
Method Article
Ein Pathway Association Study Tool für GWAS-Analysen von Stoffwechselweginformationen
In diesem Artikel
Erratum Notice
Zusammenfassung
Durch die Ausführung des Pathway Association Study Tool (PAST), entweder über die Shiny-Anwendung oder über die R-Konsole, können Forscher ein tieferes Verständnis der biologischen Bedeutung ihrer Ergebnisse einer genomweiten Assoziationsstudie (GWAS) erlangen, indem sie die beteiligten Stoffwechselwege untersuchen.
Zusammenfassung
Vor kurzem wurde eine neue Implementierung einer zuvor beschriebenen Methode zur Interpretation von GWAS-Daten (Genome-Wide Association Study) mittels Stoffwechselweganalyse entwickelt und veröffentlicht. Das Pathway Association Study Tool (PAST) wurde entwickelt, um Bedenken hinsichtlich Benutzerfreundlichkeit und langsam laufenden Analysen auszuräumen. Dieses neue benutzerfreundliche Tool wurde auf Bioconductor und Github veröffentlicht. In Tests führte PAST Analysen in weniger als einer Stunde durch, die zuvor vierundzwanzig oder mehr Stunden benötigten. In diesem Artikel stellen wir das Protokoll für die Verwendung der Shiny-Anwendung oder der R-Konsole zum Ausführen von PAST vor.
Einleitung
Genomweite Assoziationsstudien (GWAS) sind eine beliebte Methode zur Untersuchung komplexer Merkmale und der damit verbundenen genomischen Regionen1,2,3. In dieser Art von Studie werden Hunderttausende von Einzelnukleotidpolymorphismusmarkern (SNP) auf ihre Assoziation mit dem Merkmal getestet und die Bedeutung der Assoziationen bewertet. Marker-Merkmals-Assoziationen, die den Schwellenwert für die falsche Entdeckungsrate (FDR) (oder eine andere Art von Signifikanzschwelle) erfüllen, werden für die Studie beibehalten, aber wahre Assoziationen können herausgefiltert werden. Bei komplexen, polygenen Merkmalen kann die Wirkung jedes Gens gering sein (und somit herausgefiltert werden), und einige Allele werden nur unter bestimmten Bedingungen exprimiert, die in der Studie möglicherweise nicht vorhanden sind3. Während also viele SNPs als mit dem Merkmal verbunden beibehalten werden können, kann jede einen sehr geringen Effekt haben. Zu viele SNP-Aufrufe werden fehlen, und eine Interpretation der biologischen Bedeutung und der genetischen Architektur des Merkmals kann unvollständig und verwirrend sein. Die Analyse des Stoffwechselwegs kann helfen, einige dieser Probleme anzugehen, indem sie sich auf die kombinierten Wirkungen von Genenkonzentriert,die nach ihrer biologischen Funktion gruppiert sind4,5,6.
Mehrere Studien wurden mit einer früheren Implementierung der in diesem Artikel beschriebenen Methode abgeschlossen. Aflatoxinakkumulation7, Maisohrwurmresistenz8und Ölbiosynthese9 wurden alle mit der vorherigen Implementierung untersucht. Während diese Analysen erfolgreich waren, war der Analyseprozess kompliziert, zeitaufwendig und umständlich, da die Analysewerkzeuge in einer Kombination aus R, Perl und Bash geschrieben wurden und die Pipeline nicht automatisiert war. Aufgrund des Fachwissens, das erforderlich ist, um diese Methode für jede Analyse zu modifizieren, wurde nun eine neue Methode entwickelt, die mit anderen Forschern geteilt werden kann.
Das Pathway Association Study Tool (PAST)10 wurde entwickelt, um die Mängel der vorherigen Methode zu beheben, indem weniger Kenntnisse in Programmiersprachen erforderlich sind und Analysen in kürzerer Zeit ausgeführt werden. Während die Methode mit Mais getestet wurde, macht PAST keine artspezifischen Annahmen. PAST kann über die R-Konsole als Shiny-App ausgeführt werden, und eine Online-Version wird voraussichtlich bald auf MaizeGDB verfügbar sein.
Protokoll
1. Einrichtung
- Installieren Sie R, falls es noch nicht installiert ist.
HINWEIS: PAST ist in R geschrieben und erfordert daher, dass seine Benutzer R installiert haben. Zum Zeitpunkt des Schreibens dieses Artikels erfordert die Installation von PAST direkt von Bioconductor R4.0. Ältere Versionen von PAST können von Bioconductor für R3.6 installiert werden, und PAST kann von Github für Benutzer mit R3.5 installiert werden. Die R-Installationsanleitung kann unter folgendem Link heruntergeladen werden: https://www.r-project.org/. - Installieren Sie die neueste Version von RStudio Desktop oder aktualisieren Sie RStudio (optional).
HINWEIS: RStudio ist eine hilfreiche Umgebung für die Arbeit mit der Sprache R. Die Installation wird empfohlen, insbesondere für diejenigen, die PAST in der Befehlszeile und nicht über die Shiny GUI-Anwendung ausführen möchten. RStudio und seine Installationsanleitung finden Sie unter folgendem Link: https://rstudio.com/products/rstudio/. - Installieren Sie PAST von Bioconductor11, indem Sie den Anweisungen auf Bioconductor folgen.
HINWEIS: Die Installation über Bioconductor sollte die Installation der Abhängigkeiten von PAST übernehmen. Darüber hinaus kann PAST von Github12installiert werden, aber die Installation von Github installiert Abhängigkeiten nicht automatisch. - Installieren Sie PAST Shiny (optional). Laden Sie die Datei "app. R" von der Seite "Releases" des Github-Repositorys: https://github.com/IGBB/PAST/releases/ und merken Sie sich, wo sich die heruntergeladene Datei befindet.
HINWEIS: PAST kann verwendet werden, indem seine Methoden direkt mit R aufruft werden, aber Benutzer, die mit R weniger vertraut sind, können die Past Shiny-Anwendung ausführen, die eine geführte Benutzeroberfläche bereitstellt. PAST Shiny ist ein R-Skript, das im shiny_app Zweig des PAST Github-Repositorys verfügbar ist. PAST Shiny versucht, seine Abhängigkeiten während der ersten Ausführung zu installieren. - Beginnen Sie die Analyse, indem Sie die Anwendung auf eine der drei unten beschriebenen Arten starten.
- PAST Shiny mit RStudio
- Erstellen Sie mit RStudio ein neues Projekt in dem Ordner, in dem sich die App befindet. R befindet sich. Klicken Sie auf Datei | Neues Projekt und wählen Sie diesen Ordner aus.
- Sobald ein neues Projekt erstellt wurde, öffnen Sie die App. R-Datei, die zuvor heruntergeladen wurde. RStudio erkennt diese App. R ist eine Shiny-App und erstellt eine Schaltfläche App ausführen auf der Leiste über dem angezeigten Quellcode. Klicken Sie auf App ausführen. RStudio öffnet dann ein Fenster, in dem die Anwendung PAST Shiny angezeigt wird.
- PAST Shiny mit R-Konsole
- Starten Sie R und führen Sie den folgenden Code aus, um die PAST Shiny-Anwendung zu starten: shiny::runApp('path/to/folder/with/shiny/app. R". Ersetzen Sie den Text in Anführungszeichen durch den Ordner, in dem die App angezeigt wird. R wurde heruntergeladen und behalten Sie die Zitate.
- PAST ohne R Shiny
- Führen Sie library(PAST) in einer R-Konsole aus, um PAST zu laden.
- PAST Shiny mit RStudio
2. Shiny-Analyse anpassen (optional)
- Ändern Sie den Analysetitel von "Neue Analyse" in etwas, das die Art der ausgeführten Analyse besser widerspiegelt, was dazu beiträgt, den Überblick über mehrere Analysen zu behalten (siehe Abbildung 1).

Abbildung 1. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
- Ändern Sie die Anzahl der Kerne und den Modus. Legen Sie die Anzahl der Kerne auf eine beliebige Zahl zwischen 1 und der Gesamtzahl auf dem Computer fest, aber beachten Sie, dass die Zuweisen weiterer Ressourcen für PAST andere Vorgänge auf dem Computer verlangsamen kann. Legen Sie den Modus basierend auf der Beschreibung in Abschnitt 6 fest.
3. GWAS-Daten laden
HINWEIS: Stellen Sie sicher, dass die GWAS-Daten tabulatorgetrennt sind. Stellen Sie sicher, dass die Assoziationsdatei die folgenden Spalten enthält: Merkmal, Markername, Locus oder Chromosom, Position auf dem Chromosom, p-Wert undR2-Wert für den Marker. Stellen Sie sicher, dass die Effektdatei die folgenden Spalten enthält: Merkmal, Markername, Ort oder Chromosom, Position auf dem Chromosom und Effekt. Die Reihenfolge dieser Spalten ist nicht wichtig, da der Benutzer beim Laden der Daten die Namen der Spalten angeben kann. Alle zusätzlichen Spalten werden ignoriert. Tassel13 kann verwendet werden, um diese Dateien zu erzeugen.
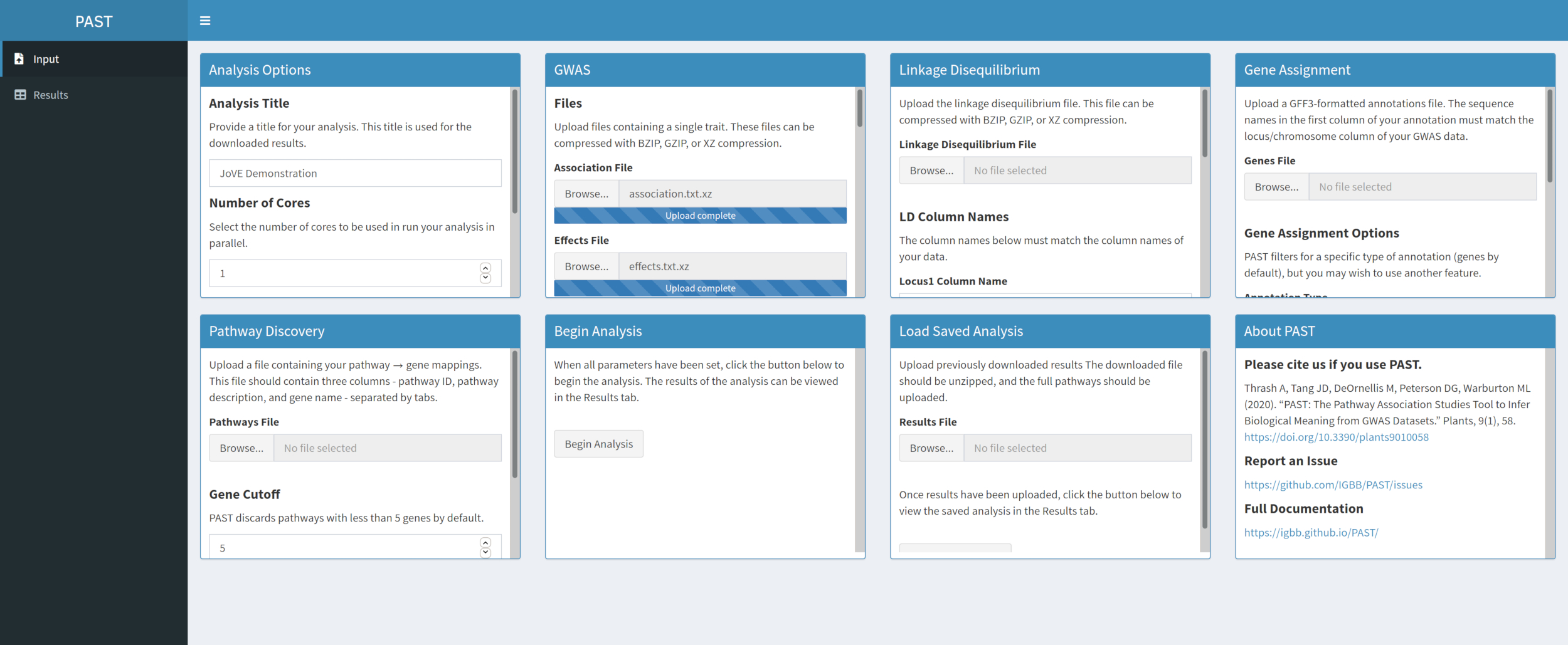
- Laden Sie GWAS-Daten mit PAST Shiny.
- Wählen Sie eine Assoziationsdatei und eine Effektdatei aus, indem Sie die Auswahlfelder Zuordnungsdatei und Effektdatei verwenden. Ändern Sie die Spaltennamen in den Eingabefeldern Spaltenname Zuordnen und Effektspaltenname unterhalb der Dateiauswahlfelder, um die Spaltennamen in den Daten widerzuspiegeln.

Abbildung 2. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
- Laden Sie GWAS-Daten mit PAST in die R-Konsole.
- Ändern Sie den folgenden Code, und führen Sie ihn aus:
gwas_data = load_GWAS_data("path/to/association_file.tsv", "path/to/effects_file.tsv", association_columns = c("Trait", "Marker", "Locus", "Site", "p", "marker_R2"), effects_columns = c("Trait", "Marker", "Locus", "Site", "Effect")
- Ändern Sie den folgenden Code, und führen Sie ihn aus:
- HINWEIS: Ändern Sie die Pfade zum tatsächlichen Speicherort der GWAS-Dateien. Die für association_columns und effects_columns angegebenen Werte sind die Standardwerte. Wenn die Namen nicht mit den Standardwerten übereinstimmen, geben Sie die Spaltennamen an. Andernfalls können diese weggelassen werden.
4. Lastverknüpfungs-Ungleichgewichtsdaten (LD)
HINWEIS: Stellen Sie sicher, dass die LD-Daten (Linkage Disequilibrium) tabulatorgetrennt sind und die folgenden Datentypen enthalten: Locus, Position1, Site1, Position2, Site2, Abstand in Basispaaren zwischen Position1 und Position2 undR2-Wert.
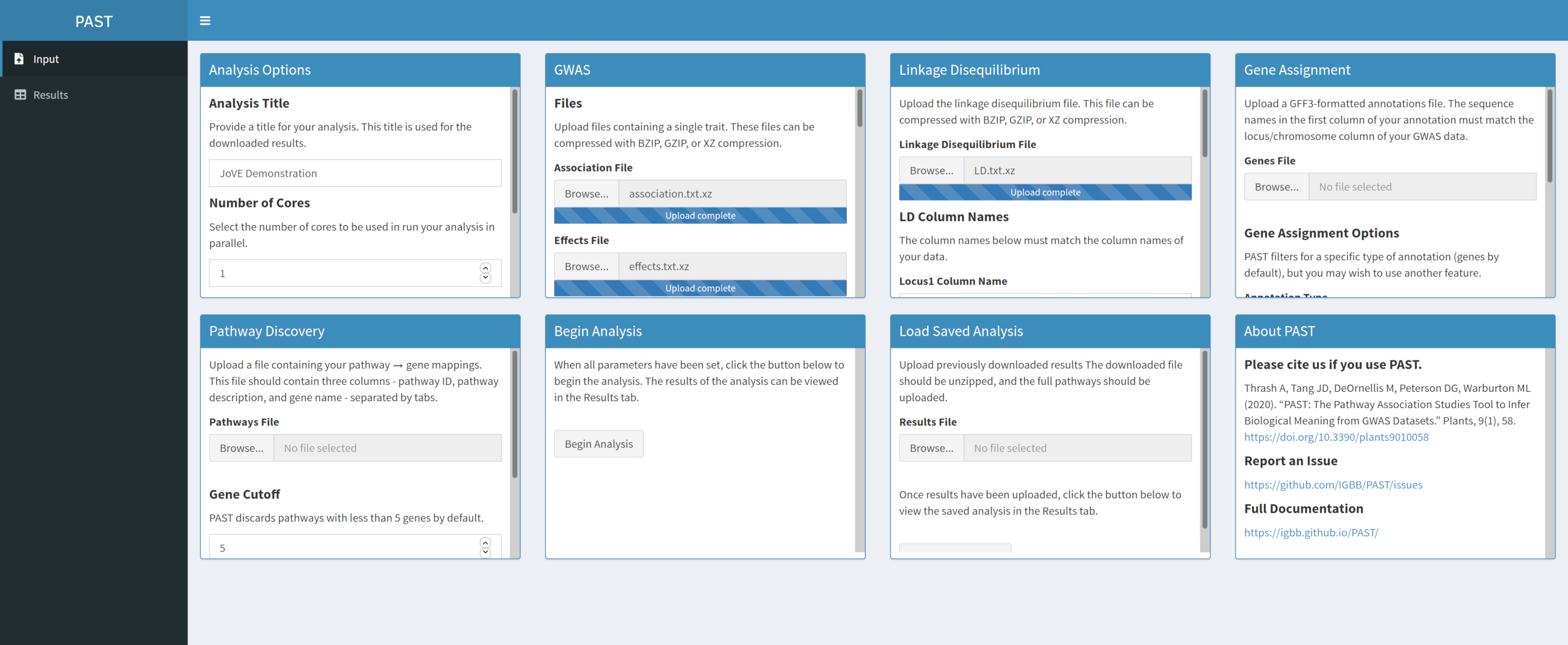
- Laden Sie LD-Daten mit PAST Shiny.
- Wählen Sie die Datei aus, die LD-Daten enthält. Ändern Sie die Spaltennamen in den Eingabefeldern LD-Spaltennamen unterhalb des Dateiauswahlfelds so, dass sie bei Bedarf mit den Spaltennamen in den LD-Daten übereinstimmen.

Abbildung 3. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
- Laden Sie LD-Daten mit PAST in die R-Konsole.
- Ändern Sie den folgenden Code, und führen Sie ihn aus, um LD-Daten zu laden:
LD = load_LD("path/to/LD.tsv", LD_columns = c("Locus1", "Position1", "Site1", "Position2", "Site2", "Dist_bp", "R.2")
HINWEIS: Ändern Sie den Pfad zum tatsächlichen Speicherort der LD-Datei. Die für LD_columns angegebenen Werte sind die Standardwerte. Wenn die Namen nicht mit diesen Standardwerten übereinstimmen, geben Sie die richtigen Namen der Spalten an. andernfalls können diese weggelassen werden.
- Ändern Sie den folgenden Code, und führen Sie ihn aus, um LD-Daten zu laden:
5. Zuweisen von SNPs zu Genen
HINWEIS: Laden Sie Anmerkungen im GFF-Format herunter oder suchen Sie sie anderweitig. Diese Anmerkungen sind oft in Online-Datenbanken für bestimmte Organismen zu finden. Seien Sie vorsichtig bei Anmerkungen von geringer Qualität, da die Qualität der Anmerkungsdaten die Qualität der Pfadanalyse beeinflusst. Vergewissern Sie sich, dass die erste Spalte dieser Anmerkungen (das Chromosom) mit dem Format des Locus/Chromosoms in den Assoziations-, Effekt- und LD-Daten übereinstimmt. Beispielsweise sollten die Anmerkungen das erste Chromosom nicht "chr1" nennen, wenn die GWAS- und LD-Datendateien das erste Chromosom "1" nennen.
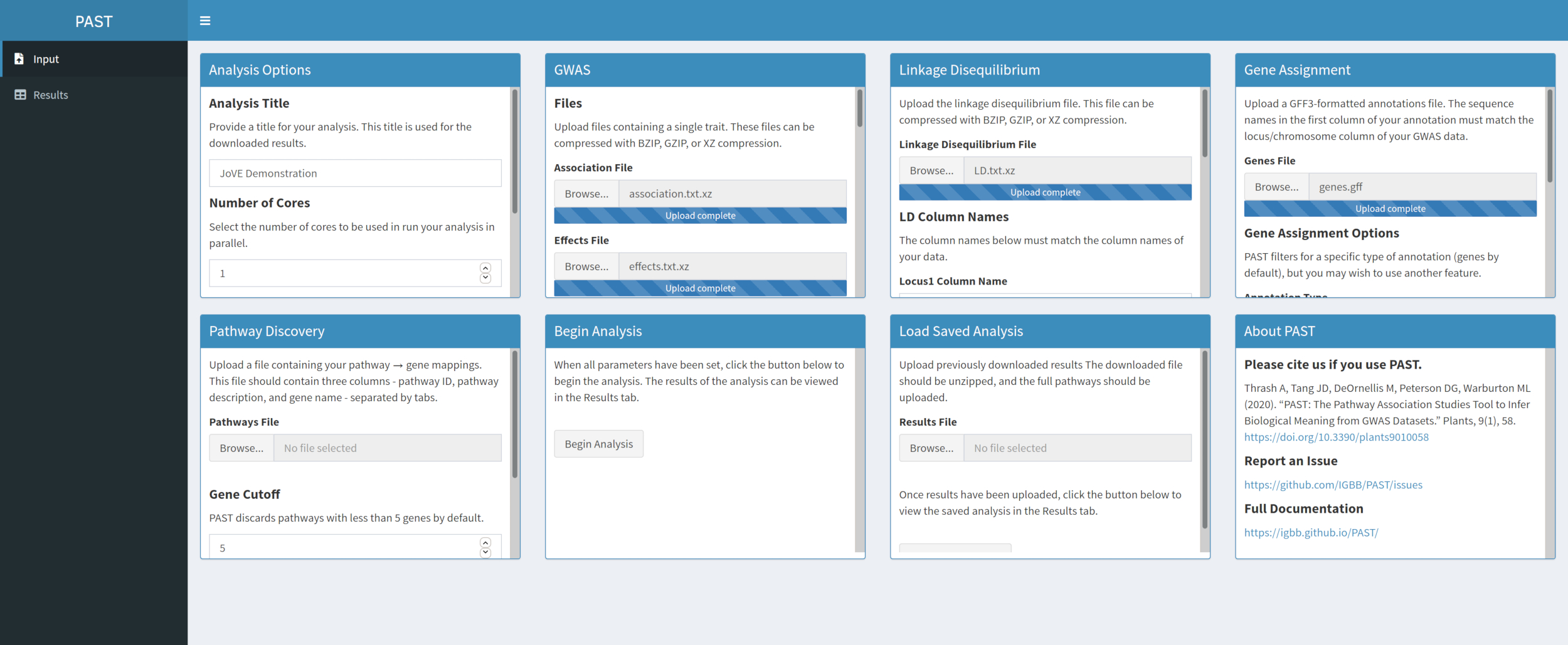
- Weisen Sie SNPs Genen mit PAST Shiny zu.
HINWEIS: Weitere Informationen zur Bestimmung eines geeignetenR2-Cutoffs finden Sie in Tang et al.6, im Abschnitt "SNP to gene algorithm for the pathway analysis".- Wählen Sie die Datei aus, die GFF-Anmerkungen enthält. Überlegen Sie, welche Fenstergröße und derR2-Cutoff für die betrachtete Art am besten geeignet sind, und ändern Sie sie, wenn die Standardwerte nicht zu den hochgeladenen Daten passen.
HINWEIS: Die Standardwerte in PAST spiegeln in erster Linie werte wider, die für Mais geeignet sind. In diesem Schritt wird die Anzahl der Kerne verwendet, die zu Beginn der PAST Shiny-Analyse (Schritt 2.2) festgelegt wurden.
- Wählen Sie die Datei aus, die GFF-Anmerkungen enthält. Überlegen Sie, welche Fenstergröße und derR2-Cutoff für die betrachtete Art am besten geeignet sind, und ändern Sie sie, wenn die Standardwerte nicht zu den hochgeladenen Daten passen.

Abbildung 4. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
- Weisen Sie Genen mit PAST in der R-Konsole SNPs zu.
- Ändern und führen Sie den folgenden Code aus, um Genen SNPs zuzuweisen:
Gene = assign_SNPs_to_genes(gwas_data, LD, "path/to/annotations.gff", c("gene"), 1000, 0.8, 2)
HINWEIS: In diesem Beispielcode werden mehrere Standardvorschläge bereitgestellt: 1000 ist die Größe des Fensters um den SNP, um nach Genen zu suchen; 0,8 ist der Grenzwert fürR2; 2 ist die Anzahl der Kerne, die für die parallele Verarbeitung verwendet werden. Der Pfad zu den Anmerkungen sollte auch in den tatsächlichen Speicherort der Anmerkungsdatei geändert werden.
- Ändern und führen Sie den folgenden Code aus, um Genen SNPs zuzuweisen:
6. Entdecken Sie wichtige Pfade
HINWEIS: Stellen Sie sicher, dass die Pathways-Datei die folgenden Daten im tabulatorgetrennten Format mit einer Zeile für jedes Gen in jedem Pathway enthält: Pathway ID - ein Bezeichner wie "PWY-6475-1"; Pfadbeschreibung - eine längere Beschreibung dessen, was die Wege tun, wie "Trans-Lycopin-Biosynthese"; Gen - ein Gen im Signalweg, das mit den in den Anmerkungen angegebenen Namen übereinstimmen sollte. Pathway-Informationen können wahrscheinlich in Online-Datenbanken für bestimmte Organismen wie MaizeGDB gefunden werden. Die zweite vom Benutzer angegebene Option ist der Modus. "Zunehmend" bezieht sich auf Phänotypen, die widerspiegeln, wann ein steigender Wert des gemessenen Merkmals wünschenswert ist, wie z. B. Ertrag, während "abnehmend" sich auf ein Merkmal bezieht, bei dem eine Abnahme der gemessenen Werte von Vorteil ist, z. B. Insektenschadenswerte. Die Signifikanz von Signalwegen wird mit den zuvorbeschriebenenMethoden4,6,14getestet.
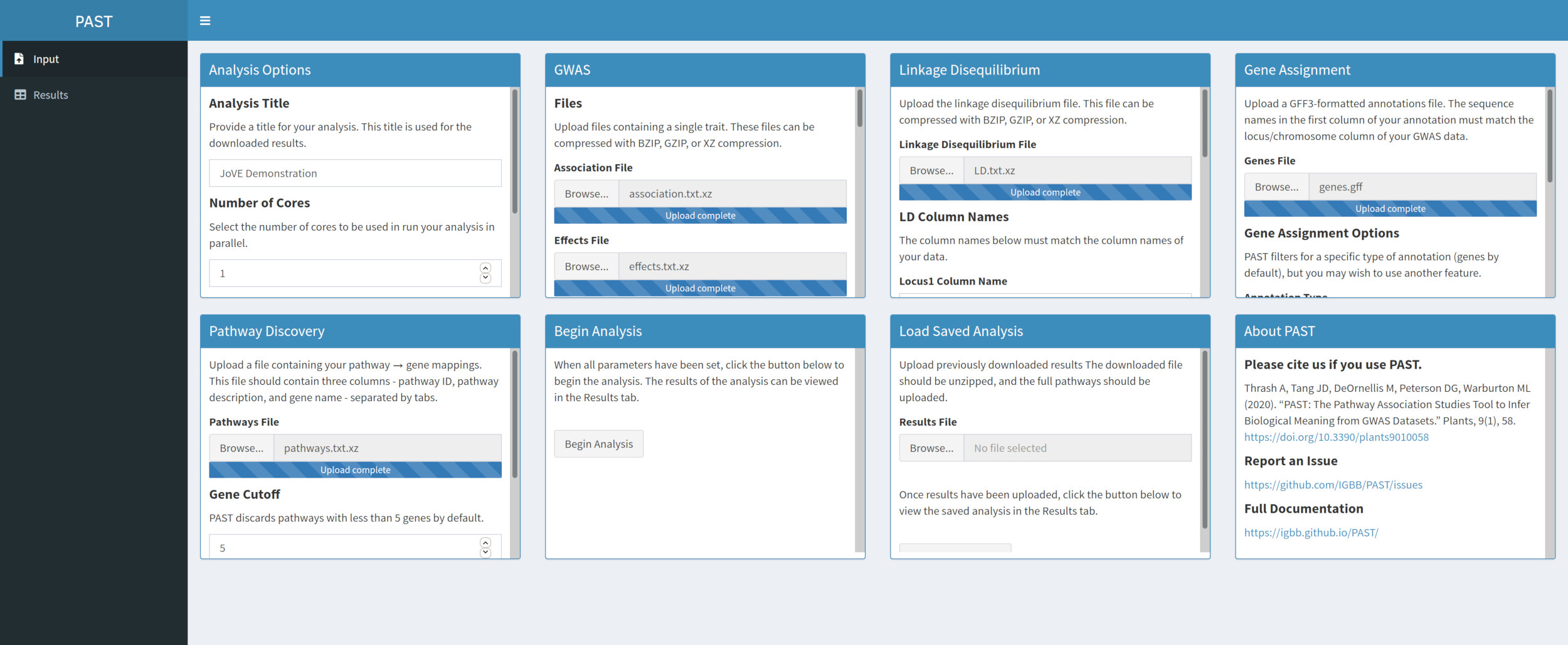
- Entdecken Sie bedeutende Wege mit PAST Shiny.
- Wählen Sie die Datei mit den Pfaddaten aus, und stellen Sie sicher, dass der Modus in den Analyseoptionen ausgewählt ist. Ändern Sie bei Bedarf die Anzahl der Gene, die sich in einem Signalweg befindet, um sie für die Analyse beizubehalten, und die Anzahl der Permutationen, die zum Erstellen der Nullverteilung verwendet werden, um die Signifikanz der Wirkung zu testen.

Abbildung 5. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
HINWEIS: In diesem Schritt wird die Anzahl der Kerne und der Modus verwendet, der zu Beginn der PAST Shiny-Analyse (Schritt 2.2) eingestellt wurde. Die Standardanzahl der Gene ist derzeit auf 5 Gene festgelegt, so dass Wege mit weniger bekannten Genen entfernt werden. Der Benutzer kann diesen Wert auf 4 oder 3 senken, um kürzere Pfade einzubeziehen, aber dadurch werden falsch positive Ergebnisse riskiert. Die Erhöhung dieses Wertes kann die Leistungsfähigkeit der Analyse erhöhen, entfernt jedoch mehr Pfade aus der Analyse. Das Ändern der Anzahl der verwendeten Permutationen erhöht und verringert die Leistung des Tests.
- Entdecken Sie wichtige Pfade mit PAST in der R Console.
- Ändern Sie den folgenden Code, und führen Sie ihn aus, um wichtige Pfade zu ermitteln:
rugplots_data <- find_pathway_significance(Gene, "path/to/pathways.tsv", 5, "increasing", 1000, 2)
HINWEIS: In diesem Beispielcode werden mehrere vorgeschlagene Standardwerte bereitgestellt. 5 ist die Mindestanzahl von Genen, die sich in einem Signalweg befindet, um den Signalweg in der Analyse zu halten, zunehmend bezieht sich auf eine zunehmende Menge des gemessenen Merkmals (es wird empfohlen, dass der Benutzer unabhängig vom Merkmal sowohl zunehmend als auch abnimmt; die Dateninterpretation unterscheidet sich jedoch für die beiden), 1000 ist die Anzahl der Male, um die Effekte zu untersuchen, um die Nullverteilung zu bestimmen, und 2 ist die Anzahl der Kerne, die für die parallele Verarbeitung verwendet werden. Ändern Sie den Pfad zum tatsächlichen Speicherort der Pfaddatei.
- Ändern Sie den folgenden Code, und führen Sie ihn aus, um wichtige Pfade zu ermitteln:
7. Rugplots ansehen
- Sehen Sie sich Rugplots mit PAST Shiny an.
- Nachdem alle Eingaben hochgeladen und festgelegt wurden, klicken Sie auf Analyse starten. Ein Fortschrittsbalken wird angezeigt und zeigt an, welcher Schritt der Analyse zuletzt abgeschlossen wurde. Wenn die Analyse abgeschlossen ist, wechselt PAST Shiny zur Registerkarte Ergebnisse. In der linken Spalte wird eine Ergebnistabelle angezeigt (mit der Bezeichnung "Pathways") und die Rugplots in der rechten Spalte (mit der Bezeichnung "plots").
- Verwenden Sie den Schieberegler, um die Filterparameter zu steuern. Wenn die Filterstufe zufriedenstellend ist, klicken Sie unten links auf die Schaltfläche Ergebnisse herunterladen, um alle Bilder und Tabellen einzeln in eine ZIP-Datei herunterzuladen, die mit dem Analysetitel benannt ist. Diese ZIP-Datei enthält die gefilterte Tabelle, die ungefilterte Tabelle und ein Bild pro Pfad in der gefilterten Tabelle.

Abbildung 6. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 7. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
- Rugplots mit PAST in der R-Konsole anzeigen
- Ändern Sie den folgenden Code, und führen Sie ihn aus, um die Ergebnisse zu speichern:
plot_pathways(rugplots_data, "Pvalue", 0,02, "steigend", "output_folder")
HINWEIS: In diesem Beispielcode werden mehrere vorgeschlagene Standardwerte bereitgestellt. pvalue stellt die Daten bereit, die zum Filtern unbedeutender Pfade verwendet werden können, nachdem der Benutzer einen Signifikanzschwellenwert ausgewählt hat. 0,02 ist der Standardwert, der bei der Filterung verwendet wird, und erhöhen bezieht sich auf eine zunehmende Menge des gemessenen Merkmals (es wird empfohlen, dass der Benutzer unabhängig vom Merkmal sowohl zunehmend als auch abnehmend ausgeht; die Dateninterpretation unterscheidet sich jedoch für die beiden); output_folder ist der Ordner, in den die Bilder und Tabellen geschrieben werden (dieser Ordner muss vor dem Ausführen der Funktion vorhanden sein). Eine Tabelle mit gefilterten Ergebnissen, die ungefilterten Ergebnisse und einzelne Bilder für jeden Pfad in den gefilterten Ergebnissen werden in diesen Ordner geschrieben.
- Ändern Sie den folgenden Code, und führen Sie ihn aus, um die Ergebnisse zu speichern:
Ergebnisse
Wenn nach einer Ausführung des PAST-Softwaretools keine Ergebnisse erzielt werden, überprüfen Sie, ob alle Eingabedateien korrekt formatiert sind. Ein erfolgreicher Lauf anhand der Beispieldaten im PAST-Paket, die auf einem Mais-GWAS kornfarbener Farbe basieren, ist in Abbildung 8 dargestellt. Diese Tabelle und das resultierende Bild können über die Schaltfläche Ergebnisse herunterladen heruntergeladen werden. Ein Beispiel für das heruntergeladene Bild ist in Abbildung 210 dargestellt. Falsche Einstellungen können zu Ergebnissen führen, die biologisch nicht sinnvoll sind, aber die Bestimmung der Unrichtigkeit muss dem Forscher überlassen werden, der die Gültigkeit der gewählten Einstellungen überprüfen und alle bekannten Beweise für das interessierende Merkmal berücksichtigen sollte.
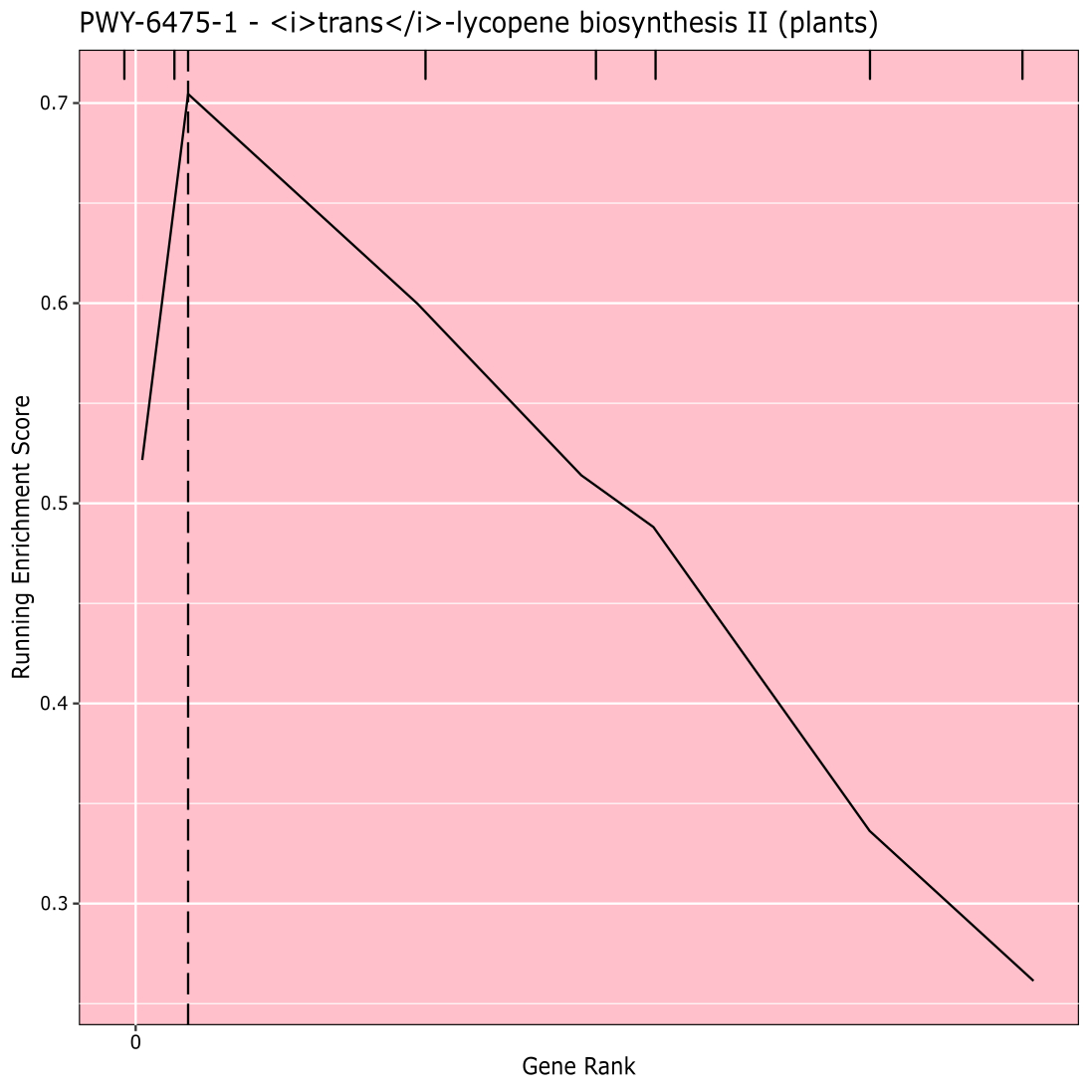
Abbildung 910 zeigt den Rugplot, der aus der Pfadanalyse der GWAS-Ergebnisse hergestellt wurde, die mit einer Maisplatte von 288 Inzuchtlinien erstellt wurden, die für die Kornfarbe phänotypisiert worden waren. Dieses vereinfachte Beispiel, bei dem die Phänotypen entweder "weiß" oder "gelb" waren, wurde verwendet, weil der Weg, der für die Schaffung der hellgelben Carotinoidpigmente verantwortlich ist, bekannt ist und für den größten Teil des Phänotyps verantwortlich sein sollte. Daher erwarteten wir, dass der Trans-Lycopin-Biosyntheseweg (der Carotinoide produziert) signifikant mit der Kornfarbe assoziiert ist, was er ist. Pfad-ID und Name sind oben im Diagramm aufgeführt. Die horizontale Achse des Diagramms ordnet alle Gene, die in die Analyse einbezogen wurden, von links nach rechts in der Reihenfolge der größten Wirkung auf das Merkmal bis zum kleinsten. Allerdings sind nur die Gene im Trans-Lycopin-Biosyntheseweg markiert (oben in der Grafik als Schraffurmarkierungen, die im Vergleich zu allen anderen Genen in der Analyse im Genrang ihrer Wirkung erscheinen). Es gibt 7 Gene in diesem Weg. Der Running Enrichment Score (ES) wird entlang der vertikalen Achse gezeichnet. Die ES für jedes Gen wird in der Reihenfolge der Wirkung in die laufende Summe aufgenommen und die Summe wird an die Anzahl der analysierten Gene angepasst. So ändert sich der Score, wenn man sich direkt entlang der horizontalen Achse bewegt und neigt dazu, zu steigen, wenn die größeren Effektgene eingeschlossen werden, aber irgendwann ist die Zunahme des Effekts kleiner als die Anpassung für das Hinzufügen eines anderen Gens, und der gesamte Score beginnt zu sinken. Die Spitze der laufenden ES-Linie ist mit einer gepunkteten vertikalen Linie markiert; Dies ist der ES für den gesamten Pathway und wird vom Programm verwendet, um festzustellen, ob der Pathway ausgewählt und als Rugplot dargestellt wird.

Abbildung 8: Abgeschlossener Lauf von PAST Shiny. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 9:Pfadbild von abgeschlossener Ausführung von PAST (oder von Shiny heruntergeladen). Diese Zahl wurde aus Thrash et al.10zitiert. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Diskussion
Ein primäres Ziel von PAST ist es, Stoffwechselweganalysen von GWAS-Daten einem breiteren Publikum zugänglich zu machen, insbesondere für nicht-menschliche und nicht-tierische Organismen. Alternative Methoden zu PAST sind oft Befehlszeilenprogramme, die sich auf Menschen oder Tiere konzentrieren. Benutzerfreundlichkeit war ein primäres Ziel bei der Entwicklung von PAST, sowohl bei der Entscheidung für die Entwicklung einer Shiny-Anwendung als auch bei der Entscheidung, R und Bioconductor zur Freigabe der Anwendung zu verwenden. Benutzer müssen nicht lernen, wie man Programme kompiliert, um PAST zu verwenden.
Wie bei den meisten Arten von Analysesoftware sind die Ergebnisse von PAST nur so gut wie die Eingabedaten; Wenn die Eingabedaten Fehler aufweisen oder falsch formatiert sind, kann PAST nicht ausgeführt werden oder führt zu uninformativen Ergebnissen. Sicherzustellen, dass die GWAS-Daten, LD-Daten, Anmerkungen und Pfaddateien korrekt formatiert sind, ist entscheidend für den Erhalt der korrekten Ausgabe von PAST. PAST analysiert nur biallele Marker und kann nur ein Merkmal für jeden Satz von Eingabedaten ausführen. Darüber hinaus ist es unwahrscheinlich, dass GWAS-Daten, die durch schlechte Genotypisierung oder falsche oder ungenaue Phänotypisierung erzeugt werden, klare oder wiederholbare Ergebnisse liefern. PAST kann bei der biologischen Interpretation von GWAS-Ergebnissen helfen, aber es ist unwahrscheinlich, dass chaotische Datensätze geklärt werden, wenn Umweltschwankungen, experimentelle Fehler oder Populationsstrukturen nicht richtig berücksichtigt wurden.
Benutzer können einige Parameter der Analyse ändern, sowohl in der Shiny-Anwendung als auch durch Übergeben dieser Parameter an die Funktionen von PAST in der R-Konsole. Diese Parameter können die von PAST gemeldeten Ergebnisse ändern, und Benutzer sollten vorsichtig sein, wenn sie diese von den Standardwerten ändern. Da LD von den Benutzern gemessen wird, in der Regel mit dem gleichen Markerdatensatz, der auch in der GWAS verwendet wurde, sind die LD-Messungen spezifisch für die Population. Für alle Studien, insbesondere für andere Arten als Mais (insbesondere selbstbestäubende, polyploide oder hochheterogene Arten), können Änderungen der Standardwerte gerechtfertigt sein.
Offenlegungen
Die Autoren haben nichts preiszugeben.
Danksagungen
Nichts.
Materialien
| Name | Company | Catalog Number | Comments |
| Computer | NA | NA | Any computer with 8GB RAM should be sufficient |
| R | R Project | NA | R 4.0 or greater is required to install from Bioconductor 3.11 |
Referenzen
- Rafalski, J. Association genetics in crop improvement. Current Opinion in Plant Biology. 13 (2), 174-180 (2010).
- Yan, J., Warburton, M., Crouch, J. Association Mapping for Enhancing Maize (Zea mays L.) Genetic Improvement. Crop Science. 51 (2), 433-449 (2011).
- Xiao, Y., Liu, H., Wu, L., Warburton, M., Yan, J. Genome-wide Association Studies in Maize: Praise and Stargaze. Molecular Plant. 10 (3), 359-374 (2017).
- Wang, K., Li, M., Bucan, M. Pathway-Based Approaches for Analysis of Genomewide Association Studies. The American Journal of Human Genetics. 81 (6), 1278-1283 (2007).
- Weng, L., et al. SNP-based pathway enrichment analysis for genome-wide association studies. BMC Bioinformatics. 12 (1), 99(2011).
- Tang, J., Perkins, A., Williams, W., Warburton, M. Using genome-wide associations to identify metabolic pathways involved in maize aflatoxin accumulation resistance. BMC Genomics. 16 (1), 673(2015).
- Warburton, M., et al. Genome-Wide Association Mapping of Aspergillus flavus and Aflatoxin Accumulation Resistance in Maize. Crop Science. 55 (5), 1857-1867 (2015).
- Warburton, M., et al. Genome-Wide Association and Metabolic Pathway Analysis of Corn Earworm Resistance in Maize. The Plant Genome. 11 (1), 170069(2018).
- Li, H., Thrash, A., Tang, J., He, L., Yan, J., Warburton, M. Leveraging GWAS data to identify metabolic pathways and networks involved in maize lipid biosynthesis. The Plant Journal. 98 (5), 853-863 (2019).
- Thrash, A., Tang, J., DeOrnellis, M., Peterson, D., Warburton, M. PAST: The Pathway Association Studies Tool to Infer Biological Meaning from GWAS Datasets. Plants. 9 (1), 58(2020).
- Adam, T., Mason, D. PAST: Pathway Association Study Tool (PAST). Bioconductor version: Release (3.10). , (2020).
- Thrash, A., DeOrnellis, M. IGBB/PAST. , at https://github.com/IGBB/PAST (2019).
- Bradbury, P., et al. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 23 (19), 2633-2635 (2007).
- Subramanian, A., et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences U.S.A. 102, 15545-15550 (2005).
Erratum
Formal Correction: Erratum: A Pathway Association Study Tool for GWAS Analyses of Metabolic Pathway Information
Posted by JoVE Editors on 10/08/2021. Citeable Link.
An erratum was issued for: A Pathway Association Study Tool for GWAS Analyses of Metabolic Pathway Information. One of the affiliations was updated.
The second affiliation was updated from:
USDA-ARS Corn Host Plant Resistance Research Unit, Mississippi State University
to:
Corn Host Plant Resistance Research Unit, USDA-ARS
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten