
Method Article
Una herramienta de estudio de asociación de vías para los análisis GWAS de la información de las vías metabólicas
En este artículo
Erratum Notice
Resumen
Al ejecutar la Herramienta de Estudio de Asociación pathway (PAST), ya sea a través de la aplicación Shiny o a través de la consola R, los investigadores pueden obtener una comprensión más profunda del significado biológico de los resultados de su estudio de asociación de todo el genoma (GWAS) al investigar las vías metabólicas involucradas.
Resumen
Recientemente, se ha desarrollado y publicado una nueva implementación de un método previamente descrito para interpretar los datos del estudio de asociación de todo el genoma (GWAS) utilizando el análisis de la vía metabólica. La Herramienta de Estudio de la Asociación Pathway (PAST) se desarrolló para abordar las preocupaciones con la facilidad de uso y los análisis de ejecución lenta. Esta nueva herramienta fácil de usar ha sido lanzada en Bioconductor y Github. En las pruebas, PAST realizó análisis en menos de una hora que anteriormente requerían veinticuatro o más horas. En este artículo, presentamos el protocolo para usar la aplicación Shiny o la consola R para ejecutar PAST.
Introducción
Los estudios de asociación de todo el genoma (GWAS) son un método popular para estudiar rasgos complejos y las regiones genómicas asociadas con ellos1,2,3. En este tipo de estudio, se prueban cientos de miles de marcadores de polimorfismo de un solo nucleótido (SNP) para determinar su asociación con el rasgo, y se evalúa la importancia de las asociaciones. Las asociaciones marcador-rasgo que cumplen con el umbral de la tasa de descubrimiento falso (FDR) (o algún otro tipo de umbral de significación) se conservan para el estudio, pero las asociaciones verdaderas pueden filtrarse. Para rasgos poligénicos complejos, el efecto de cada gen puede ser pequeño (y por lo tanto filtrado), y algunos alelos solo se expresan en condiciones específicas que podrían no estar presentes en el estudio3. Por lo tanto, si bien muchos SNP pueden conservarse como asociados con el rasgo, cada uno puede tener un efecto muy pequeño. Faltarán demasiadas llamadas SNP, y una interpretación del significado biológico y la arquitectura genética del rasgo puede ser incompleta y confusa. El análisis de la vía metabólica puede ayudar a abordar algunos de estos problemas centrándose en los efectos combinados de los genes agrupados según su función biológica4,5,6.
Se completaron varios estudios utilizando una implementación previa del método descrito en este artículo. La acumulación de aflatoxinas7,la resistencia al gusano de la mazorca de maíz8y la biosíntesis de aceite9 se estudiaron con la implementación anterior. Si bien estos análisis tuvieron éxito, el proceso de análisis fue complicado, lento y engorroso, porque las herramientas de análisis se escribieron en una combinación de R, Perl y Bash, y la canalización no se automatizó. Debido al conocimiento especializado requerido para modificar este método para cada análisis, ahora se ha desarrollado un nuevo método que se puede compartir con otros investigadores.
La Herramienta de Estudio de la Asociación Pathway (PAST)10 fue diseñada para abordar las deficiencias del método anterior al requerir menos conocimiento de los lenguajes de programación y al ejecutar análisis en un período más corto. Si bien el método se probó con maíz, PAST no hace suposiciones específicas de la especie. PAST se puede ejecutar a través de la consola R, como una aplicación Shiny, y se espera que pronto esté disponible una versión en línea en MaizeGDB.
Protocolo
1. Configuración
- Instale R, si aún no está instalado.
NOTA: PAST está escrito en R y, por lo tanto, requiere que sus usuarios tengan R instalado. En el momento de escribir este artículo, la instalación de PAST directamente desde Bioconductor requiere R4.0. Las versiones anteriores de PAST se pueden instalar desde Bioconductor para R3.6, y PAST se puede instalar desde Github para usuarios con R3.5. Las instrucciones de instalación de R se pueden descargar desde el siguiente enlace: https://www.r-project.org/. - Instale la versión más reciente de RStudio Desktop o actualice RStudio (opcional).
NOTA: RStudio es un entorno útil para trabajar con el lenguaje R. Se recomienda su instalación, especialmente para aquellos que eligen ejecutar PAST en la línea de comandos en lugar de a través de la aplicación Shiny GUI. RStudio y sus instrucciones de instalación se pueden encontrar en el siguiente enlace: https://rstudio.com/products/rstudio/. - Instale PAST desde Bioconductor11 siguiendo las instrucciones de Bioconductor.
NOTA: La instalación a través de Bioconductor debe manejar la instalación de las dependencias de PAST. Además, PAST se puede instalar desde Github12,pero la instalación desde Github no instalará dependencias automáticamente. - Instale PAST Shiny (opcional). Descargue el archivo "app. R" de la página Releases del repositorio de Github: https://github.com/IGBB/PAST/releases/ y recuerda dónde se encuentra el archivo descargado.
NOTA: PAST se puede utilizar llamando a sus métodos directamente con R, pero los usuarios que están menos familiarizados con R pueden ejecutar la aplicación PAST Shiny, que proporciona una interfaz de usuario guiada. PAST Shiny es un script de R disponible en la rama shiny_app del repositorio past Github. PAST Shiny intentará instalar sus dependencias durante la primera ejecución. - Comience el análisis iniciando la aplicación de una de las tres formas que se describen a continuación.
- PAST Shiny con RStudio
- Con RStudio, cree un nuevo proyecto en la carpeta donde se encuentra la aplicación. R se encuentra. Haga clic en | de archivo Nuevo proyecto y seleccione esa carpeta.
- Una vez que se haya creado un nuevo proyecto, abra la aplicación. Archivo R descargado anteriormente. RStudio reconoce esa aplicación. R es una aplicación Shiny y crea un botón Ejecutar aplicación en la barra sobre el código fuente mostrado. Haga clic en Ejecutar aplicación. RStudio iniciará una ventana que muestra la aplicación PAST Shiny.
- PAST Shiny con R Console
- Inicie R y ejecute el siguiente código para iniciar la aplicación PAST Shiny: shiny::runApp('path/to/folder/with/shiny/app. R'. Reemplace el texto entre comillas con la carpeta a la que se encuentra la aplicación. R fue descargado, y mantener las comillas.
- PASADO sin R Shiny
- Ejecute library(PAST) en una consola de R para cargar PAST.
- PAST Shiny con RStudio
2. Personaliza el análisis brillante (opcional)
- Cambie el título del análisis de "Nuevo análisis" a algo que refleje mejor el tipo de análisis que se está ejecutando, lo que ayuda a realizar un seguimiento de múltiples análisis (consulte la Figura 1).

Figura 1. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
- Modifique el número de núcleos y el modo. Establezca el número de núcleos en cualquier número entre 1 y el número total en la máquina, pero tenga en cuenta que dedicar más recursos a PAST puede ralentizar otras operaciones en la máquina. Establezca el modo en función de la descripción de la sección 6.
3. Cargar datos GWAS
NOTA: Compruebe que los datos de GWAS están delimitados por tabulaciones. Asegúrese de que el archivo de asociación contiene las siguientes columnas: rasgo, nombre del marcador, locus o cromosoma, posición en el cromosoma, valor p y valor R2 para el marcador. Asegúrese de que el archivo de efectos contenga las siguientes columnas: rasgo, nombre del marcador, locus o cromosoma, posición en el cromosoma y efecto. El orden de estas columnas no es importante, ya que el usuario puede especificar los nombres de las columnas al cargar los datos. Se ignoran las columnas adicionales. TASSEL13 se puede utilizar para producir estos archivos.
- Cargue datos GWAS con PAST Shiny.
- Seleccione un archivo de asociación y un archivo de efectos mediante los cuadros de selección Archivo de asociación y Archivo de efectos. Cambie los nombres de columna en los cuadros de entrada Nombre de columna de asociación y Efectos Nombre de columna debajo de los cuadros de selección de archivos para reflejar los nombres de columna en los datos.

Figura 2. Haga clic aquí para ver una versión más grande de esta figura.
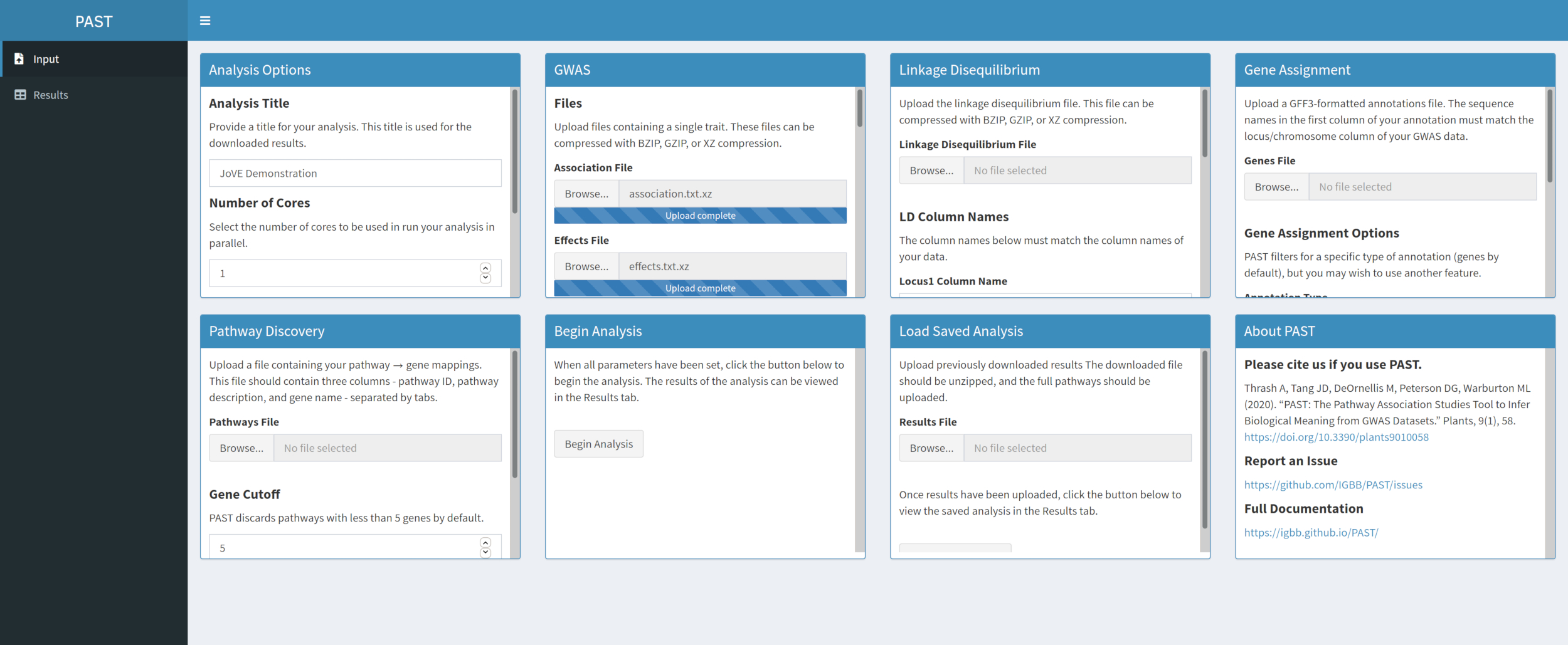{kind=link}
- Cargue datos GWAS con PAST en R Console.
- Modifique y ejecute el código siguiente:
gwas_data = load_GWAS_data("path/to/association_file.tsv", "path/to/effects_file.tsv", association_columns = c("Trait", "Marker", "Locus", "Site", "p", "marker_R2"), effects_columns = c("Trait", "Marker", "Locus", "Site", "Effect")
- Modifique y ejecute el código siguiente:
- NOTA: Cambie las rutas a la ubicación real de los archivos GWAS. Los valores proporcionados para association_columns y effects_columns son los valores predeterminados. Si los nombres no coinciden con los valores predeterminados, especifique los nombres de columna. De lo contrario, estos pueden omitirse.
4. Datos de desequilibrio de enlace de carga (LD)
NOTA: Compruebe que los datos de desequilibrio de enlace (LD) están delimitados por tabulación y contienen los siguientes tipos de datos: Locus, Position1, Site1, Position2, Site2, Distance en pares de bases entre Position1 y Position2, y R2 value.
- Cargue datos LD con PAST Shiny.
- Seleccione el archivo que contiene los datos LD. Cambie los nombres de columna en los cuadros de entrada Nombres de columna LD debajo del cuadro de selección de archivos para que coincidan con los nombres de columna en los datos LD si es necesario.

Figura 3. Haga clic aquí para ver una versión más grande de esta figura.
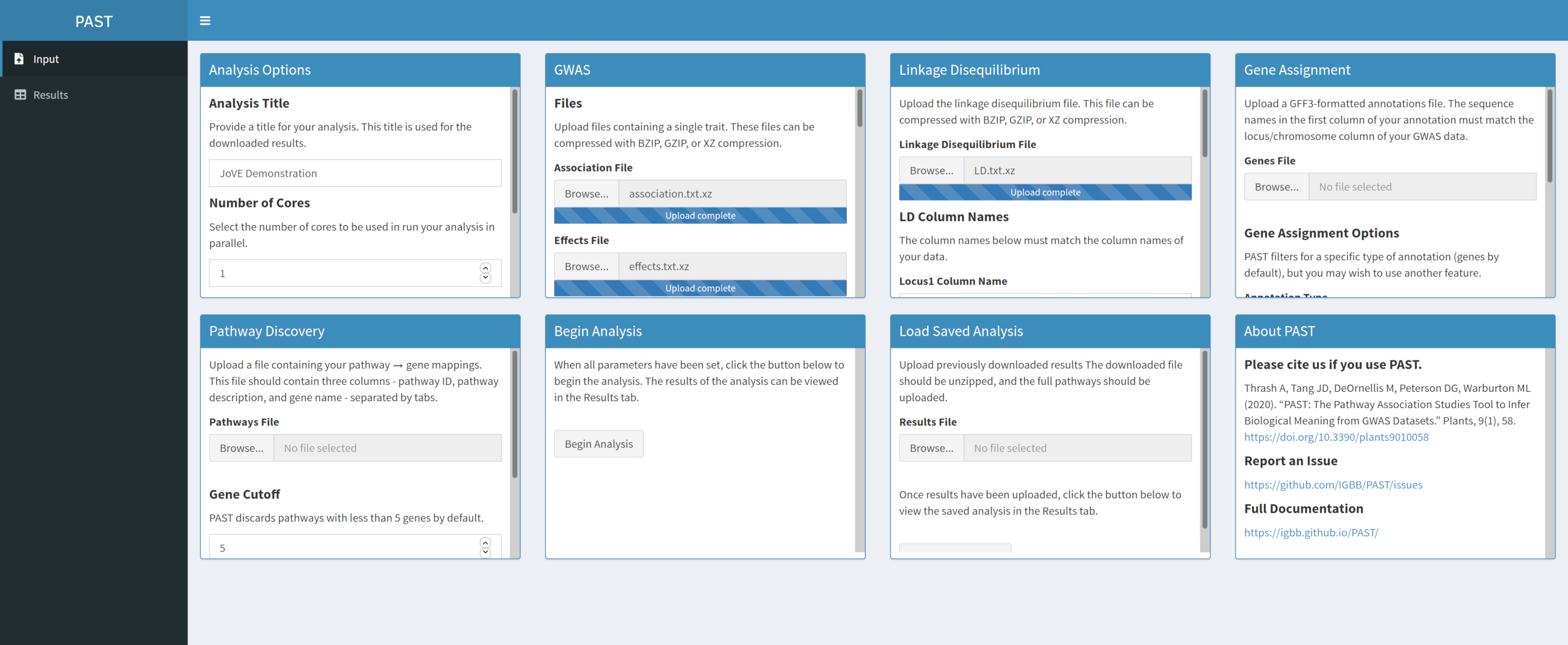{kind=link}
- Cargue datos LD con PAST en la consola de R.
- Modifique y ejecute el código siguiente para cargar datos LD:
LD = load_LD("path/to/LD.tsv", LD_columns = c("Locus1", "Position1", "Site1", "Position2", "Site2", "Dist_bp", "R.2")
Nota : cambie la ruta de acceso a la ubicación real del archivo LD. Los valores proporcionados para LD_columns son los valores predeterminados. Si los nombres no coinciden con estos valores predeterminados, especifique los nombres correctos de las columnas; de lo contrario, se pueden omitir.
- Modifique y ejecute el código siguiente para cargar datos LD:
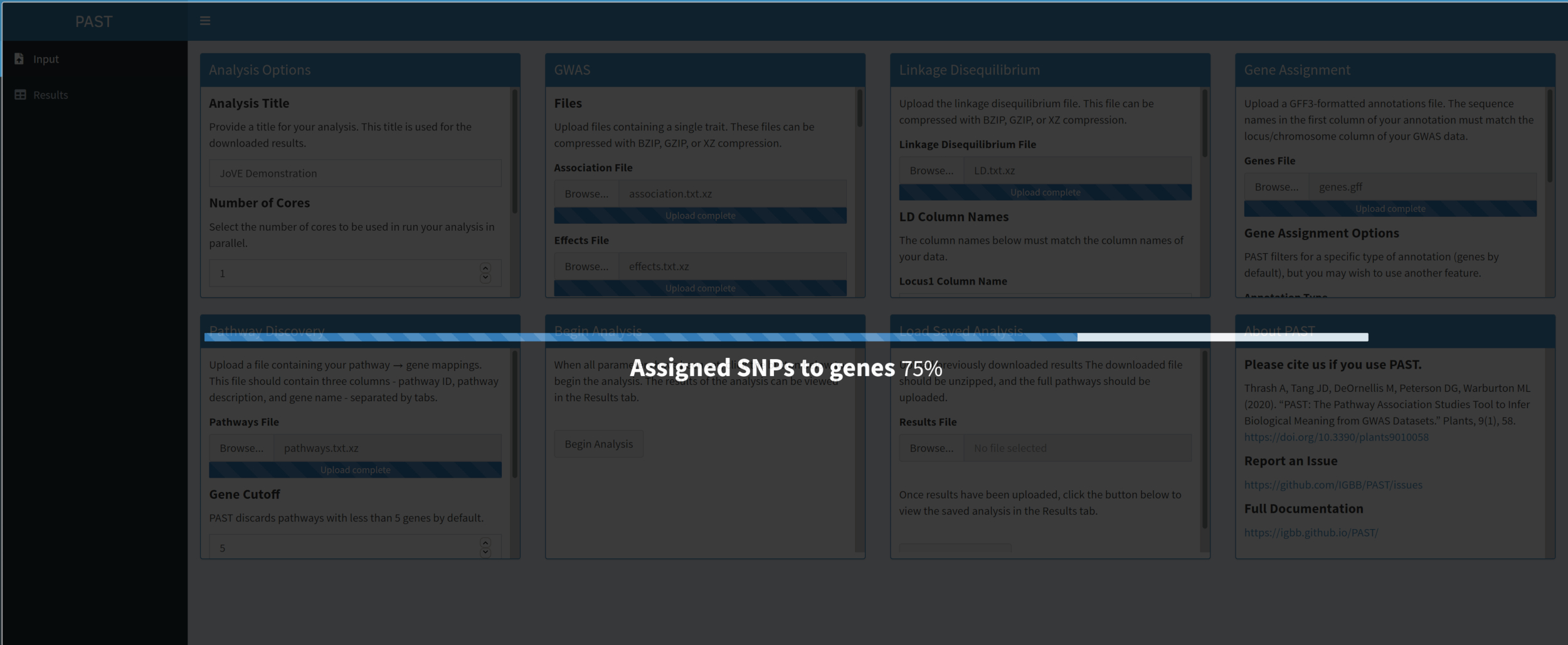
5. Asignar SNP a los genes
NOTA: Descargue o localice de otro modo las anotaciones en formato GFF. Estas anotaciones a menudo se pueden encontrar en bases de datos en línea para organismos específicos. Tenga cuidado con las anotaciones de baja calidad, ya que la calidad de los datos de las anotaciones afectará la calidad del análisis de la vía. Confirme que la primera columna de estas anotaciones (el cromosoma) coincide con el formato del locus/cromosoma en los datos de asociación, efectos y DA. Por ejemplo, las anotaciones no deben llamar al primer cromosoma "chr1" si los archivos de datos GWAS y LD llaman al primer cromosoma "1".
- Asigne SNP a genes con PAST Shiny.
NOTA: Se puede encontrar más información sobre la determinación de un límite R2 apropiado en Tang et al.6,en la sección llamada "SNP to gene algorithm for the pathway analysis".- Seleccione el archivo que contiene anotaciones GFF. Considere qué tamaño de ventana y corte R2 son los más adecuados para la especie que se está considerando y modifíquelo si los valores predeterminados no se adaptan a los datos cargados.
NOTA: Los valores predeterminados en past reflejan principalmente los valores apropiados para el maíz. El número de núcleos establecidos al principio del análisis PAST Shiny (Paso 2.2) se utiliza en este paso.
- Seleccione el archivo que contiene anotaciones GFF. Considere qué tamaño de ventana y corte R2 son los más adecuados para la especie que se está considerando y modifíquelo si los valores predeterminados no se adaptan a los datos cargados.

Figura 4. Haga clic aquí para ver una versión más grande de esta figura.
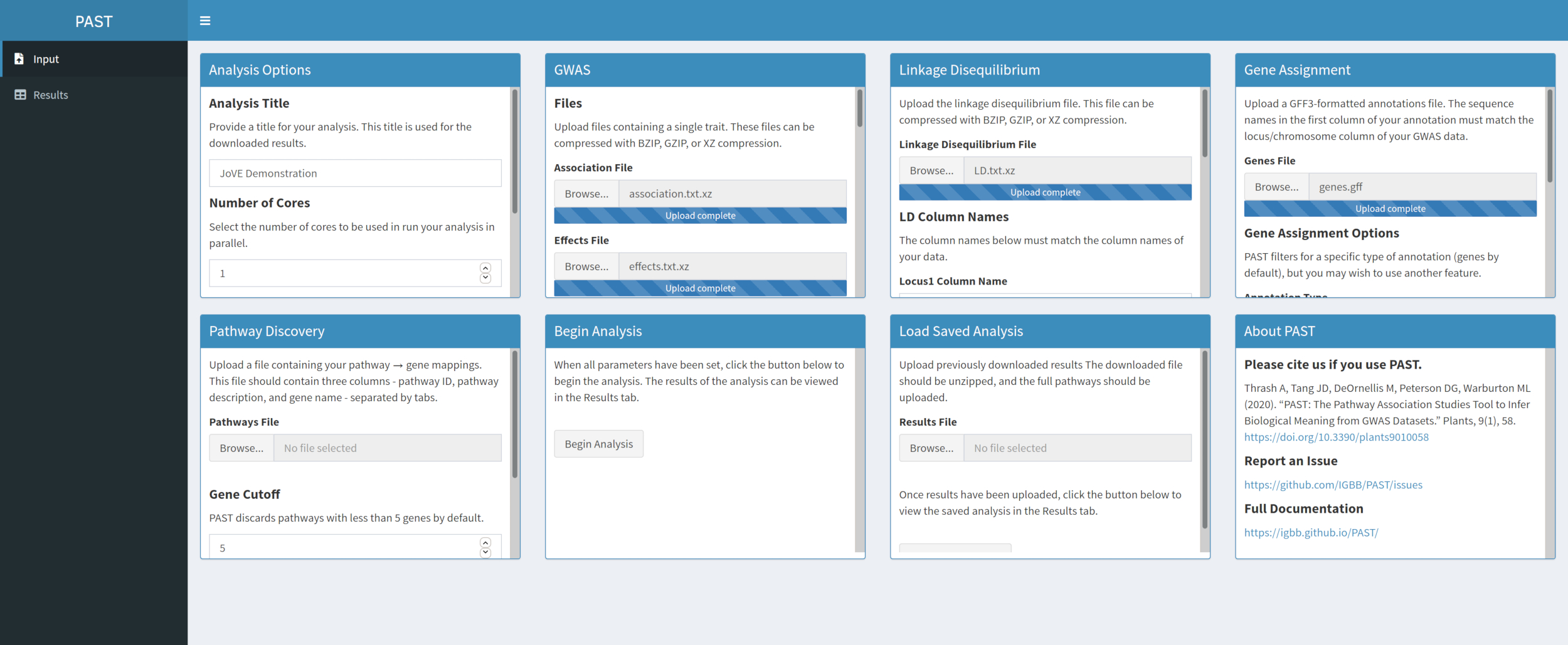{kind=link}
- Asigne SNP a genes con PAST en la consola de R.
- Modifique y ejecute el código siguiente para asignar SNP a los genes:
genes = assign_SNPs_to_genes(gwas_data, LD, "path/to/annotations.gff", c("gene"), 1000, 0.8, 2)
Nota : en este código de ejemplo, se proporcionan varias sugerencias predeterminadas: 1000 es el tamaño de la ventana alrededor del SNP para buscar genes; 0,8 es el valor de corte para R2; 2 es el número de núcleos utilizados para el procesamiento paralelo. La ruta de acceso a las anotaciones también debe cambiarse a la ubicación real del archivo de anotaciones.
- Modifique y ejecute el código siguiente para asignar SNP a los genes:
6. Descubre vías significativas
NOTA: Verifique que el archivo de vías contenga los siguientes datos en formato delimitado por tabuladores, con una línea para cada gen en cada vía: ID de vía - un identificador como "PWY-6475-1"; descripción de la vía: una descripción más larga de lo que hacen las vías, como la "biosíntesis de trans-licopeno"; gen - un gen en la vía, que debe coincidir con los nombres proporcionados en las anotaciones. Es probable que la información de la vía se pueda encontrar en bases de datos en línea para organismos específicos, como MaizeGDB. La segunda opción especificada por el usuario es el modo. "Aumentar" se refiere a fenotipos que reflejan cuándo es deseable un valor creciente del rasgo medido, como el rendimiento, mientras que "disminuir" se refiere a un rasgo donde una disminución en los valores medidos es beneficiosa, como las calificaciones de daño por insectos. La importancia de las vías se prueba utilizando métodos descritos anteriormente4,6,14.
- Descubre vías significativas con PAST Shiny.
- Seleccione el archivo que contiene los datos de las rutas y asegúrese de que el modo está seleccionado en las opciones de análisis. Si es necesario, cambie el número de genes que deben estar en una vía para retenerlo para el análisis y el número de permutaciones utilizadas para crear la distribución nula para probar la significación del efecto.

Figura 5. Haga clic aquí para ver una versión más grande de esta figura.
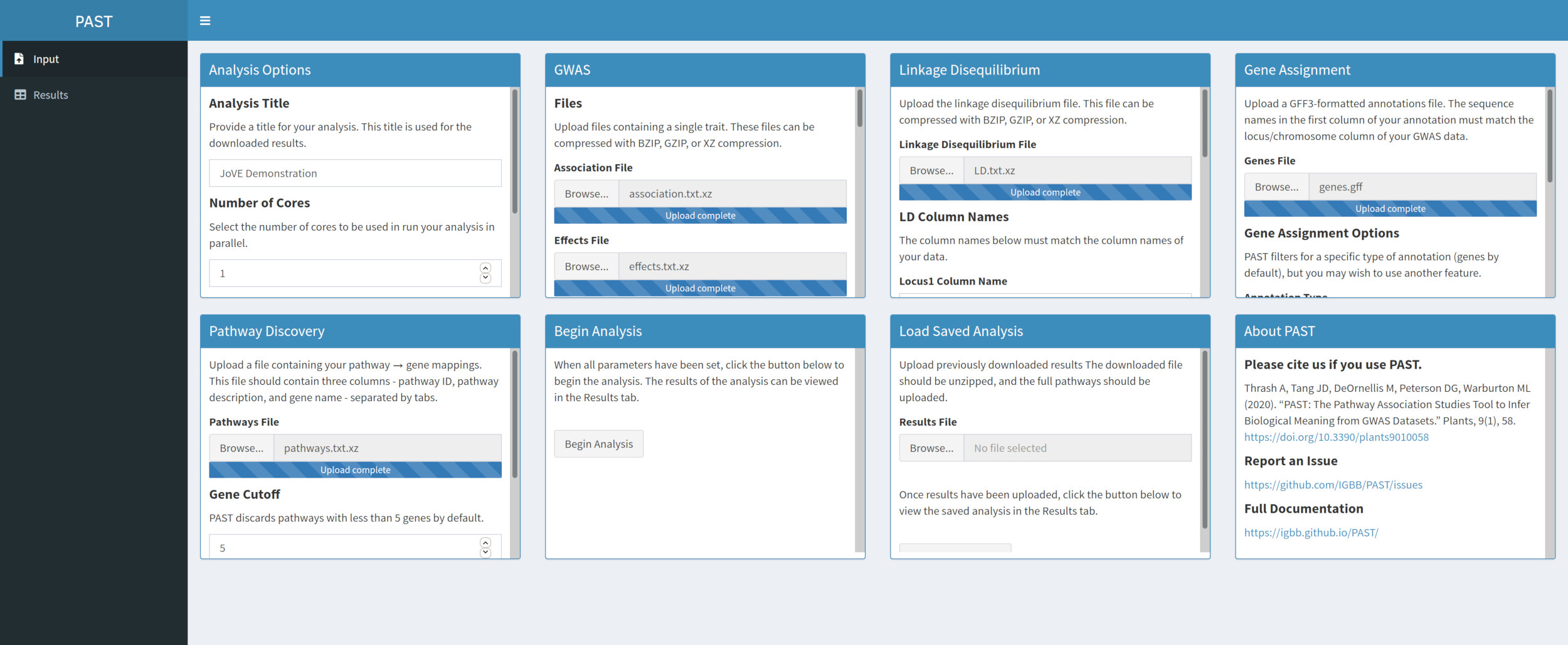{kind=link}
NOTA: El número de núcleos y el modo establecido al principio del análisis PAST Shiny (Paso 2.2) se utiliza en este paso. El número predeterminado de genes se establece actualmente en 5 genes, por lo que se eliminarán las vías con menos genes conocidos. El usuario puede reducir este valor a 4 o 3, para incluir vías más cortas, pero hacerlo se arriesgará a resultados falsos positivos. Aumentar este valor puede aumentar el poder del análisis, pero eliminará más vías del análisis. Cambiar el número de permutaciones utilizadas aumenta y disminuye la potencia de la prueba.
- Descubra vías importantes con PAST en R Console.
- Modifique y ejecute el código siguiente para descubrir rutas importantes:
rugplots_data <- find_pathway_significance(genes, "path/to/pathways.tsv", 5, "increasing", 1000, 2)
Nota : en este código de ejemplo, se proporcionan varios valores predeterminados sugeridos. 5 es el número mínimo de genes que deben estar en una vía para mantener la vía en el análisis, aumentar se refiere a una cantidad creciente del rasgo medido (se recomienda que el usuario ejecute tanto aumentando como disminuyendo, independientemente del rasgo; la interpretación de los datos diferirá para los dos, sin embargo), 1000 es el número de veces que muestrea los efectos para determinar la distribución nula, y 2 es el número de núcleos utilizados para el procesamiento paralelo. Cambie la ruta de acceso a la ubicación real del archivo de rutas.
- Modifique y ejecute el código siguiente para descubrir rutas importantes:
7. Ver Rugplots
- Ver Rugplots con PAST Shiny.
- Una vez que todas las entradas se cargan y se establecen, haga clic en Comenzar análisis. Aparecerá una barra de progreso e indicará qué paso del análisis se completó por última vez. Cuando se complete el análisis, PAST Shiny cambiará a la pestaña Resultados. Se mostrará una tabla de resultados en la columna izquierda (etiquetada como "caminos") y los Rugplots se mostrarán en la columna derecha (etiquetados como "gráficos").
- Utilice el control deslizante para controlar los parámetros de filtrado. Cuando el nivel de filtrado sea satisfactorio, haga clic en el botón Descargar resultados en la parte inferior izquierda para descargar todas las imágenes y tablas individualmente a un archivo ZIP que se nombra con el título del análisis. Este archivo ZIP contiene la tabla filtrada, la tabla sin filtrar y una imagen por ruta en la tabla filtrada.

Figura 6. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 7. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
- Ver Rugplots con PAST en la consola R
- Modifique y ejecute el código siguiente para guardar los resultados:
plot_pathways(rugplots_data, "pvalue", 0.02, "creciente", "output_folder")
Nota : en este código de ejemplo, se proporcionan varios valores predeterminados sugeridos. pvalue proporciona los datos que se pueden utilizar para filtrar vías insignificantes después de que el usuario elija un umbral de significación; 0.02 es el valor predeterminado utilizado en el filtrado, y aumentar se refiere a una cantidad creciente del rasgo medido (se recomienda que el usuario ejecute tanto el aumento como la disminución, independientemente del rasgo; sin embargo, la interpretación de los datos diferirá para los dos); output_folder es la carpeta donde se escribirán las imágenes y tablas (esta carpeta debe existir antes de ejecutar la función). En esta carpeta se escribe una tabla de resultados filtrados, los resultados sin filtrar y las imágenes individuales para cada ruta de los resultados filtrados.
- Modifique y ejecute el código siguiente para guardar los resultados:
Resultados
Si los resultados no se producen después de una ejecución de la herramienta de software PAST, asegúrese de que todos los archivos de entrada estén formateados correctamente. Una ejecución exitosa utilizando los datos de ejemplo en el paquete PAST, que se basan en un GWAS de maíz de color grano, se muestra en la Figura 8. Esta tabla y la imagen resultante se pueden descargar utilizando el botón Descargar resultados. Un ejemplo de la imagen descargada se muestra en la Figura 210. Los ajustes incorrectos pueden conducir a resultados que no tienen sentido biológico, pero la determinación de la incorrección debe ser responsabilidad del investigador, quien debe verificar la validez de los ajustes elegidos y considerar toda la evidencia conocida con respecto al rasgo de interés.
La Figura 910 muestra la rugplot producida a partir del análisis de la vía de los resultados de GWAS creado con un panel de maíz de 288 líneas endogámicas que habían sido fenotipadas para el color del grano. Este ejemplo simplista, donde los fenotipos eran "blancos" o "amarillos", se utilizó porque la vía responsable de crear los pigmentos carotenoides de color amarillo brillante es conocida y debería ser responsable de la mayor parte del fenotipo. Por lo tanto, esperábamos ver que la vía de biosíntesis de trans-licopeno (que produce carotenoides) se asociaba significativamente con el color del grano, que lo es. El ID y el nombre de la ruta se enumeran en la parte superior del gráfico. El eje horizontal del gráfico clasifica todos los genes que se incluyeron en el análisis, dispuestos de izquierda a derecha en orden de mayor efecto sobre el rasgo al más pequeño. Sin embargo, solo los genes en la vía de biosíntesis trans-licopeno están marcados (en la parte superior del gráfico, como marcas de eclosión, que aparecen en el rango genético de su efecto en comparación con todos los demás genes en el análisis). Hay 7 genes en esta vía. La puntuación de enriquecimiento en ejecución (ES) se traza a lo largo del eje vertical. El ES para cada gen se agrega al total en ejecución en orden de efecto y el total se ajusta al número de genes analizados. Por lo tanto, la puntuación cambia a medida que uno se mueve a lo largo del eje horizontal y tiende a aumentar a medida que se incluyen los genes de efecto más grandes, pero en algún momento, el aumento en el efecto es menor que el ajuste por haber agregado otro gen, y toda la puntuación comienza a disminuir. El vértice de la línea ES en funcionamiento está marcado con una línea vertical punteada; este es el ES para toda la vía y es utilizado por el programa para determinar si la vía se elige y se presenta como una alfombra.

Figura 8: Ejecución completada de PAST Shiny. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 9:Imagen de la ruta de la ejecución completada de PAST (o descargada de Shiny). Esta cifra ha sido citada de Thrash et al.10. Haga clic aquí para ver una versión más grande de esta figura.
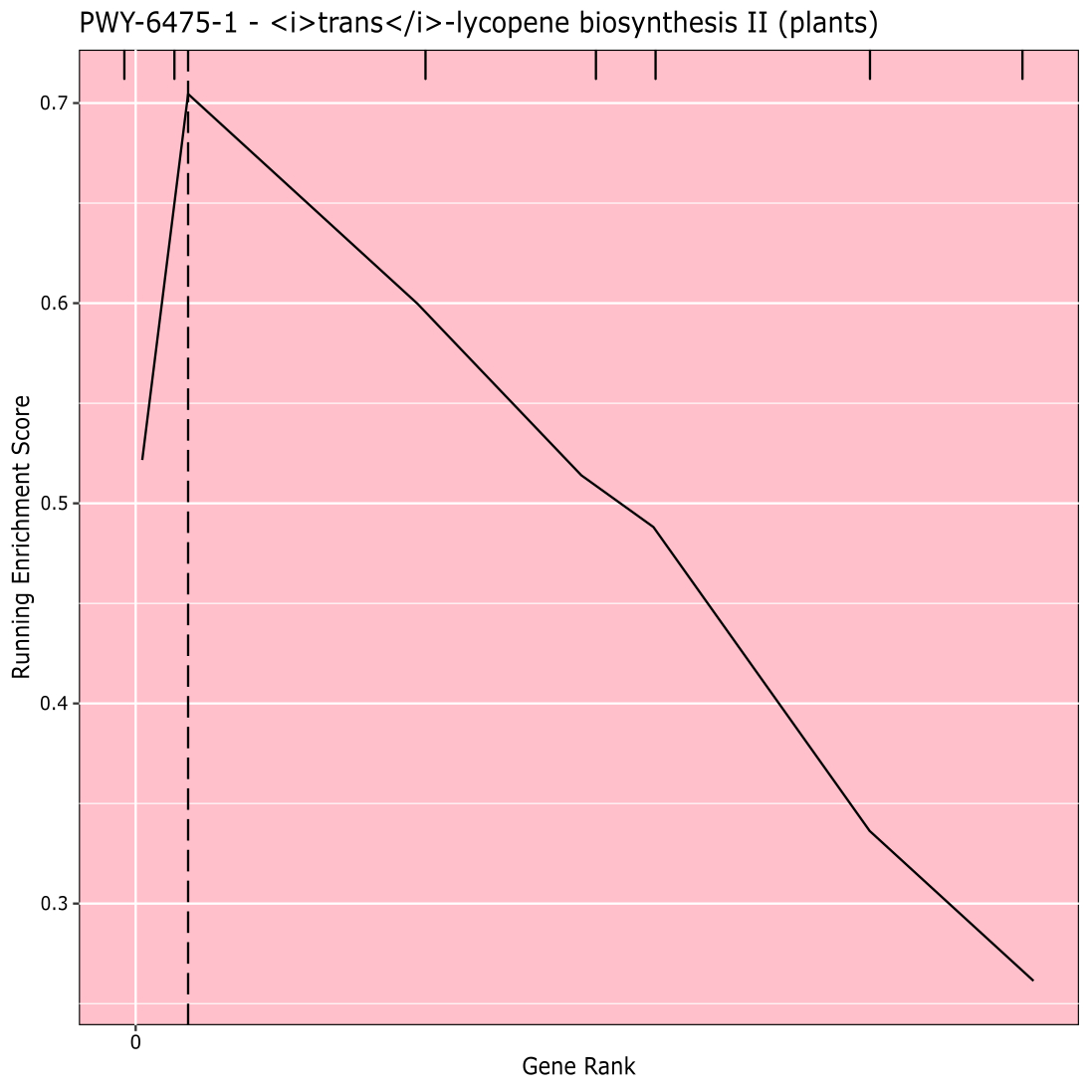{kind=link}
Discusión
Un objetivo principal de PAST es llevar los análisis de la vía metabólica de los datos de GWAS a un público más amplio, especialmente para organismos no humanos y no animales. Los métodos alternativos a PAST son a menudo programas de línea de comandos que se centran en humanos o animales. La facilidad de uso fue un objetivo principal en el desarrollo de PAST, tanto en la elección de desarrollar una aplicación Shiny como en la elección de usar R y Bioconductor para lanzar la aplicación. Los usuarios no necesitan aprender a compilar programas para usar PAST.
Al igual que con la mayoría de los tipos de software de análisis, los resultados de PAST son tan buenos como los datos de entrada; si los datos de entrada tienen errores o tienen un formato incorrecto, PAST no se ejecutará o producirá resultados poco informativos. Asegurarse de que los datos GWAS, los datos LD, las anotaciones y los archivos de rutas estén correctamente formateados es fundamental para recibir la salida correcta de PAST. PAST solo analiza marcadores bialélicos y solo puede ejecutar un rasgo para cada conjunto de datos de entrada. Además, los datos de GWAS producidos por un genotipado deficiente o un fenotipado incorrecto o impreciso tampoco es probable que produzcan resultados claros o repetibles. PAST puede ayudar en la interpretación biológica de los resultados de GWAS, pero es poco probable que aclare conjuntos de datos caóticos si la variación ambiental, el error experimental o la estructura de la población no se contabilizaron adecuadamente.
Los usuarios pueden optar por cambiar algunos parámetros del análisis, tanto en la aplicación Shiny como pasando esos parámetros a las funciones de PAST en la consola de R. Estos parámetros pueden cambiar los resultados reportados por PAST, y los usuarios deben tener cuidado al modificarlos desde los valores predeterminados. Debido a que la LD es medida por los usuarios, generalmente utilizando el mismo conjunto de datos de marcadores que también se utilizó en el GWAS, las mediciones de LD son específicas de la población. Para todos los estudios, especialmente para especies distintas del maíz (en particular las especies autopolinizantes, poliploides o altamente heterogéneas), pueden justificarse cambios en los valores predeterminados.
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Ninguno.
Materiales
| Name | Company | Catalog Number | Comments |
| Computer | NA | NA | Any computer with 8GB RAM should be sufficient |
| R | R Project | NA | R 4.0 or greater is required to install from Bioconductor 3.11 |
Referencias
- Rafalski, J. Association genetics in crop improvement. Current Opinion in Plant Biology. 13 (2), 174-180 (2010).
- Yan, J., Warburton, M., Crouch, J. Association Mapping for Enhancing Maize (Zea mays L.) Genetic Improvement. Crop Science. 51 (2), 433-449 (2011).
- Xiao, Y., Liu, H., Wu, L., Warburton, M., Yan, J. Genome-wide Association Studies in Maize: Praise and Stargaze. Molecular Plant. 10 (3), 359-374 (2017).
- Wang, K., Li, M., Bucan, M. Pathway-Based Approaches for Analysis of Genomewide Association Studies. The American Journal of Human Genetics. 81 (6), 1278-1283 (2007).
- Weng, L., et al. SNP-based pathway enrichment analysis for genome-wide association studies. BMC Bioinformatics. 12 (1), 99(2011).
- Tang, J., Perkins, A., Williams, W., Warburton, M. Using genome-wide associations to identify metabolic pathways involved in maize aflatoxin accumulation resistance. BMC Genomics. 16 (1), 673(2015).
- Warburton, M., et al. Genome-Wide Association Mapping of Aspergillus flavus and Aflatoxin Accumulation Resistance in Maize. Crop Science. 55 (5), 1857-1867 (2015).
- Warburton, M., et al. Genome-Wide Association and Metabolic Pathway Analysis of Corn Earworm Resistance in Maize. The Plant Genome. 11 (1), 170069(2018).
- Li, H., Thrash, A., Tang, J., He, L., Yan, J., Warburton, M. Leveraging GWAS data to identify metabolic pathways and networks involved in maize lipid biosynthesis. The Plant Journal. 98 (5), 853-863 (2019).
- Thrash, A., Tang, J., DeOrnellis, M., Peterson, D., Warburton, M. PAST: The Pathway Association Studies Tool to Infer Biological Meaning from GWAS Datasets. Plants. 9 (1), 58(2020).
- Adam, T., Mason, D. PAST: Pathway Association Study Tool (PAST). Bioconductor version: Release (3.10). , (2020).
- Thrash, A., DeOrnellis, M. IGBB/PAST. , at https://github.com/IGBB/PAST (2019).
- Bradbury, P., et al. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 23 (19), 2633-2635 (2007).
- Subramanian, A., et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences U.S.A. 102, 15545-15550 (2005).
Erratum
Formal Correction: Erratum: A Pathway Association Study Tool for GWAS Analyses of Metabolic Pathway Information
Posted by JoVE Editors on 10/08/2021. Citeable Link.
An erratum was issued for: A Pathway Association Study Tool for GWAS Analyses of Metabolic Pathway Information. One of the affiliations was updated.
The second affiliation was updated from:
USDA-ARS Corn Host Plant Resistance Research Unit, Mississippi State University
to:
Corn Host Plant Resistance Research Unit, USDA-ARS
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados