
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Pulitura del DNA microsatellite e citometria di flusso Ploidy Analisi dei tessuti molari Molari Molari Molar fissati di paraffino fissati in parina
In questo articolo
Riepilogo
Le talpe idatidiformi sono gravidanze umane anomale con aeziologie eterogenee che possono essere classificate in base alle loro caratteristiche morfologiche e al contributo dei genitori ai genomi molare. Qui, i protocolli di genotipizzazione del DNA microsatellite multiplex e citometria di flusso dei tessuti molari incorporati in paraffina fissati in formalina sono descritti in dettaglio, insieme all'interpretazione e all'integrazione dei risultati.
Abstract
La talpa idatidiforme (HM) è una gravidanza umana anomala caratterizzata da un'eccessiva proliferazione trofoblastica e da uno sviluppo embrionale anormale. Esistono due tipi di HM basati sulla valutazione morfologica microscopica, HM completo (CHM) e HM parziale (PHM). Questi possono essere ulteriormente suddivisi in base al contributo dei genitori ai genomi molare. Tale caratterizzazione dell'HM, mediante morfologia e analisi del genotipo, è fondamentale per la gestione del paziente e per la comprensione fondamentale di questa patologia intrigante. È ben documentato che l'analisi morfologica dell'HM è soggetta ad ampia variabilità interosservante e non è sufficiente da sola per classificare con precisione HM in CHM e PHM e distinguerli dagli aborti idropici non molare. L'analisi della genotipizzazione viene eseguita principalmente su DNA e tessuti da prodotti di concezione incorporati in paraffina fissata in formalina (FFPE), che hanno una qualità inferiore a quella ottimale e possono quindi portare a conclusioni errate. In questo articolo vengono forniti protocolli dettagliati per la genotipizzazione multiplex e l'analisi della citometria di flusso dei tessuti molari FFPE, insieme all'interpretazione dei risultati di questi metodi, alla loro risoluzione dei problemi e all'integrazione con la valutazione morfologica , l'immunosofochimicap57 KIP2 e l'ibridazione in situ (FISH) della fluorescenza in situ per raggiungere una diagnosi corretta e robusta. Qui, gli autori condividono i metodi e le lezioni appresi negli ultimi 10 anni dall'analisi di circa 400 prodotti di concepimento.
Introduzione
Una talpa idatidiforme (HM) è una gravidanza umana anomala caratterizzata da sviluppo embrionale anormale, iperproliferazione del trofoblasto e degenerazione ipicca di villi cirionico (CV). Storicamente, HM era diviso in due tipi, HM completo (CHM) e HM parziale (PHM) basato solo sulla valutazione morfologica1. Tuttavia, è stato dimostrato che la valutazione morfologica da sola non è sufficiente per classificare HM nei due sottotipi (CHM e PHM) e distinguerli da aborti non molori2,3,4.
Poiché CHM e PHM hanno diverse propensioni alle neoplasie, è quindi importante determinare con precisione il tipo genotypic di HM per fornire un adeguato follow-up e gestione ai pazienti. Di conseguenza, negli ultimi decenni, diverse metodologie sono state sviluppate ed evolute allo scopo di identificare il contributo dei genitori ai tessuti molari e raggiungere una corretta classificazione dell'HM. Questi includono l'analisi del cariotipo, il polimorfismo delle bande cromosomiche, la tipiagrafia sierologica dell'antigene leucocito umano (HLA), il polimorfismo della lunghezza del frammento di restrizione, il numero variabile di ripetizioni tandem, la genotipizzazione dei microsatelliti, la citometria di flusso e il p57 immunohistochimica KIP2. Ciò ha permesso una suddivisione accurata delle concezioni HM in base al contributo dei genitori al loro genoma, come segue: CHM, che sono diploidi androgenetici o diploidi dispermiati, e PHM, che sono triploidi, dispermiche nel 99% e monospermico nell'1% dei casi5,6,7,8. Inoltre, c'è un altro tipo genotypic di HM che è emerso negli ultimi due decenni, che è biparentalo diploide. Quest'ultimo è per lo più ricorrente e può interessare un singolo membro della famiglia (casi simplex) o almeno due membri della famiglia (casi familiari). Queste talpe biparentali diploidi sono per lo più causate da mutazioni recessive in NLRP7 o KHDC3L nei pazienti9,10,11,12. Diploid biparentalHM in pazienti con mutazioni recessive in NLRP7 può essere diagnosticato come CHM o PHM da analisi morfologica e questo sembra essere associato con la gravità delle mutazioni nei pazienti13,14. Oltre alla classificazione della HM in base ai loro genotipi, l'introduzione e l'uso di diversi metodi di genotipizzazione hanno permesso la distinzione delle varie entità molare da aborti spontanei non molari, come le concezioni biparentali diploidi aneuploidi e altri tipi di concezioni5,15. Tali concezioni possono avere una certa proliferazione di trofoblasti e una morfologia villosa anormale che imitano, in una certa misura, alcune caratteristiche morfologiche di HM.
Lo scopo di questo articolo è quello di fornire protocolli dettagliati per la genotipizzazione multiplex e la citometria di flusso dei tessuti incorporati in paraffina fissata alla formalina (FFPE), nonché analisi complete dei risultati di questi metodi e della loro integrazione con altri metodi per diagnosi corretta e conclusiva dei tessuti molari.
Protocollo
Questo studio di ricerca è stato approvato dal McGill Institutional Review Board. Tutti i pazienti hanno fornito il consenso scritto per partecipare allo studio e per avere i loro prodotti FFPE di concepimento (POC) recuperati da vari reparti di patologia.
NOT: Mentre ci sono diversi metodi per la genotipizzazione e la determinazione ploidia per citometria di flusso, i protocolli qui forniti descrivono un metodo di analisi utilizzando una piattaforma per ciascuno.
1. Genotipizzazione
-
Selezione del miglior blocco FFPE
- Per ogni prodotto FFPE del concepimento (POC), preparare 4 sezioni colorate di ematossia e ematosina (H&E) spesse a 4 m di spessore e di eosina (H&E), come descritto nelle sezioni 1.2 e 1.3, una per ogni blocco disponibile, per la valutazione morfologica mediante microscopia.
- Utilizzando i vetrini H&E e un microscopio luminoso, selezionare il blocco FFPE che ha la maggiore quantità di villi corionico (CV), e se possibile, il blocco che ha CV separato e non mescolato con, tessuti materni.
-
Taglio
- Posizionare il blocco scelto sul ghiaccio per 15 min per facilitare il sezionamento.
- Regolare il microtoma per tagliare sezioni spesse 4 m per la valutazione morfologica microscopica e spesse 10 m per l'estrazione del DNA.
- Posizionare il blocco freddo nel microtome e tagliare una sezione da ogni blocco per la colorazione H&E e 10-30 sezioni dal blocco scelto, a seconda della quantità di CV nel blocco, per l'estrazione del DNA.
NOT: Per i blocchi pieni di CV, 10 sezioni sono sufficienti per l'estrazione del DNA. Se solo circa il 10% del blocco contiene CV mentre il resto sono tessuti materni, allora sono necessarie 20-30 sezioni per garantire una quantità sufficiente di DNA. - Utilizzando le pinze, trasferire ogni sezione a un bagno d'acqua di 45 gradi centigradi. Raccogliere la sezione dal bagno d'acqua con una diapositiva carica positivamente (Tabella dei materiali) che è precedentemente etichettato con il numero di identificazione del campione utilizzando una matita.
- Posizionare i vetrini che contengono le sezioni in forno a 65 gradi centigradi per consentire alle sezioni di aderire ai vetrini. Tenere i vetrini per H&E in forno per 25 min.
NOT: Il tempo di incubazione più breve rende i tessuti leggermente meno aderenti alle diapositive e di conseguenza facilita la rimozione dei tessuti materni.
-
Colorazione H&E
- Lasciare raffreddare gli scivoli a temperatura ambiente (10 min).
- Preparazione del reagente
- Preparare la soluzione di lavoro Eosin Y (0,25%) come da tabella 1. Mescolare bene e conservare a temperatura ambiente.
- Preparare la soluzione di ematossilina funzionante diluindo la soluzione stock di ematossilina 5x in acqua (cioè mescolare 80 mL di acqua con 20 mL di ematossillina).
NOT: Avvolgere la soluzione stock di ematossialina in lamina per lo stoccaggio.
- Preparare i vasetti di colorazione con i reagenti corretti sotto un cofano di fumi secondo la tabella 2.
- Eseguire la colorazione H&E immergendo i vetrini nei vasetti di colorazione appropriati per il periodo di tempo corretto in base alla tabella 2.
- Montare le 4 sezioni m per l'analisi morfologica con supporto di montaggio e coverslip con coperchi in vetro (Tabella dei materiali).
NOT: Le 10 sezioni di 10 m per la genotipizzazione non devono essere ritagliate. - Lasciare le sezioni da 10 m sotto il cofano fumatore per un minimo di 3 h affinché gli odori xileni tossici si dissipano.
ATTENZIONE: Tutte le fasi di colorazione devono essere eseguite sotto un cappuccio di fumi. I prodotti Xylene devono essere tenuti sempre sotto il cofano perché gli odori di xilene sono tossici. Inoltre, lo xilene e l'ematossialina devono essere scartati in appositi contenitori. Una volta che questi contenitori sono pieni, devono essere scartati come raccomandato dall'organizzazione di sicurezza del laboratorio.
| Reagente | quantità |
| Soluzione azionaria Eosin Y (1%) | 250 mL |
| 80% Etanolo | 750 mL |
| Acido acetico glaciale (concentrato) | 5 mL |
Tabella 1: Soluzione di lavoro Eosin Y (0,25%) preparazione.
| Reagente utilizzato (100 mL per contenitore) | durata |
| 1) Sleonina | 5 min |
| 2) Sylene | 5 min |
| 3) 100% Etanolo | 2 min |
| 4) 95% Etanolo | 2 min |
| 5) 70% Etanolo | 2 min |
| 6) 50% Etanolo | 2 min |
| 7) Acqua distillata | 5 min |
| 8) Ematossina | 4 min |
| 9) Acqua distillata | 5 min |
| 10) Eosina | 1 min |
| 11) 95% Etanolo | 5 min |
| 12) 100% Etanolo | 5 min |
| 13) Slene | 5 min |
| 14) Xilene | 5 min |
Tabella 2: Reagenti e durate per il protocollo di colorazione H&E.
-
Isolamento del CV
- Sotto uno stereoscopio leggero, utilizzare pinze e piccoli pezzi di salviette di carta idratate per l'acqua (Tabella dei materiali) per raschiare via i tessuti materni indesiderati da sezioni di 10 m di spessore H&E.
NOT: L'obiettivo finale è quello di mantenere nient'altro che CV o membrane fetali (se presenti) sulle diapositive e quindi rimuovere tutti gli altri tessuti. Questo passaggio può richiedere un sacco di tempo e pazienza, a seconda del blocco, in quanto richiede un'attenzione meticolosa ai dettagli. - Chiedi a una seconda persona di controllare i vetrini dopo la pulizia per assicurarti che siano privi di tessuti materni.
- Scattare foto delle diapositive pulite o documentare quanto segue per aiutare con l'interpretazione dei dati: 1) se il tessuto era difficile da pulire, emorragico, o molto pulito, 2) il numero di sezioni utilizzate, e 3) la quantità approssimativa di tessuti puliti.
NOTA: La figura 1 fornisce un esempio di una diapositiva facile da pulire. Per un blocco contenente all'incirca questa quantità di CV, 10 sezioni sono sufficienti per l'estrazione del DNA. Lo slittamento in Figura 2 ha pochissimi CV che si mescolano con i tessuti materni, rendendo molto difficile e richiede molto tempo per pulire. Per un blocco contenente all'incirca questa quantità di CV, sono necessarie 30 sezioni per l'estrazione del DNA. - Raccogliere il CV utilizzando piccoli pezzi inumiditi di salviette di carta. Usando le pinze, strappa un pezzetto dalle salviette di carta inumidite e usalo per raccogliere il CV.
- Posizionare i pezzi di carta con il loro CV attaccato in un tubo etichettato da 1,5 mL.
- Ridurre al minimo la quantità di salviette della carta utilizzate in questo passaggio, in quanto troppo può ostruire la colonna di estrazione del DNA e di conseguenza ridurre la quantità finale di DNA raccolto. In media, mirare a utilizzare meno di sette piccoli pezzi di salviette di carta per campione. Se ciò non è possibile a causa della presenza di grandi quantità di CV, dividere il campione tra due tubi per facilitare l'estrazione.
- Sotto uno stereoscopio leggero, utilizzare pinze e piccoli pezzi di salviette di carta idratate per l'acqua (Tabella dei materiali) per raschiare via i tessuti materni indesiderati da sezioni di 10 m di spessore H&E.

Figura 1: diapositiva rappresentativa per la genotipizzazione. In alto: uno scivolo che deve essere "pulito" per liberarsi dei tessuti materni. In basso: la stessa diapositiva mostrata dopo che è stata pulita e ora non contiene altro che CV per l'estrazione del DNA. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2: diapositiva rappresentativa per la genotipizzazione. In alto: uno scivolo che deve essere "pulito" per liberarsi dei tessuti materni. In basso: la stessa diapositiva mostrata dopo che è stata pulita e ora non contiene altro che CV per l'estrazione del DNA. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
- Seguire il protocollo dell'estrazione del DNA dal kit FFPE (Tabella dei materiali) per eseguire l'estrazione del DNA.
NOT: Alcuni kit raccomandano di utilizzare un buffer di eluizione da 15-20 gradi per l'eluizione finale. Per esperienza, l'eluizione con 15 l di tampone di eluizione funziona bene per la maggior parte dei campioni. Le diluizioni possono essere preparate dal DNA dello stock in base alle esigenze.
-
Quantificazione del DNA
- Utilizzando un dispositivo spettrofotometro di laboratorio, caricare 1 ll di DNA e misurare l'assorbimento a 260 nm per la quantificazione.
- Caricare 1 l l di DNA su un gel di agarose del 2% ed eseguire l'elettroforesi del gel ad una tensione di 80-100 V per una valutazione qualitativa.
- Sulla base dei risultati dei passaggi 1.6.1 e 1.6.2, scegliere il volume del DNA da utilizzare nell'amplificazione della reazione a catena di polimerasi (PCR) a ripetizione lunga tandem (STR) multiplax. Obiettivo di utilizzare un minimo di 1000 ng di DNA nell'amplificazione PCR che segue.
NOTA: Figura 3 mostra esempi rappresentativi di gel insieme alle concentrazioni del DNA (in base ai risultati dello spettrofotometro) e al volume della soluzione di DNA consigliata per la MULTIPLEx STR PCR che segue.

Figura 3: Gel rappresentativo per la quantificazione del DNA. Sono incluse le concentrazioni di ogni DNA, misurate utilizzando uno spettrofotometro, e le quantità utilizzate per la PCR multiplex. Fare clic qui per visualizzare una versione più grande di questa figura.
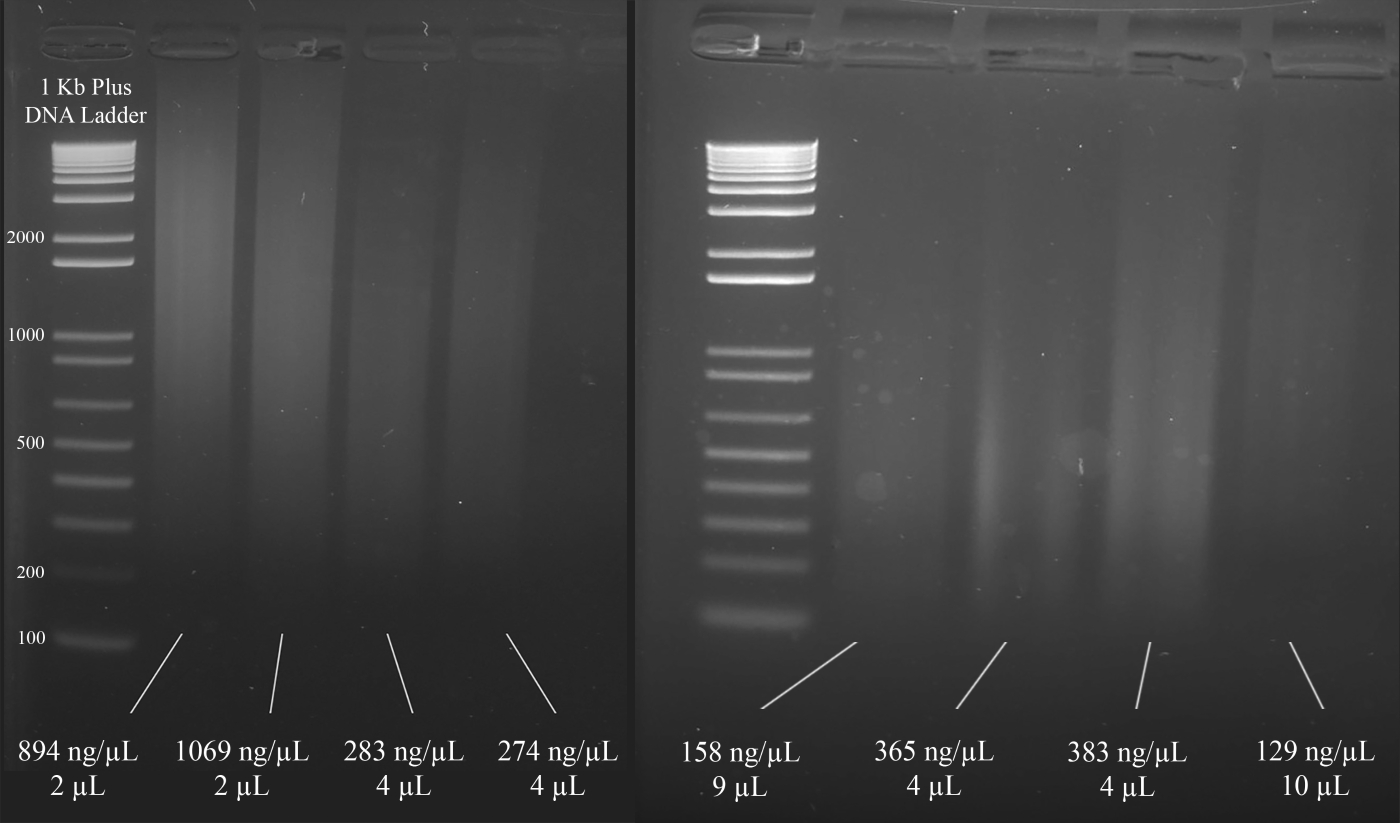{kind=link}
-
Amplificazione PCR
- Eseguire la genotipizzazione microsatellite fluorescente utilizzando un sistema multiplex STR (Tabella dei materiali).
- Utilizzare le condizioni PCR illustrate nella Figura 4 per l'amplificazione PCR utilizzando il sistema multiplex STR ( Tabelladei materiali).
NOT: In questo sistema multiplex STR vengono utilizzati i seguenti primer: D18S51, D21S11, TH01, D3S1358, Penta E, FGA, TPOX, D8S1179, vWA, Amelogenin, CSF1PO, D16S539, D7S820, D13S317, D5S818 e D.

Figura 4: condizioni del ciclo PCR per il sistema multiplex STR. Fare clic qui per visualizzare una versione più grande di questa figura.
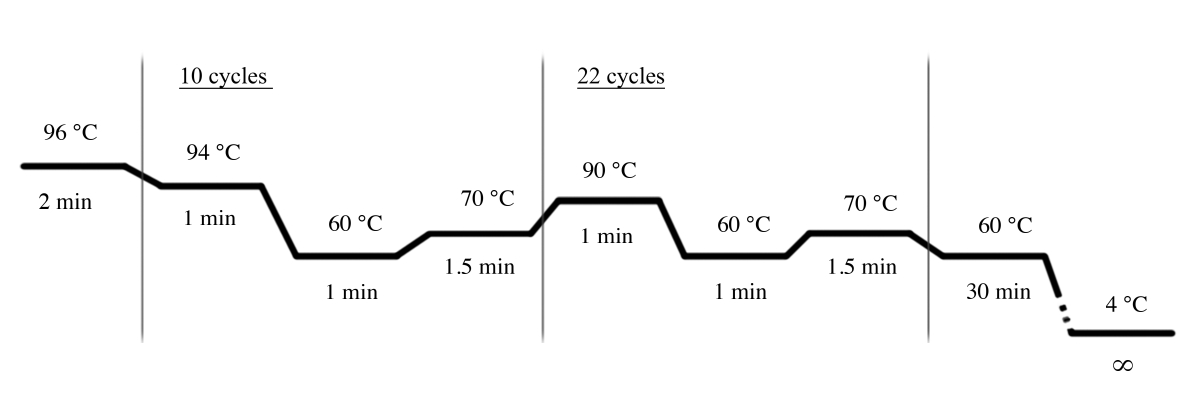{kind=link}
-
Risolvere i prodotti PCR mediante elettroforesi capillare.
- Sospendere 1 l di ogni campione amplificato in 0,5 litri della corsia standard interna del sistema multiplex e 9,5 l di formamide altamente dionizzata (Tabella dei materiali).
- Eseguire i campioni attraverso uno strumento di elettroforesi capillare (Tabella dei materiali) utilizzando una matrice di separazione appropriata ( Tabella deimateriali) per lo strumento e il set di tinture del sistema multiplex.
- Analisi dei dati
- Analizzare i dati con un software di analisi del frammento di DNA e confrontare gli alleli POC con gli alleli dei genitori per determinarne l'origine.
- Impostare uno standard di dimensioni.
NOT: Ciò consente al software di riconoscere la scala utilizzata nel sistema multiplex STR e di assegnare le basi agli amplificatori in base alla scala. I seguenti passaggi sono per un software specifico (Tabella dei materiali) ma può essere di aiuto per la configurazione di altri tipi di software pure.- Aprire il software. Fare clic su Avvia nuovo progetto e quindi su Nuovo standard di dimensione.
- Assegnare un nome allo standard di dimensione (ad esempio, ABI_600).
- Nella casella Immetterela nuova definizione standard di dimensioni : immettere quanto segue: 60, 80, 100, 120, 140, 160, 180, 200, 225, 250, 275, 300, 375, 400, 425, 450, 475, 500, 550, 600. Quindi fare clic su Aggiungi dimensioni.
NOTA: i numeri immessi appariranno sotto la casella a destra, denominata Definizione standard dimensioni correnti (vedere la figura 5). - Fare clic su Salva.
- Per importare e analizzare un file, fare clic su Aggiungi filee scegliere il file fsa da analizzare. Fare clic su Aggiungi file selezionati e quindi su OK. Quindi attenersi alla seguente procedura:
- Individuare la colonna Size Standard e scegliere ABI_600 (o qualsiasi nome assegnato allo standard di dimensione).
- In Metodo di analisi, fare clic su Ridimensionamento predefinito - NPP e quindi fare clic sul pulsante verde Analizza.
- Il file è ora pronto per la visualizzazione. Regolare le opzioni di visualizzazione per visualizzare i dati come desiderato.
- Risoluzione dei problemi - metodo di analisiTroubleshooting - analysis method
NOT: Il software a volte potrebbe non riuscire a identificare i picchi e allinearli correttamente. Questo accade quando i picchi sono troppo bassi o troppo alti. I due metodi di analisi seguenti possono correggersi e devono essere provati prima che un campione venga sottoposto a test.- Metodo di analisi 1 per picchi elevati:
- Fare clic su Nuovo metodo di analisi e denominarlo High Peaks (o un altro nome in base alle preferenze personali).
- Fare clic su Intervallo e quindi su Intervallo parziale per l'analisi e il dimensionamento. Digitare quindi 100 per Il punto iniziale e la dimensione iniziale.
- Per il puntodi arresto , immettere 10.000. Per Dimensione arresto, immettere 1000.
- Quindi fare clic su Altezze massime di picco e modificare i numeri in modo che la soglia di picco per i colori è la seguente: Blu: 50; Verde: 50; Giallo: 20; Rosso: 100; Arancione: 5000.
- Salvare il nuovo metodo di analisi.
- Metodo di analisi 2 per picchi bassi:
- Fare clic su Nuovo metodo di analisi e denominarlo Low Peaks (o un altro nome in base alle preferenze personali).
- Fare clic su Intervallo e quindi su Intervallo parziale per l'analisi e il dimensionamento. Digitare quindi 100 per Il punto iniziale e la dimensione iniziale.
- Per il puntodi arresto , immettere 10.000. Per Dimensione arresto, immettere 1000.
- Quindi fare clic su Bandiere di qualità e modificare l'intervallo di passaggio in modo che si legge Da 0.5 a 1. Modificare l'intervallo di qualità basso in modo che venga letto da 0.0 a 0.0. Modificare Presupporre Linearità al seguente: da (bp) 100.0 a (bp) 800.0.
- Salvare il nuovo metodo di analisi.
NOT: È ora possibile rianalizzare un file scegliendo Low Peaks o High Peaks in Analysis Method e quindi facendo clic sul pulsante verde Analizza.
- Metodo di analisi 1 per picchi elevati:

Figura 5: Schermata che mostra l'Editor standard delle dimensioni. Fare clic qui per visualizzare una versione più grande di questa figura.
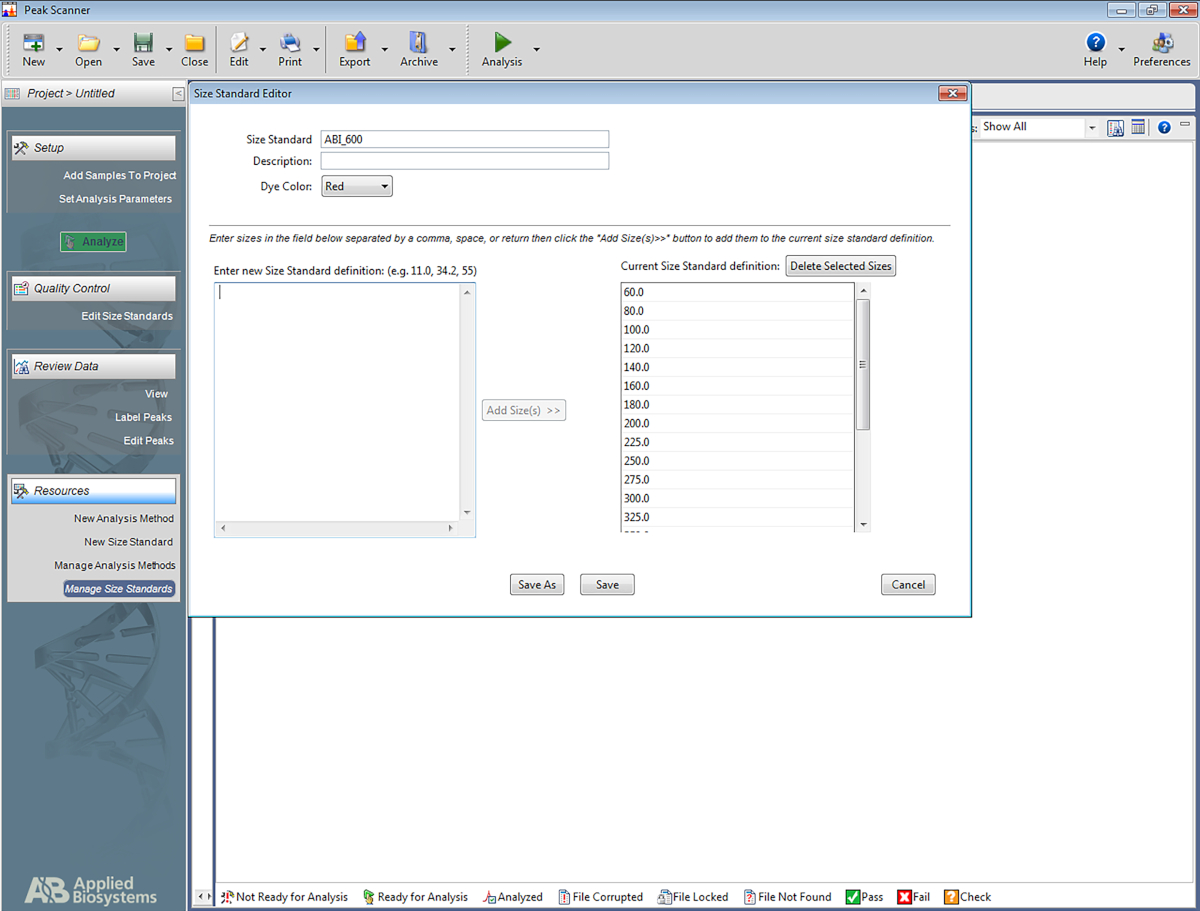{kind=link}
2. Citometria di flusso
-
Scelta del blocco FFPE ideale
- Utilizzando i vetrini H&E e un microscopio luminoso, selezionare un blocco FFPE che ha circa il 50-70% dei suoi tessuti composti da CV.
NOTA: la figura 6 è un esempio rappresentativo di un blocco appropriato per l'analisi della citometria di flusso, in quanto è composto da circa il 50% di CV (metà destra della sezione) e 50% tessuti materni (metà sinistra). La presenza di tessuti materni è importante perché servono come controllo interno per il picco diploide. - Per i blocchi che non hanno la quantità ideale di CV, arricchisci per CV mentre viene eseguita la sezionamento. Per farlo, identifica quale lato delle sezioni appena tagliate contiene più CV in base alla corrispondente diapositiva H&E. Sulla base di ciò, utilizzare una lama per tagliare l'altra metà che deve essere scartato al fine di arricchire per CV.
NOTA: la figura 7 mostra un blocco che non dispone di CV sufficiente per l'analisi della citometria di flusso. Per blocchi come questo, le sezioni devono essere tagliate in modo che la metà che contiene meno CV venga scartata al fine di aumentare la quantità di CV rispetto ai tessuti materni, come mostrato nella figura. Assicurarsi di tagliare più sezioni per compensare ciò che viene scartato.
- Utilizzando i vetrini H&E e un microscopio luminoso, selezionare un blocco FFPE che ha circa il 50-70% dei suoi tessuti composti da CV.

Figura 6: sezione H&E che rappresenta un blocco POC ideale per la citometria di flusso. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 7: sezione H&E che rappresenta un blocco più difficile per la citometria di flusso. Questa sezione h&E rappresentativa mostra che solo la metà inferiore di questa sezione deve essere utilizzata per l'analisi della citometria di flusso, con l'obiettivo di arricchire per il CV. L'area delineata, denominata "CV", è composta per lo più da CV.
{kind=link}
- Taglio
- Lasciare i blocchi sul ghiaccio per 15 min per facilitare la sezionamento.
- Utilizzando il miglior blocco FFPE possibile, tagliare quattro sezioni spesse 50 m (o due sezioni spesse 100 m) utilizzando un microtoma.
NOT: Per la citometria a flusso è preferibile avere sezioni più spesse. - Nel caso in cui non sia disponibile un blocco FFPE ideale, mira a mantenere comunque il rapporto tra CV e tessuto materno. Ad esempio, se solo il 30% del blocco è costituito da CV mentre il resto ha tessuti materni, rimuovere almeno la metà della sezione che contiene i tessuti materni e utilizzare più sezioni per compensare (vedere Figura 7).
- Posizionare le sezioni in tubi da 15 mL etichettati.
NOT: Assicurarsi di sovrapinserire le etichette perché i reagenti organici utilizzati nel passaggio successivo possono sciogliere e rimuovere l'inchiostro.
- Protocollo di citometria di flusso dai tessuti FFPE
- Deparaffinazione e reidratazione
- Eseguire i seguenti lavamenti (Tabella 3) sotto un cofano di fumi.
- Riempire il tubo da 15 mL con 6 mL del reagente appropriato, seguendo l'ordine presentato nella Tabella 3, lasciare le sezioni nel reagenti per la rispettiva durata, quindi rimuovere il reagente utilizzando l'aspirazione a vuoto e una pipetta Pasteur in vetro.
- Tra ogni passo, immergere la pipetta Pasteur prima nel 70% di etanolo, poi in acqua distillata, e poi procedere alla fase successiva.
- Fare molta attenzione a non rimuovere pezzi di tessuto insieme al reagente. Inclinare il tubo di 15 mL ad un angolo di 60 gradi per facilitare l'aspirazione del reagente liquido senza disegnare i tessuti.
ATTENZIONE: I liquidi scartati contengono xilene e devono essere smaltiti in contenitori di rifiuti di xilene.
- Preparazione della soluzione
- Preparare la soluzione citrate sciogliendo 2 g di acido citrico in 1 L di acqua doppia distillata. Portare il pH a 6. Conservare a 4 gradi centigradi.
- Preparare la soluzione di pepsina sciogliendo 0,01 g di pepsina in 2 mL di 9 parti per mille NaCl, pH 1,64. Questo è per un campione.
ATTENZIONE: La pepsina è tossica e può facilmente disperdersi e diventare trasportata dall'aria. Indossare una maschera durante la manipolazione della pepsina nella sua forma di polvere e pulire tutta l'area di lavoro dopo averlo usato. - Propidium Iodide (PI)-ribonuclease Preparazione di una soluzione per un campione.
- Mescolare 50 - L di PI con 450 l di PBS (per diluire 10x).
- Aggiungere 50 - L di ribonuclease A (1 mg/mL) alla miscela. Tenere sempre avvolto nella pellicola.
- Digestione e colorazione
- Aggiungete la soluzione di 4 gradi centigradi ai tubi da 15 mL, quindi collocate in un bagno d'acqua di 80 gradi per 2 ore.
- Lasciare raffreddare la soluzione a temperatura ambiente (15 min). Rimuovere la soluzione citrate.
- Aggiungete 6 mL di 1x PBS, vortice, e aspettate 1/2 min per permettere ai tessuti di depositarsi sul fondo. Rimuovere il 1x PBS utilizzando aspirazione sottovuoto e una pipetta Pasteur in vetro.
- Aggiungere 1 mL di soluzione di pepsina (preriscaldata a 37 gradi centigradi) e mettere in un bagno asciutto a 37 gradi per 30 min.
- Aggiungete 6 mL di 1x PBS, vortice, e aspettate 1/2 min per permettere ai tessuti di depositarsi sul fondo. Rimuovere il 1x PBS utilizzando aspirazione sottovuoto e una pipetta Pasteur in vetro.
- Aggiungere 550 - L della soluzione PI-ribonuclease A e mettere i campioni in un bagno secco a 37 gradi per 30 min.
NOT: A questo punto, i campioni possono essere avvolti in un foglio e lasciati durante la notte a 4 gradi centigradi fino alla mattina successiva. - Filtrare la soluzione attraverso una rete filtrante da 48 m. Raccogliere il filtrato in tubi rotondi in polistirolo, che possono essere utilizzati con il citometro di flusso. Utilizzare pinze per posizionare un pezzo di 5 cm per 5 cm di rete filtrante nella parte superiore del tubo, in modo che il liquido può essere convogliato attraverso la rete e nel tubo.
NOT: I campioni sono ora pronti per essere eseguiti con il citometro di flusso. Tenerli avvolti nella pellicola fino a quando non sono pronti per essere eseguiti.
- Esegui campioni con un citometro di flusso con l'aiuto del tecnico della piattaforma di citometria di flusso dell'organizzazione.
NOT: Il canale PE viene utilizzato per rilevare il DNA macchiato di PI e la portata deve essere impostata su Slow durante l'acquisizione. Assicurarsi che la tensione sia scelta in modo tale che il picco diploide sia approssimativamente a 200 lungo l'asse XPE-A per facilitare l'analisi e l'interpretazione. Obiettivo di registrare un minimo di 20.000 eventi per campione.
- Deparaffinazione e reidratazione
| Reagente utilizzato (6 mL ciascuno) | durata |
| 1) Sleonina | 2 x 10 min |
| 2) 100% Etanolo | 2 x 10 min |
| 3) 95% Etanolo | 10 min |
| 4) 70% Etanolo | 10 min |
| 5) 50% Etanolo | 10 min |
| 6) Acqua distillata | 2 x 10 min |
Tabella 3: Reagenti e durate per deparaffinazione e reidratazione.
-
Analisi dei dati della citometria di flusso
- Analizzare i dati con un software di analisi della citometria di flusso (Tabella dei materiali).
NOT: I seguenti passaggi sono per un software specifico (Tabella dei materiali) ma può essere di aiuto per la configurazione di altri tipi di software pure.- Dopo aver eseguito gli esempi su un citometro di flusso, scaricare i file FCS 2.0 per l'analisi.
- Aprire il software di analisi della citometria di flusso, fare clic su File Nuovo documento.
- Fare clic sull'icona Istogramma (
 ), quindi trascinare il puntatore per creare un rettangolo.
), quindi trascinare il puntatore per creare un rettangolo. - Cercare il file FCS e quindi fare clic su Apri. Lungo l'asse x, fare clic su FCS-A, quindi selezionare PE-A.
- Fare clic sull'icona
 Grafico a punti ( ) e trascinare il puntatore per creare un altro rettangolo sotto il grafico dell'istogramma. Quindi cercare lo stesso file FCS selezionato per l'istogramma.
Grafico a punti ( ) e trascinare il puntatore per creare un altro rettangolo sotto il grafico dell'istogramma. Quindi cercare lo stesso file FCS selezionato per l'istogramma. - Modificare l'asse x del grafico a punti in PE-A e l'asse y in PE-W.
NOTA: Figura 8A dimostra l'aspetto dei grafici a questo punto. - Fare clic sull'icona
 Regione ( ) e disegnare una casella sul grafico a punti che inizia prima del picco diploide (circa 100 sull'asse x in Figura 8B) e che termina intorno a 700 sull'asse x, come mostrato nel grafico a punti in Figura 8 B.
Regione ( ) e disegnare una casella sul grafico a punti che inizia prima del picco diploide (circa 100 sull'asse x in Figura 8B) e che termina intorno a 700 sull'asse x, come mostrato nel grafico a punti in Figura 8 B.
NOT: Il picco diploide nella Figura 8 è all'incirca a 200 sull'asse x. Questo viene scelto arbitrariamente come i campioni vengono registrati attraverso il citometro di flusso, semplicemente per facilitare l'analisi e l'interpretazione dei risultati. - Fare clic su Stampa . Modificare Regioni/Gates, quindi digitare R0 nella cella accanto alla cella G0 in Strategia. Quindi fare clic su Chiudi.
- Fare clic in un punto qualsiasi dell'istogramma, quindi su Grafico. Formato stampa/sovrapposizione. In Porta, selezionare G0 e R0, quindi fare clic su OK.
NOT: Questo è il passo di gating che permette di visualizzare meglio i picchi ploidi. L'istogramma dovrebbe ora essere simile all'istogramma in Figura 8B. È possibile giocare con il cancello creato (spostando la casella disegnata al passo 2.4.1.7) al fine di concentrarsi su specifiche regioni del grafico a punti. - Per etichettare i grafici, fare clic
 sull'icona Area di testo ( ), quindi trascinare il puntatore per creare una casella nella parte superiore del documento, quindi digitare le seguenti informazioni: ID paziente, ID POC e blocco utilizzato (poiché potrebbero essere presenti diversi blocchi per un POC) , percentuale CV presente sul blocco, tensione utilizzata per eseguire il campione e data.
sull'icona Area di testo ( ), quindi trascinare il puntatore per creare una casella nella parte superiore del documento, quindi digitare le seguenti informazioni: ID paziente, ID POC e blocco utilizzato (poiché potrebbero essere presenti diversi blocchi per un POC) , percentuale CV presente sul blocco, tensione utilizzata per eseguire il campione e data.
- Analizzare i dati con un software di analisi della citometria di flusso (Tabella dei materiali).

Figura 8: Schermata che mostra un istogramma e un grafico a punti di un campione rappresentativo che è ungated (A) e gated (B). Fare clic qui per visualizzare una versione più grande di questa figura.
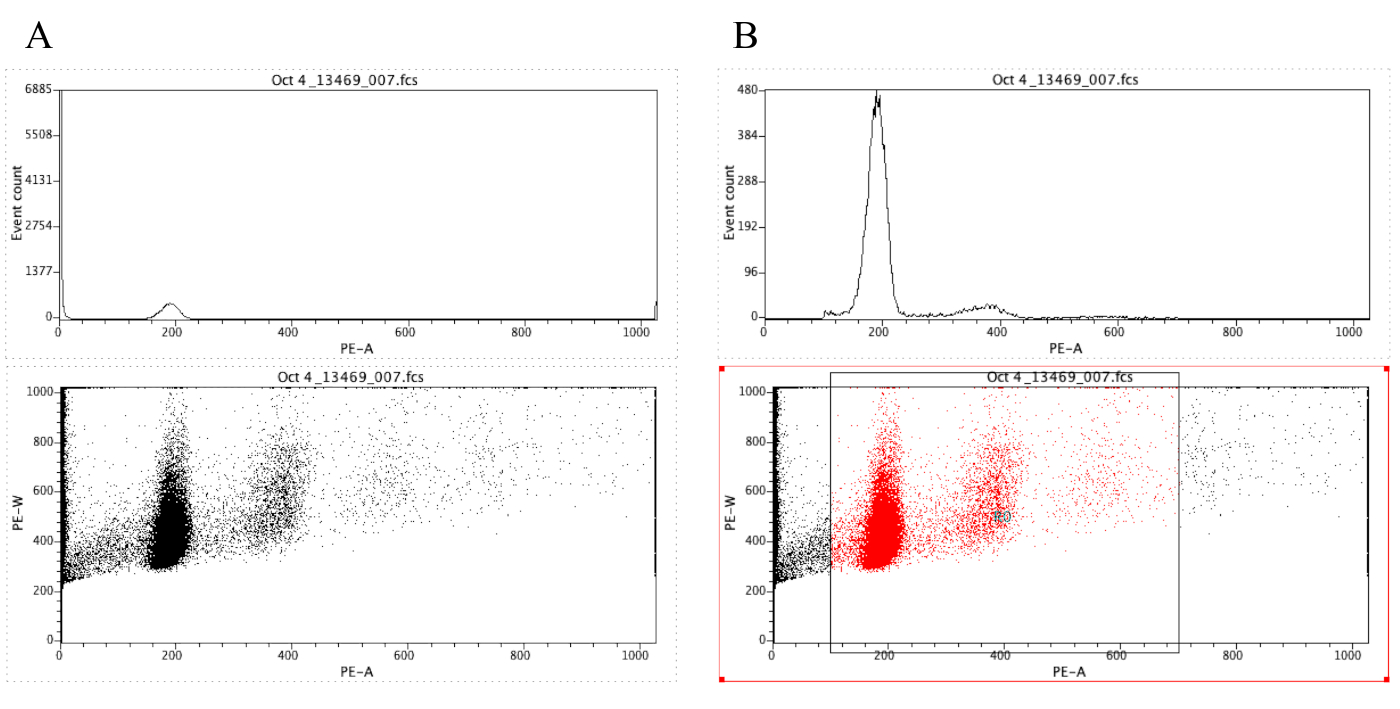{kind=link}
Risultati
La complessità dei tessuti molari e il fatto che possano avere vari genotipi richiede un'analisi rigorosa e l'uso di diversi metodi come la valutazione morfologica, l'immunohistochimica p57, la genotipizzazione microsatellite, la citometria di flusso e la FISH. Ad esempio, un paziente (1790) è stato riferito con due PHM che sono stati trovati triploide dall'analisi microarray solo dei POC. Al paziente è stato quindi diagnosticato un PHM ricorrente. La genotipizzazione microsatellite de...
Discussione
Le HM sono gravidanze umane anormali con eziologie eterogenee e hanno diversi tipi istologici e genotypic, il che rende la loro classificazione e diagnosi accurate impegnative. La valutazione morfologica istopatologica è stata spesso dimostrata imprecisa ed è quindi inaffidabile da sola per classificare HM in CHM e PHM e distinguerli da aborti non molari. Pertanto, una diagnosi accurata di HM richiede l'uso di altri metodi come la genotipizzazione del DNA microsatellite multiplex, l'analisi della ploidia per citometria...
Divulgazioni
Gli autori non hanno nulla da rivelare.
Riconoscimenti
Gli autori ringraziano Sophie Patrier e Marianne Parésy per aver condiviso il protocollo originale della citometria di flusso, e Promega e Qiagen per la fornitura di rifornimenti e reagenti. Questo lavoro è stato sostenuto dal Réseau Québécois en Reproduction e dal Canadian Institute for Health Research (MOP-130364) a R.S.
Materiali
| Name | Company | Catalog Number | Comments |
| BD FACS Canto II | BD BioSciences | 338960 | |
| Capillary electrophoresis instrument: Genomes Applied Biosystems 3730xl DNA Analyzer | Applied biosystems | 313001R | Service offered by the Centre for Applied Genomics (http://www.tcag.ca) |
| Citric acid | Sigma | 251275 | |
| Cytoseal 60, histopathology mounting medium | Fisher | 23244257 | |
| Eosin Y stock solution (1%) | Fisher | SE23-500D | |
| FCSalyzer - flow cytometry analysis software | SourceForge | - | https://sourceforge.net/projects/fcsalyzer/ |
| FFPE Qiagen kit | Qiagen | 80234 | |
| Forceps | Fine Science Tools | 11295-51 | For sectioning and for the cleaning process |
| Glacial Acetic Acid (Concentrated) | Sigma | A6283-500mL | |
| Glass coverslips: Cover Glass | Fisher | 12-541a | |
| Hematoxylin | Fisher | CS401-1D | |
| Highly deionized formamide: Hi-Di Formamide | Thermofisher | 4311320 | |
| IHC platform: Benchmark Ultra | Roche | - | |
| Kimwipes | Ultident | 30-34120 | |
| Microtome | Leica | RM2135 | |
| Microtome blades | Fisher | 12-634-1C | |
| Nitex filtering mesh, 48 μm | Filmar | 74011 | http://www.filmar.qc.ca/index.php?filet=produits&id=51&lang=en ; any other filter is suitable, but this is an inexpensive and effective option from a non-research company |
| p57 antibody | Cell Marque | 457M | |
| Pasteur pipette | VWR | 53499-632 | |
| PCR machine | Perkin Elmer, Applied Biosystems | GeneAmp PCR System 9700 | |
| PeakScanner 1.0 | Applied Biosystems | 4381867 | Software for genotyping analysis. |
| Pepsin from porcine gastric mucosa | Sigma | P7012 | |
| Polystyrene round-bottom tubes | BD Falcon | 352058 | |
| Positively charged slides: Superfrost Plus 25x75mm | Fisher | 1255015 | |
| PowerPlex 16 HS System | Promega Corporation | DC2102 | |
| Propidium Iodide | Sigma | P4864 | |
| Ribonuclease A from bovine pancreas | Sigma | R4875 | |
| Separation matrix: POP-7 Polymer | Thermofisher | 4352759 | |
| UltraPure Agarose | Fisher | 16500-500 | |
| Xylene | Fisher | X3P1GAL |
Riferimenti
- Szulman, A. E., Surti, U. The syndromes of hydatidiform mole. II. Morphologic evolution of the complete and partial mole. American Journal of Obstetrics & Gynecology. 132 (1), 20-27 (1978).
- Fukunaga, M., et al. Interobserver and intraobserver variability in the diagnosis of hydatidiform mole. The American Journal of Surgical Pathology. 29 (7), 942-947 (2005).
- Gupta, M., et al. Diagnostic reproducibility of hydatidiform moles: ancillary techniques (p57 immunohistochemistry and molecular genotyping) improve morphologic diagnosis for both recently trained and experienced gynecologic pathologists. The American Journal of Surgical Pathology. 36 (12), 1747-1760 (2012).
- Howat, A. J., et al. Can histopathologists reliably diagnose molar pregnancy?. Journal of Clinical Pathology. 46 (7), 599-602 (1993).
- Banet, N., et al. Characteristics of hydatidiform moles: analysis of a prospective series with p57 immunohistochemistry and molecular genotyping. Modern Pathology. 27 (2), 238-254 (2014).
- Lipata, F., et al. Precise DNA genotyping diagnosis of hydatidiform mole. Obstetrics & Gynecology. 115 (4), 784-794 (2010).
- Buza, N., Hui, P. Partial hydatidiform mole: histologic parameters in correlation with DNA genotyping. International Journal of Gynecologic Pathology. 32 (3), 307-315 (2013).
- Fisher, R. A., et al. Frequency of heterozygous complete hydatidiform moles, estimated by locus-specific minisatellite and Y chromosome-specific probes. Human Genetics. 82 (3), 259-263 (1989).
- Murdoch, S., et al. Mutations in NALP7 cause recurrent hydatidiform moles and reproductive wastage in humans. Nature Genetics. 38 (3), 300-302 (2006).
- Parry, D. A., et al. Mutations causing familial biparental hydatidiform mole implicate c6orf221 as a possible regulator of genomic imprinting in the human oocyte. American Journal of Human Genetics. 89 (3), 451-458 (2011).
- Nguyen, N. M., Slim, R. Genetics and Epigenetics of Recurrent Hydatidiform Moles: Basic Science and Genetic Counselling. Current Obstetrics and Gynecology Reports. 3, 55-64 (2014).
- Sebire, N. J., Savage, P. M., Seckl, M. J., Fisher, R. A. Histopathological features of biparental complete hydatidiform moles in women with NLRP7 mutations. Placenta. 34 (1), 50-56 (2013).
- Nguyen, N. M., et al. Comprehensive genotype-phenotype correlations between NLRP7 mutations and the balance between embryonic tissue differentiation and trophoblastic proliferation. Journal of Medical Genetics. 51 (9), 623-634 (2014).
- Brown, L., et al. Recurrent pregnancy loss in a woman with NLRP7 mutation: not all molar pregnancies can be easily classified as either "partial" or "complete" hydatidiform moles. International Journal of Gynecologic Pathology. 32 (4), 399-405 (2013).
- Colgan, T. J., Chang, M. C., Nanji, S., Kolomietz, E. A Reappraisal of the Incidence of Placental Hydatidiform Mole Using Selective Molecular Genotyping. The International Journal of Gynecological Cancer. 26 (7), 1345-1350 (2016).
- Murphy, K. M., McConnell, T. G., Hafez, M. J., Vang, R., Ronnett, B. M. Molecular genotyping of hydatidiform moles: analytic validation of a multiplex short tandem repeat assay. The Journal of Molecular Diagnostics. 11 (6), 598-605 (2009).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati