kit | Qiagen | 80234 | |
| Forceps | Fine Science Tools | 11295-51 | For sectioning and for the cleaning process |
| Glacial Acetic Acid (Concentrated) | Sigma | A6283-500mL | |
| Glass coverslips: Cover Glass | Fisher | 12-541a | |
| Hematoxylin | Fisher | CS401-1D | |
| Highly deionized formamide: Hi-Di Formamide | Thermofisher | 4311320 | |
| IHC platform: Benchmark Ultra | Roche | - | |
| Kimwipes | Ultident | 30-34120 | |
| Microtome | Leica | RM2135 | |
| Microtome blades | Fisher | 12-634-1C | |
| Nitex filtering mesh, 48 μm | Filmar | 74011 | http://www.filmar.qc.ca/index.php?filet=produits&id=51&lang=en ; any other filter is suitable, but this is an inexpensive and effective option from a non-research company |
| p57 antibody | Cell Marque | 457M | |
| Pasteur pipette | VWR | 53499-632 | |
| PCR machine | Perkin Elmer, Applied Biosystems | GeneAmp PCR System 9700 | |
| PeakScanner 1.0 | Applied Biosystems | 4381867 | Software for genotyping analysis. |
| Pepsin from porcine gastric mucosa | Sigma | P7012 | |
| Polystyrene round-bottom tubes | BD Falcon | 352058 | |
| Positively charged slides: Superfrost Plus 25x75mm | Fisher | 1255015 | |
| PowerPlex 16 HS System | Promega Corporation | DC2102 | |
| Propidium Iodide | Sigma | P4864 | |
| Ribonuclease A from bovine pancreas | Sigma | R4875 | |
| Separation matrix: POP-7 Polymer | Thermofisher | 4352759 | |
| UltraPure Agarose | Fisher | 16500-500 | |
| Xylene | Fisher | X3P1GAL |
Referências
- Szulman, A. E., Surti, U. The syndromes of hydatidiform mole. II. Morphologic evolution of the complete and partial mole. American Journal of Obstetrics & Gynecology. 132 (1), 20-27 (1978).
- Fukunaga, M., et al. Interobserver and intraobserver variability in the diagnosis of hydatidiform mole. The American Journal of Surgical Pathology. 29 (7), 942-947 (2005).
- Gupta, M., et al. Diagnostic reproducibility of hydatidiform moles: ancillary techniques (p57 immunohistochemistry and molecular genotyping) improve morphologic diagnosis for both recently trained and experienced gynecologic pathologists. The American Journal of Surgical Pathology. 36 (12), 1747-1760 (2012).
- Howat, A. J., et al. Can histopathologists reliably diagnose molar pregnancy? Journal of Clinical Pathology. 46 (7), 599-602 (1993).
- Banet, N., et al. Characteristics of hydatidiform moles: analysis of a prospective series with p57 immunohistochemistry and molecular genotyping. Modern Pathology. 27 (2), 238-254 (2014).
- Lipata, F., et al. Precise DNA genotyping diagnosis of hydatidiform mole. Obstetrics & Gynecology. 115 (4), 784-794 (2010).
- Buza, N., Hui, P. Partial hydatidiform mole: histologic parameters in correlation with DNA genotyping. International Journal of Gynecologic Pathology. 32 (3), 307-315 (2013).
- Fisher, R. A., et al. Frequency of heterozygous complete hydatidiform moles, estimated by locus-specific minisatellite and Y chromosome-specific probes. Human Genetics. 82 (3), 259-263 (1989).
- Murdoch, S., et al. Mutations in NALP7 cause recurrent hydatidiform moles and reproductive wastage in humans. Nature Genetics. 38 (3), 300-302 (2006).
- Parry, D. A., et al. Mutations causing familial biparental hydatidiform mole implicate c6orf221 as a possible regulator of genomic imprinting in the human oocyte. American Journal of Human Genetics. 89 (3), 451-458 (2011).
- Nguyen, N. M., Slim, R. Genetics and Epigenetics of Recurrent Hydatidiform Moles: Basic Science and Genetic Counselling. Current Obstetrics and Gynecology Reports. 3, 55-64 (2014).
- Sebire, N. J., Savage, P. M., Seckl, M. J., Fisher, R. A. Histopathological features of biparental complete hydatidiform moles in women with NLRP7 mutations. Placenta. 34 (1), 50-56 (2013).
- Nguyen, N. M., et al. Comprehensive genotype-phenotype correlations between NLRP7 mutations and the balance between embryonic tissue differentiation and trophoblastic proliferation. Journal of Medical Genetics. 51 (9), 623-634 (2014).
- Brown, L., et al. Recurrent pregnancy loss in a woman with NLRP7 mutation: not all molar pregnancies can be easily classified as either "partial" or "complete" hydatidiform moles. International Journal of Gynecologic Pathology. 32 (4), 399-405 (2013).
- Colgan, T. J., Chang, M. C., Nanji, S., Kolomietz, E. A Reappraisal of the Incidence of Placental Hydatidiform Mole Using Selective Molecular Genotyping. The International Journal of Gynecological Cancer. 26 (7), 1345-1350 (2016).
- Murphy, K. M., McConnell, T. G., Hafez, M. J., Vang, R., Ronnett, B. M. Molecular genotyping of hydatidiform moles: analytic validation of a multiplex short tandem repeat assay. The Journal of Molecular Diagnostics. 11 (6), 598-605 (2009).
Access restricted. Please log in or start a trial to view this content.
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados