
Method Article
Uno strumento di studio della Pathway Association per le analisi GWAS delle informazioni sulle vie metaboliche
In questo articolo
Erratum Notice
Riepilogo
Eseguendo il Pathway Association Study Tool (PAST), attraverso l'applicazione Shiny o attraverso la console R, i ricercatori possono acquisire una comprensione più profonda del significato biologico dei risultati del loro studio di associazione genome-wide (GWAS) studiando le vie metaboliche coinvolte.
Abstract
Recentemente, è stata sviluppata e rilasciata una nuova implementazione di un metodo precedentemente descritto per interpretare i dati dello studio di associazione genome-wide (GWAS) utilizzando l'analisi delle vie metaboliche. Il Pathway Association Study Tool (PAST) è stato sviluppato per affrontare le preoccupazioni relative alla facilità d'uso e alle analisi a esecuzione lenta. Questo nuovo strumento user-friendly è stato rilasciato su Bioconductor e Github. Nei test, PAST ha eseguito analisi in meno di un'ora che in precedenza richiedevano ventiquattro o più ore. In questo articolo, presentiamo il protocollo per l'utilizzo dell'applicazione Shiny o della console R per eseguire PAST.
Introduzione
Gli studi di associazione genome-wide (GWAS) sono un metodo popolare per studiare tratti complessi e le regioni genomiche ad essi associate1,2,3. In questo tipo di studio, centinaia di migliaia di marcatori di polimorfismo a singolo nucleotide (SNP) vengono testati per la loro associazione con il tratto e viene valutato il significato delle associazioni. Le associazioni marcatore-tratto che soddisfano la soglia del tasso di falsa scoperta (FDR) (o qualche altro tipo di soglia di significatività) vengono mantenute per lo studio, ma le associazioni vere possono essere filtrate. Per tratti complessi e poligenici, l'effetto di ciascun gene potrebbe essere piccolo (e quindi filtrato), e alcuni alleli sono espressi solo in condizioni specifiche che potrebbero non essere presenti nello studio3. Pertanto, mentre molti SNP possono essere mantenuti come associati al tratto, ognuno può avere un effetto molto piccolo. Mancheranno troppe chiamate SNP e un'interpretazione del significato biologico e dell'architettura genetica del tratto potrebbe essere incompleta e confusa. L'analisi delle vie metaboliche può aiutare ad affrontare alcuni di questi problemi concentrandosi sugli effetti combinati di geni raggruppati in base alla loro funzione biologica4,5,6.
Diversi studi sono stati completati utilizzando una precedente implementazione del metodo descritto in questo articolo. L'accumulo di aflatossina7,la resistenza del verme dell'orecchio del mais8e la biosintesi dell'olio9 sono stati tutti studiati con l'implementazione precedente. Mentre queste analisi hanno avuto successo, il processo di analisi è stato complicato, dispendioso in termini di tempo e ingombrante, perché gli strumenti di analisi sono stati scritti in una combinazione di R, Perl e Bash e la pipeline non è stata automatizzata. A causa delle conoscenze specialistiche necessarie per modificare questo metodo per ogni analisi, è stato ora sviluppato un nuovo metodo che può essere condiviso con altri ricercatori.
Il Pathway Association Study Tool (PAST)10 è stato progettato per affrontare le carenze del metodo precedente richiedendo una minore conoscenza dei linguaggi di programmazione ed eseguendo analisi in un periodo più breve. Mentre il metodo è stato testato con il mais, PAST non fa ipotesi specie-specifiche. PAST può essere eseguito attraverso la console R, come app Shiny, e una versione online dovrebbe essere presto disponibile su MaizeGDB.
Protocollo
1. Configurazione
- Installare R, se non è già installato.
NOTA: PAST è scritto in R e, pertanto, richiede che i suoi utenti abbiano installato R. Al momento della stesura di questo documento, l'installazione di PAST direttamente da Bioconductor richiede R4.0. Le versioni precedenti di PAST possono essere installate da Bioconductor per R3.6 e PAST può essere installato da Github per gli utenti con R3.5. Le istruzioni per l'installazione di R possono essere scaricate dal seguente link: https://www.r-project.org/. - Installare la versione più recente di RStudio Desktop o aggiornare RStudio (facoltativo).
NOTA: RStudio è un ambiente utile per lavorare con il linguaggio R. La sua installazione è consigliata, soprattutto per coloro che scelgono di eseguire PAST nella riga di comando piuttosto che attraverso l'applicazione Shiny GUI. RStudio e le sue istruzioni di installazione sono disponibili al seguente link: https://rstudio.com/products/rstudio/. - Installare PAST da Bioconductor11 seguendo le istruzioni su Bioconductor.
NOTA: l'installazione tramite Bioconductor dovrebbe gestire l'installazione delle dipendenze di PAST. Inoltre, PAST può essere installato da Github12, ma l'installazione da Github non installerà automaticamente le dipendenze. - Installare PAST Shiny (opzionale). Scarica il file "app. R" dalla pagina Rilasci del repository Github: https://github.com/IGBB/PAST/releases/ e ricorda dove si trova il file scaricato.
NOTA: PAST può essere utilizzato chiamando i suoi metodi direttamente con R, ma gli utenti che hanno meno familiarità con R possono eseguire l'applicazione PAST Shiny, che fornisce un'interfaccia utente guidata. PAST Shiny è uno script R disponibile nel ramo shiny_app del repository Github PAST. PAST Shiny tenterà di installare le sue dipendenze durante la prima esecuzione. - Iniziare l'analisi avviando l'applicazione in uno dei tre modi descritti di seguito.
- PAST Shiny con RStudio
- Utilizzando RStudio, crea un nuovo progetto nella cartella in cui si trova l'app. R si trova. Fare clic su File | Nuovo progetto e selezionare la cartella.
- Una volta creato un nuovo progetto, apri l'app. R scaricato in precedenza. RStudio riconosce l'app. R è un'app Shiny e crea un pulsante Esegui app sulla barra sopra il codice sorgente visualizzato. Fare clic su Esegui app. RStudio avvierà quindi una finestra che visualizza l'applicazione PAST Shiny.
- PAST Shiny con console R
- Avvia R ed esegui il seguente codice per avviare l'applicazione PAST Shiny: shiny::runApp('path/to/folder/with/shiny/app. R'. Sostituisci il testo tra virgolette con la cartella in cui l'app. R è stato scaricato e conserva le citazioni.
- PASSATO senza R Shiny
- Eseguire library(PAST) in una console R per caricare PAST.
- PAST Shiny con RStudio
2. Personalizza l'analisi Shiny (opzionale)
- Modificare il titolo dell'analisi da "Nuova analisi" a qualcosa che rifletta meglio il tipo di analisi in esecuzione che aiuta a tenere traccia di più analisi (vedere la Figura 1).

Figura 1. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
- Modificare il numero di core e la modalità. Impostare il numero di core su qualsiasi numero compreso tra 1 e il numero totale sulla macchina, ma tenere presente che dedicare più risorse a PAST può rallentare altre operazioni sulla macchina. Impostare la modalità in base alla descrizione nella sezione 6.
3. Carica i dati GWAS
NOTA: verificare che i dati GWAS siano delimitati da tabulazioni. Assicurarsi che il file di associazione contenga le seguenti colonne: tratto, nome del marcatore, locus o cromosoma, posizione sul cromosoma, valore p e valore R2 per il marcatore. Assicurarsi che il file degli effetti contenga le seguenti colonne: tratto, nome del marcatore, locus o cromosoma, posizione sul cromosoma ed effetto. L'ordine di queste colonne non è importante, in quanto l'utente può specificare i nomi delle colonne durante il caricamento dei dati. Eventuali colonne aggiuntive vengono ignorate. TASSEL13 può essere utilizzato per produrre questi file.
- Carica i dati GWAS con PAST Shiny.
- Selezionate un file di associazione e un file di effetti utilizzando le caselle di selezione File di associazione e File di effetti. Modificare i nomi delle colonne nelle caselle di input Nome colonna di associazione e Nome colonne effetti sotto le caselle di selezione dei file in modo da riflettere i nomi delle colonne nei dati.

Figura 2. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
- Caricare i dati GWAS con PAST nella console R.
- Modificare ed eseguire il codice seguente:
gwas_data = load_GWAS_data("path/to/association_file.tsv", "path/to/effects_file.tsv", association_columns = c("Trait", "Marker", "Locus", "Site", "p", "marker_R2"), effects_columns = c("Trait", "Marker", "Locus", "Site", "Effect")
- Modificare ed eseguire il codice seguente:
- NOTA: modificare i percorsi della posizione effettiva dei file GWAS. I valori forniti per association_columns e effects_columns sono i valori predefiniti. Se i nomi non corrispondono ai valori predefiniti, specificare i nomi delle colonne. Altrimenti, questi possono essere omessi.
4. Dati di squilibrio del collegamento di carico (LD)
NOTA: verificare che i dati di squilibrio di collegamento (LD) siano delimitati da tabulazioni e contengano i seguenti tipi di dati: Locus, Position1, Site1, Position2, Site2, Distance in base pairs between Position1 e Position2 e R2 value.
- Carica i dati LD con PAST Shiny.
- Selezionare il file contenente i dati LD. Modificare i nomi delle colonne nelle caselle di input Nomi colonna LD sotto la casella di selezione dei file in modo che corrispondano ai nomi delle colonne nei dati LD, se necessario.

Figura 3. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
- Caricare i dati LD con PAST nella console R.
- Modificare ed eseguire il codice seguente per caricare i dati LD:
LD = load_LD("path/to/LD.tsv", LD_columns = c("Locus1", "Position1", "Site1", "Position2", "Site2", "Dist_bp", "R.2")
NOTA: modificare il percorso del percorso effettivo del file LD. I valori forniti per LD_columns sono i valori predefiniti. Se i nomi non corrispondono a questi valori predefiniti, specificare i nomi corretti delle colonne; in caso contrario, questi possono essere omessi.
- Modificare ed eseguire il codice seguente per caricare i dati LD:
5. Assegna SNP ai geni
NOTA: scaricare o individuare in altro modo le annotazioni in formato GFF. Queste annotazioni possono spesso essere trovate in database online per organismi specifici. Prestare attenzione alle annotazioni di bassa qualità, poiché la qualità dei dati delle annotazioni influenzerà la qualità dell'analisi del percorso. Verificare che la prima colonna di queste annotazioni (il cromosoma) corrisponda al formato del locus/cromosoma nei dati di associazione, effetti e LD. Ad esempio, le annotazioni non dovrebbero chiamare il primo cromosoma "chr1" se i file di dati GWAS e LD chiamano il primo cromosoma "1".
- Assegna SNP ai geni con PAST Shiny.
NOTA: Maggiori informazioni sulla determinazione di un cutoff R2 appropriato possono essere trovate in Tang et al.6, nella sezione chiamata "Algoritmo SNP to gene per l'analisi del percorso".- Selezionare il file contenente le annotazioni GFF. Considera quali dimensioni della finestra e il cutoff R2 sono più adatti per la specie considerata e modifica se i valori predefiniti non si adattano ai dati caricati.
NOTA: i valori predefiniti in PAST riflettono principalmente i valori appropriati per il mais. In questo passaggio viene utilizzato il numero di core impostato all'inizio dell'analisi PAST Shiny (Passaggio 2.2).
- Selezionare il file contenente le annotazioni GFF. Considera quali dimensioni della finestra e il cutoff R2 sono più adatti per la specie considerata e modifica se i valori predefiniti non si adattano ai dati caricati.

Figura 4. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
- Assegna SNP ai geni con PAST nella console R.
- Modificare ed eseguire il codice seguente per assegnare SNP ai geni:
genes = assign_SNPs_to_genes(gwas_data, LD, "path/to/annotations.gff", c("gene"), 1000, 0.8, 2)
NOTA: in questo codice di esempio vengono forniti diversi suggerimenti predefiniti: 1000 è la dimensione della finestra intorno all'SNP per la ricerca dei geni; 0,8 è il valore limite per R2; 2 è il numero di core utilizzati per l'elaborazione parallela. Anche il percorso delle annotazioni deve essere modificato nella posizione effettiva del file delle annotazioni.
- Modificare ed eseguire il codice seguente per assegnare SNP ai geni:
6. Scopri percorsi significativi
NOTA: verificare che il file pathways contenga i seguenti dati in formato delimitato da tabulazioni, con una riga per ogni gene in ogni pathway: pathway ID - un identificatore come "PWY-6475-1"; descrizione del percorso - una descrizione più lunga di ciò che i percorsi fanno come la "biosintesi trans-licopene"; gene - un gene nel percorso, che dovrebbe corrispondere ai nomi forniti nelle annotazioni. Le informazioni sul percorso possono probabilmente essere trovate in database online per organismi specifici, come MaizeGDB. La seconda opzione specificata dall'utente è la modalità. "Crescente" si riferisce a fenotipi che riflettono quando è desiderabile un valore crescente del tratto misurato, come la resa, mentre "decrescente" si riferisce a un tratto in cui una diminuzione dei valori misurati è benefica, come le valutazioni dei danni agli insetti. Il significato dei percorsi viene testato utilizzando i metodi precedentemente descritti4,6,14.
- Scopri percorsi significativi con PAST Shiny.
- Selezionate il file contenente i dati dei percorsi e assicuratevi che la modalità sia selezionata nelle opzioni di analisi. Se necessario, modificare il numero di geni che devono essere in un percorso per conservarlo per l'analisi e il numero di permutazioni utilizzate per creare la distribuzione nulla per testare il significato dell'effetto.

Figura 5. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
NOTA: in questo passaggio vengono utilizzati il numero di core e la modalità impostata all'inizio dell'analisi PAST Shiny (passaggio 2.2). Il numero predefinito di geni è attualmente impostato su 5 geni, quindi i percorsi con meno geni noti verranno rimossi. L'utente può abbassare questo valore a 4 o 3, per includere percorsi più brevi, ma così facendo rischierà risultati falsi positivi. L'aumento di questo valore può aumentare la potenza dell'analisi, ma rimuoverà più percorsi dall'analisi. La modifica del numero di permutazioni utilizzate aumenta e diminuisce la potenza del test.
- Scopri percorsi significativi con PAST nella console R.
- Modificare ed eseguire il codice seguente per individuare percorsi significativi:
rugplots_data <- find_pathway_significance(geni, "path/to/pathways.tsv", 5, "crescente", 1000, 2)
Nota : in questo codice di esempio, vengono fornite diverse impostazioni predefinite suggerite. 5 è il numero minimo di geni che devono essere in un percorso per mantenere il percorso nell'analisi, aumentando si riferisce a una quantità crescente del tratto misurato (si raccomanda che l'utente esegua sia aumentando che diminuendo, indipendentemente dal tratto; l'interpretazione dei dati sarà diversa per i due, tuttavia), 1000 è il numero di volte per campionare gli effetti per determinare la distribuzione nulla, e 2 è il numero di core utilizzati per l'elaborazione parallela. Modificare il percorso del percorso effettivo del file dei percorsi.
- Modificare ed eseguire il codice seguente per individuare percorsi significativi:
7. Visualizza Rugplots
- Visualizza Rugplots con PAST Shiny.
- Una volta caricati e impostati tutti gli input, fare clic su Inizia analisi. Verrà visualizzata una barra di avanzamento che indicherà quale fase dell'analisi è stata completata l'ultima volta. Al termine dell'analisi, PAST Shiny passa alla scheda Risultati. Una tabella dei risultati verrà visualizzata nella colonna di sinistra (etichettata "pathways") e i Rugplots verranno visualizzati nella colonna di destra (etichettati "plots").
- Utilizzare il dispositivo di scorrimento per controllare i parametri di filtro. Quando il livello di filtraggio è soddisfacente, fare clic sul pulsante Scarica risultati in basso a sinistra per scaricare tutte le immagini e le tabelle singolarmente in un file ZIP denominato con il titolo dell'analisi. Questo file ZIP contiene la tabella filtrata, la tabella non filtrata e un'immagine per percorso nella tabella filtrata.

Figura 6. Fare clic qui per visualizzare una versione più grande di questa figura.
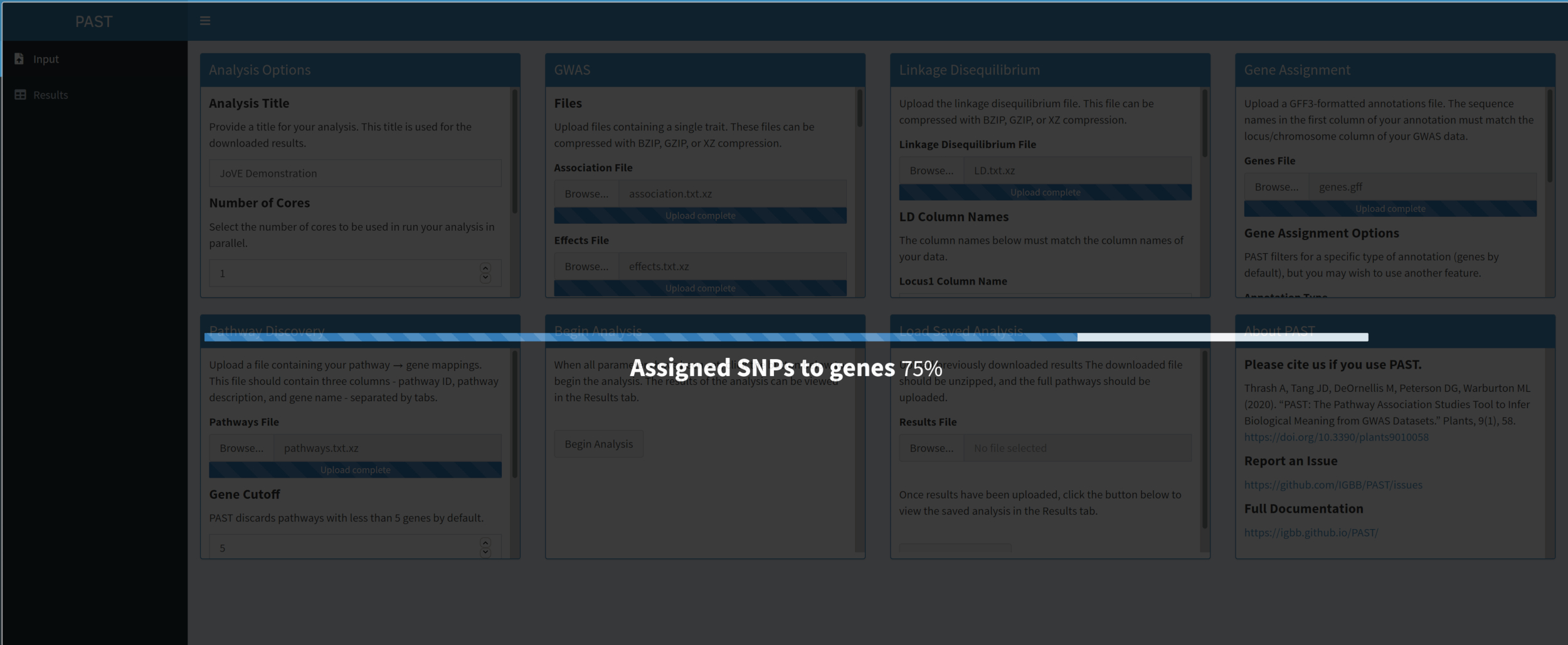{kind=link}

Figura 7. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
- Visualizza i tappeti con PAST nella console R
- Modificare ed eseguire il codice seguente per salvare i risultati:
plot_pathways(rugplots_data, "pvalue", 0.02, "crescente", "output_folder")
Nota : in questo codice di esempio, vengono fornite diverse impostazioni predefinite suggerite. pvalue fornisce i dati che possono essere utilizzati per filtrare percorsi insignificanti dopo che una soglia di significatività è stata scelta dall'utente; 0.02 è il valore predefinito utilizzato nel filtraggio e l'aumento si riferisce a una quantità crescente del tratto misurato (si consiglia all'utente di eseguire sia aumentando che diminuendo, indipendentemente dal tratto; l'interpretazione dei dati sarà diversa per i due, tuttavia); output_folder è la cartella in cui verranno scritte le immagini e le tabelle (questa cartella deve esistere prima di eseguire la funzione). In questa cartella vengono scritti una tabella di risultati filtrati, i risultati non filtrati e le singole immagini per ogni percorso nei risultati filtrati.
- Modificare ed eseguire il codice seguente per salvare i risultati:
Risultati
Se i risultati non vengono prodotti a seguito di un'esecuzione dello strumento software PAST, verificare che tutti i file di input siano formattati correttamente. Un'esecuzione riuscita utilizzando i dati di esempio nel pacchetto PAST, che si basano su un GWAS di mais di colore granuloso, è mostrata nella Figura 8. Questa tabella e l'immagine risultante possono essere scaricate utilizzando il pulsante Scarica risultati. Un esempio dell'immagine scaricata è mostrato nella Figura 210. Impostazioni errate potrebbero portare a risultati che non hanno senso biologico, ma determinare l'inesattezza deve spettare al ricercatore, che dovrebbe ricontrollare la validità delle impostazioni scelte e considerare tutte le prove conosciute riguardanti il tratto di interesse.
La Figura 910 mostra il tappeto prodotto dall'analisi del percorso dei risultati GWAS creati con un pannello di mais di 288 linee inbred che erano state fenotipizzate per il colore del grano. Questo esempio semplicistico, in cui i fenotipi erano "bianchi" o "gialli", è stato usato perché il percorso responsabile della creazione dei pigmenti carotenoidi gialli brillanti è noto e dovrebbe essere responsabile della maggior parte del fenotipo. Pertanto, ci aspettavamo di vedere la via della biosintesi trans-licopene (che produce carotenoidi) essere significativamente associata al colore del grano, che è. L'ID e il nome del percorso sono elencati nella parte superiore del grafico. L'asse orizzontale del grafico classifica tutti i geni che sono stati inclusi nell'analisi, disposti da sinistra a destra in ordine di effetto maggiore sul tratto al più piccolo. Tuttavia, solo i geni nella via di biosintesi trans-licopene sono marcati (nella parte superiore del grafico, come segni di tratteggio, che appaiono nel rango genico del loro effetto rispetto a tutti gli altri geni nell'analisi). Ci sono 7 geni in questo percorso. Il punteggio di arricchimento in esecuzione (ES) viene tracciato lungo l'asse verticale. L'ES per ciascun gene viene aggiunto al totale corrente in ordine di effetto e il totale viene regolato in base al numero di geni analizzati. Pertanto, il punteggio cambia man mano che ci si muove lungo l'asse orizzontale e tende ad aumentare man mano che vengono inclusi i geni dell'effetto più grandi, ma ad un certo punto, l'aumento dell'effetto è inferiore all'aggiustamento per aver aggiunto un altro gene e l'intero punteggio inizia a diminuire. L'apice della linea ES in esecuzione è contrassegnato da una linea verticale tratteggiata; questo è l'ES per l'intero percorso e viene utilizzato dal programma per determinare se il percorso viene scelto e presentato come un tappeto.

Figura 8: Esecuzione completata di PAST Shiny. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 9: Immagine del percorso dall'esecuzione completata di PAST (o scaricata da Shiny). Questa cifra è stata citata da Thrash et al.10. Fare clic qui per visualizzare una versione più grande di questa figura.
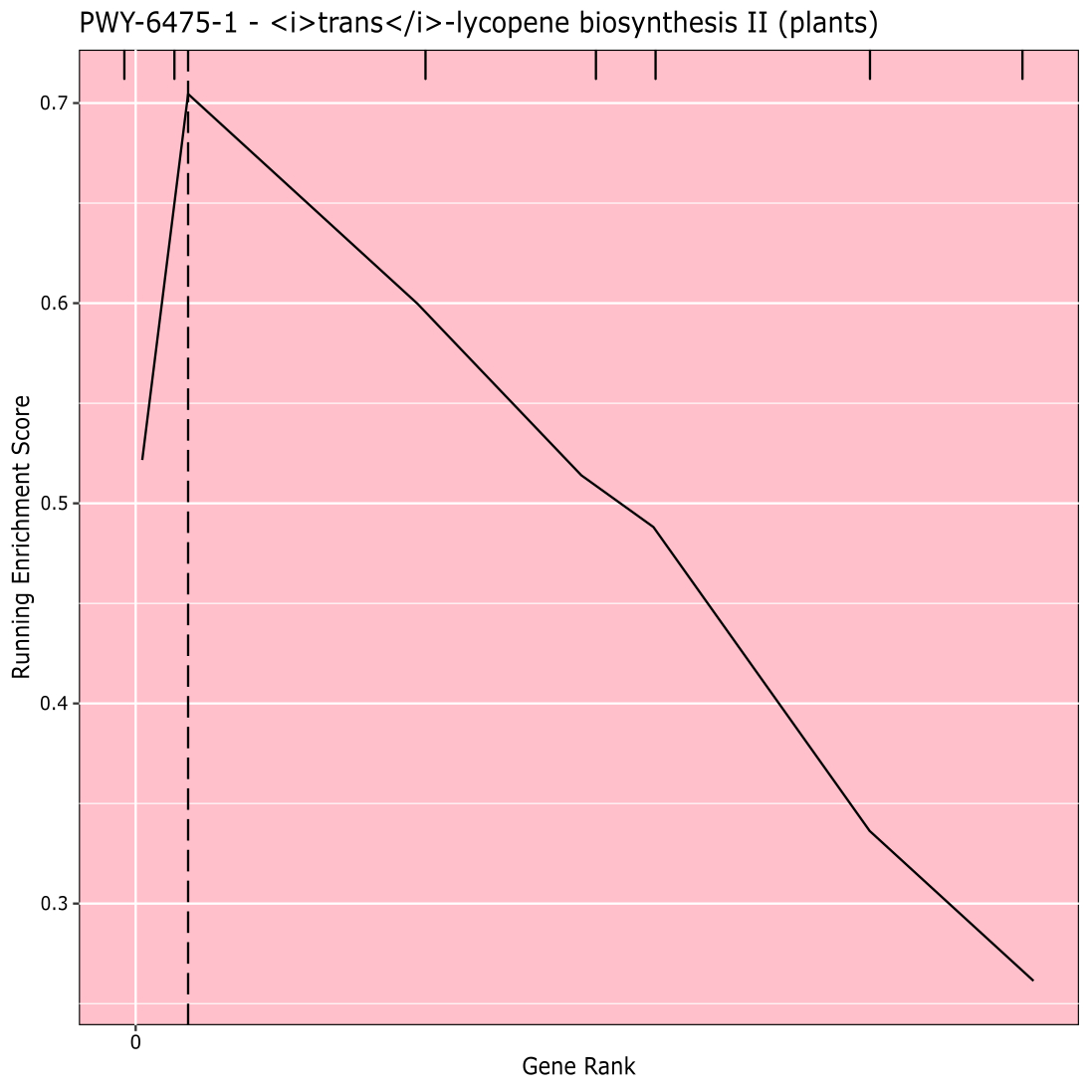{kind=link}
Discussione
Un obiettivo primario di PAST è quello di portare le analisi delle vie metaboliche dei dati GWAS a un pubblico più ampio, in particolare per gli organismi non umani e non animali. I metodi alternativi a PAST sono spesso programmi a riga di comando che si concentrano su esseri umani o animali. La facilità d'uso è stato un obiettivo primario nello sviluppo di PAST, sia nella scelta di sviluppare un'applicazione Shiny sia nella scelta di utilizzare R e Bioconductor per rilasciare l'applicazione. Gli utenti non hanno bisogno di imparare a compilare programmi per utilizzare PAST.
Come con la maggior parte dei tipi di software di analisi, i risultati di PAST sono buoni solo quanto i dati di input; se i dati di input ingessero errori o fossero formattati in modo errato, PAST non verrà eseguito o produrrà risultati non informativi. Garantire che i dati GWAS, i dati LD, le annotazioni e i file pathways siano formattati correttamente è fondamentale per ricevere l'output corretto da PAST. PAST analizza solo i marcatori biallici e può eseguire un solo tratto per ogni set di dati di input. Inoltre, i dati GWAS prodotti da una genotipizzazione scadente o da una fenotipizzazione errata o imprecisa non sono suscettibili di produrre risultati chiari o ripetibili. PAST può aiutare nell'interpretazione biologica dei risultati GWAS, ma è improbabile che chiarisca set di dati caotici se la variazione ambientale, l'errore sperimentale o la struttura della popolazione non sono stati adeguatamente contabilati.
Gli utenti possono scegliere di modificare alcuni parametri dell'analisi, sia nell'applicazione Shiny che passando tali parametri alle funzioni di PAST nella console R. Questi parametri possono modificare i risultati riportati da PAST e gli utenti devono fare attenzione quando li modificano dai valori predefiniti. Poiché LD viene misurato dagli utenti, in genere utilizzando lo stesso set di dati marcatori utilizzato anche nel GWAS, le misurazioni LD sono specifiche per la popolazione. Per tutti gli studi, in particolare per le specie diverse dal mais (in particolare le specie autoimpollinanti, poliploidi o altamente eterogenee), possono essere giustificati cambiamenti nelle impostazioni predefinite.
Divulgazioni
Gli autori non hanno nulla da rivelare.
Riconoscimenti
Nessuno.
Materiali
| Name | Company | Catalog Number | Comments |
| Computer | NA | NA | Any computer with 8GB RAM should be sufficient |
| R | R Project | NA | R 4.0 or greater is required to install from Bioconductor 3.11 |
Riferimenti
- Rafalski, J. Association genetics in crop improvement. Current Opinion in Plant Biology. 13 (2), 174-180 (2010).
- Yan, J., Warburton, M., Crouch, J. Association Mapping for Enhancing Maize (Zea mays L.) Genetic Improvement. Crop Science. 51 (2), 433-449 (2011).
- Xiao, Y., Liu, H., Wu, L., Warburton, M., Yan, J. Genome-wide Association Studies in Maize: Praise and Stargaze. Molecular Plant. 10 (3), 359-374 (2017).
- Wang, K., Li, M., Bucan, M. Pathway-Based Approaches for Analysis of Genomewide Association Studies. The American Journal of Human Genetics. 81 (6), 1278-1283 (2007).
- Weng, L., et al. SNP-based pathway enrichment analysis for genome-wide association studies. BMC Bioinformatics. 12 (1), 99(2011).
- Tang, J., Perkins, A., Williams, W., Warburton, M. Using genome-wide associations to identify metabolic pathways involved in maize aflatoxin accumulation resistance. BMC Genomics. 16 (1), 673(2015).
- Warburton, M., et al. Genome-Wide Association Mapping of Aspergillus flavus and Aflatoxin Accumulation Resistance in Maize. Crop Science. 55 (5), 1857-1867 (2015).
- Warburton, M., et al. Genome-Wide Association and Metabolic Pathway Analysis of Corn Earworm Resistance in Maize. The Plant Genome. 11 (1), 170069(2018).
- Li, H., Thrash, A., Tang, J., He, L., Yan, J., Warburton, M. Leveraging GWAS data to identify metabolic pathways and networks involved in maize lipid biosynthesis. The Plant Journal. 98 (5), 853-863 (2019).
- Thrash, A., Tang, J., DeOrnellis, M., Peterson, D., Warburton, M. PAST: The Pathway Association Studies Tool to Infer Biological Meaning from GWAS Datasets. Plants. 9 (1), 58(2020).
- Adam, T., Mason, D. PAST: Pathway Association Study Tool (PAST). Bioconductor version: Release (3.10). , (2020).
- Thrash, A., DeOrnellis, M. IGBB/PAST. , at https://github.com/IGBB/PAST (2019).
- Bradbury, P., et al. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 23 (19), 2633-2635 (2007).
- Subramanian, A., et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences U.S.A. 102, 15545-15550 (2005).
Erratum
Formal Correction: Erratum: A Pathway Association Study Tool for GWAS Analyses of Metabolic Pathway Information
Posted by JoVE Editors on 10/08/2021. Citeable Link.
An erratum was issued for: A Pathway Association Study Tool for GWAS Analyses of Metabolic Pathway Information. One of the affiliations was updated.
The second affiliation was updated from:
USDA-ARS Corn Host Plant Resistance Research Unit, Mississippi State University
to:
Corn Host Plant Resistance Research Unit, USDA-ARS
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati