
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
FLIM-FRET Mediciones de interacciones proteína-proteína en bacterias vivas.
En este artículo
Resumen
Aquí describimos un protocolo para caracterizar las interacciones proteína-proteína entre dos proteínas muy diferentes en Pseudomonas aeruginosa viva utilizando mediciones FLIM-FRET. El protocolo incluye construcciones de cepas de bacterias, inmovilización de bacterias, imágenes y rutinas de análisis de datos post-imágenes.
Resumen
Las interacciones proteína-proteína (IPP) controlan varios procesos clave en las células. La microscopía de imágenes de por vida de fluorescencia (FLIM) combinada con la transferencia de energía por resonancia de F.R. (FRET) proporciona información precisa sobre los IBP en células vivas. FLIM-FRET se basa en la medición de la descomposición de por vida de la fluorescencia de un donante FRET en cada píxel de la imagen FLIM, proporcionando información cuantitativa y precisa sobre los IBP y sus organizaciones celulares espaciales. Proponemos aquí un protocolo detallado para las mediciones FLIM-FRET que aplicamos para monitorear los IBP en Pseudomonas aeruginosa en vivo en el caso particular de dos proteínas que interactúan expresadas con números de copia muy diferentes para demostrar la calidad y robustez de la técnica para revelar características críticas de los IBP. Este protocolo describe en detalle todos los pasos necesarios para la caracterización de PPI, desde construcciones mutantes bacterianas hasta el análisis final utilizando herramientas desarrolladas recientemente que proporcionan posibilidades de visualización avanzadas para una interpretación directa de datos complejos de FLIM-FRET.
Introducción
Las interacciones proteína-proteína (IPP) controlan varios procesos clave en las células1. Las funciones de los IBP difieren en función de la composición de proteínas, las funciones de afinidades y las ubicaciones en las células2. Los IBP se pueden investigar a través de diferentes técnicas3. Por ejemplo, la co-inmunoprecipitación es una herramienta relativamente simple, robusta y económica de uso común para identificar o confirmar los IBP. Sin embargo, estudiar los IBP puede ser difícil cuando las proteínas que interactúan tienen niveles de expresión bajos o cuando las interacciones son transitorias o relevantes solo en entornos específicos. El estudio de los IBP que se produce entre las diferentes enzimas de la vía de la pyoverdina en P. aeruginosa requiere que la represión del represor general cofactorado del hierro Fur se libere para permitir que la expresión de todas las proteínas de la vía pyoverdina se exprese en la célula4,5,6. Esta regulación común para todas las proteínas de la vía resulta en expresiones oportunas en la célula que se espera que promuevan sus interacciones. La diversidad en términos de tamaño, naturaleza, niveles de expresión y el número de proteínas de esta vía metabólica dificultan el estudio en sistemas reconstituidos6. Por lo tanto, explorar los IBP en su entorno celular es fundamental para comprender mejor las funciones biológicas de las proteínas en su contexto nativo.
Sólo unos pocos métodos, incluyendo la fluorescencia, permiten explorar los IBP en las células vivas7. Entre los diferentes parámetros de fluorescencia que se pueden medir, la vida útil de la fluorescencia (es decir, el tiempo promedio que un fluoróforo permanece en su estado excitado antes de emitir un fotón) es probablemente uno de los parámetros más interesantes para explorar en las células vivas. La vida útil de la fluorescencia de un fluoróforo es altamente sensible a su entorno y, por lo tanto, FLIM puede proporcionar información química o física sobre el entorno del fluoróforo8. Esto incluye la presencia de la transferencia de energía por resonancia de la resonancia (FRET) que puede ocurrir en presencia de un "aceptador" de fluorescencia situado a una corta distancia de un "donante" de fluorescencia. La transferencia de energía da como resultado un acortamiento significativo de la vida útil de la fluorescencia del donante(Figura 1A),haciendo de la Microscopía de Imagen de Por Vida de Fluorescencia (FLIM) un enfoque poderoso para explorar las interacciones proteína-proteína directamente en las células vivas. FLIM también puede proporcionar información espacial sobre dónde tienen lugar las interacciones en las celdas7,8. Este enfoque es extremadamente eficaz para investigar los IBP en situaciones en las que es posible etiquetar con fluoróforos de los dos socios que interactúan.
Para que se produzca FRET - se requieren condiciones críticas en la distancia entre dos fluoróforos8,9. Los dos fluoróforos no deben estar distantes entre sí por más de 10 nm. Por lo tanto, se deben tomar precauciones al diseñar experimentos FLIM-FRET para asegurar que el donante y el aceptador de la fluorescencia tengan la oportunidad de ubicarse cerca uno del otro en el complejo que interactúa. Si bien esto puede parecer restricnte, de hecho es una verdadera ventaja, ya que la dependencia a distancia de FRET garantiza que dos proteínas etiquetadas sometidas a FRET tienen que interactuar físicamente (Figura 1A). Por lo tanto, las dificultades para obtener respuestas claras sobre el IBP en experimentos de colocación (dos proteínas colocadas pueden no interactuar necesariamente) no son un problema al usar FLIM-FRET.

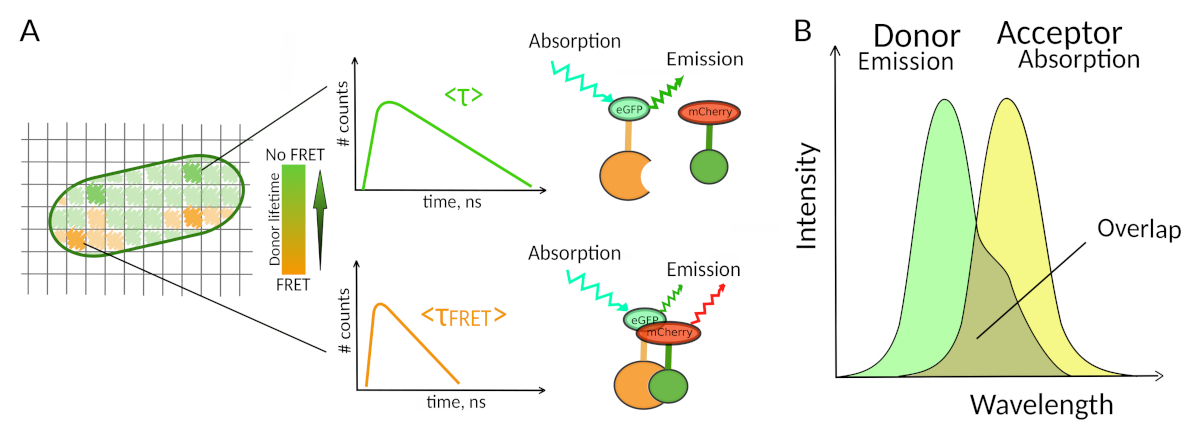
Figura 1: Principio de análisis FLIM-FRET. Cada píxel de la imagen multidimensional FLIM-FRET contiene información sobre la descomposición de la fluorescencia registrada en esta ubicación en particular (#counts número de fotones detectados en el canal t). (A) La representación clásica de la imagen FLIM suele ser una imagen 2D codificada de duración de color falso (izquierda). Una disminución en la vida media de fluorescencia del donante - como se ve por un cambio en la escala de color - se puede observar en presencia de FRET y es informativo sobre la presencia de IBP en esta área espacial. (B) La superposición entre el espectro de emisión del donante y el espectro de absorción del aceptador es necesaria para que se produzca el FRET. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Un segundo requisito para el FRET es que el espectro de emisión del donante y los espectros de absorción del aceptador se superpongana 8 (Figura 1B). La excitación fluorescencia del donante debe estar en longitudes de onda que contribuyan muy poco a la excitación directa de fluorescencia del aceptador. No todas las combinaciones de fluoróforos son posibles y además recomendamos utilizar preferentemente donantes con decaimientos monoexponenciales de fluorescencia para facilitar las interpretaciones FLIM-FRET10. Varias parejas de proteínas de fluorescencia cumplen con estos requisitos, incluyendo la popular pareja eGFP-mCherry11 (para una revisión sobre la paleta de pares de proteínas fluorescentes disponibles FRET ver12,13).
FLIM-FRET permite medir la descomposición de por vida de la fluorescencia de un donante FRET en cada píxel de una imagen FLIM (Figura 1A). Hay dos técnicas principales para determinar la vida útil de la fluorescencia que difieren en la adquisición y el análisis: dominio de frecuencia (FD)14 y dominio de tiempo (TD). TD FLIM es más extendido y se realiza utilizando una iluminación pulsada combinada con diferentes configuraciones de detección posibles, incluidos los métodos de medición15,la cámara de rayas16 o las técnicas de recuento de fotones individuales (TCSPC) correlacionadas en el tiempo8. Para las técnicas de DF y TD, la vida útil de la fluorescencia no se mide directamente, pero requiere un análisis de los datos medidos para estimar la duración o la presencia de interacciones. Para las técnicas TCSPC, el análisis más utilizado se basa en el ajuste de los decaimientos con funciones exponenciales únicas o múltiples utilizando re-convoluciones iterativas menos cuadradas que minimizan la suma ponderada de los residuos.
Por último, FLIM-FRET se puede realizar mediante excitaciones de un solo fotón o multifotón. Los últimos tienen varias ventajas como reducir la autofluorescencia y fotodago fuera del plano focal. Las excitaciones multifotofoto permiten también una profundidad de excitación más larga si se trabaja en muestras 3D gruesas8. Por el contrario, la excitación de un solo fotón suele ser más eficiente, ya que las secciones transversales de absorción de dos fotones de las proteínas fluorescentes están limitadas17.
Aquí, proponemos un protocolo para las mediciones FLIM-FRET de los IBP en vivo P. aeruginosa en el caso particular de dos proteínas que interactúan (PvdA y PvdL) expresadas con un número muy diferente de copias para demostrar la calidad y robustez de la técnica para revelar características críticas de los IBP. Las proteínas PvdA y PvdL están implicadas en la biosíntesis de la pioverdina. PvdA es una L-ornitina N5-oxigenasa y sintetiza la L-N5-formyl-N5-hidroxiornitina de L-ornitina por hidroxilación (PvdA) y formilación (PvdF)18. PvdL es una enzima de síntesis de péptidos no ribosomales (NRPS) compuesta por cuatro módulos. El primer módulo cataliza la acilación del ácido mirístico. El segundo módulo cataliza la activación de L-Glu y su condensación al mirístico-coA. Entonces, el tercer módulo condensa un aminoácido L-Tyr que luego se isomeriza en D-Tyr. Finalmente, el cuarto módulo une un aminoácido L-Dab (ácido diaminobutírico) para formar el tripéptido acilado L-Glu/D-Tyr/L-Dab6. PvdL es así responsable de la síntesis de los tres primeros aminoácidos del precursor de la pyoverdina. La interacción de la proteína PvdA con PvdL es sorprendente ya que PvdL, al contrario de PvdI y PvdJ, no lleva un módulo específico para la L-N5-formyl-N5-hidroxiornitina. Esta interacción sugiere que todas las enzimas responsables de la biosíntesis precursora de la pyoverdina están dispuestas en grandes complejos multienzimáticos transitorios y dinámicos19,20.
En este informe explicamos en detalle cómo construir las cepas bacterianas expresando de forma nativa las dos proteínas eGFP y mCherry etiquetadas de forma nativa. También describimos la preparación de muestras y las condiciones para obtener imágenes celulares FLIM-FRET eficientes. Por último, proponemos un tutorial paso a paso para el análisis de imágenes que incluye una herramienta desarrollada recientemente que ofrece posibilidades de visualización avanzadas para la interpretación directa de datos complejos de FLIM-FRET. Con este informe, nos gustaría convencer no sólo a los aventureros, sino a la mayoría de los biólogos de que FRET-FLIM es una técnica accesible y poderosa capaz de abordar sus preguntas sobre los IBP directamente en el entorno celular nativo.
Protocolo
1. Construcción de Plásmido
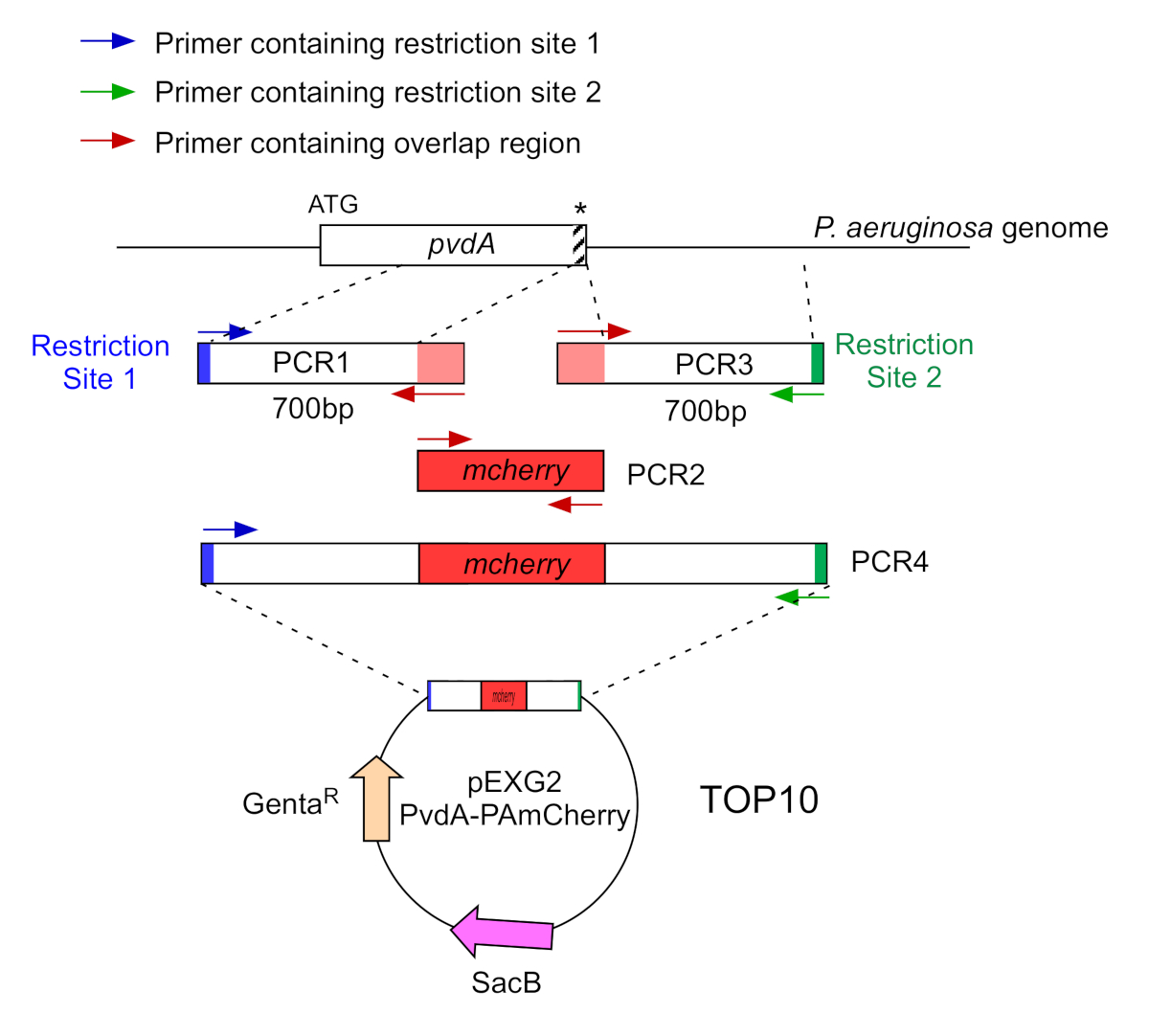
- Amplificar por dos PCR (PCR1 y 3) las secuencias de ADN (utilizar ADN genómico de P. aeruginosa PAO1) de los 700 pares base aguas arriba y aguas abajo de las regiones correspondientes al sitio de inserción en el genoma de P. aeruginosa con polimerasa de ADN de alta fidelidad. Agregue sitios de restricción a los imprimadores en azul y verde y agregue una secuencia superpuesta con mCherry a los imprimadores en rojo (Figura 2).
- Para PvdA etiquetado en el C-terminus con eGFP, amplificar la región de 700 bp aguas arriba en relación con el codón de parada por los imprimadores en azul, y amplificar la región aguas abajo de 700 bp que contiene el codón de parada con las imprimaciones en verde.
- Para PvdL etiquetado en el N-terminus con mCherry, amplificar la región de 700 bp aguas arriba al gen PvdL, incluyendo el codón de inicio, por las imprimaciones en azul, y amplificar la región descendente de 700 bp con las imprimaciones en verde.

Figura 2: Visión general de la estrategia de PCR y la construcción de plásmidos utilizados para la construcción de PvdA-mCherry. Ver texto para más detalles - pvdA codifica una enzima implicada en la biosíntesis de la sioverdina siderofore, un metabolito secundario implicado en la adquisición de hierro. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
- Amplifique el ADN de codificación eGFP (sin los codones de inicio y parada) con imprimaciones en rojo con polimerasa de ADN de alta fidelidad.
- Purificar los productos PCR en una columna de limpieza de PCR(Tabla de materiales).
- Mezclar productos de PCR superpuestos en relación equimolar y realizar una segunda PCR utilizando imprimaciones con el sitio de restricción utilizado para pcR 1 y 3 (verde y azul en la figura 2).
- Migre el producto PCR en gel TAE (Tris-Base Acetate EDTA pH 8.0), corte la banda correspondiente y extraiga el amplicon con un kit de limpieza PCR (Tabla de materiales).
NOTA: El protocolo se puede pausar aquí. - Digest PCR amplicon y plásmido pEXG2 utilizando las enzimas de restricción correspondientes21.
- Plásmido ligato e inserción con ligasa de ADN T4 utilizando 90 ng de plásmido y relación molecular 1:1 (plásmido:inserto).
- Transforme la construcción de plásmidos en células químicamente competentes E. coli TOP10 células mezclando el producto de ligadura y 100 l de TOP10. Incubar la mezcla de bacterias/plásmidos competentes sobre hielo durante 30 minutos antes de proceder con un choque térmico de 42oC durante 60 s. Luego, ponga el tubo en hielo durante 10 minutos.
- Añadir 1 ml de caldo de lysogeny (LB) a las bacterias e incubar a 37oC durante 1 h.
- Placa de bacterias de 100 l en agar LB que contiene 15 g/ml de gentamicina.
- Incubar durante la noche a 37oC.
- Examinar la presencia de la plaquita por colonia PCR: de una colonia transformadora aislada, recoger una cantidad minúscula de bacterias que se añadirán a una mezcla de PCR que contiene imprimaciones que se hibrida en el plásmido de tal manera que la presencia del amplión podría ser detectada ejecutando el producto en un gel de agarosa (DNA polimerasa). De la misma colonia utilizada para la PCR, transfiera una pequeña cantidad de bacterias en una placa fresca que contenga 15 g/ml de gentamicina para ser aislada y utilizada para la extracción de plásmido. Por último, aísle y purifique el plásmido(Tabla de Materiales)y verifique la plaquita por secuenciación.
- Almacene las bacterias TOP10 que contengan el plásmido en LB con un 20 % de glicerol en microtubo de 1,5 ml a -80 oC y el plásmido purificado a -20 oC en un tubo de 1,5 ml.
NOTA: El protocolo se puede pausar aquí
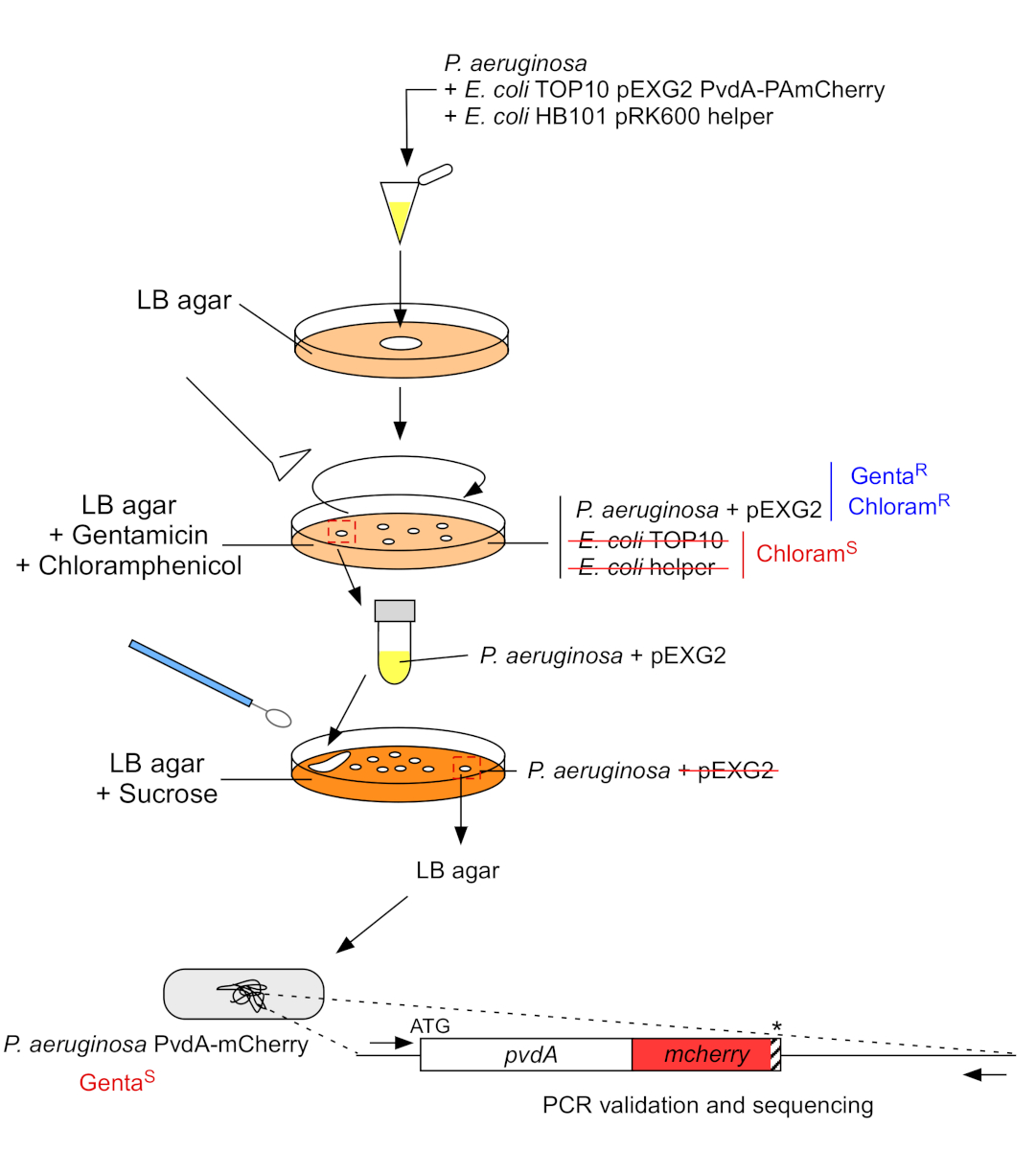
2. Inserción de etiquetas fluorescentes en el genoma cromosómico de P. aeruginosa (Figura 3)
- Cultivar P. aeruginosa,TOP10 y E. coli bacterias auxiliares, cada una en 5 ml de LB sin antibiótico a 30 oC bajo temblor orbital durante la noche22. Generar la inserción de etiquetas fluorescentes en el genoma de P. aeruginosa transfiriendo el plásmido de E. coli TOP10 a la cepa PAO1 e integrando el plásmido en el genoma mediante una recombinación homóloga. Un segundo evento de cruce que excita el vector genera el mutante correspondiente.
- Mida la densidad óptica a 600 nm (OD600 nm)del cultivo bacteriano y mezcle una cantidad igual de P. aeruginosa (500 l, OD600 nm a 1,0) con E. coli TOP10 pEXG2 (500 l, OD600 nm a 1,0) y E. coli HB101 pRK600 helper (500 l, OD600 nm a 1,0) en microtube de 1,5 mL.
- Centrifugar 5 min a 9.300 x g para peletizar las bacterias.
NOTA: Las herramientas en línea se pueden utilizar para convertir la fuerza g centrífuga a rotación por minuto (rpm) para ajustar la velocidad de la centrífuga. - Conservar el pellet bacteriano y desechar el sobrenadante.
- Resuspender el pellet que contiene bacterias en 50 l de LB.
- Encuado un punto (50 oL) de la mezcla en el centro del agar LB (precalentamiento a 37 oC) e incubar 5 h a 37 oC.
- Desguace el punto con un lazo de inoculación estéril y resuspend en 1 ml de LB.
- Placa 100 l de esta suspensión bacteriana en agar LB que contiene 10 g/ml de cloranfenicol para eliminar E. coli (E. coli TOP10 pEXG2 y E. coli HB101 pRK600 helper son sensibles al cloramphenicol, pero P. aeruginosa es naturalmente resistente) y 30 g/mL gentamicin e incubar 2 días a 37 oC.
- Resuspender una colonia en 1 ml de LB e incubar a 37 oC bajo temblor orbital 4h.
- Centrifugar 3 min a 9.300 x g y deseche 950 ml de sobrenadante. Resuspender el pellet en 50 l de LB y aislar la mezcla en agar LB que contiene sacarosa y sin NaCl.
- Incubar durante la noche a 30oC.
- Detectar colonias aisladas en el agar LB y el agar LB que contiene 15 mg/ml de gentamicina con el fin de comprobar la sensibilidad a la gentamicina.
- Verifique la inserción de eGFP o mCherry por colonias de PCR (DNA polimerasa) y la secuenciación utilizando imprimaciones específicas.

Figura 3: Protocolo de construcción de cepas P. aeruginosa mediante inserción de etiquetas fluorescentes. Consulte el texto para obtener más información. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
3. Medición de pyoverdina
- Cultivar bacterias en 5 ml de LB a 30 oC bajo temblor orbital durante la noche.
- Las bacterias del pellet por centrifugación, lavarlas y cultivarlas en 5 ml de SM (Succinate Medium, composición: 6 g-L1 K2HPO4, 3 g-L-1 KH2PO4, 1 g-L-1 (NH4)2 SO4, 0,2 g-L1 MgSO4, 7 H2O y 4g-L-1 succinato de sodio con el pH ajustado a 7.0 añadiendo NaOH) a 30 oC bajo temblor orbital durante la noche. SM es un medio de hierro privado - la ausencia de hierro activará la expresión de las proteínas de la vía de la pyoverdina normalmente reprimida en presencia de hierro.
- Mida OD600 nm y diluya de nuevo las bacterias en un medio SM fresco a600 nm a 0,1 Y escátelas a 30 oC bajo temblor orbital durante la noche.
- Mida OD600 nm para determinar la cantidad de bacterias en cada muestra.
- Preparar una cubeta de cuarzo que contenga 100 l de cultivo de P. aeruginosa y completa a 1 ml de SM (900 l). Preparar una cubeta de cuarzo que contenga 1 ml de medio SM (en blanco).
- Usando un espectrofotómetro visible UV, mida la absorbancia al máximo del pico de absorción. A pH 7,0, el máximo de absorción de pyoverdina se producirá a 400 nm. Determinar la concentración de pyoverdina (forma apo) en la muestra utilizando la ley Beer-Lambert utilizando un coeficiente de extinción molar a 400 nm de e a 19 000 M-1cm-1.
NOTA: Pyoverdine se puede cuantificar en el rango de absorbancia de 0,1 a 1 (dependiendo del espectrofotómetro UV-Visible) en el que la absorbancia aumenta linealmente con la concentración.
4. Cultivo de bacterias y condiciones para que las células expresen PvdA, PvdL y PvdJ
- El día 1, inocular un tubo con 5 ml de LB de la población de bacterias de glicerol apropiada y cultivar bacterias durante la noche a 30 oC a 200 rpm en una incubadora de agitador orbital.
- En el día 2, las células de pellets por centrifugación a 3.000 x g durante 3 min y desechar el sobrenadante.
- Resuspender las células en 10 ml de SM.
- Repita los pasos 4.2-4.3 una vez y haga crecer las bacterias en SM durante la noche a 30 oC 200 rpm.
- En el día 3, diluir 1/10 el cultivo de bacterias en SM fresco.
- Crecer bacterias diluidas de nuevo durante la noche en las mismas condiciones.
NOTA: La presencia de pyoverdina se puede detectar visualmente a medida que colorea en amarillo-verde los medios en crecimiento. Muestra que se ha activado la expresión de las proteínas de la vía de la pioverdina y que las enzimas de interés se expresan en las células.
5. Preparación de la almohadilla de agarosa (Figura 4)
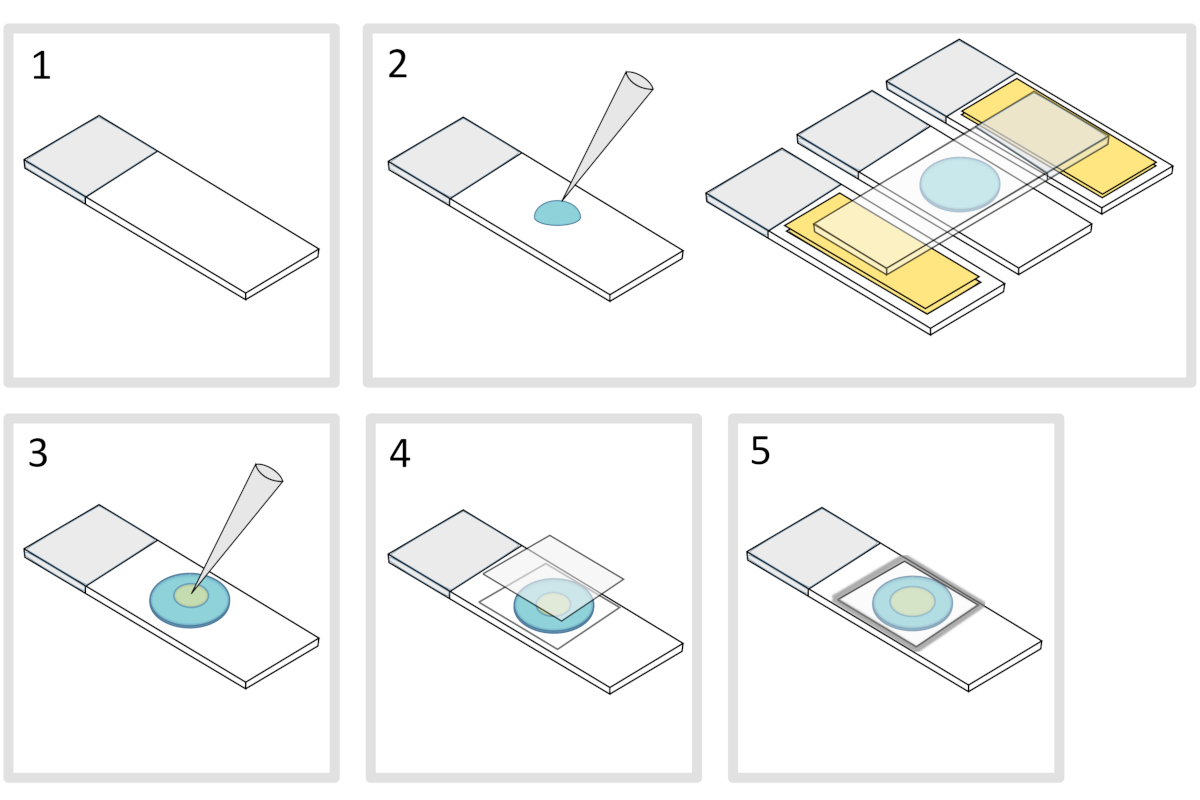
- Coloque un deslizamiento de vidrio del microscopio sobre una superficie horizontal plana. Coloque dos diapositivas de vidrio rematadas con dos capas de cinta adhesiva a cada lado de la diapositiva inicial.
NOTA: Mantenga un espacio de 1-2 mm entre las tres diapositivas alineadas para evitar que la agarosa meleta se propague finalmente en las diapositivas con cinta adhesiva. - Pipetear y verter una gota de 70 ml de 1% de agarosa derretida en el tobogán de vidrio. Añade una cuarta diapositiva en la parte superior para aplanar la gota de agarosa y presiona suavemente hacia abajo. Espera un minuto.
- Quítese el portaobjetos superior y suelte con una pipeta de aproximadamente 3 l de bacterias en 3 a 4 puntos en diferentes lugares de la almohadilla de agarosa.
- Cubra con un cubreobjetos de vidrio de microscopía (por ejemplo, un espesor de 22x22 mm #1,5).
NOTA: La planitud y el grosor de los cubreobjetos son importantes para trabajar con excitaciones de dos fotones. Los cubreobjetos de precisión con planitud uniforme controlada y baja autofluorescencia suelen ser una buena opción. - Fije el cubreobjetos con parafina fundida para sellar el cubreobjetos en el portaobjetos de vidrio. Comience fijando las cuatro esquinas del cubreobjetos.

Figura 4: Preparación de la almohadilla de agarosa. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
6. Imágenes con una configuración de microscopía de dos fotones
NOTA: Estamos utilizando un microscopio invertido de escaneo de excitación de dos fotones casero con un objetivo de inmersión en agua de 60x 1.2NA que funciona en modo de recolección de fluorescencia desencandada. La longitud de onda de excitación de dos fotones se establece en 930 nm. Es proporcionado por un láser Ti:Sapphire (tasa de repetición de 80 MHz, ≈ 70 fs de ancho de pulso) que funciona a 10-20 mW. Los fotones de fluorescencia se recogieron a través de un filtro de paso corto de 680 nm y un filtro de paso de banda de 525/50 nm antes de ser dirigidos a un fotodida de avalancha acoplada de fibra conectado a un módulo de recuento de fotones único (TCSPC) correlacionado con el tiempo. El microscopio también está equipado con una lámpara de fluorescencia de transmisión. Varios microscopios FLIM-FRET están ahora disponibles comercialmente y muchas instalaciones de imágenes están equipadas con configuraciones capaces de realizar mediciones FLIM-FRET.
- Utilice la lámpara de fluorescencia para centrar el objetivo en la monocapa de bacterias en la muestra y regiones de interés selectas.
- Compruebe que el obturador láser de excitación esté cerrado y que la luz infrarroja procedente del láser esté bloqueada y no entre en el microscopio.
Precaución: Se debe prestar atención cuidadosa y vigilancia constante trabajando con láseres pulsados IR, ya que la luz láser no puede ser vista por los ojos, pero cualquier exposición directa transitoria o reflexión láser puede ser extremadamente dañina y crear daños oculares irreversibles. Consulte los procedimientos locales de seguridad del láser y el entrenamiento antes de utilizar las configuraciones de microscopía. - Coloque la diapositiva de la microscopía en el escenario con los cubreobjetos frente al objetivo.
- Compruebe que la lámpara de fluorescencia esté encendida.
- Gire la torreta del cubo del filtro para seleccionar el cubo eGFP y abra el obturador de la lámpara de fluorescencia.
- Envíe la luz de fluorescencia hacia el ocular del microscopio.
Precaución: Asegúrese de que los filtros apropiados se desechan en el camino de la luz para descartar la luz de excitación directa procedente de la lámpara de fluorescencia que puede dañar los ojos. - Enfoque el objetivo en bacterias utilizando la perilla del microscopio.
- Seleccione una región de interés en la muestra traduciéndola utilizando el joystick que controla la etapa motorizada
NOTA: El enfoque es más fácil con una muestra altamente fluorescente que permite ver la fluorescencia directamente con los ojos. - Cambie la excitación del láser 2PE para mediciones FLIM-FRET.
- Envíe la ruta de emisión de fluorescencia hacia el detector.
- Gire hacia atrás la torreta del cubo de filtro para seleccionar el cubo dicroico para el láser de 930 nm.
- Ajuste la potencia del láser a 20 mW.
- Establezca el tamaño de la región de interés en 30 m. Esta operación ajusta la tensión que opera los espejos galvo y define el rango de sus movimientos (Figura 5).
- Encienda el detector y comience a escanear la muestra - los botones de inicio y parada que controlan el escaneo también controlan la apertura y el cierre del obturador láser tanto por razones de seguridad como para limitar el fotoblanquido de la muestra (Figura 5).

Figura 5: Representación esquemática de la interfaz del software de control del microscopio. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
- Si es necesario, ajuste el enfoque moviendo ligeramente la perilla de enfoque fino del microscopio.
- Elija el campo de visión de la imagen moviendo finamente el escenario desde la interfaz del ordenador. Esto se puede hacer en la configuración moviendo la cruz en la imagen en el software de control del microscopio (Figura 5) que definirá el nuevo centro de la imagen y presionando Mover etapa. Un buen campo de visión para la adquisición corresponde a una imagen con 10-30 bacterias inmóviles, todas correctamente enfocadas (todas las bacterias están en el mismo plano). Si está interesado en extraer datos FLIM-FRET de celdas individuales, asegúrese de que las bacterias estén bien individualizadas (la segmentación de imágenes será mucho más fácil).
- Abra el software SPCM (software comercial para la adquisición de datos) y compruebe que la tasa de recuento de fotones no es demasiado alta para evitar el efecto de acumulación que puede afectar a las mediciones de por vida. Si es necesario, baje la intensidad del láser para mantener la tasa de recuento de fotones baja (alrededor del 1% de la tasa de repetición láser).
NOTA: El efecto Pile-Up describe los efectos de los fotones perdidos a altas tasas de recuento de fotones debido al tiempo muerto de los dispositivos de recuento de fotones individuales (TCSPC) correlacionados por el tiempo. Si se produce Pile-Up, la vida media medida se acorta artificialmente con posiblemente un componente más corto adicional que puede aparecer en la descomposición debido al sobremuestreo de los fotones de emisión rápida. - Ajuste los parámetros de adquisición, incluido el tiempo de recogida de la adquisición (normalmente se requieren entre 60 s y 180 s para recopilar suficientes fotones).
- Pulse el botón Inicio y espere a que finalice la adquisición.
- Guarde los datos.
- Deje de escanear la muestra y apague el detector.
- Seleccione otro campo de visión en la muestra y repita los pasos 6.14-6.22 o imagine una nueva diapositiva de microscopía repitiendo los pasos 6.1-6.22.
NOTA: P. aeruginosa puede vivir y dividir hasta 6-8 horas a temperatura ambiente en la almohadilla de agarosa (correspondiente a por fin 4 veces el tiempo de duplicación a 20 oC). Idealmente, no espere demasiado tiempo para realizar la medición FLIM-FRET para evitar observar una almohadilla completamente cubierta de bacterias.
7. Análisis de datos

Figura 6: Panel principal de la ventana de análisis de datos del software SPCImage. Imagen de intensidad (caja azul), imagen de por vida (caja púrpura), histograma de por vida (arriba a la derecha), curva de decaimiento en la posición seleccionada (caja verde) y parámetros de decaimiento en la posición seleccionada (caja cian) de una descomposición pvdA-eGFP representativa registrada en vivo P. aeruginosa utilizando una tarjeta de adquisición bh SPC830 en una configuración de dos fotones de dos fotones de origen de tipo local.FLIM-FRET. La curva de descomposición experimental del píxel apuntado en la imagen anterior, su ajuste mono-exponencial (curva roja) desconvolucionando la decadencia de su función de respuesta instrumental calculada (curva verde) se puede ver en el panel verde. Haga clic aquí para ver una versión más grande de esta figura.
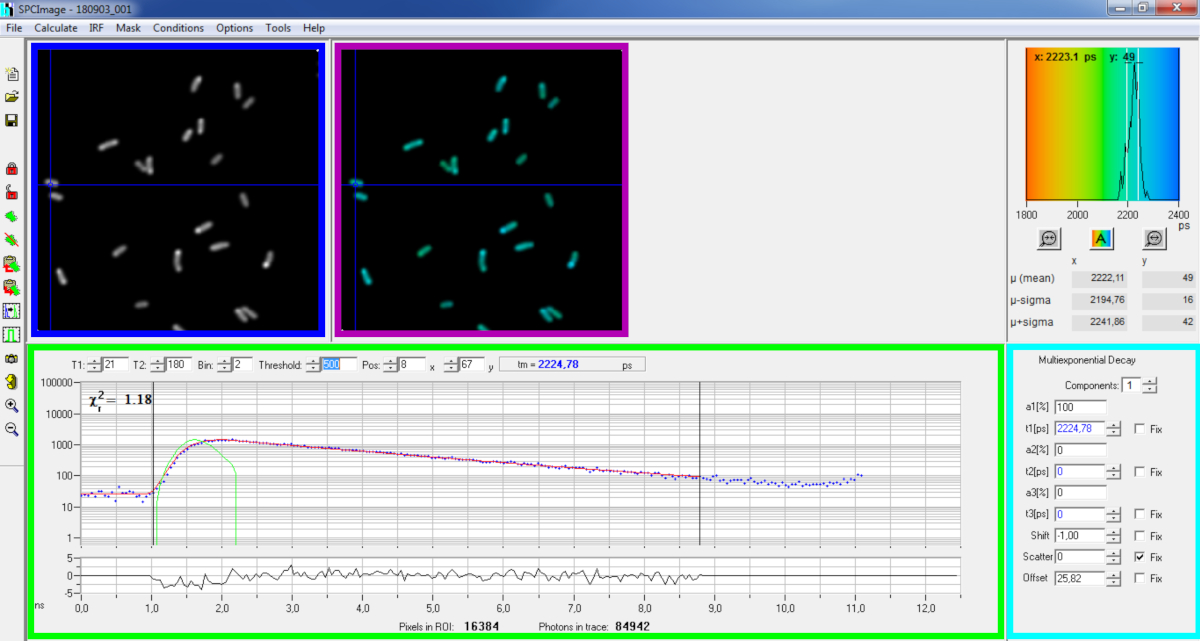{kind=link}
- Análisis básico
- Ejecute el software SPCImage.
- Importe el archivo SPCM guardado. La imagen de intensidad se muestra en el panel superior izquierdo del software(Figura 6 caja azul).
- Examine la ventana de curva de decaimiento (cuadro verdefigura 6) que muestra los datos de decaimiento correspondientes al píxel seleccionado en la imagen de intensidad (cuadro azulfigura 6). Los números de fotones de cada canal de tiempo se muestran como puntos azules y el ajuste de la descomposición se dibuja como una línea roja. Tenga en cuenta que después de cargar los datos, el software muestra el píxel más brillante de la imagen. Mueva la cruz azul a través de la imagen para examinar los píxeles con menor intensidad. La ventana de descomposición se actualizará automáticamente en cada nueva posición de píxel.
NOTA: Medir la función de respuesta instrumental (IRF) de un sistema de escaneo láser es muy difícil. Un IRF calculado a partir del borde ascendente de las curvas de decaimiento de fluorescencia en los datos FLIM se puede utilizar para la desconvolución de descomposición. Esta es la opción realizada de forma predeterminada en SPCimage(Figura 6 curva verde). - Ajuste el rango de ajuste moviendo los canales inicial y final de la caja de ajuste (T1 y T2 en el cuadro verde). T1 debe comenzar en los primeros canales de la descomposición creciente y T2 definir el último canal al final de la decadencia y se puede elegir como uno de los últimos canales de la decadencia con un número de fotones por encima del desplazamiento de recuento de fotones (es decir, los niveles de fotones contados antes de que la decadencia se levante).
- Elija el binning cambiando el valor Bin. La decaimiento de la curva integra el recuento de fotones del píxel seleccionado junto con un área de i píxeles alrededor de la posición del cursor definida por el parámetro bin (aumentar el binning aumentará el número de fotones en la decadencia y puede ser útil alcanzar los recuentos de fotones requeridos para los modelos multi exponenciales).
- Ajuste el valor de Umbral. Los píxeles que no tengan al menos un canal con un número de fotones superior al valor de umbral no se incluirán en el procedimiento de ajuste. Por supuesto, cuanto mayor sea el número de píxeles que se ajusten, más largo será el análisis.
NOTA: Los datos FLIM pueden contener un gran número de píxeles y canales de tiempo. Las últimas versiones del software permiten el uso de GPU (Unidad de procesador de gráficos) para procesar un gran número de píxeles en paralelo, lo que reduce masivamente los tiempos de procesamiento. Puede ser interesante ajustar los parámetros de binning y umbral utilizando imágenes correspondientes a construcciones bacterianas que presentan la menor intensidad de fluorescencia (por ejemplo, con cepas bacterianas con los niveles de expresión más bajos). Esto garantizará que los decaimientos pertinentes observados en estas muestras cumplan los criterios de filtrado y se incluirán en el análisis. Estos parámetros se pueden utilizar para todas las imágenes. - Ajuste, si es necesario, los parámetros de decaimiento (caja cian). Deje que el desplazamiento varíe, la mayor parte del tiempo dispersión y desplazamiento se puede fijar a cero si un vistazo a las funciones de descomposición muestra que su contribución es insignificante. El desplazamiento se puede estimar mirando los primeros canales de la caries - tenga en cuenta que la imagen durante mucho tiempo debido a la baja fluorescencia en la muestra generalmente resulta en desplazamiento distinto de cero. La dispersión se produce principalmente en muestras gruesas y puede considerarse insignificante de lo contrario.
- Antes de ejecutar el ajuste, seleccione el algoritmo de ajuste. Abra la ventana de configuración del algoritmo en Mostrar/ocultar opciones de modelo. Seleccione el algoritmo de estimación de máxima verosimilitud (MLE) (Figura 7A).
- Ejecute el ajuste de la imagen haciendo clic en Calcular | matriz de descomposición. Una vez completada, la imagen FLIM codificada de por vida aparece en el panel de imágenes de por vida(figura 6 caja púrpura).
NOTA: En la ventana de curva de descomposición (cuadro verdefigura 6) es posible ver el valor de duración que corresponde a cada píxel de la imagen moviendo la cruz azul.
NOTA: para procesar un gran número de archivos de datos FLIM similares automáticamente, se puede utilizar un modo de procesamiento por lotes. - Compruebe la calidad del ajuste mirando los residuos (idealmente distribuidos aleatoriamente alrededor de 0) y un valor cuadrado de Chi cercano a 1.
- Los datos ajustados se pueden exportar en diferentes formatos. Para exportar archivos en archivos txt, vaya a Archivo | Exportar. En la ventana Opciones de exportación (Figura 7B), elija Seleccionar todo y, a continuación, haga clic en Exportar.
- Finalmente guarde el archivo de análisis. Los archivos de análisis se guardan como archivos *.img y se pueden volver a abrir directamente en SPCImage.
NOTA: En casos particulares de cantidades de donantes/receptores desequilibradas, FLIM-FRET puede revelar subpúmelos en una mezcla de complejos proteicos que interactúan, en particular cuando las concentraciones de los dos socios son muy diferentes, lo que resulta en mezclas de especies complejas y libres. Las especies no interactuantes (caracterizadas por una caries muy similar a la única caries del donante) pueden ser discriminadas por las interactúas asumiendo una invariancia espacial de los componentes de la vida útil del donante en todo el conjunto de datos. Del mismo modo, pueden formarse complejos que interactúen no estequiométricos con más donantes o más aceptadores de fluorescencia. Las descomposiciones de fluorescencia de tales complejos suelen ser difíciles de interpretar. Una gráfica de diagrama FLIM se puede utilizar para proporcionar información crítica sobre la estequiometría y el modo de enlace de los IBP20,23. La gráfica de diagrama FLIM es una representación gráfica del componente de vida útil más corta en función de su amplitud. Se puede utilizar para visualizar píxeles con firmas de decaimiento similares. Para dibujar tales representaciones, las decaimientos experimentales de fluorescencia tienen que ser equipadas con un modelo exponencial de dos. Los siguientes pasos pueden ser una guía a través de este proceso. - Comience analizando los datos de la única construcción del donante. Permitirá determinar el valor de por vida del donante. Idealmente, mida este valor en varias imágenes grabadas en las mismas condiciones que las construcciones de donante/aceptador para recuperar un valor sólido de por vida para el donante.
- Una vez determinado, ajuste las caries de fluorescencia de las construcciones de donante/aceptador con un modelo de dos exponenciales. En el cuadro de parámetro de decaimiento cian (Figura 6), establezca el número de componentes en 2. Corrija el parámetro t2(ps) al valor de vida útil robusto del donante determinado en el paso 1 y marque la casilla para fijar este parámetro.
NOTA: Es importante fijar la vida útil de larga duración de 2 para limitar el ajuste excesivo, mejorar la convergencia de ajuste y obtener parámetros de ajuste birádica más fiables24,25,26. - Guarde el archivo *img y exporte los datos como archivos *.asc como en el paso 7.1.11.

Figura 7: (A) Configuración del algoritmo para ajustar los decaimientos con modelos exponenciales. Selección de MLE (algoritmo de máxima verosimilitud o estimación de máxima verosimilitud, MLE) como modelo de ajuste y ventana de opciones de exportación (B). Haga clic aquí para ver una versión más grande de esta figura.
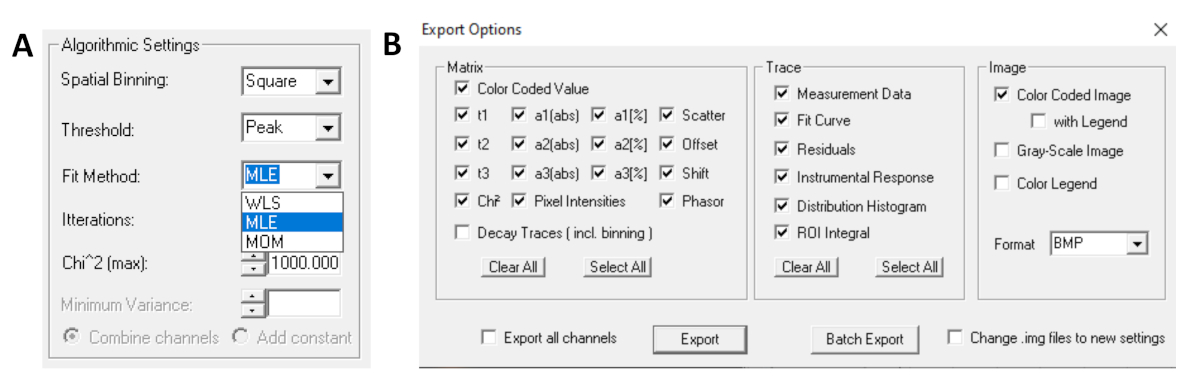{kind=link}
- Análisis avanzado de las imágenes FLIM en R
- Instale R (https://cran.r-project.org) y RStudio (https://rstudio.com) si es necesario.
- Abra RStudio y cree un nuevo proyecto.
- Mueva todo el archivo *.asc de análisis en una carpeta denominada "datos" en la carpeta principal del proyecto.
- Abra un nuevo archivo de script (o abra el script complementario FLIM_analysis. R).
- Instale el paquete flimDiagRam dedicado para el análisis de datos en https://github.com/jgodet/flimDiagRam. Llame al paquete en el área de trabajo. (Véase el aviso HowTo_FlimDiagRam)
NOTA: La instalación de paquetes debe realizarse una sola vez. Una vez instalados, los paquetes se pueden llamar desde cualquier nueva sesión de R. La descarga de paquetes de R desde github requiere la instalación del paquete 'devtools'. La instalación de 'devtools' puede tardar varios minutos. El paquete flimDiagRam se puede utilizar para representar los parámetros y distribuciones de los datos FLIM, para extraer datos FLIM a nivel de celdas individualizadas individualizadas individualizadas únicas, para comparar los resultados de FLIM entre condiciones o cepas y para explorar los datos FLIM utilizando herramientas de visualización avanzadas como la gráfica del diagrama FLIM. - Utilice el código comentado paso a paso y los datos se ponen a disposición para reproducir de forma independiente todas las sub cifras presentadas en la sección Resultados representativos a continuación. Este tutorial se puede encontrar en el aviso HowTo_FlimDiagRam en https://github.com/jgodet/flimDiagRam/blob/master/HowTo.pdf. El código se puede transponer fácilmente para analizar los datos.
Resultados
Las funciones de distribución acumulativa empírica (ecdf) de la vida útil de la fluorescencia medidas para las diferentes cepas bacterianas se muestran en la Figura 8. Si se produce FRET, los ecdfs se desplazan hacia la vida útil más corta(Figura 8A,8B). Tenga en cuenta que cuando la interacción de las dos proteínas da como resultado una larga distancia entre los dos fluoróforos, no puede ocurrir fret (Figura 8C
Discusión
FLIM-FRET ofrece algunas ventajas clave sobre las imágenes FRET basadas en la intensidad. La vida útil de la fluorescencia es un parámetro intrínseco del fluoróforo. Como consecuencia, no depende de las concentraciones locales de fluoróforos ni de la intensidad de la excitación de la luz. La vida útil de la fluorescencia también se ve mal afectada por el foto-blanqueo. Es particularmente interesante evidenciar IBP en células donde las concentraciones de proteínas locales pueden ser altamente heterogéneas en t...
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Reconocemos al Dr. Ludovic Richert por su valiosa asistencia en la adquisición de datos FLIM y por el mantenimiento técnico y desarrollo de la configuración de FLIM. Esta obra fue financiada por subvenciones de la Fundación para la Recherche en Chimie (https://icfrc.fr/). VN está financiado por la Fondation pour la Recherche Médicale (FRM-SPF201809006906). YM agradece al Institut Universitaire de France (IUF) por su apoyo y por su tiempo adicional para dedicarse a la investigación. IJS y JG reconocen al Instituto de Entrega de Drogas de Estrasburgo por su apoyo financiero.
Materiales
| Name | Company | Catalog Number | Comments |
| 525/50 nm band-pass filter | F37-516, AHF, Germany | ||
| 680 nm short pass filter | F75-680, AHF, Germany | ||
| Agarose | Sigma-Aldrich | A9539 | |
| Ammonium Sulfate (NH4)2SO4 | Sigma-Aldrich | A4418 | |
| DreamTaq DNA polymerase 5U/μL | ThermoFisher Scientific | EP0714 | |
| E. coli TOP10 | Invitrogen | C404010 | |
| Fiber-coupled avalanche photo-diode | SPCM-AQR-14- FC, Perkin Elmer | ||
| Glass coverslips (Thickness No. 1.5, 20×20mm | Knitel glass | MS0011 | |
| High-Fidelity DNA polymerase Phusion 2U/μL | ThermoFisher Scientific | F530S | |
| Lysogeny broth (LB) | Millipore | 1.10285 | |
| Magnesium Sulfate Heptahydrate (MgSO4 . 7H2O) | Sigma-Aldrich | 10034-99-8 | |
| Microscope slides (25×75mm) | Knitel glass | MS0057 | |
| NucleoSpin Gel and PCR Clean-up | Macherey-Nagel | 740609.50 | |
| NucleoSpin Plasmid | Macherey-Nagel | 740588.10 | |
| Potassium Phosphate Dibasic (K2HPO4) | Sigma-Aldrich | RES20765 | |
| Potassium Phosphate Monobasic (KH2PO4) | Sigma-Aldrich | P5655 | |
| Sodium Succinate (Disodium) | Sigma-Aldrich | 14160 | |
| SPCImage, SPCM software | Becker & Hickl | ||
| Sterile inoculating loop | Nunc | 7648-1PAK | |
| T4 DNA ligase 1U/μL | ThermoFisher Scientific | 15224017 | |
| TCSPC module | SPC830, Becker & Hickl, Germany | ||
| Ti:Sapphire laser | Insight DeepSee, Spectra Physics | ||
| Tubes 50mL | Falcon | 352070 |
Referencias
- Braun, P., Gingras, A. C. History of protein-protein interactions: From egg-white to complex networks. Proteomics. 12, 1478-1498 (2012).
- Nooren, I. M. A., Thornton, J. M. Structural characterisation and functional significance of transient protein-protein interactions. Journal of Molecular Biology. 325, 991-1018 (2003).
- Hayes, S., Malacrida, B., Kiely, M., Kiely, P. A. Studying protein-protein interactions: Progress, pitfalls and solutions. Biochemical Society Transactions. 44, 994-1004 (2016).
- Guillon, L., Altenburger, S., Graumann, P. L., Schalk, I. J. Deciphering protein dynamics of the siderophore pyoverdine pathway in Pseudomonas aeruginosa. PLoS ONE. 8, 1-9 (2013).
- Ringel, M. T., Brüser, T. The biosynthesis of pyoverdines. Microbial Cell. 5, 424-437 (2018).
- Schalk, I. J., Rigouin, C., Godet, J. An overview of siderophore biosynthesis among fluorescent Pseudomonads and new insights into their complex cellular organization. Environmental Microbiology. 22, 1447-1466 (2020).
- Cui, Y., et al. Techniques for detecting protein-protein interactions in living cells: principles, limitations, and recent progress. Science China Life Sciences. , (2019).
- Day, R. N., Mazumder, N., Sun, Y., Christopher, K. G. FRET microscopy: Basics, issues and advantages of FLIM-FRET imaging. Springer Series in Chemical Physics. 111, 249-276 (2015).
- Bastiaens, P. I. H., Squire, A. Fluorescence lifetime imaging microscopy: Spatial resolution of biochemical processes in the cell. Trends in Cell Biology. 9, 48-52 (1999).
- Yasuda, R. Imaging spatiotemporal dynamics of neuronal signaling using fluorescence resonance energy transfer and fluorescence lifetime imaging microscopy. Current Opinion in Neurobiology. 16, 551-561 (2006).
- Tramier, M., Zahid, M., Mevel, J. -. C., Masse, M. -. J., Coppey-Moisan, M. Sensitivity of CFP/YFP and GFP/mCherry Pairs to Donor Photobleaching on FRET Determination by Fluorescence Lifetime Imaging Microscopy in Living Cells. Microscopy Research and Technique. 71, 146-157 (2008).
- Bajar, B. T., Wang, E. S., Zhang, S., Lin, M. Z., Chu, J. A guide to fluorescent protein FRET pairs. Sensors (Switzerland). 16, 1-24 (2016).
- Piston, D. W., Kremers, G. J. Fluorescent protein FRET: the good, the bad and the ugly. Trends in Biochemical Sciences. 32, 407-414 (2007).
- Leray, A., et al. Optimized protocol of a frequency domain fluorescence lifetime imaging microscope for fret measurements. Microscopy Research and Technique. 72, 371-379 (2009).
- Elson, D. S., et al. Real-time time-domain fluorescence lifetime imaging including single-shot acquisition with a segmented optical image intensifier. New Journal of Physics. 6, 1-13 (2004).
- Rajoria, S., Zhao, L., Intes, X., Barroso, M. FLIM-FRET for Cancer Applications. Current Molecular Imaging. 3, 144-161 (2014).
- Drobizhev, M., Makarov, N. S., Tillo, S. E., Hughes, T. E., Rebane, A. Two-photon absorption properties of fluorescent proteins. Nature Methods. 8, 393-399 (2011).
- Visca, P., Ciervo, A., Orsi, N. Cloning and nucleotide sequence of the pvdA gene encoding the pyoverdin biosynthetic enzyme L-ornithine N5-oxygenase in Pseudomonas aeruginosa. Journal of Bacteriology. 176, 1128-1140 (1994).
- Imperi, F., Visca, P. Subcellular localization of the pyoverdine biogenesis machinery of Pseudomonas aeruginosa: A membrane-associated 'siderosome'. FEBS Letters. 587, 3387-3391 (2013).
- Gasser, V., et al. In cellulo FRET-FLIM and single molecule tracking reveal the supra-molecular organization of the pyoverdine bio-synthetic enzymes in Pseudomonas aeruginosa. Quarterly Reviews of Biophysics. , 1-11 (2019).
- Rietsch, A., Mekalanos, J. J. Metabolic regulation of type III secretion gene expression in Pseudomonas aeruginosa. Molecular Microbiology. 59, 807-820 (2006).
- Herrero, M., De Lorenzo, V., Timmis, K. N. Transposon vectors containing non-antibiotic resistance selection markers for cloning and stable chromosomal insertion of foreign genes in gram-negative bacteria. Journal of Bacteriology. 172, 6557-6567 (1990).
- Godet, J., Mély, Y. Exploring protein-protein interactions with large differences in protein expression levels using FLIM-FRET. Methods and Applications in Fluorescence. 8, 014007 (2019).
- El Meshri, S. E., et al. Role of the nucleocapsid domain in HIV-1 gag oligomerization and trafficking to the plasma membrane: A fluorescence lifetime imaging microscopy investigation. Journal of Molecular Biology. 427, 1480-1494 (2015).
- Becker, W. The bh TCSPC Handbook. Scanning. , 1 (2010).
- Richert, L., Didier, P., de Rocquigny, H., Mély, Y. Monitoring HIV-1 protein oligomerization by FLIM FRET microscopy. Springer Series in Chemical Physics. , 111 (2015).
- Fereidouni, F., Blab, G. A., Gerritsen, H. C. Phasor based analysis of FRET images recorded using spectrally resolved lifetime imaging. Methods and Applications in Fluorescence. 2, (2014).
- Fereidouni, F., Gorpas, D., Ma, D., Fatakdawala, H., Marcu, L. Rapid fluorescence lifetime estimation with modified phasor approach and Laguerre deconvolution: a comparative study. Methods and Applications in Fluorescence. 5, 035003 (2017).
- Margineanu, A., et al. Screening for protein-protein interactions using Förster resonance energy transfer (FRET) and fluorescence lifetime imaging microscopy (FLIM). Scientific Reports. 6, (2016).
- Guzmán, C., Oetken-Lindholm, C., Abankwa, D. Automated High-Throughput Fluorescence Lifetime Imaging Microscopy to Detect Protein-Protein Interactions. Journal of Laboratory Automation. 21, 238-245 (2016).
- Liu, W., Cui, Y., Ren, W., Irudayaraj, J. Epigenetic biomarker screening by FLIM-FRET for combination therapy in ER+ breast cancer. Clinical Epigenetics. 11, 1-9 (2019).
- Liu, X., et al. Fast fluorescence lifetime imaging techniques: A review on challenge and development. Journal of Innovative Optical Health Sciences. 12, 1-27 (2019).
- Padilla-Parra, S., Auduge, N., Coppey-Moisan, M., Tramier, M. Non fitting based FRET-FLIM analysis approaches applied to quantify protein-protein interactions in live cells. Biophysical Reviews. 3, 63-70 (2011).
- Padilla-Parra, S., Audugé, N., Coppey-Moisan, M., Tramier, M. Quantitative FRET analysis by fast acquisition time domain FLIM at high spatial resolution in living cells. Biophysical Journal. 95, 2976-2988 (2008).
- Stringari, C., et al. Phasor approach to fluorescence lifetime microscopy distinguishes different metabolic states of germ cells in a live tissue. Proceedings of the National Academy of Sciences of the United States of America. 108, 13582-13587 (2011).
- Digman, M. A., Caiolfa, V. R., Zamai, M., Gratton, E. The phasor approach to fluorescence lifetime imaging analysis. Biophysical Journal. 94, 14-16 (2008).
- Liang, Z., Lou, J., Scipioni, L., Gratton, E., Hinde, E. Quantifying nuclear wide chromatin compaction by phasor analysis of histone Förster resonance energy transfer (FRET) in frequency domain fluorescence lifetime imaging microscopy (FLIM) data. Data in Brief. 30, 105401 (2020).
- Grimm, J. B., Heckman, L. M., Lavis, L. D. The chemistry of small-molecule fluorogenic probes. Progress in Molecular Biology and Translational Science. 113, (2013).
- Li, L., Sun, H. Next Generation of Small-Molecule Fluorogenic Probes for Bioimaging. Biochemistry. 59, 216-217 (2020).
- Yao, R., Ochoa, M., Yan, P., Intes, X. Net-FLICS: fast quantitative wide-field fluorescence lifetime imaging with compressed sensing - deep learning approach. Light: Science and Applications. 8, 1-7 (2019).
- Smith, J. T., et al. Fast fit-free analysis of fluorescence lifetime imaging via deep learning. Proceedings of the National Academy of Sciences of the United States of America. 116, 24019-24030 (2019).
- Yao, R., Ochoa, M., Intes, X., Yan, P. Deep compressive macroscopic fluorescence lifetime imaging. Proceedings - International Symposium on Biomedical Imaging. 2018, 908-911 (2018).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados