
Method Article
Un outil d’étude d’association de voies pour les analyses GWAS de l’information sur les voies métaboliques
Dans cet article
Erratum Notice
Résumé
En exécutant l’outil d’étude pathway association (PAST), soit via l’application Shiny, soit via la console R, les chercheurs peuvent mieux comprendre la signification biologique des résultats de leur étude d’association à l’échelle du génome (GWAS) en étudiant les voies métaboliques impliquées.
Résumé
Récemment, une nouvelle mise en œuvre d’une méthode décrite précédemment pour interpréter les données d’étude d’association à l’échelle du génome (GWAS) à l’aide de l’analyse des voies métaboliques a été développée et publiée. L’outil d’étude pathway association (PAST) a été développé pour répondre aux préoccupations concernant la convivialité et les analyses lentes. Ce nouvel outil convivial a été publié sur Bioconductor et Github. Lors des tests, PAST a exécuté des analyses en moins d’une heure qui nécessitaient auparavant vingt-quatre heures ou plus. Dans cet article, nous présentons le protocole permettant d’utiliser l’application Shiny ou la console R pour exécuter PAST.
Introduction
Les études d’association à l’échelle du génome (GWAS) sont une méthode populaire d’étude des traits complexes et des régions génomiques qui leur sont associées1,2,3. Dans ce type d’étude, des centaines de milliers de marqueurs de polymorphisme nucléotidique unique (SNP) sont testés pour leur association avec le trait, et la signification des associations est évaluée. Les associations marqueurs-traits qui atteignent le seuil du taux de fausse découverte (FDR) (ou un autre type de seuil de signification) sont conservées pour l’étude, mais les associations vraies peuvent être filtrées. Pour les caractères polygéniques complexes, l’effet de chaque gène peut être faible (et donc filtré), et certains allèles ne sont exprimés que dans des conditions spécifiques qui pourraient ne pas être présentes dans l’étude3. Ainsi, bien que de nombreux SLP puissent être conservés comme associés au trait, chacun peut avoir un très faible effet. Trop d’appels SNP seront manquants, et une interprétation de la signification biologique et de l’architecture génétique du trait peut être incomplète et déroutante. L’analyse des voies métaboliques peut aider à résoudre certains de ces problèmes en se concentrant sur les effets combinés des gènes regroupés en fonction de leur fonction biologique4,5,6.
Plusieurs études ont été réalisées à l’aide d’une implémentation antérieure de la méthode décrite dans cet article. L’accumulation d’aflatoxines7,la résistance au ver de l’oreille du maïs8et la biosynthèse de l’huile9 ont toutes été étudiées avec la mise en œuvre précédente. Bien que ces analyses aient été couronnées de succès, le processus d’analyse était compliqué, long et fastidieux, car les outils d’analyse étaient écrits dans une combinaison de R, Perl et Bash, et le pipeline n’était pas automatisé. En raison des connaissances spécialisées nécessaires pour modifier cette méthode pour chaque analyse, une nouvelle méthode a maintenant été développée qui peut être partagée avec d’autres chercheurs.
Le Pathway Association Study Tool (PAST)10 a été conçu pour remédier aux lacunes de la méthode précédente en exigeant moins de connaissances en langages de programmation et en exécutant des analyses dans un délai plus court. Bien que la méthode ait été testée avec du maïs, PAST ne fait aucune hypothèse spécifique à l’espèce. PAST peut être exécuté via la console R, en tant qu’application Shiny, et une version en ligne devrait bientôt être disponible sur MaizeGDB.
Protocole
1. Configuration
- Installez R, s’il n’est pas déjà installé.
REMARQUE: PAST est écrit en R et, par conséquent, nécessite que ses utilisateurs aient installé R. Au moment d’écrire ces lignes, l’installation de PAST directement à partir de Bioconductor nécessite R4.0. Les anciennes versions de PAST peuvent être installées à partir de Bioconductor pour R3.6, et PAST peut être installé à partir de Github pour les utilisateurs de R3.5. Les instructions d’installation de R peuvent être téléchargées à partir du lien suivant : https://www.r-project.org/. - Installez la dernière version de RStudio Desktop ou mettez à jour RStudio (facultatif).
REMARQUE: RStudio est un environnement utile pour travailler avec le langage R. Son installation est recommandée, en particulier pour ceux qui choisissent d’exécuter PAST dans la ligne de commande plutôt que via l’application Shiny GUI. RStudio et ses instructions d’installation peuvent être trouvés sur le lien suivant: https://rstudio.com/products/rstudio/. - Installez PAST à partir de Bioconductor11 en suivant les instructions sur Bioconductor.
REMARQUE: L’installation via Bioconductor doit gérer l’installation des dépendances de PAST. En outre, PAST peut être installé à partir de Github12, mais l’installation à partir de Github n’installera pas automatiquement les dépendances. - Installez PAST Shiny (facultatif). Téléchargez le fichier « app. R » à partir de la page Versions du référentiel Github : https://github.com/IGBB/PAST/releases/ et rappelez-vous où se trouve le fichier téléchargé.
REMARQUE : PAST peut être utilisé en appelant ses méthodes directement avec R, mais les utilisateurs qui sont moins familiers avec R peuvent exécuter l’application PAST Shiny, qui fournit une interface utilisateur guidée. PAST Shiny est un script R disponible dans la branche shiny_app du référentiel GITHUB PAST. PAST Shiny tentera d’installer ses dépendances lors de la première exécution. - Commencez l’analyse en démarrant l’application de l’une des trois manières décrites ci-dessous.
- PAST Shiny avec RStudio
- À l’aide de RStudio, créez un nouveau projet dans le dossier où se trouve l’application. R est situé. Cliquez sur | fichier Nouveau projet et sélectionnez ce dossier.
- Une fois qu’un nouveau projet a été créé, ouvrez l’application. R téléchargé précédemment. RStudio reconnaît cette application. R est une application Shiny et crée un bouton Exécuter l’application sur la barre au-dessus du code source affiché. Cliquez sur Exécuter l’application. RStudio lancera alors une fenêtre qui affiche l’application PAST Shiny.
- PAST Shiny avec console R
- Lancez R et exécutez le code suivant pour démarrer l’application PAST Shiny : shiny::runApp('path/to/folder/with/shiny/app. R'. Remplacez le texte entre guillemets par le dossier dans lequel l’application. R a été téléchargé et conservez les citations.
- PASSÉ sans R Shiny
- Exécutez library(PAST) dans une console R pour charger PAST.
- PAST Shiny avec RStudio
2. Personnaliser l’analyse Shiny (facultatif)
- Remplacez le titre de l’analyse de « Nouvelle analyse » par quelque chose qui reflète mieux le type d’analyse en cours d’exécution, ce qui permet de suivre plusieurs analyses (voir la figure 1).

Figure 1. Veuillez cliquer ici pour l’agrandir.
{kind=link}
- Modifiez le nombre de cœurs et le mode. Définissez le nombre de cœurs sur n’importe quel nombre compris entre 1 et le nombre total sur la machine, mais sachez que consacrer plus de ressources à PAST peut ralentir d’autres opérations sur la machine. Définissez le mode en fonction de la description de la section 6.
3. Charger les données GWAS
Remarque : Vérifiez que les données GWAS sont délimitées par des tabulations. Assurez-vous que le fichier d’association contient les colonnes suivantes : trait, nom du marqueur, locus ou chromosome, position sur le chromosome, valeur p et valeur R2 pour le marqueur. Assurez-vous que le fichier d’effets contient les colonnes suivantes : trait, nom du marqueur, locus ou chromosome, position sur le chromosome et effet. L’ordre de ces colonnes n’est pas important, car l’utilisateur peut spécifier les noms des colonnes lors du chargement des données. Toutes les colonnes supplémentaires sont ignorées. TASSEL13 peut être utilisé pour produire ces fichiers.
- Chargez les données GWAS avec PAST Shiny.
- Sélectionnez un fichier d’association et un fichier d’effets à l’aide des zones de sélection Fichier d’association et Fichier d’effets. Modifiez les noms des colonnes dans les zones de saisie Nom de la colonne d’association et Nom des colonnes d’effets sous les zones de sélection de fichier pour refléter les noms de colonne dans les données.

Figure 2. Veuillez cliquer ici pour l’agrandir.
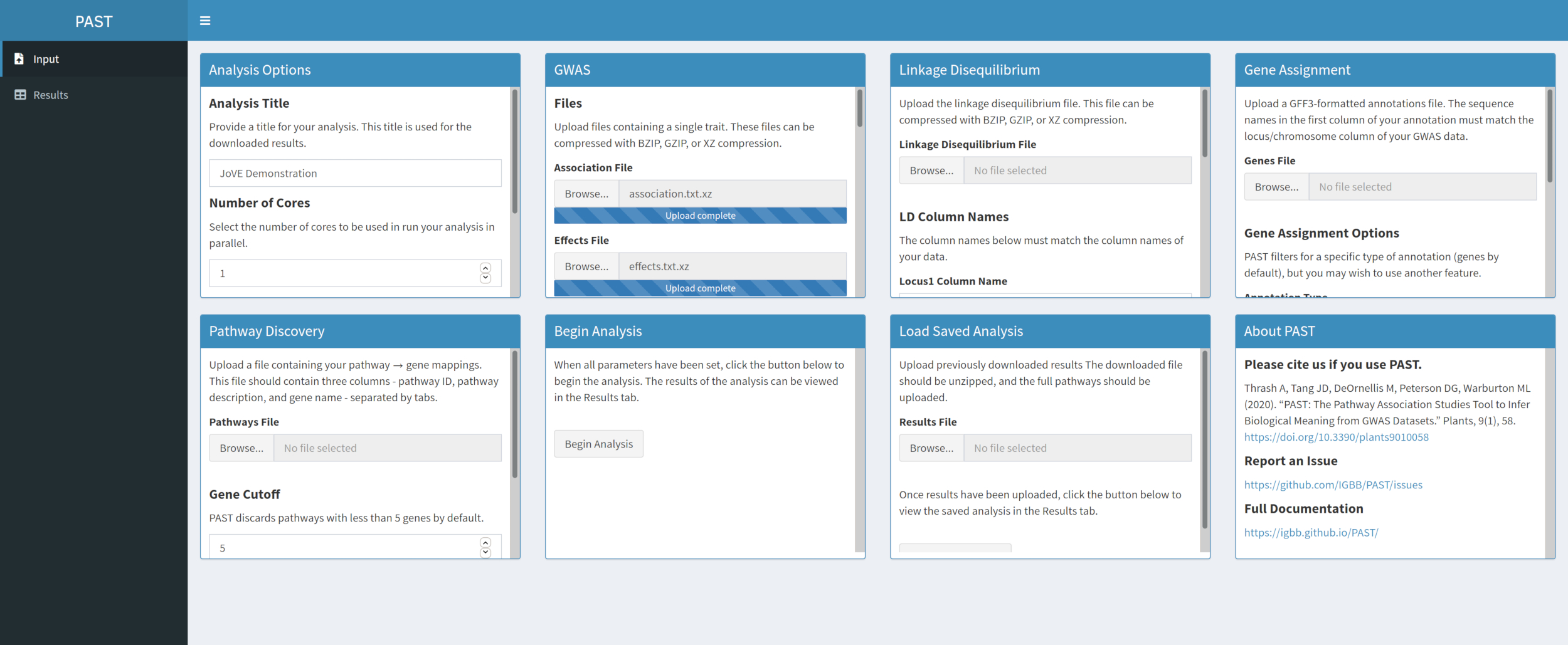{kind=link}
- Chargez les données GWAS avec PAST dans la console R.
- Modifiez et exécutez le code suivant :
gwas_data = load_GWAS_data(« path/to/association_file.tsv », « path/to/effects_file.tsv », association_columns = c(« Trait », « Marker », « Locus », « Site », « p », « marker_R2 »), effects_columns = c(« Trait », « Marker », « Locus », « Site », « Effect »)
- Modifiez et exécutez le code suivant :
- REMARQUE : Modifiez les chemins d’accès à l’emplacement réel des fichiers GWAS. Les valeurs fournies pour association_columns et effects_columns sont les valeurs par défaut. Si les noms ne correspondent pas aux valeurs par défaut, spécifiez les noms des colonnes. Sinon, ceux-ci peuvent être omis.
4. Données sur le déséquilibre de liaison de charge (LD)
Remarque : Vérifiez que les données de déséquilibre de liaison (LD) sont délimitées par des tabulations et contiennent les types de données suivants : Locus, Position1, Site1, Position2, Site2, Distance dans les paires de bases entre Position1 et Position2 et valeur R2.
- Chargez les données LD avec PAST Shiny.
- Sélectionnez le fichier contenant les données LD. Modifiez les noms des colonnes dans les zones de saisie Noms des colonnes LD sous la zone de sélection de fichier pour qu’elles correspondent aux noms des colonnes dans les données LD si nécessaire.

Figure 3. Veuillez cliquer ici pour l’agrandir.
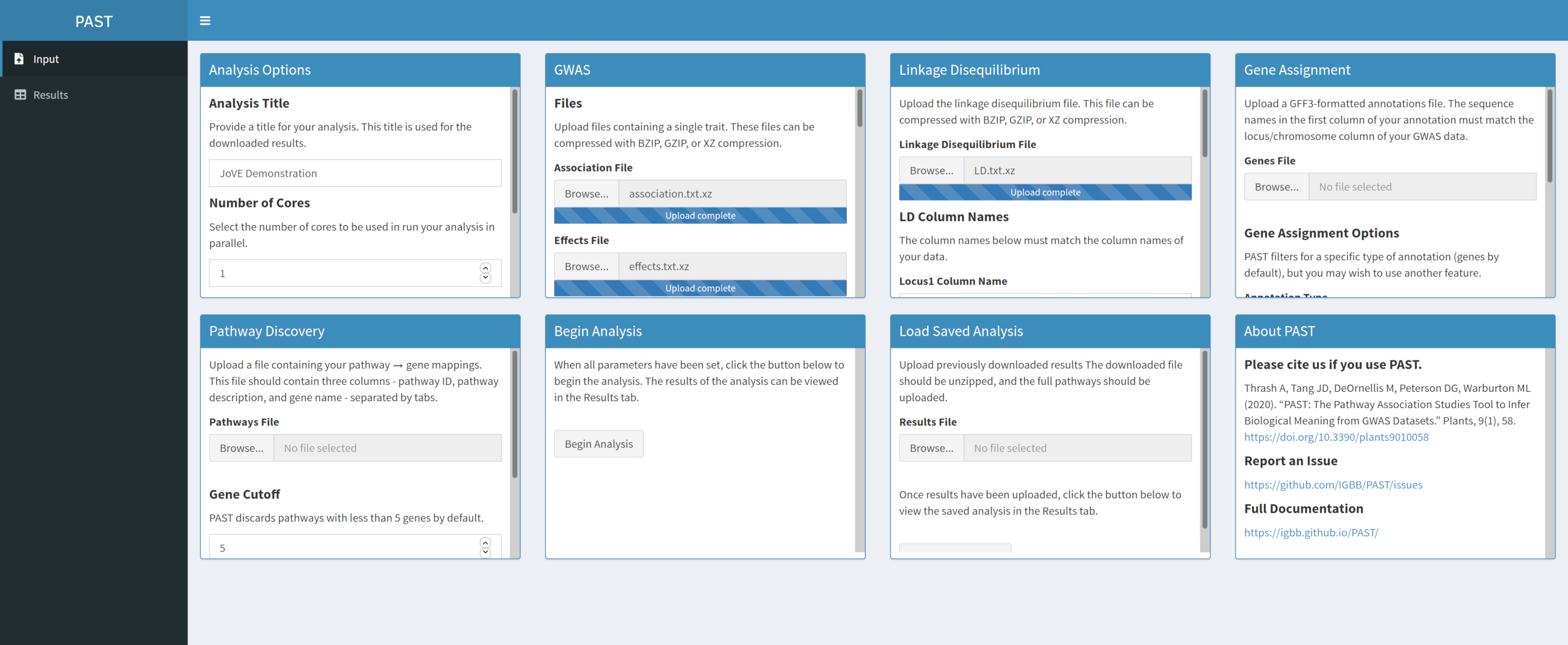{kind=link}
- Chargez LD Data avec PAST dans la console R.
- Modifiez et exécutez le code suivant pour charger les données LD :
LD = load_LD(« path/to/LD.tsv », LD_columns = c(« Locus1 », « Position1 », « Site1 », « Position2 », « Site2 », « Dist_bp », « R.2 »)
Remarque : Modifiez le chemin d’accès à l’emplacement réel du fichier LD. Les valeurs fournies pour LD_columns sont les valeurs par défaut. Si les noms ne correspondent pas à ces valeurs par défaut, spécifiez les noms corrects des colonnes ; sinon, ceux-ci peuvent être omis.
- Modifiez et exécutez le code suivant pour charger les données LD :
5. Assigner des SLP aux gènes
REMARQUE: Téléchargez ou recherchez des annotations au format GFF. Ces annotations peuvent souvent être trouvées dans des bases de données en ligne pour des organismes spécifiques. Soyez prudent avec les annotations de faible qualité, car la qualité des données d’annotations affectera la qualité de l’analyse des voies. Vérifiez que la première colonne de ces annotations (le chromosome) correspond au format du locus/chromosome dans les données d’association, d’effets et de LD. Par exemple, les annotations ne doivent pas appeler le premier chromosome « chr1 » si les fichiers de données GWAS et LD appellent le premier chromosome « 1 ».
- Attribuez des SLP aux gènes avec PAST Shiny.
REMARQUE: Plus d’informations sur la détermination d’un seuil R2 approprié peuvent être trouvées dans Tang et al.6, dans la section intitulée « SNP to gene algorithm for the pathway analysis ».- Sélectionnez le fichier contenant les annotations GFF. Considérez quelle taille de fenêtre et quelle coupure R2 conviennent le mieux à l’espèce considérée et modifiez-la si les valeurs par défaut ne conviennent pas aux données téléchargées.
REMARQUE: Les valeurs par défaut dans PAST reflètent principalement les valeurs appropriées pour le maïs. Le nombre de cœurs défini au début de l’analyse PAST Shiny (étape 2.2) est utilisé dans cette étape.
- Sélectionnez le fichier contenant les annotations GFF. Considérez quelle taille de fenêtre et quelle coupure R2 conviennent le mieux à l’espèce considérée et modifiez-la si les valeurs par défaut ne conviennent pas aux données téléchargées.

Figure 4. Veuillez cliquer ici pour l’agrandir.
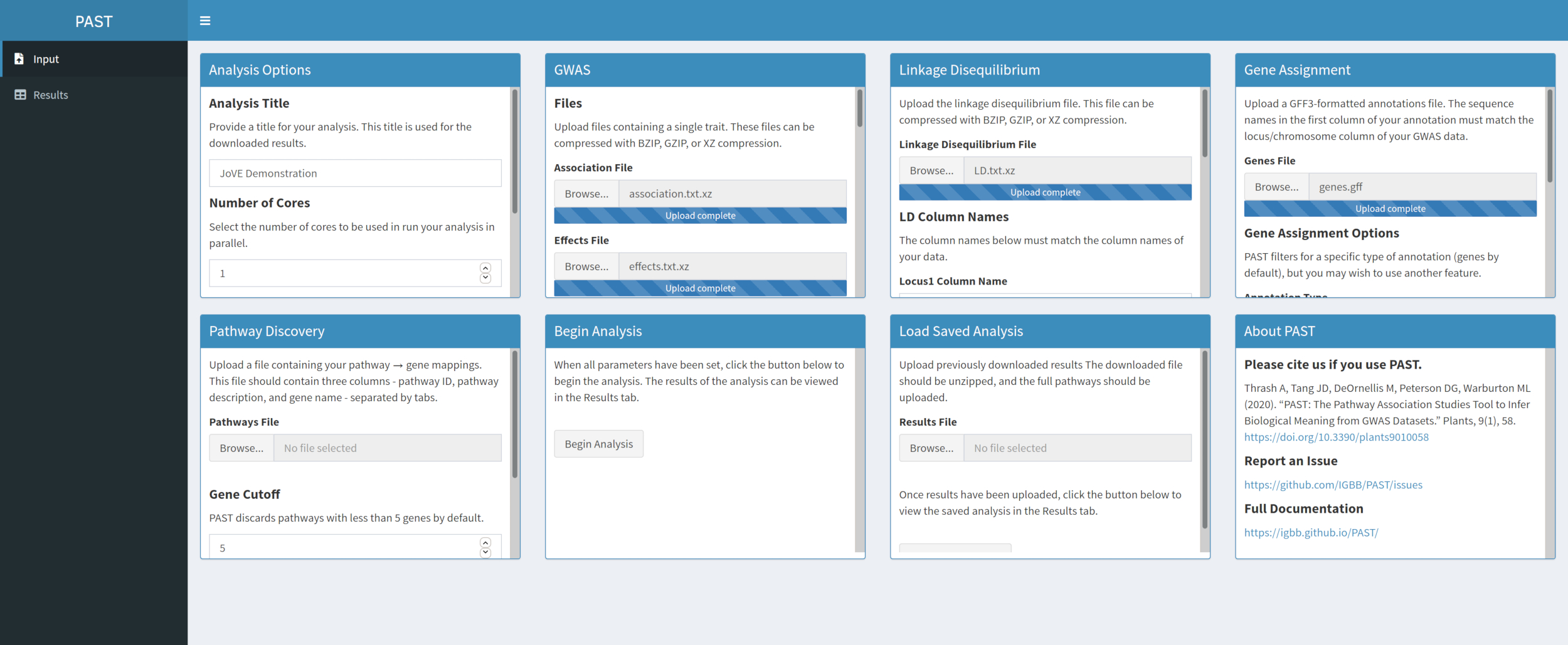{kind=link}
- Attribuez des SLP aux gènes avec PAST dans la console R.
- Modifiez et exécutez le code suivant pour affecter des SMP aux gènes :
gènes = assign_SNPs_to_genes(gwas_data, LD, « path/to/annotations.gff », c(« gene »), 1000, 0.8, 2)
REMARQUE: Dans cet exemple de code, plusieurs suggestions par défaut sont fournies: 1000 est la taille de la fenêtre autour du SNP pour rechercher des gènes; 0,8 est la valeur seuil pour R2; 2 est le nombre de cœurs utilisés pour le traitement parallèle. Le chemin d’accès aux annotations doit également être remplacé par l’emplacement réel du fichier d’annotations.
- Modifiez et exécutez le code suivant pour affecter des SMP aux gènes :
6. Découvrez des voies importantes
REMARQUE: Vérifiez que le fichier de voies contient les données suivantes dans un format délimité par des tabulations, avec une ligne pour chaque gène dans chaque voie: ID de voie - un identificateur tel que « PWY-6475-1 »; description de la voie - une description plus longue de ce que font les voies, comme la « biosynthèse trans-lycopène »; gène - un gène dans la voie, qui doit correspondre aux noms fournis dans les annotations. Les informations sur les voies d’accès peuvent probablement être trouvées dans des bases de données en ligne pour des organismes spécifiques, tels que MaizeGDB. La deuxième option spécifiée par l’utilisateur est le mode. « Croissant » fait référence aux phénotypes qui reflètent quand une valeur croissante du trait mesuré est souhaitable, comme le rendement, tandis que « décroissant » fait référence à un trait où une diminution des valeurs mesurées est bénéfique, comme les cotes de dommages causés par les insectes. L’importance des voies est testée à l’aide des méthodesdécritesprécédemment 4,6,14.
- Découvrez des voies importantes avec PAST Shiny.
- Sélectionnez le fichier contenant les données des chemins d’accès et assurez-vous que le mode est sélectionné dans les options d’analyse. Si nécessaire, modifiez le nombre de gènes qui doivent se présenter dans une voie pour le conserver pour l’analyse et le nombre de permutations utilisées pour créer la distribution nulle afin de tester la signification de l’effet.

Graphique 5. Veuillez cliquer ici pour voir une version agrandie de cette figure.
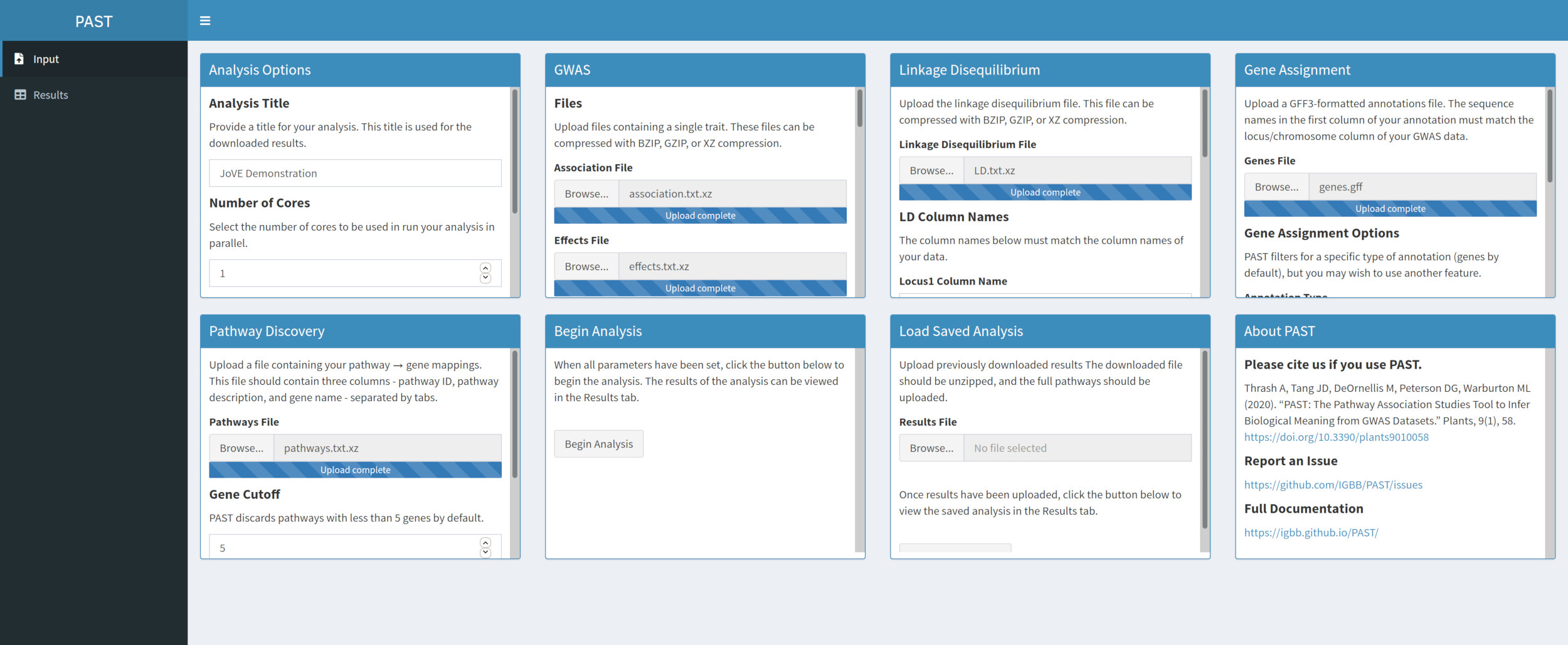{kind=link}
REMARQUE : Le nombre de cœurs et le mode défini au début de l’analyse PAST Shiny (étape 2.2) sont utilisés dans cette étape. Le nombre par défaut de gènes est actuellement fixé à 5 gènes, de sorte que les voies avec moins de gènes connus seront supprimées. L’utilisateur peut abaisser cette valeur à 4 ou 3, pour inclure des voies plus courtes, mais cela risque de fausses résultats positifs. L’augmentation de cette valeur peut augmenter la puissance de l’analyse, mais supprimera davantage de voies de l’analyse. Changer le nombre de permutations utilisées augmente et diminue la puissance du test.
- Découvrez des voies importantes avec PAST dans la console R.
- Modifiez et exécutez le code suivant pour découvrir des chemins d’accès significatifs :
rugplots_data <- find_pathway_significance(gènes, « path/to/pathways.tsv », 5, « increasing », 1000, 2)
Remarque : Dans cet exemple de code, plusieurs valeurs par défaut suggérées sont fournies. 5 est le nombre minimum de gènes qui doivent être dans une voie afin de maintenir la voie dans l’analyse, l’augmentation fait référence à une quantité croissante du trait mesuré (il est recommandé que l’utilisateur exécute à la fois en augmentation et en diminution, quel que soit le trait; l’interprétation des données différera pour les deux, cependant), 1000 est le nombre de fois pour échantillonner les effets pour déterminer la distribution nulle, et 2 est le nombre de cœurs utilisés pour le traitement parallèle. Modifiez le chemin d’accès à l’emplacement réel du fichier de chemins d’accès.
- Modifiez et exécutez le code suivant pour découvrir des chemins d’accès significatifs :
7. Voir Rugplots
- Voir Rugplots avec PAST Shiny.
- Une fois toutes les entrées téléchargées et définies, cliquez sur Commencer l’analyse. Une barre de progression apparaîtra et indiquera quelle étape de l’analyse a été terminée pour la dernière fois. Une fois l’analyse terminée, PAST Shiny passe à l’onglet Résultats. Un tableau des résultats sera affiché dans la colonne de gauche (intitulée « chemins ») et les Rugplots seront affichés dans la colonne de droite (intitulée « tracés »).
- Utilisez le curseur pour contrôler les paramètres de filtrage. Lorsque le niveau de filtrage est satisfaisant, cliquez sur le bouton Télécharger les résultats en bas à gauche pour télécharger toutes les images et tous les tableaux individuellement dans un fichier ZIP nommé avec le titre de l’analyse. Ce fichier ZIP contient la table filtrée, la table non filtrée et une image par chemin d’accès dans la table filtrée.

Figure 6. Veuillez cliquer ici pour l’agrandir.
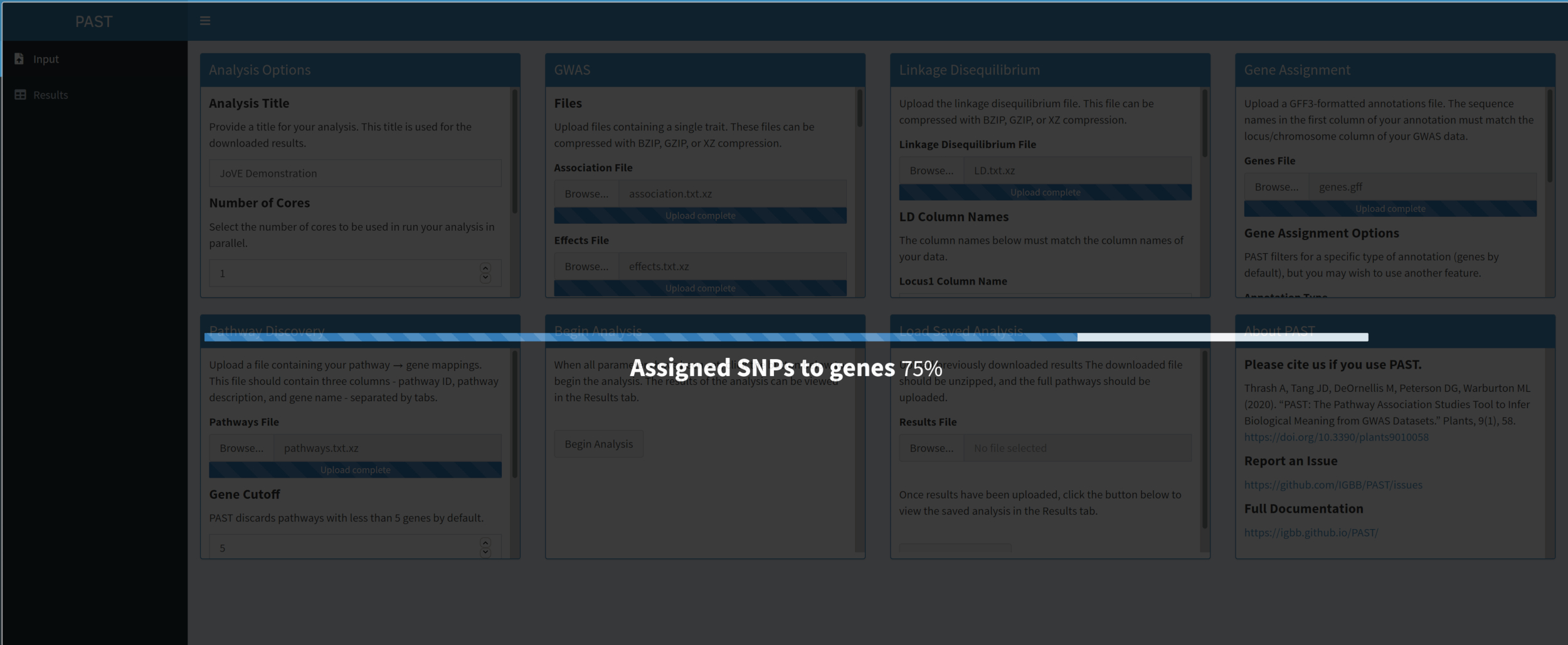{kind=link}

Figure 7. Veuillez cliquer ici pour l’agrandir.
{kind=link}
- Afficher rugplots avec PAST dans la console R
- Modifiez et exécutez le code suivant pour enregistrer les résultats :
plot_pathways(rugplots_data, « pvalue », 0,02, « augmentation », « output_folder »)
Remarque : Dans cet exemple de code, plusieurs valeurs par défaut suggérées sont fournies. pvalue fournit les données qui peuvent être utilisées pour filtrer les voies insignifiantes après qu’un seuil de signification a été choisi par l’utilisateur; 0,02 est la valeur par défaut utilisée dans le filtrage, et l’augmentation fait référence à une quantité croissante du trait mesuré (il est recommandé que l’utilisateur exécute à la fois en augmentation et en diminution, quel que soit le trait; l’interprétation des données différera pour les deux, cependant); output_folder est le dossier dans lequel les images et les tables seront écrites (ce dossier doit exister avant d’exécuter la fonction). Un tableau des résultats filtrés, des résultats non filtrés et des images individuelles pour chaque chemin d’accès dans les résultats filtrés sont écrits dans ce dossier.
- Modifiez et exécutez le code suivant pour enregistrer les résultats :
Résultats
Si les résultats ne sont pas produits après l’exécution de l’outil logiciel PAST, vérifiez que tous les fichiers d’entrée sont correctement formatés. Une exécution réussie à l’aide des exemples de données du package PAST, qui sont basées sur un GWAS de maïs de couleur de grain, est illustrée à la figure 8. Ce tableau et l’image résultante peuvent être téléchargés à l’aide du bouton Télécharger les résultats. Un exemple de l’image téléchargée est illustré à la Figure 210. Des paramètres incorrects peuvent conduire à des résultats qui n’ont pas de sens biologique, mais la détermination de l’incorrect doit être du choix du chercheur, qui doit vérifier la validité des paramètres choisis et prendre en compte toutes les preuves connues concernant le trait d’intérêt.
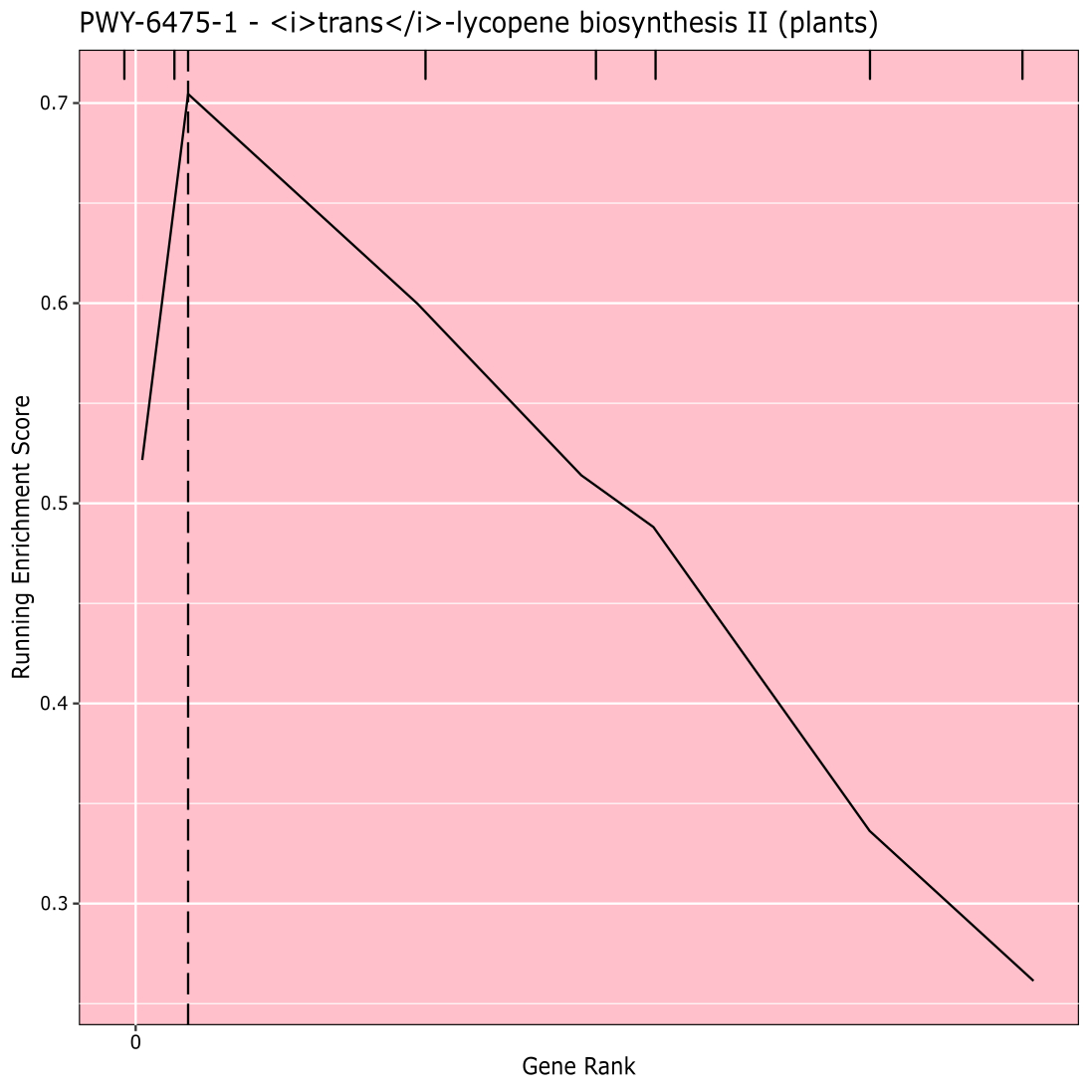
La figure 910 montre le rugplot produit à partir de l’analyse des voies des résultats GWAS créés avec un panneau de maïs de 288 lignées consanguines qui avaient été phénotypées pour la couleur des grains. Cet exemple simpliste, où les phénotypes étaient soit « blancs » soit « jaunes », a été utilisé parce que la voie responsable de la création des pigments caroténoïdes jaune vif est connue et devrait être responsable de la plupart des phénotypes. Ainsi, nous nous attendions à ce que la voie de biosynthèse trans-lycopène (qui produit des caroténoïdes) soit significativement associée à la couleur du grain, ce qui est le cas. L’ID et le nom du chemin d’accès sont répertoriés en haut du graphique. L’axe horizontal du graphique classe tous les gènes qui ont été inclus dans l’analyse, disposés de gauche à droite dans l’ordre du plus grand effet sur le trait au plus petit. Cependant, seuls les gènes de la voie de biosynthèse du trans-lycopène sont marqués (en haut du graphique, sous forme de marques d’éclosion, apparaissant dans le rang génétique de leur effet par rapport à tous les autres gènes de l’analyse). Il y a 7 gènes dans cette voie. Le score d’enrichissement en cours d’exécution (ES) est tracé le long de l’axe vertical. L’ES pour chaque gène est ajouté au total courant par ordre d’effet et le total est ajusté au nombre de gènes analysés. Ainsi, le score change à mesure que l’on se déplace le long de l’axe horizontal et tend à augmenter à mesure que les gènes à effet plus importants sont inclus, mais à un moment donné, l’augmentation de l’effet est plus petite que l’ajustement pour avoir ajouté un autre gène, et le score entier commence à diminuer. Le sommet de la ligne ES en cours d’exécution est marqué d’une ligne verticale pointillée; il s’agit de l’ES pour l’ensemble du parcours et est utilisé par le programme pour déterminer si le chemin est choisi et présenté comme un rugplot.

Figure 8: Exécution terminée de PAST Shiny. Veuillez cliquer ici pour afficher une version agrandie de cette figure.
{kind=link}

Figure 9: Image du chemin d’accès à partir de l’exécution terminée de PAST (ou téléchargée à partir de Shiny). Ce chiffre a été cité dans Thrash et al.10. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Discussion
L’un des principaux objectifs de PAST est d’apporter des analyses des voies métaboliques des données GWAS à un public plus large, en particulier pour les organismes non humains et non animaux. Les méthodes alternatives à PAST sont souvent des programmes en ligne de commande qui se concentrent sur les humains ou les animaux. La convivialité était l’un des principaux objectifs du développement de PAST, à la fois en choisissant de développer une application Shiny et en choisissant d’utiliser R et Bioconductor pour libérer l’application. Les utilisateurs n’ont pas besoin d’apprendre à compiler des programmes pour utiliser PAST.
Comme avec la plupart des types de logiciels d’analyse, les résultats de PAST ne sont aussi bons que les données d’entrée; si les données d’entrée présentent des erreurs ou sont mal formatées, PAST ne s’exécutera pas ou ne produira pas de résultats non informatifs. Il est essentiel de s’assurer que les données GWAS, les données LD, les annotations et les fichiers de chemins d’accès sont correctement formatés est essentiel pour recevoir une sortie correcte de PAST. PAST n’analyse que les marqueurs bi-alléliques et ne peut exécuter qu’un seul trait pour chaque ensemble de données d’entrée. En outre, les données GWAS produites par un mauvais génotypage ou un phénotypage incorrect ou imprécis ne sont pas non plus susceptibles de produire des résultats clairs ou reproductibles. PAST peut aider à l’interprétation biologique des résultats du GWAS, mais il est peu probable qu’il clarifie les ensembles de données chaotiques si la variation environnementale, l’erreur expérimentale ou la structure de la population n’ont pas été correctement prises en compte.
Les utilisateurs peuvent choisir de modifier certains paramètres de l’analyse, à la fois dans l’application Shiny et en transmettant ces paramètres aux fonctions de PAST dans la console R. Ces paramètres peuvent modifier les résultats rapportés par PAST, et les utilisateurs doivent faire attention lorsqu’ils les modifient à partir des valeurs par défaut. Étant donné que la LD est mesurée par les utilisateurs, généralement à l’aide du même ensemble de données de marqueurs qui a également été utilisé dans le GWAS, les mesures de LD sont spécifiques à la population. Pour toutes les études, en particulier pour les espèces autres que le maïs (en particulier les espèces autopolinisatrices, polyploïdes ou très hétérogènes), des changements dans les valeurs par défaut peuvent être justifiés.
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Aucun.
matériels
| Name | Company | Catalog Number | Comments |
| Computer | NA | NA | Any computer with 8GB RAM should be sufficient |
| R | R Project | NA | R 4.0 or greater is required to install from Bioconductor 3.11 |
Références
- Rafalski, J. Association genetics in crop improvement. Current Opinion in Plant Biology. 13 (2), 174-180 (2010).
- Yan, J., Warburton, M., Crouch, J. Association Mapping for Enhancing Maize (Zea mays L.) Genetic Improvement. Crop Science. 51 (2), 433-449 (2011).
- Xiao, Y., Liu, H., Wu, L., Warburton, M., Yan, J. Genome-wide Association Studies in Maize: Praise and Stargaze. Molecular Plant. 10 (3), 359-374 (2017).
- Wang, K., Li, M., Bucan, M. Pathway-Based Approaches for Analysis of Genomewide Association Studies. The American Journal of Human Genetics. 81 (6), 1278-1283 (2007).
- Weng, L., et al. SNP-based pathway enrichment analysis for genome-wide association studies. BMC Bioinformatics. 12 (1), 99(2011).
- Tang, J., Perkins, A., Williams, W., Warburton, M. Using genome-wide associations to identify metabolic pathways involved in maize aflatoxin accumulation resistance. BMC Genomics. 16 (1), 673(2015).
- Warburton, M., et al. Genome-Wide Association Mapping of Aspergillus flavus and Aflatoxin Accumulation Resistance in Maize. Crop Science. 55 (5), 1857-1867 (2015).
- Warburton, M., et al. Genome-Wide Association and Metabolic Pathway Analysis of Corn Earworm Resistance in Maize. The Plant Genome. 11 (1), 170069(2018).
- Li, H., Thrash, A., Tang, J., He, L., Yan, J., Warburton, M. Leveraging GWAS data to identify metabolic pathways and networks involved in maize lipid biosynthesis. The Plant Journal. 98 (5), 853-863 (2019).
- Thrash, A., Tang, J., DeOrnellis, M., Peterson, D., Warburton, M. PAST: The Pathway Association Studies Tool to Infer Biological Meaning from GWAS Datasets. Plants. 9 (1), 58(2020).
- Adam, T., Mason, D. PAST: Pathway Association Study Tool (PAST). Bioconductor version: Release (3.10). , (2020).
- Thrash, A., DeOrnellis, M. IGBB/PAST. , at https://github.com/IGBB/PAST (2019).
- Bradbury, P., et al. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 23 (19), 2633-2635 (2007).
- Subramanian, A., et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences U.S.A. 102, 15545-15550 (2005).
Erratum
Formal Correction: Erratum: A Pathway Association Study Tool for GWAS Analyses of Metabolic Pathway Information
Posted by JoVE Editors on 10/08/2021. Citeable Link.
An erratum was issued for: A Pathway Association Study Tool for GWAS Analyses of Metabolic Pathway Information. One of the affiliations was updated.
The second affiliation was updated from:
USDA-ARS Corn Host Plant Resistance Research Unit, Mississippi State University
to:
Corn Host Plant Resistance Research Unit, USDA-ARS
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.