
Method Article
Uma ferramenta de estudo da Associação de Caminhos para análises gwas de informações de via metabólica
Neste Artigo
Erratum Notice
Resumo
Ao executar a Ferramenta de Estudo da Associação pathway (PAST), seja através do aplicativo Shiny ou através do console R, os pesquisadores podem obter uma compreensão mais profunda do significado biológico de seus resultados de estudo de associação genoma -em todo o genoma (GWAS) investigando as vias metabólicas envolvidas.
Resumo
Recentemente, uma nova implementação de um método descrito anteriormente para interpretar dados do estudo de associação genoma (GWAS) utilizando a análise metabólica da via foi desenvolvida e liberada. A Ferramenta de Estudo da Associação pathway (PAST) foi desenvolvida para abordar preocupações com a simpatia do usuário e análises de execução lenta. Esta nova ferramenta fácil de usar foi lançada no Bioconductor e no Github. Em testes, o PAST realizou análises em menos de uma hora que antes exigia 24 ou mais horas. Neste artigo, apresentamos o protocolo para usar o aplicativo Shiny ou o console R para executar PAST.
Introdução
Os estudos de associação genoma (GWAS) são um método popular de estudo de traços complexos e as regiões genômicas associadas a eles1,2,3. Neste tipo de estudo, centenas de milhares de marcadores de polimorfismo de nucleotídeos únicos (SNP) são testados para sua associação com o traço, e a significância das associações é avaliada. Associações de características marcadoras que atendam ao limiar da taxa de descoberta falsa (FDR) (ou algum outro tipo de limiar de significância) são mantidas para o estudo, mas associações verdadeiras podem ser filtradas. Para traços complexos e poligênicos, o efeito de cada gene pode ser pequeno (e, portanto, filtrado), e alguns alelos são expressos apenas em condições específicas que podem não estar presentes no estudo3. Assim, enquanto muitos SNPs podem ser retidos como associados à característica, cada um pode ter um efeito muito pequeno. Muitas chamadas SNP estarão faltando, e uma interpretação do significado biológico e da arquitetura genética do traço pode ser incompleta e confusa. A análise da via metabólica pode ajudar a resolver algumas dessas questões, focando nos efeitos combinados dos genes agrupados de acordo com sua função biológica4,5,6.
Vários estudos foram concluídos utilizando-se uma implementação prévia do método descrito neste artigo. Acúmulo deaflatoxina 7,resistência à minhoca de milho8e biossíntese de óleo9 foram todos estudados com a implementação anterior. Embora essas análises tenham sido bem sucedidas, o processo de análise foi complicado, demorado e complicado, pois as ferramentas de análise foram escritas em uma combinação de R, Perl e Bash, e o pipeline não foi automatizado. Devido ao conhecimento especializado necessário para modificar esse método para cada análise, foi desenvolvido agora um novo método que pode ser compartilhado com outros pesquisadores.
A Ferramenta de Estudo da Associação de Caminhos (PAST)10 foi projetada para resolver as deficiências do método anterior, exigindo menos conhecimento das linguagens de programação e executando análises em um período mais curto. Embora o método tenha sido testado com milho, o PAST não faz suposições específicas de espécies. Past pode ser executado através do console R, como um aplicativo Shiny, e espera-se que uma versão on-line esteja disponível em breve no MaizeGDB.
Protocolo
1. Configuração
- Instale R, se ainda não estiver instalado.
NOTA: PAST é escrito em R e, portanto, requer que seus usuários tenham R instalado. No momento desta redação, a instalação do PAST diretamente do Bioconductor requer R4.0. Versões mais antigas do PAST podem ser instaladas a partir do Bioconductor para R3.6, e o PAST pode ser instalado no Github para usuários com R3.5. As instruções de instalação R podem ser baixadas no seguinte link: https://www.r-project.org/. - Instale a versão mais recente do RStudio Desktop ou atualize o RStudio (opcional).
NOTA: O RStudio é um ambiente útil para trabalhar com a linguagem R. Sua instalação é recomendada, especialmente para aqueles que optarem por executar PAST na linha de comando e não através do aplicativo Shiny GUI. O RStudio e suas instruções de instalação podem ser encontrados no seguinte link: https://rstudio.com/products/rstudio/. - Instale o PAST do Bioconductor11 seguindo as instruções do Bioconductor.
NOTA: A instalação através do Bioconductor deve lidar com a instalação das dependências do PAST. Além disso, o PAST pode ser instalado a partir do Github12,mas a instalação do Github não instalará dependências automaticamente. - Instale PAST Shiny (opcional). Baixe o arquivo "app. R" da página Lançamentos do repositório do Github: https://github.com/IGBB/PAST/releases/ e lembre-se onde o arquivo baixado está localizado.
NOTA: O PAST pode ser usado ligando diretamente para seus métodos com R, mas os usuários que estão menos familiarizados com R podem executar o aplicativo PAST Shiny, que fornece uma interface de usuário guiada. PAST Shiny é um script R disponível na filial shiny_app do repositório PAST Github. Past Shiny tentará instalar suas dependências durante a primeira execução. - Inicie a análise iniciando a aplicação em uma das três maneiras descritas abaixo.
- PASSADO Brilhante com RStudio
- Usando o RStudio, crie um novo projeto na pasta onde o aplicativo. R está localizado. Clique em | de arquivos Novo Projeto e selecione essa pasta.
- Uma vez criado um novo projeto, abra o aplicativo. Arquivo R baixado mais cedo. RStudio reconhece esse aplicativo. R é um aplicativo Shiny e cria um botão Executar App na barra acima do código-fonte exibido. Clique em Executar app. Em seguida, o RStudio iniciará uma janela que exibe o aplicativo PAST Shiny.
- PASSADO brilhante com console R
- Inicie R e execute o seguinte código para iniciar o aplicativo PAST Shiny: brilhante::runApp('path/to/folder/with/shiny/app. R'. Substitua o texto entre aspas pela pasta para qual aplicativo. R foi baixado, e manter as cotações.
- PASSADO sem R Shiny
- Execute a biblioteca (PAST) em um console R para carregar PAST.
- PASSADO Brilhante com RStudio
2. Personalize a análise brilhante (opcional)
- Alterar o título de análise de "Nova Análise" para algo que melhor reflita o tipo de análise que está sendo executada, o que ajuda a acompanhar as múltiplas análises (ver Figura 1).

Figura 1. Por favor, clique aqui para ver uma versão maior desta figura.
{kind=link}
- Modifique o número de núcleos e o modo. Defina o número de núcleos para qualquer número entre 1 e o número total na máquina, mas esteja ciente de que dedicar mais recursos ao PAST pode retardar outras operações na máquina. Defina o modo com base na descrição da seção 6.
3. Carregar dados gwas
NOTA: Verifique se os dados do GWAS estão delimitados. Certifique-se de que o arquivo de associação contém as seguintes colunas: característica, nome do marcador, lócus ou cromossomo, posição no cromossomo, valor p e valor R2 para o marcador. Certifique-se de que o arquivo de efeitos contém as seguintes colunas: característica, nome do marcador, lócus ou cromossomo, posição no cromossomo e efeito. A ordem dessas colunas não é importante, pois o usuário pode especificar os nomes das colunas ao carregar os dados. Quaisquer colunas adicionais são ignoradas. TASSEL13 pode ser usado para produzir esses arquivos.
- Carregue os dados do GWAS com o PAST Shiny.
- Selecione um arquivo de associação e um arquivo de efeitos usando as caixas de seleção de arquivos e arquivos de arquivos de arquivos da associação. Alterar os nomes das colunas nas colunas Nome e Efeitos da Associação Nomear caixas de entrada abaixo das caixas de seleção de arquivos para refletir os nomes das colunas nos dados.

Figura 2. Clique aqui para ver uma versão maior desta figura.
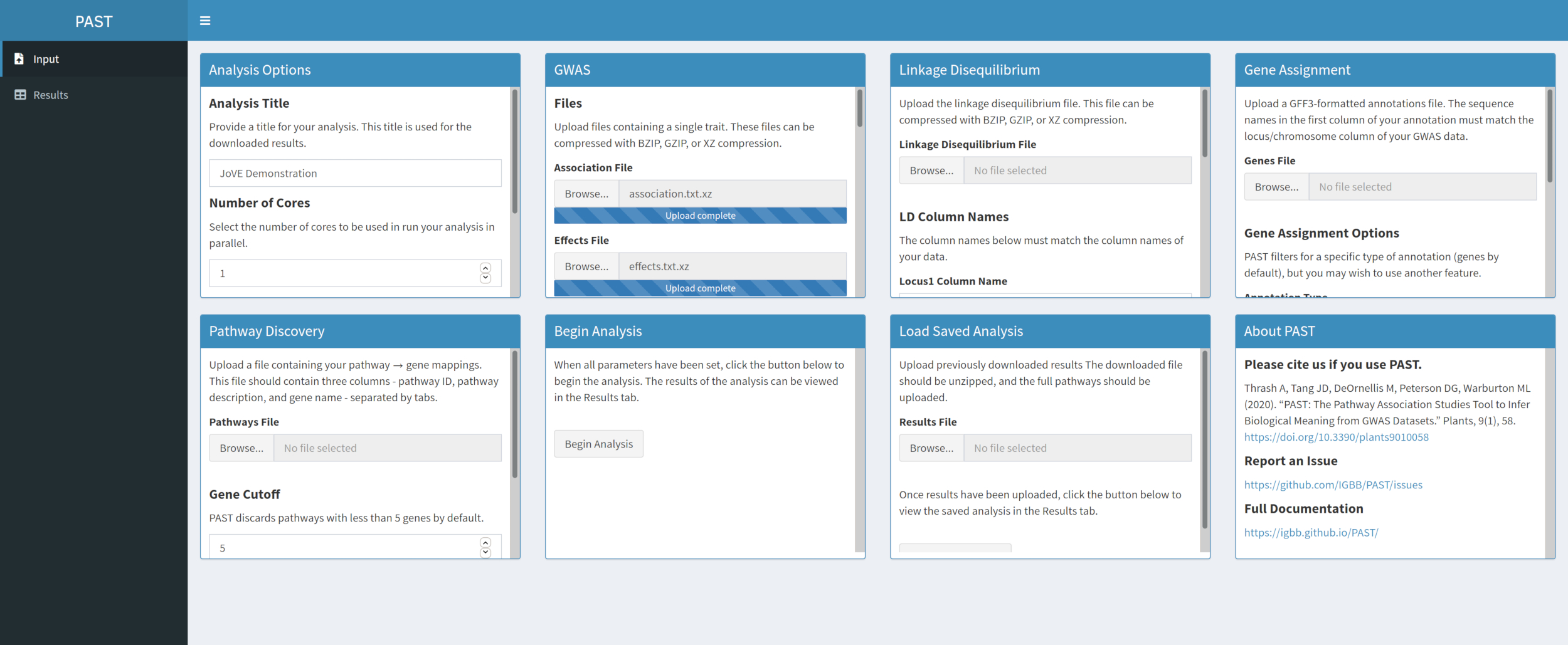{kind=link}
- Carregue os dados GWAS com PAST no console R.
- Modifique e execute o seguinte código:
gwas_data = load_GWAS_data("path/to/association_file.tsv", "path/to/effects_file.tsv", association_columns = c("Trait", "Marker", "Locus", "Site", "p", "marker_R2"), effects_columns = c("Trait", "Marker", "Locus", "Site", "Effect")
- Modifique e execute o seguinte código:
- NOTA: Altere os caminhos para a localização real dos arquivos GWAS. Os valores previstos para association_columns e effects_columns são os valores padrão. Se os nomes não corresponderem aos valores padrão, especifique os nomes das colunas. Caso contrário, estes podem ser omitidos.
4. Dados de desequilíbrio de ligação de carga (LD)
NOTA: Verifique se os dados de desequilíbrio de ligação (LD) são delimitados por guias e contém os seguintes tipos de dados: Locus, Posição1, Site1, Posição2, Site2, Distância em pares de base entre o valor posição1 e posição2 e R2.
- Carregue os dados LD com o PAST Shiny.
- Selecione o arquivo que contém dados LD. Alterar os nomes das colunas LD Nomes caixas de entrada abaixo da caixa de seleção de arquivos para corresponder aos nomes da coluna nos dados LD, se necessário.

Figura 3. Clique aqui para ver uma versão maior desta figura.
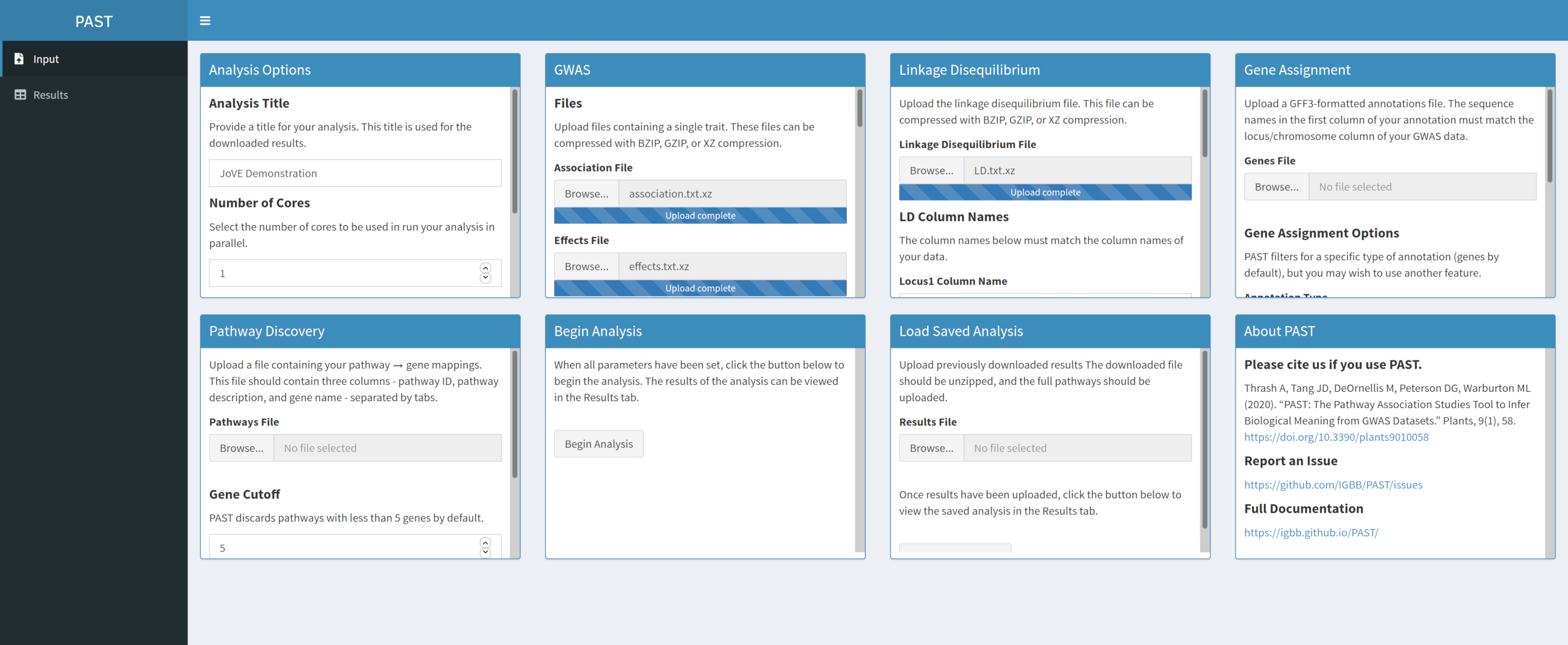{kind=link}
- Carregue dados LD com PAST no console R.
- Modifique e execute o seguinte código para carregar dados LD:
LD = load_LD("path/to/LD.tsv", LD_columns = c("Locus1", "Posição1", "Site1", "Posição2", "Site2", "Dist_bp", "R.2")
NOTA: Altere o caminho para a localização real do arquivo LD. Os valores previstos para LD_columns são os valores padrão. Se os nomes não corresponderem a esses padrões, especifique os nomes corretos das colunas; caso contrário, estes podem ser omitidos.
- Modifique e execute o seguinte código para carregar dados LD:
5. Atribuir SNPs aos genes
NOTA: Baixe ou localize anotações em formato GFF. Essas anotações podem ser encontradas frequentemente em bancos de dados online para organismos específicos. Tenha cuidado com as anotações de baixa qualidade, pois a qualidade dos dados das anotações afetará a qualidade da análise da via. Confirme que a primeira coluna dessas anotações (o cromossomo) corresponde ao formato do lócus/cromossomo na associação, efeitos e dados LD. Por exemplo, as anotações não devem chamar o primeiro cromossomo de "chr1" se os arquivos de dados GWAS e LD chamarem o primeiro cromossomo de "1".
- Atribua SNPs a genes com PAST Shiny.
NOTA: Mais informações sobre a determinação de um corte R2 apropriado podem ser encontradas em Tang et al.6, na seção chamada "SNP to gene algorithm for the pathway analysis".- Selecione o arquivo contendo anotações GFF. Considere qual o tamanho da janela e o corte R2 são mais adequados para as espécies que estão sendo consideradas e modificar se os padrões não se adequarem aos dados carregados.
NOTA: Os valores padrão no PASSADO refletem principalmente os valores apropriados para o milho. O número de núcleos definidos no início da análise PAST Shiny (Passo 2.2) é usado nesta etapa.
- Selecione o arquivo contendo anotações GFF. Considere qual o tamanho da janela e o corte R2 são mais adequados para as espécies que estão sendo consideradas e modificar se os padrões não se adequarem aos dados carregados.

Figura 4. Clique aqui para ver uma versão maior desta figura.
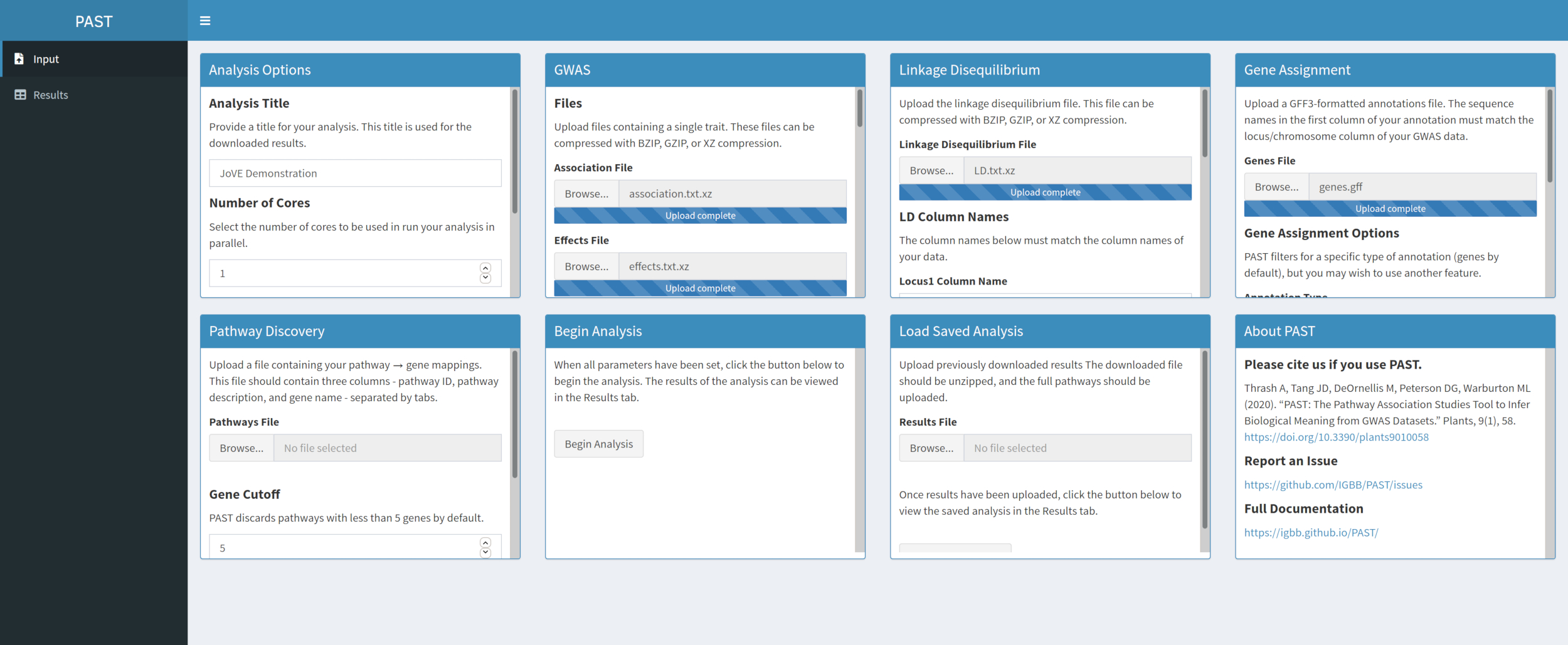{kind=link}
- Atribua SNPs a genes com PAST no console R.
- Modifique e execute o seguinte código para atribuir SNPs a genes:
genes = assign_SNPs_to_genes(gwas_data, LD, "path/to/anotações.gff", c("gene"), 1000, 0,8, 2)
NOTA: Neste código de amostra, várias sugestões padrão são fornecidas: 1000 é o tamanho da janela ao redor do SNP para procurar genes; 0.8 é o valor de corte para R2; 2 é o número de núcleos utilizados para processamento paralelo. O caminho para as anotações também deve ser alterado para a localização real do arquivo de anotações.
- Modifique e execute o seguinte código para atribuir SNPs a genes:
6. Descubra caminhos significativos
NOTA: Verifique se o arquivo pathways contém os seguintes dados em formato delimitado por guia, com uma linha para cada gene em cada caminho: Pathway ID - um identificador como "PWY-6475-1"; descrição do caminho - uma descrição mais longa do que os caminhos fazem, como "biossíntese trans-liocopeno"; gene - um gene na via, que deve coincidir com os nomes fornecidos nas anotações. As informações do caminho provavelmente podem ser encontradas em bancos de dados on-line para organismos específicos, como o MaizeGDB. A segunda opção especificada pelo usuário é o modo. "Aumentar" refere-se a fenótipos que refletem quando um valor crescente do traço medido é desejável, como o rendimento, enquanto "diminuir" refere-se a um traço onde uma diminuição nos valores medidos é benéfica, como classificações de danos de insetos. A significância das vias é testada utilizando métodos descritos anteriormente4,6,14.
- Descubra caminhos significativos com o PAST Shiny.
- Selecione o arquivo contendo dados de caminhos e certifique-se de que o modo está selecionado nas opções de análise. Se necessário, altere o número de genes que devem estar em uma via de retê-lo para a análise e o número de permutações usadas para criar a distribuição nula para testar o significado do efeito.

Figura 5. Clique aqui para ver uma versão maior desta figura.
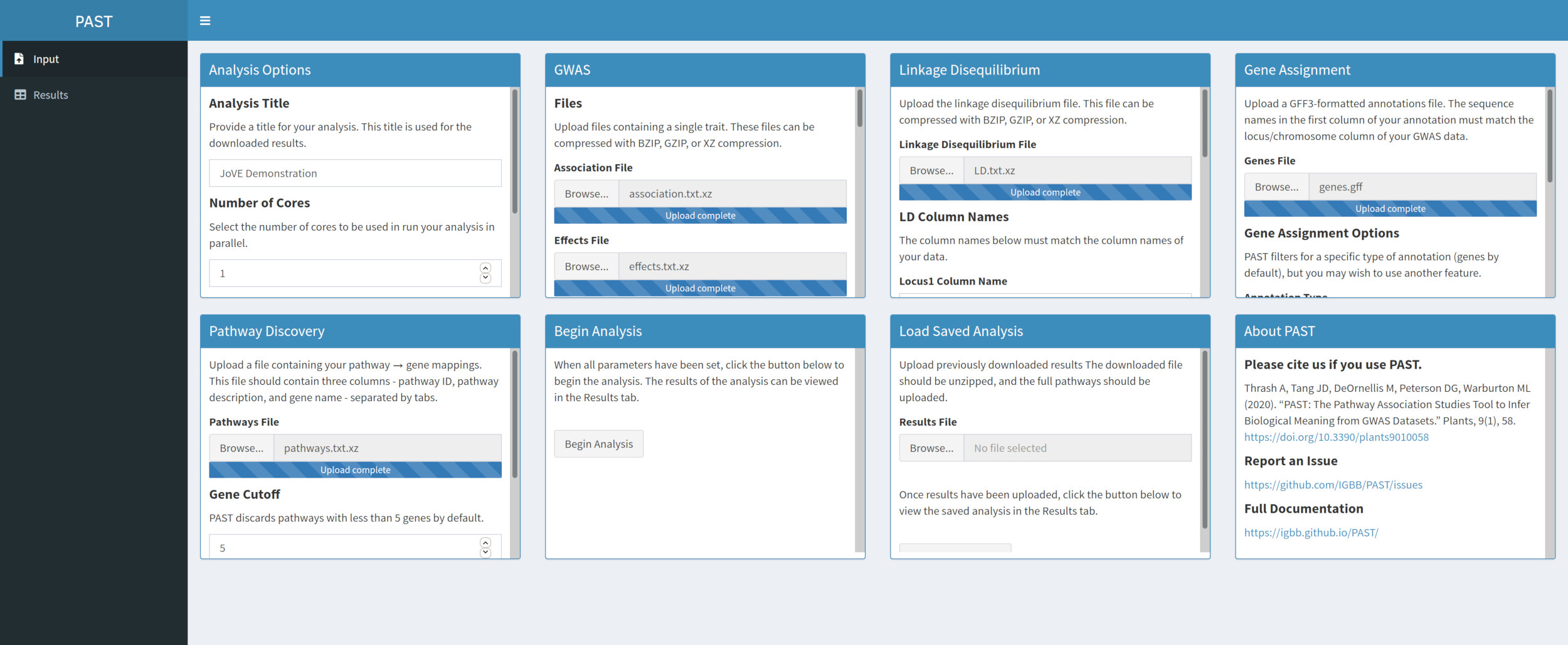{kind=link}
NOTA: O número de núcleos e o modo definido no início da análise PAST Shiny (Passo 2.2) são usados nesta etapa. O número padrão de genes é atualmente definido em 5 genes, de modo que caminhos com menos genes conhecidos serão removidos. O usuário pode reduzir esse valor para 4 ou 3, para incluir caminhos mais curtos, mas isso correrá o risco de resultados falsos positivos. O aumento desse valor pode aumentar o poder da análise, mas removerá mais caminhos da análise. A alteração do número de permutações utilizadas aumenta e diminui a potência do teste.
- Descubra caminhos significativos com o PAST no Console R.
- Modifique e execute o seguinte código para descobrir caminhos significativos:
rugplots_data <- find_pathway_significance(genes, "path/to/pathways.tsv", 5, "increasing", 1000, 2)
NOTA: Neste código de amostra, vários padrões sugeridos são fornecidos. 5 é o número mínimo de genes que devem estar em um caminho para manter o caminho na análise, aumentando refere-se a uma quantidade crescente do traço medido (recomenda-se que o usuário execute tanto aumentando quanto diminuindo, independentemente da característica; a interpretação dos dados difere para os dois, no entanto), 1000 é o número de vezes para provar os efeitos para determinar a distribuição nula, e 2 é o número de núcleos usados para processamento paralelo. Altere o caminho para a localização real do arquivo pathways.
- Modifique e execute o seguinte código para descobrir caminhos significativos:
7. Veja rugplots
- Veja Rugplots com PAST Shiny.
- Uma vez que todas as entradas sejam carregadas e definidas, clique em Iniciar Análise. Uma barra de progresso aparecerá e indicará qual etapa da análise foi concluída pela última vez. Quando a análise for concluída, o PAST Shiny mudará para a guia Resultados. Uma tabela de resultados será exibida na coluna esquerda (rotulada como "caminhos") e os Rugplots serão exibidos na coluna direita (rotulados como "parcelas").
- Use o controle deslizante para controlar os parâmetros de filtragem. Quando o nível de filtragem for satisfatório, clique no botão Resultados de Download na parte inferior esquerda para baixar todas as imagens e tabelas individualmente para um arquivo ZIP que é nomeado com o título de análise. Este arquivo ZIP contém a tabela filtrada, a tabela não filtrada e uma imagem por caminho na tabela filtrada.

Figura 6. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 7. Clique aqui para ver uma versão maior desta figura.
{kind=link}
- Ver rugplots com PAST no console R
- Modifique e execute o seguinte código para salvar os resultados:
plot_pathways(rugplots_data, "pvalue", 0,02, "aumentando", "output_folder")
NOTA: Neste código de amostra, vários padrões sugeridos são fornecidos. pvalue fornece os dados que podem ser usados para filtrar caminhos insignificantes após um limiar de significância ser escolhido pelo usuário; 0.02 é o valor padrão utilizado na filtragem, e o aumento refere-se a uma quantidade crescente do traço medido (recomenda-se que o usuário execute tanto aumentando quanto diminuindo, independentemente da característica; a interpretação dos dados difere para os dois, no entanto); output_folder é a pasta onde as imagens e tabelas serão escritas (esta pasta deve existir antes de executar a função). Uma tabela de resultados filtrados, os resultados não filtrados e imagens individuais para cada caminho nos resultados filtrados são escritos para esta pasta.
- Modifique e execute o seguinte código para salvar os resultados:
Resultados
Se os resultados não forem produzidos após uma execução da ferramenta de software PAST, verifique se todos os arquivos de entrada estão corretamente formatados. Uma execução bem-sucedida usando os dados de exemplo no pacote PAST, que são baseados em um GWAS de milho de cor de grão, é mostrado na Figura 8. Esta tabela e a imagem resultante podem ser baixadas usando o botão Download Results. Um exemplo da imagem baixada é mostrado na Figura 210. Configurações incorretas podem levar a resultados que não fazem sentido biológico, mas determinar a incorreção de incorreção deve ser do pesquisador, que deve verificar a validade das configurações escolhidas e considerar todas as evidências conhecidas sobre o traço de interesse.
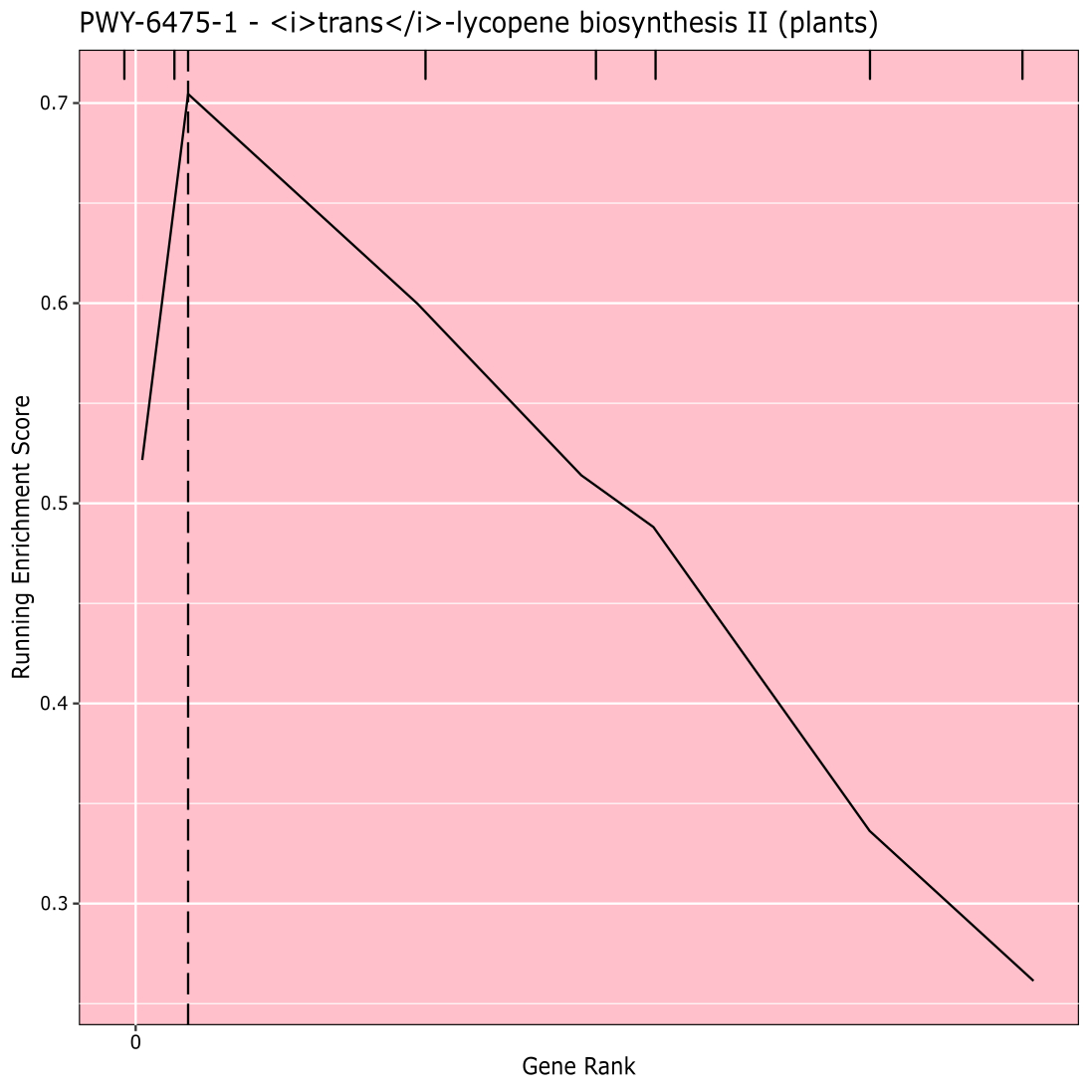
A Figura 910 mostra o rugplot produzido a partir da análise de caminhos dos resultados gwas criados com um painel de milho de 288 linhas de raça que haviam sido fenotipadas para a cor do grão. Este exemplo simplista, onde os fenótipos eram "brancos" ou "amarelos", foi utilizado porque o caminho responsável pela criação dos pigmentos carotenoides amarelos brilhantes é conhecido e deve ser responsável pela maior parte do fenótipo. Assim, esperávamos ver que a via de biossíntese trans-liocopeno (que produz carotenoides) está significativamente associada à cor do grão, que é. O ID do caminho e o nome estão listados no topo do gráfico. O eixo horizontal do gráfico classifica todos os genes que foram incluídos na análise, dispostos da esquerda para a direita na ordem de maior efeito sobre o traço para o menor. No entanto, apenas os genes na via de biossíntese trans-liocopeno são marcados (no topo do gráfico, como marcas de escotilha, aparecendo no grau genético de seu efeito em comparação com todos os outros genes da análise). Há 7 genes neste caminho. A pontuação de enriquecimento em execução (ES) é plotada ao longo do eixo vertical. O ES para cada gene é adicionado ao total em execução por ordem de efeito e o total é ajustado ao número de genes analisados. Assim, a pontuação muda à medida que se move ao longo do eixo horizontal e tende a aumentar à medida que os genes de efeito maior são incluídos, mas em algum momento, o aumento do efeito é menor do que o ajuste por ter adicionado outro gene, e toda a pontuação começa a diminuir. O ápice da linha ES em execução é marcado com uma linha vertical pontilhada; este é o ES para todo o caminho e é usado pelo programa para determinar se o caminho é escolhido e apresentado como um rugplot.

Figura 8: Execução completa do PAST Shiny. Por favor, clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 9: Imagem de caminho a partir de execução completa de PAST (ou baixado de Shiny). Este valor foi citado por Thrash et al.10. Clique aqui para ver uma versão maior desta figura.
{kind=link}
Discussão
Um objetivo primário do PAST é levar as análises metabólicas das vias metabólicas dos dados do GWAS para um público mais amplo, especialmente para organismos não humanos e não animais. Métodos alternativos ao PAST são frequentemente programas de linha de comando que se concentram em humanos ou animais. A simpatia do usuário foi um objetivo principal no desenvolvimento do PAST, tanto na escolha de desenvolver um aplicativo Shiny quanto na escolha de usar R e Bioconductor para liberar o aplicativo. Os usuários não precisam aprender a compilar programas para usar o PAST.
Como na maioria dos tipos de software de análise, os resultados do PAST são tão bons quanto os dados de entrada; se os dados de entrada tiver erros ou forem formatados incorretamente, o PAST falhará em executar ou produzir resultados não informativos. Garantir que os dados, dados LD, anotações e caminhos do GWAS sejam corretamente formatados é fundamental para receber a saída correta do PAST. O PAST analisa apenas marcadores bieélicos e pode executar apenas uma característica para cada conjunto de dados de entrada. Além disso, os dados GWAS produzidos por mau genotipagem ou fenotipagem incorreta ou imprecisa também não produzem resultados claros ou repetíveis. O PAST pode auxiliar na interpretação biológica dos resultados do GWAS, mas é improvável que esclareça conjuntos de dados caóticos se a variação ambiental, o erro experimental ou a estrutura populacional não foram devidamente contabilizados.
Os usuários podem optar por alterar alguns parâmetros da análise, tanto no aplicativo Shiny quanto passando esses parâmetros para as funções do PAST no console R. Esses parâmetros podem alterar os resultados relatados pelo PAST, e os usuários devem tomar cuidado ao modificá-los dos padrões. Como o LD é medido pelos usuários, normalmente usando o mesmo conjunto de dados marcador que também foi usado no GWAS, as medidas de LD são específicas para a população. Para todos os estudos, especialmente para espécies que não sejam o milho, (particularmente espécies auto-polinizadora, poliploide ou altamente heterogênios), mudanças nos padrões podem ser justificadas.
Divulgações
Os autores não têm nada a revelar.
Agradecimentos
Nenhum.
Materiais
| Name | Company | Catalog Number | Comments |
| Computer | NA | NA | Any computer with 8GB RAM should be sufficient |
| R | R Project | NA | R 4.0 or greater is required to install from Bioconductor 3.11 |
Referências
- Rafalski, J. Association genetics in crop improvement. Current Opinion in Plant Biology. 13 (2), 174-180 (2010).
- Yan, J., Warburton, M., Crouch, J. Association Mapping for Enhancing Maize (Zea mays L.) Genetic Improvement. Crop Science. 51 (2), 433-449 (2011).
- Xiao, Y., Liu, H., Wu, L., Warburton, M., Yan, J. Genome-wide Association Studies in Maize: Praise and Stargaze. Molecular Plant. 10 (3), 359-374 (2017).
- Wang, K., Li, M., Bucan, M. Pathway-Based Approaches for Analysis of Genomewide Association Studies. The American Journal of Human Genetics. 81 (6), 1278-1283 (2007).
- Weng, L., et al. SNP-based pathway enrichment analysis for genome-wide association studies. BMC Bioinformatics. 12 (1), 99(2011).
- Tang, J., Perkins, A., Williams, W., Warburton, M. Using genome-wide associations to identify metabolic pathways involved in maize aflatoxin accumulation resistance. BMC Genomics. 16 (1), 673(2015).
- Warburton, M., et al. Genome-Wide Association Mapping of Aspergillus flavus and Aflatoxin Accumulation Resistance in Maize. Crop Science. 55 (5), 1857-1867 (2015).
- Warburton, M., et al. Genome-Wide Association and Metabolic Pathway Analysis of Corn Earworm Resistance in Maize. The Plant Genome. 11 (1), 170069(2018).
- Li, H., Thrash, A., Tang, J., He, L., Yan, J., Warburton, M. Leveraging GWAS data to identify metabolic pathways and networks involved in maize lipid biosynthesis. The Plant Journal. 98 (5), 853-863 (2019).
- Thrash, A., Tang, J., DeOrnellis, M., Peterson, D., Warburton, M. PAST: The Pathway Association Studies Tool to Infer Biological Meaning from GWAS Datasets. Plants. 9 (1), 58(2020).
- Adam, T., Mason, D. PAST: Pathway Association Study Tool (PAST). Bioconductor version: Release (3.10). , (2020).
- Thrash, A., DeOrnellis, M. IGBB/PAST. , at https://github.com/IGBB/PAST (2019).
- Bradbury, P., et al. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 23 (19), 2633-2635 (2007).
- Subramanian, A., et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences U.S.A. 102, 15545-15550 (2005).
Erratum
Formal Correction: Erratum: A Pathway Association Study Tool for GWAS Analyses of Metabolic Pathway Information
Posted by JoVE Editors on 10/08/2021. Citeable Link.
An erratum was issued for: A Pathway Association Study Tool for GWAS Analyses of Metabolic Pathway Information. One of the affiliations was updated.
The second affiliation was updated from:
USDA-ARS Corn Host Plant Resistance Research Unit, Mississippi State University
to:
Corn Host Plant Resistance Research Unit, USDA-ARS
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados