
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
FLIM-FRET Messungen von Protein-Protein-Wechselwirkungen in lebenden Bakterien.
In diesem Artikel
Zusammenfassung
Wir beschreiben hier ein Protokoll zur Charakterisierung von Protein-Protein-Wechselwirkungen zwischen zwei stark unterschiedlich exprimierenden Proteinen in lebenden Pseudomonas aeruginosa mit FLIM-FRET-Messungen. Das Protokoll umfasst Bakterienstammkonstruktionen, Bakterienimmobilisierung, Bildgebung und Nach-Imaging-Datenanalyseroutinen.
Zusammenfassung
Protein-Protein-Wechselwirkungen (PPIs) steuern verschiedene Schlüsselprozesse in Zellen. Fluoreszenz-Lebensdauer-Bildgebungsmikroskopie (FLIM) in Kombination mit Förster Resonanzenergietransfer (FRET) liefern genaue Informationen über PPI in lebenden Zellen. FLIM-FRET stützt sich auf die Messung des Fluoreszenz-Lebensdauerzerfalls eines FRET-Spenders an jedem Pixel des FLIM-Bildes und liefert quantitative und genaue Informationen über PPI und ihre räumlichen zellulären Organisationen. Wir schlagen hier ein detailliertes Protokoll für FLIM-FRET-Messungen vor, das wir zur Überwachung von PPI in lebenden Pseudomonas aeruginosa im speziellen Fall von zwei interagierenden Proteinen angewendet haben, die mit sehr unterschiedlichen Kopiernummern ausgedrückt werden, um die Qualität und Robustheit der Technik bei der Aufdeckung kritischer Merkmale von PPI zu demonstrieren. Dieses Protokoll beschreibt detailliert alle notwendigen Schritte für die PPI-Charakterisierung - von bakteriellen Mutantenkonstruktionen bis zur Endanalyse mit neu entwickelten Tools, die erweiterte Visualisierungsmöglichkeiten für eine einfache Interpretation komplexer FLIM-FRET-Daten bieten.
Einleitung
Protein-Protein-Wechselwirkungen (PPIs) steuern verschiedene Schlüsselprozesse in Zellen1. Die Rollen von PPI unterscheiden sich je nach Proteinzusammensetzung, Affinitätsfunktionen und Positionen in Zellen2. PPI können mit verschiedenen Techniken untersucht werden3. Beispielsweise ist Die Co-Immunpräzipitation ein relativ einfaches, robustes und kostengünstiges, häufig verwendetes Werkzeug zur Identifizierung oder Bestätigung von PPI. Das Studium von PPI kann jedoch eine Herausforderung sein, wenn die interagierenden Proteine einen niedrigen Expressionsgrad aufweisen oder wenn die Wechselwirkungen nur in bestimmten Umgebungen vorübergehend oder relevant sind. Die Untersuchung von PPIs, die zwischen den verschiedenen Enzymen des Pyoverdin-Signalwegs in P. aeruginosa auftreten, erfordert, dass die Unterdrückung des allgemeinen eisenkofaktorierten Repressors Fur entlastet wird, damit die Expression aller Proteine des Pyoverdinwegs in der Zelle4,5,6ausgedrückt werden kann. Diese gemeinsame Regulierung für alle Proteine des Pfades führt zu rechtzeitigen Ausdrücken in der Zelle, von der erwartet wird, dass sie ihre Wechselwirkungen fördert. Die Vielfalt in Umfang, Natur, Expressionsniveaus und die Anzahl der Proteine dieses Stoffwechselwegs erschweren die Untersuchung in rekonstituierten Systemen6. Die Erforschung von PPI in ihrer zellulären Umgebung ist daher entscheidend, um die biologischen Funktionen von Proteinen in ihrem nativen Kontext besser zu verstehen.
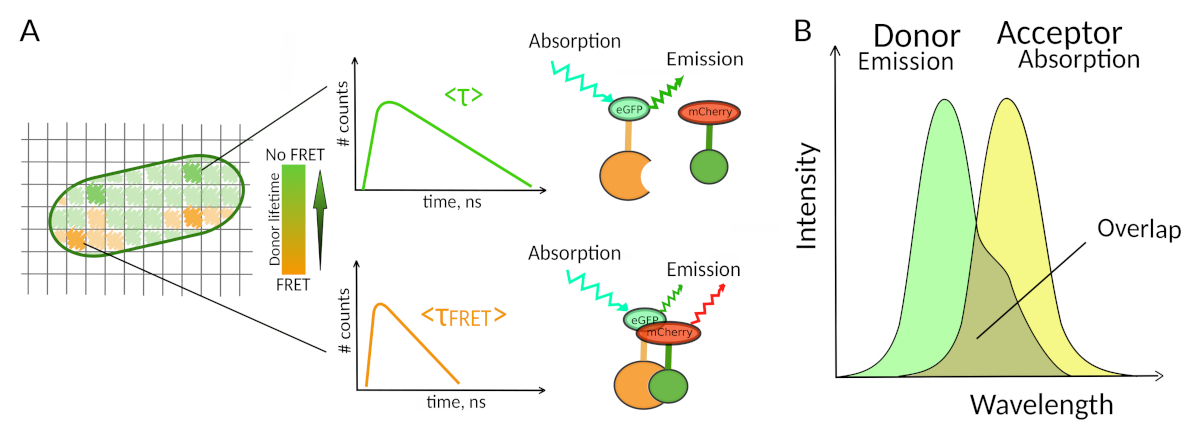
Nur wenige Methoden, einschließlich Fluoreszenz, erlauben die Untersuchung von PPI in lebenden Zellen7. Unter den verschiedenen Fluoreszenzparametern, die gemessen werden können, ist die Fluoreszenzlebensdauer (d. h. die durchschnittliche Zeit, in der ein Fluorophor in seinem angeregten Zustand verbleibt, bevor ein Photon emittiert wird) wahrscheinlich einer der interessantesten Parameter, um in lebenden Zellen zu erforschen. Die Fluoreszenzlebensdauer eines Fluorophors ist sehr umweltempfindlich und FLIM kann daher chemische oder physikalische Informationen über die Fluorophorumgebung8liefern. Dazu gehört auch das Vorhandensein von Förster Resonanzenergietransfer (FRET), die in Gegenwart eines "Akzeptors" der Fluoreszenz in kurzer Entfernung eines Fluoreszenz-"Spenders" auftreten kann. Energietransfer führt zu einer signifikanten Verkürzung der Spenderfluoreszenzlebensdauer (Abbildung 1A), was die Fluoreszenz-Lebensdauer-Bildgebungsmikroskopie (FLIM) zu einem leistungsstarken Ansatz zur Erforschung von Protein-Protein-Wechselwirkungen direkt in lebenden Zellen macht. FLIM kann zusätzlich räumliche Informationen darüber bereitstellen, wo die Wechselwirkungen in den Zellen7,8stattfinden. Dieser Ansatz ist äußerst leistungsfähig für die Untersuchung von PPI in Situationen, in denen die Kennzeichnung mit Fluorophoren der beiden interagierenden Partner möglich ist.
Damit FRET auftritt - kritische Bedingungen auf dem Abstand zwischen zwei Fluorophoren sinderforderlich 8,9. Die beiden Fluorophore sollten nicht um mehr als 10 nm voneinander entfernt sein. Daher ist bei der Entwicklung von FLIM-FRET-Experimenten Vorsicht geboten, um sicherzustellen, dass der Spender und der Fluoreszenzak eine Chance haben, sich im interagierenden Komplex nahe beieinander zu befinden. Obwohl dies einschränkend erscheinen mag, ist es in der Tat ein echter Vorteil, da die Entfernungsabhängigkeit von FRET sicherstellt, dass zwei markierte Proteine, die SICH FRET unterziehen, physisch interagieren müssen (Abbildung 1A). Die Schwierigkeiten, klare Antworten über PPI in Kolokalisierungsexperimenten zu erhalten (zwei kolokalisierte Proteine können nicht unbedingt interagieren) sind daher kein Problem mit FLIM-FRET.

Abbildung 1: FLIM-FRET-Analyseprinzip. Jedes Pixel des mehrdimensionalen FLIM-FRET-Bildes enthält Informationen über den fluoreszenzzerfall, der an dieser bestimmten Stelle aufgezeichnet wurde (#counts = Anzahl der erkannten Photonen im Kanal t). (A) Die klassische Darstellung des FLIM-Bildes ist in der Regel ein falschfarbiges, kodiertes 2D-Bild (links). Eine Abnahme der mittleren Fluoreszenzlebensdauer des Spenders - wie durch eine Änderung der Farbskala gesehen - kann in Gegenwart von FRET beobachtet werden und ist informativ über das Vorhandensein von PPI in diesem räumlichen Bereich. (B) Für FRET ist eine Überlappung zwischen dem Emissionsspektrum des Gebers und dem Absorptionsspektrum des Gebers erforderlich. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Eine zweite Voraussetzung für FRET ist, dass sich das Emissionsspektrum des Spenders und die Absorptionsspektren des Akzeptors überlappen 8 (Abbildung 1B). Die Fluoreszenzerregung des Spenders sollte bei Wellenlängen erfolgen, die sehr wenig zur direkten Fluoreszenzerregung des Akzeptors beitragen. Nicht alle Kombinationen von Fluorophoren sind möglich und wir empfehlen zusätzlich, bevorzugt Spender mit monoexponentiellen Fluoreszenzzerfallen zu verwenden, um FLIM-FRET-Interpretationen zu erleichtern10. Mehrere Paare von Fluoreszenzproteinen erfüllen diese Anforderungen, einschließlich des beliebten eGFP-mCherry-Paares11 (für einen Überblick über die Palette des verfügbaren fluoreszierenden Proteins FRET-Paare siehe12,13).
FLIM-FRET ermöglicht die Messung des Fluoreszenz-Lebensdauerzerfalls eines FRET-Spenders an jedem Pixel eines FLIM-Bildes (Abbildung 1A). Es gibt zwei Haupttechniken zur Bestimmung der Fluoreszenzlebensdauer, die sich bei der Erfassung und Analyse unterscheiden: Frequency-Domain (FD)14 und Time-Domain (TD). TD FLIM ist weiter verbreitet und wird mit einer gepulsten Beleuchtung in Kombination mit verschiedenen möglichen Detektionskonfigurationen durchgeführt, einschließlich Der Gating-Methoden15, Streifenkamera16 oder zeitkorrelierten Single Photon Counting (TCSPC) Techniken8. Sowohl bei FD- als auch bei TD-Techniken wird die Fluoreszenzlebensdauer nicht direkt gemessen, sondern erfordert eine Analyse der gemessenen Daten, um die Lebensdauer(en) oder das Vorhandensein von Wechselwirkungen zu schätzen. Bei TCSPC-Techniken beruht die am weitesten verbreitete Analyse auf der Anpassung der Zerfälle mit einzelnen oder multiexponentiellen Funktionen unter Verwendung der am wenigsten quadratischen iterativen Revolutionen, die die gewichtete Summe der Residuen minimieren.
Schließlich kann FLIM-FRET sowohl mit einzelnen Photonen- als auch mit Multiphotonen-Erregungen durchgeführt werden. Die neuesten haben mehrere Vorteile wie die Reduzierung von Autofluoreszenz und Photoschaden aus dem Brennflugzeug. Multiphotonen-Erregungen ermöglichen auch eine längere Anregungstiefe, wenn sie in dicken 3D-Probenarbeiten 8. Im Gegenteil, einzelne Photonenanregung ist in der Regel effizienter, da die zwei Photonen Absorption spirationsübergreifende von fluoreszierenden Proteinen begrenzt sind17.
Hier schlagen wir ein Protokoll für FLIM-FRET-Messungen von PPI in Live-P. aeruginosa im speziellen Fall von zwei interagierenden Proteinen (PvdA und PvdL) vor, die mit sehr unterschiedlicher Anzahl von Kopien ausgedrückt werden, um die Qualität und Robustheit der Technik bei der Aufdeckung kritischer Merkmale von PPI zu demonstrieren. PvdA- und PvdL-Proteine sind an der Pyoverdin-Biosynthese beteiligt. PvdA ist eine L-Ornithin-N5-Sauerstoffase und synthetisiert das L-N5-Formyl-N5-Hydroxyornithin aus L-Ornithin durch Hydroxylierung (PvdA) und Formylierung (PvdF)18. PvdL ist ein nicht-ribosomales Peptidsyntheseenzym (NRPS), das aus vier Modulen besteht. Das erste Modul katalysiert die Acylation von Myristinsäure. Das zweite Modul katalysiert die Aktivierung von L-Glu und dessen Kondensation auf das myristische CoA. Dann kondensiert das dritte Modul eine L-Tyr-Aminosäure, die dann in D-Tyr isomerisiert wird. Schließlich bindet das vierte Modul eine L-Dab (Diaminobuttersäure) Aminosäure zu dem acyatierten Tripeptid L-Glu/D-Tyr/L-Dab6. PvdL ist somit für die Synthese der drei ersten Aminosäuren des Pyoverdinvorläufers verantwortlich. Die Interaktion von PvdA-Protein mit PvdL ist überraschend, da PvdL im Gegensatz zu PvdI und PvdJ kein modulspezifisches Modul für das L-N5-Formyl-N5-Hydroxyornithin enthält. Diese Wechselwirkung legt nahe, dass alle Enzyme, die für die Pyoverdin-Vorläufer-Biosynthese verantwortlich sind, in großen transienten und dynamischen multienzymatischen Komplexen angeordnet sind19,20.
In diesem Bericht erklären wir ausführlich, wie man die Bakterienstämme konstruieren kann, die nativ die beiden interagierenden eGFP- und mCherry-markierten Proteine exzieren. Wir beschreiben auch die Probenvorbereitung und die Bedingungen für eine effiziente FLIM-FRET-Zellbildgebung. Schließlich schlagen wir ein Schritt-für-Schritt-Tutorial für die Bildanalyse vor, einschließlich eines kürzlich entwickelten Tools, das erweiterte Visualisierungsmöglichkeiten für die einfache Interpretation komplexer FLIM-FRET-Daten bietet. Mit diesem Bericht möchten wir nicht nur abenteuerlustige, sondern auch die meisten Biologen davon überzeugen, dass FRET-FLIM eine zugängliche und leistungsfähige Technik ist, die in der Lage ist, ihre Fragen zu PPI direkt in der nativen zellulären Umgebung zu beantworten.
Protokoll
1. Plasmidkonstruktion
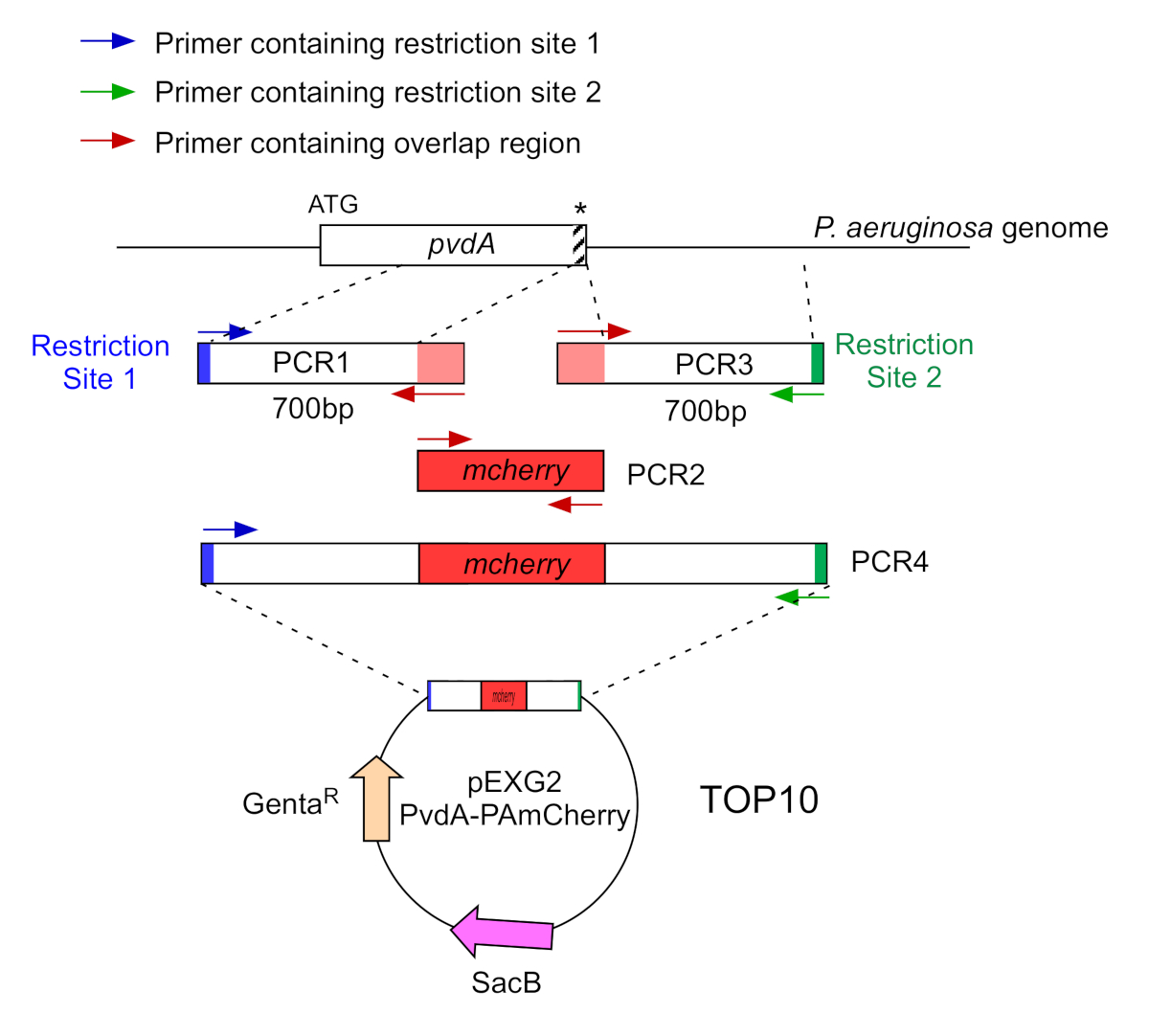
- Amplify by two PCR (PCR1 and 3) the DNA sequences (use genomic DNA of P. aeruginosa PAO1) of the 700 base pairs up und downstream of the regions corresponding to the insertion site in P. aeruginosa genome with high-fidelity DNA polymerase. Fügen Sie Restriktionssites zu Primern in Blau und Grün hinzu, und fügen Sie eine überlappende Sequenz mit mCherry zu Primern in Rot hinzu (Abbildung 2).
- Für PvdA, das am C-Terminus mit eGFP gekennzeichnet ist, verstärken Sie die 700 bp-Region stromaufwärts relativ zum Stop-Codon durch die Primer in Blau, und verstärken Sie den 700 bp downstream-Bereich, der den Stop-Codon enthält, mit den Primern in Grün.
- Für PvdL, das am N-terminus mit mCherry gekennzeichnet ist, verstärken Sie die 700 bp-Region vor dem PvdL-Gen, einschließlich des Startcodons, durch die Primer in Blau und verstärken Sie die 700 bp downstream Region mit den Primern in Grün.

Abbildung 2: Übersicht über die PCR-Strategie und die Plasmidkonstruktion, die für den Bau von PvdA-mCherry verwendet werden. Siehe Text für Details - pvdA kodiert ein Enzym, das an der Biosynthese des Siderophorpyoverdins beteiligt ist, einem sekundären Metaboliten, der an der Eisengewinnung beteiligt ist. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
- Verstärken Sie die eGFP-Kodierungs-DNA (ohne Start- und Stopp-Codons) mit Primern in Rot mit High-Fidelity-DNA-Polymerase.
- Reinigen Sie die PCR-Produkte auf einer PCR-Bereinigungssäule (Tabelle der Materialien).
- Mischen Sie überlappende PCR-Produkte im Äquimolarenverhältnis und führen Sie eine zweite PCR mit Primern mit Restriktionssite für PCR 1 und 3 aus (grün und blau in Abbildung 2).
- Migrieren Sie das PCR-Produkt in Agarose-1x TAE (Tris-Base Acetate EDTA pH 8.0) Gel, schneiden Sie das entsprechende Band und extrahieren Sie das Amplicon mit einem PCR-Reinigungskit (Materialtabelle).
HINWEIS: Das Protokoll kann hier angehalten werden. - Digest PCR Amplicon und PEXG2 Plasmid mit den entsprechenden Restriktionsenzymen21.
- Ligate-Plasmid und Insert mit T4-DNA-Ligase mit 90 ng Plasmid und Molekularverhältnis 1:1 (Plasmid:Insert).
- TransformPlasmide Konstruktion in chemisch kompetenten Zellen E. coli TOP10 Zellen durch Mischen Ligation Produkt und 100 L TOP10. Inkubieren Sie das kompetente Bakterien/Plasmid-Gemisch 30 min auf Eis, bevor Sie mit einem 42 °C-Wärmeschock für 60 s fortfahren. Dann legen Sie die Röhre auf Eis für 10 min.
- 1 ml Lysogenybrühe (LB) zu den Bakterien geben und bei 37 °C für 1 h brüten.
- Platte 100 L Bakterien auf LB-Agar, die 15 g/ml Gentamicin enthalten.
- Über Nacht bei 37 °C inkubieren.
- Screen das Vorhandensein des Inserts durch Kolonie PCR: von einer isolierten transformierenden Kolonie, nehmen Sie eine winzige Menge von Bakterien zu einem PCR-Mix hinzugefügt werden Primer Hybridisierung auf dem Plasmid in einer Weise, dass das Vorhandensein des Amplicon konnte durch Ausführen des Produkts auf einem Agarose-Gel (DNA-Polymerase) nachgewiesen werden. Übertragen Sie aus der gleichen Kolonie, die für PCR verwendet wird, eine kleine Menge bakterien auf eine frische Platte, die 15 g/ml Gentamicin enthält, die isoliert und für die Plasmidextraktion verwendet werden soll. Schließlich isolieren und reinigen Sie das Plasmid (Materialtabelle) und überprüfen Sie den Einsatz durch Sequenzierung.
- Top10-Bakterien, die das Plasmid in LB enthalten, mit 20 % Glycerin in 1,5 ml Mikrorohr bei -80 °C und das gereinigte Plasmid bei -20 °C in 1,5 ml Tube lagern.
ANMERKUNG: Das Protokoll kann hier angehalten werden
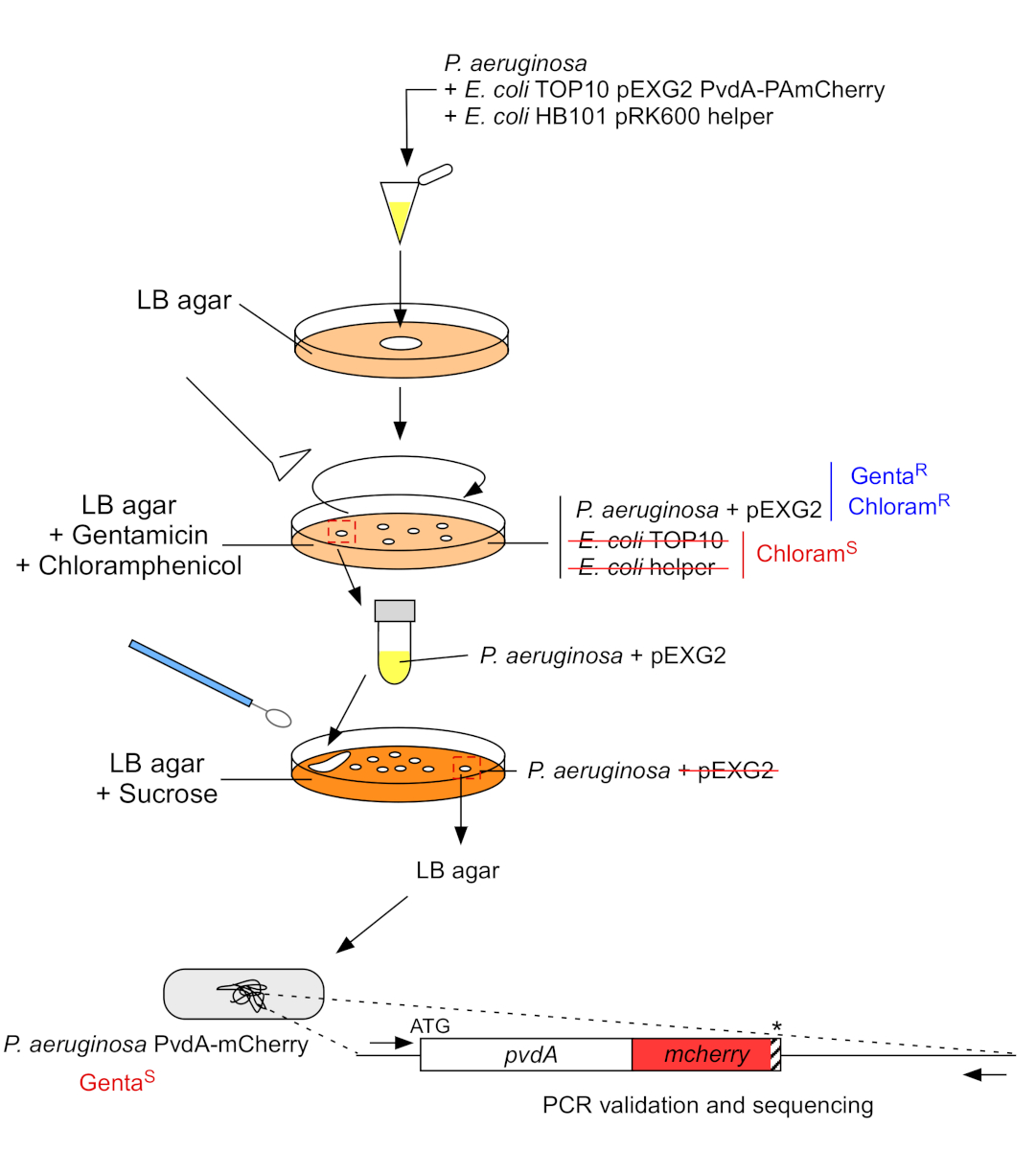
2. Fluoreszierende Tag-Insertion in das chromosomale Genom von P. aeruginosa (Abbildung 3)
- Wachsen Sie P. aeruginosa, TOP10 und E. coli Helfer Bakterien, jedes in 5 ml LB ohne Antibiotikum bei 30 °C unter Orbitalschütteln über Nacht22. Generieren Sie eine fluoreszierende Tag-Insertion in das Genom von P. aeruginosa, indem Sie das Plasmid von E. coli TOP10 in den PAO1-Stamm übertragen und das Plasmid durch homologe Rekombination in das Genom integrieren. Ein zweites Überquerungsereignis, das den Vektor ausschaltet, erzeugt den entsprechenden Mutant.
- Messen Sie die optische Dichte bei 600 nm (OD600 nm) der Bakterienkultur und mischen Sie eine gleiche Menge von P. aeruginosa (500 l, OD600 nm = 1,0) mit E. coli TOP10 pEXG2 (500 l, OD600 nm = 1,0) und E. coli HB101 pRK600 Helfer (500 l, OD600 nm = 1,0) in 1,5 ml Mikrotube.
- Zentrifuge 5 min bei 9.300 x g, um die Bakterien zu pellet.
HINWEIS: Online-Werkzeuge können verwendet werden, um zentrifugale g-Kraft in Drehung pro Minute (Rpm) umzuwandeln, um die Zentrifugengeschwindigkeit anzupassen. - Bewahren Sie das bakterielle Pellet auf und entsorgen Sie den Überstand.
- Setzen Sie das bakterielle Pellet in 50 l LB wieder aus.
- Einen Punkt (ca. 50 l) des Gemischs auf der Mitte des LB-Agars (Vorwärmung bei 37 °C) und 5 h bei 37 °C inkubieren.
- Verschrotten Sie den Punkt mit einer sterilen Impfschleife und resuspendieren Sie in 1 ml LB.
- Die Platte 100 l dieser bakteriellen Suspension auf LB-Agar, die 10 g/ml Chloramphenicol enthält, um E. coli (E. coli TOP10 pEXG2 und E. coli HB101 pRK600 Helfer zu eliminieren, sind empfindlich gegenüber Chloramphenicol, aber P. aeruginosa ist natürlich resistent) und 30 g/mL Gentamicin und inkubieren 2 Tage bei 37 °C.
- Setzen Sie eine Kolonie in 1 ml LB aus und brüten Sie bei 37 °C unter Orbitalschütteln 4h.
- Zentrifuge 3 min bei 9.300 x g und entsorgen 950 l Überstand. Setzen Sie das Pellet in 50 l LB wieder auf und isolieren Sie das Gemisch auf LB-Agar, der Saccharose enthält, und ohne NaCl.
- Über Nacht bei 30 °C inkubieren.
- Entdecken Sie isolierte Kolonien auf LB-Agar und LB-Agar mit 15 mg/ml Gentamicin, um die Gentamicin-Empfindlichkeit zu überprüfen.
- Überprüfen Sie die eGFP- oder mCherry-Einfügung durch PCR-Kolonien (DNA-Polymerase) und DieSequenzierung mit spezifischen Primern.

Abbildung 3: Protokoll der Konstruktion von P. aeruginosa-Stämmen durch fluoreszierende Tag-Einfügung. Weitere Informationen finden Sie im Text. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
3. Pyudin-Messung
- Wachsen Sie Bakterien in 5 ml LB bei 30 °C unter Orbitalschütteln über Nacht.
- Pelletbakterien durch Zentrifugieren, waschen und züchten sie in 5 ml SM (Succinat Medium, Zusammensetzung: 6 g L1 K2HPO4, 3 g-L-1 KH2PO4, 1 g-L1 (NH4)2 SO4, 0,2 g-L-1 MgSO4, 7H2O und 4 g-L-1 Natriumsuccinat mit dem pH-Wert auf 7,0 durch Zugabe von NaOH) bei 30 °C unter Orbitalschütteln über Nacht. SM ist ein eisenberaubtes Medium - das Fehlen von Eisen wird die Expression der Proteine des Pyoverdin-Wegs aktivieren, der normalerweise in Gegenwart von Eisen unterdrückt wird.
- OD600 nm messen und Bakterien in frischem SM-Medium bei OD600 nm = 0,1 wieder verdünnen und bei 30 °C unter Orbitalschütteln über Nacht anbauen.
- Messen Sie OD600 nm, um die Bakterienmenge in jeder Probe zu bestimmen.
- Bereiten Sie eine Quarzküvette mit 100 l P. aeruginosa Kultur und vollständig auf 1 ml SM (900 l). Bereiten Sie eine Quarzküvette mit 1 ml SM Medium (leer) vor.
- Messen Sie mit einem UV-sichtbaren Spektralphotometer die Absorption am Maximum der Absorptionsspitze. Bei pH 7,0 tritt die maximale Absorption von Pyoverdin bei 400 nm auf. Bestimmen Sie die Pyoverdinkonzentration (Apo-Form) in der Probe unter Verwendung des Beer-Lambert-Gesetzes mit einem Molaren-Aussterbekoeffizienten bei 400 nm e = 19 000 M-1cm-1.
HINWEIS: Pyuddin kann im Absorptionsbereich von 0,1 bis 1 (abhängig vom UV-sichtbaren Spektralphotometer) quantifiziert werden, bei dem die Absorption linear mit der Konzentration zunimmt.
4. Bakterienkultur und Bedingungen für Zellen zum Ausdruck bringen PvdA, PvdL und PvdJ
- Am ersten Tag impfen Sie eine Röhre mit 5 ml LB aus dem entsprechenden Glycerinbestand von Bakterien und wachsen Bakterien über Nacht bei 30 °C bei 200 U/min in einem Orbital-Shaker-Inkubator.
- Am 2. Tag Pelletzellen durch Zentrifugation bei 3.000 x g für 3 min und entsorgen Sie den Überstand.
- Setzen Sie die Zellen in 10 ml SM wieder aus.
- Wiederholen Sie die Schritte 4.2-4.3 einmal und wachsen Bakterien in SM über Nacht bei 30 °C 200 Rpm.
- Verdünnen Sie am 3. Tag 1/10 die Bakterienkultur in frischem SM.
- Wachsen Sie verdünnte Bakterien wieder über Nacht zu den gleichen Bedingungen.
HINWEIS: Das Vorhandensein von Pyoverdin kann visuell erkannt werden, da es in Gelb-Grün die wachsenden Medien färbt. Es zeigt, dass die Expression der Proteine des Pyobdin-Signalwegs aktiviert wurde und dass Enzyme von Interesse in den Zellen exprimiert werden.
5. Herstellung des Agarose-Pads (Abbildung 4)
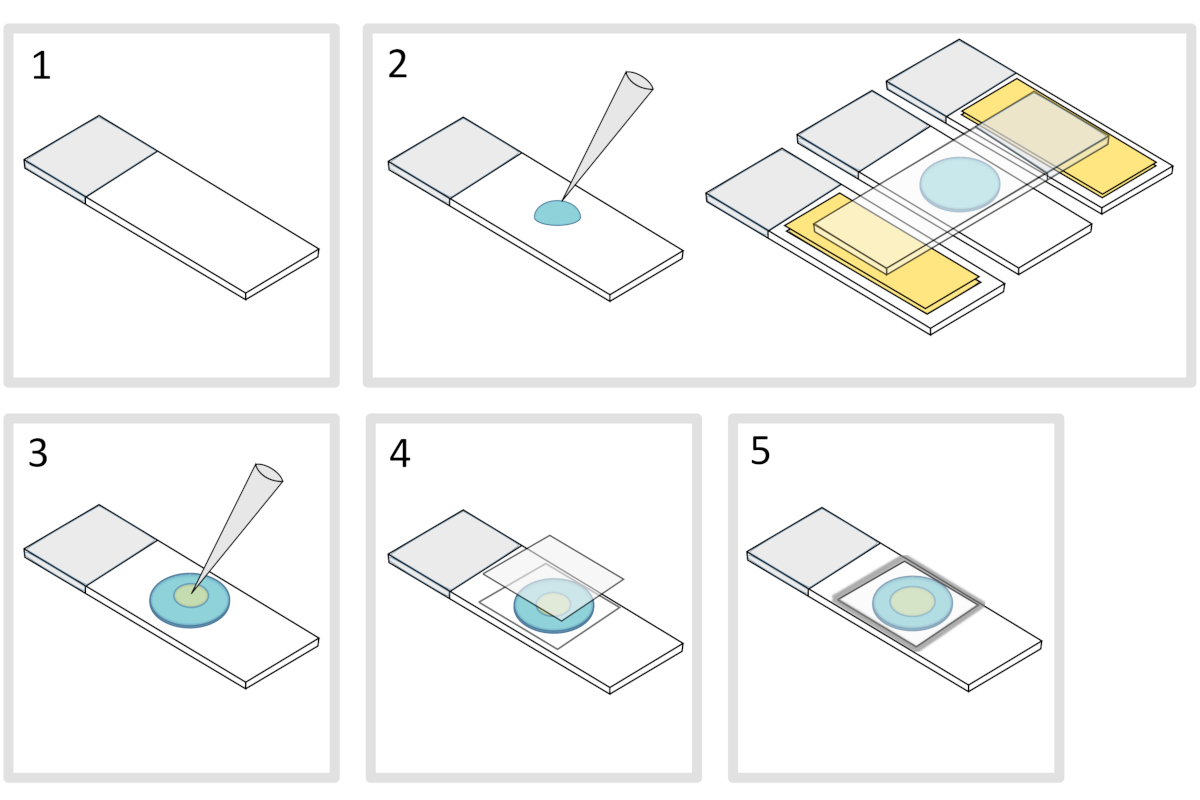
- Legen Sie ein Mikroskop-Glas-Dia auf eine flache horizontale Oberfläche. Ordnen Sie zwei Glasschlitten mit zwei Schichten Klebeband auf jeder Seite des ursprünglichen Schlittens an.
HINWEIS: Halten Sie einen Abstand von 1-2 mm zwischen den drei ausgerichteten Dias, um zu verhindern, dass sich die meletierte Agarose schließlich mit Klebeband auf den Dias ausbreitet. - Pipette und gießen Sie ein Tröpfchen von 70 l von 1% geschmolzener Agarose auf die Glasrutsche. Fügen Sie eine vierte Rutsche auf der Oberseite hinzu, um das Agarose-Tröpfchen abzuflachen und vorsichtig nach unten zu drücken. Warten Sie etwa eine Minute.
- Nehmen Sie die obere Rutsche ab und lassen Sie mit einer Pipette ca. 3 l Bakterien an verschiedenen Stellen auf das Agarose-Pad in 3 bis 4 Stellen fallen.
- Abdeckung mit mikroskopischem Glasdeckel (z.B. eine 22x22 mm #1,5 Dicke).
HINWEIS: Flachheit und Dicke der Abdeckungen sind wichtig für die Arbeit mit zweiphotonen Anregungen. Präzisionsabdeckungen mit kontrollierter gleichmäßiger Ebenheit und geringer Autofluoreszenz sind in der Regel eine gute Wahl. - Befestigen Sie den Deckelrutsch mit geschmolzenem Paraffin, um den Deckelrutsch auf dem Glasschlitten zu versiegeln. Beginnen Sie mit der Befestigung der vier Ecken des Deckels.

Abbildung 4: Agarose-Pad-Vorbereitung. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
6. Bildgebung mit zwei Photonenmikroskopie-Setup
HINWEIS: Wir verwenden ein selbstgebautes Zwei-Photonen-Anregungs-Invertiertes Mikroskop mit einem 60x 1.2NA Wasser-Immersion-Objektiv, das im degescannten Fluoreszenz-Sammlungsmodus arbeitet. Die Wellenlänge der Zweiphotonen-Erregung wird auf 930 nm eingestellt. Es wird von einem Ti:Sapphire-Laser (80 MHz Wiederholungsrate, ≈ 70 fs Pulsbreite) mit 10-20 mW zur Verfügung gestellt. Fluoreszenzphotonen wurden über einen 680 nm Kurzpassfilter und einen 525/50 nm Bandpassfilter gesammelt, bevor sie zu einer fasergekoppelten Lawinenphotodiode geleitet wurden, die mit einem zeitkorrelierten Einzelphotonenzählmodul (TCSPC) verbunden ist. Das Mikroskop ist auch mit einer Transmissionsfluoreszenzlampe ausgestattet. Mehrere FLIM-FRET-Mikroskope sind jetzt im Handel erhältlich und viele Bildgebungseinrichtungen sind mit Setups ausgestattet, die FLIM-FRET-Messungen durchführen können.
- Verwenden Sie die Fluoreszenzlampe, um das Ziel auf die Monoschicht von Bakterien in der Probe und ausgewählte Bereiche von Interesse zu konzentrieren.
- Prüfen Sie, ob der Anregungslaserverschluss geschlossen ist und dass das vom Laser kommende Infrarotlicht blockiert ist und nicht in das Mikroskop gelangt.
Achtung: Bei der Arbeit mit IR-gepulsten Lasern sollte auf sorgfältige Aufmerksamkeit und ständige Wachsamkeit geachtet werden, da das Laserlicht nicht von den Augen gesehen werden kann, aber jede vorübergehende direkte Exposition oder Laserreflexion extrem schädlich sein und irreversible Augenschäden verursachen kann. Bitte beachten Sie die lokalen Lasersicherheitsverfahren und Schulungen, bevor Sie Mikroskopie-Setups verwenden. - Platzieren Sie das Mikroskop-Dia auf der Bühne mit den Abdeckungen, die dem Objektiv zugewandt sind.
- Überprüfen Sie, ob die Leuchtstofflampe eingeschaltet ist.
- Drehen Sie den Filterwürfelrevolver, um den eGFP-Würfel auszuwählen, und öffnen Sie den Fluoreszenzlampenverschluss.
- Senden Sie das Fluoreszenzlicht in Richtung des Okulars des Mikroskops.
Achtung: Stellen Sie sicher, dass geeignete Filter im Lichtpfad angeordnet sind, um direktes Anregungslicht der Leuchtstofflampe zu entsorgen, das die Augen beschädigen kann. - Konzentrieren Sie das Ziel mit dem Mikroskopknopf auf Bakterien.
- Wählen Sie einen Bereich aus, der für die Probe von Interesse ist, indem Sie sie mit dem Joystick übersetzen, der die motorisierte Stufe steuert.
HINWEIS: Die Fokussierung ist mit hochfluoreszierenden Proben einfacher, so dass die Fluoreszenz direkt mit den Augen zu sehen ist. - Schalten Sie die Anregung für den 2PE-Laser für FLIM-FRET-Messungen.
- Senden Sie den Fluoreszenz-Emissionspfad zurück zum Detektor.
- Drehen Sie den Filterwürfelrevolver zurück, um den dichroitischen Würfel für den 930 nm-Laser auszuwählen.
- Stellen Sie die Laserleistung auf 20 mW ein.
- Legen Sie die Größe der Interessenregion auf 30 m fest. Dieser Vorgang passt die Spannung an, die die Galvo-Spiegel bedient, und definiert den Bereich ihrer Bewegungen (Abbildung 5).
- Schalten Sie den Detektor ein und beginnen Sie mit dem Scannen der Probe - die Start- und Stopptasten, die das Scannen steuern, steuern auch das Öffnen und Schließen des Laserverschlusses sowohl aus Sicherheitsgründen als auch um die Photobleiche der Probe zu begrenzen (Abbildung 5).

Abbildung 5: Schematische Darstellung der Schnittstelle der Mikroskopsteuerungssoftware. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
- Stellen Sie bei Bedarf den Fokus ein, indem Sie den Feinfokusknopf des Mikroskops leicht bewegen.
- Wählen Sie das Sichtfeld für die Bildgebung aus, indem Sie die Bühne fein von der Computeroberfläche aus bewegen. Dies kann auf dem Setup durch Verschieben des Kreuzes auf dem Bild in derMikroskop-Steuerungssoftware( Abbildung 5 ) erfolgen, die die neue Mitte des Bildes definiert und Move Stagedrückt. Ein gutes Sichtfeld zur Erfassung entspricht einem Bild mit 10-30 unbeweglichen Bakterien, die alle richtig fokussiert sind (alle Bakterien befinden sich auf derselben Ebene). Wenn Sie an der Extraktion von Einzelzellen FLIM-FRET-Daten interessiert sind, stellen Sie sicher, dass Bakterien gut individualisiert sind (Bildsegmentierung wird viel einfacher).
- Öffnen Sie die SPCM-Software (kommerzielle Software für die Datenerfassung) und überprüfen Sie, ob die Anzahl der Photonen nicht zu hoch ist, um Einen Aufgusseffekt zu vermeiden, der sich auf Lebensdauermessungen auswirken kann. Senken Sie bei Bedarf die Laserintensität, um die Photonenanzahl niedrig zu halten (ca. 1% der Laserwiederholungsrate).
HINWEIS: Der Pile-Up-Effekt beschreibt die Auswirkungen von Photonen, die aufgrund der Totzeit der Time Correlated Single Photon Counting (TCSPC)-Geräte bei hohen Photonenzahlen verloren gehen. Tritt Pile-Up auf, wird die gemessene durchschnittliche Lebensdauer künstlich kürzer, mit möglicherweise einer zusätzlichen kürzeren Komponente, die im Zerfall aufgrund der Überstichprobe der schnell emittierenden Photonen auftreten kann. - Passen Sie Erfassungsparameter einschließlich der Erfassungszeit an (in der Regel sind 60 s bis 180 s erforderlich, um genügend Photonen zu sammeln).
- Drücken Sie die Starttaste, und warten Sie, bis die Erfassung abgeschlossen ist.
- Speichern Sie die Daten.
- Beenden Sie das Scannen der Probe und schalten Sie den Detektor aus.
- Wählen Sie ein anderes Sichtfeld im Beispiel aus, und wiederholen Sie die Schritte 6.14-6.22, oder stellen Sie sich ein neues Mikroskop-Dia vor, indem Sie die Schritte 6.1-6.22 wiederholen.
HINWEIS: P. aeruginosa kann bis zu 6-8 Stunden bei Raumtemperatur auf dem Agarose-Pad leben und teilen (entsprechend der letzten Verdoppelungszeit von 4 Beifallzeit bei 20 °C). Im Idealfall warten Sie nicht zu lange, um FLIM-FRET-Messungen durchzuführen, um zu vermeiden, dass ein Pad vollständig mit Bakterien bedeckt ist.
7. Datenanalyse

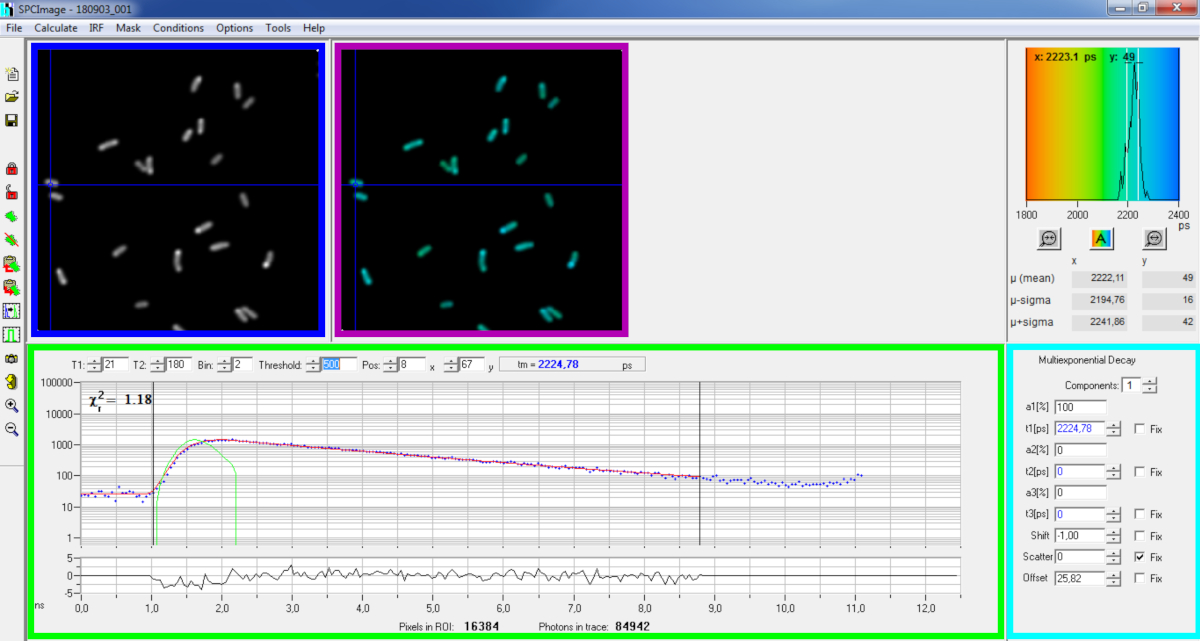
Abbildung 6: Hauptpanel des Datenanalysefensters der SPCImage-Software. Intensitätsbild (blauebox), Lebensdauerbild (lila Box), Lifetime Histogramm (oben rechts), Zerfallskurve an ausgewählter Position (grünes Feld) und Zerfallsparameter an ausgewählter Position (Cyan-Box) eines repräsentativen PvdA-eGFP-Zerfalls, der in Live-P. aeruginosa mit einer bh SPC830-Erfassungskarte auf einem hausgemachten Two-Photon Excitation-FLIM-FRET-Setup aufgezeichnet wurde. Die experimentelle Zerfallskurve des im obigen Bild gezeigten Pixels, dessen mono-exponentielle Passung (rote Kurve) den Zerfall von seiner berechneten instrumentellen Reaktionsfunktion (grüne Kurve) abkonvuuiert, kann im grünen Panel gesehen werden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
- Grundlegende Analyse
- Führen Sie die SPCImage-Software aus.
- Importieren Sie die gespeicherte SPCM-Datei. Das Intensitätsbild wird auf dem linken oberen Bildschirm der Software angezeigt(Abbildung 6 blaue Box).
- Untersuchen Sie das Fenster der Zerfallskurve(Abbildung 6 grünes Feld), in dem die Zerfallsdaten angezeigt werden, die dem im Intensitätsbild ausgewählten Pixel entsprechen(Abbildung 6 blaues Feld). Die Photonennummern jedes Zeitkanals werden als blaue Punkte angezeigt und die Passform des Zerfalls wird als rote Linie gezeichnet. Beachten Sie, dass die Software nach dem Laden der Daten das hellste Pixel des Bildes anzeigt. Verschieben Sie das blaue Kreuz über das Bild, um Pixel mit geringerer Intensität zu untersuchen. Das Zerfallsfenster wird automatisch an jeder neuen Pixelposition aktualisiert.
HINWEIS: Die Messung der Instrumental Response Function (IRF) eines Laser-Scanning-Systems ist sehr schwierig. Ein IRF, der aus der steigenden Kante der Fluoreszenzzerfallskurven in den FLIM-Daten berechnet wird, kann für die Zerfallsdekonvolution verwendet werden. Dies ist die Option, die standardmäßig in SPCimage ausgeführt wird(Abbildung 6 grüne Kurve). - Passen Sie den Anpassungsbereich an, indem Sie die Start- und Endkanäle der Fitting-Box (T1 und T2 im grünen Kasten) verschieben. T1 sollte bei den ersten Kanälen des steigenden Zerfalls beginnen und T2 definieren den letzten Kanal am Ende des Zerfalls und kann als einer der letzten Kanäle des Zerfalls mit einer Reihe von Photonen über dem Photonen zählen Offset gewählt werden (d.h. die Ebenen der Photonen gezählt, bevor der Zerfall steigt).
- Wählen Sie die Binning, indem Sie den Bin-Wert ändern. Der Kurvenzerfall integriert die Photonenzählung des ausgewählten Pixels zusammen mit einer Fläche von i Pixeln um die Cursorposition, die durch den bin-Parameter definiert wird (die Erhöhung des Binnings erhöht die Anzahl der Photonen im Zerfall und kann hilfreich sein, um die Photonenanzahl zu erreichen, die für Multi-Exponentialmodelle erforderlich ist).
- Passen Sie den Schwellenwertwert an. Pixel, die nicht über mindestens einen Kanal mit einer Anzahl von Photonen verfügen, die über dem Schwellenwert liegen, werden nicht in das Anpassungsverfahren einbezogen. Je höher die Anzahl der Pixel passt, desto länger ist die Analyse natürlich.
HINWEIS: FLIM-Daten können eine enorme Anzahl von Pixeln und Zeitkanälen enthalten. Die letzten Versionen der Software ermöglichen es, mit GPU (Graphics Processor Unit) eine große Anzahl von Pixeln parallel zu verarbeiten, was die Verarbeitungszeiten massiv reduziert. Es kann interessant sein, die Binning- und Schwellenwertparameter mithilfe von Bildern anzupassen, die Bakterienkonstruktionen entsprechen, die die niedrigste Fluoreszenzintensität aufweisen (z. B. bei Bakterienstämmen mit den niedrigsten Expressionswerten). Dadurch wird sichergestellt, dass die in diesen Proben beobachteten relevanten Zerfälle die Filterkriterien erfüllen und in die Analyse einbezogen werden. Diese Parameter können dann für alle Bilder verwendet werden. - Passen Sie ggf. die Zerfallsparameter (Cyan-Box) an. Lassen Sie die Verschiebung variieren, die meiste Zeit Streuung und Offset kann auf Null korrigiert werden, wenn ein Blick auf die Zerfallsfunktionen zeigen, dass ihr Beitrag vernachlässigbar ist. Der Offset kann geschätzt werden, wenn man die ersten Kanäle des Zerfalls betrachtet - beachten Sie, dass die Bildgebung für eine lange Zeit aufgrund der niedrigen Fluoreszenz in der Probe in der Regel zu einem Nicht-Null-Offset führt. Streuung tritt meist in dicken Proben auf und kann ansonsten als vernachlässigbar angesehen werden.
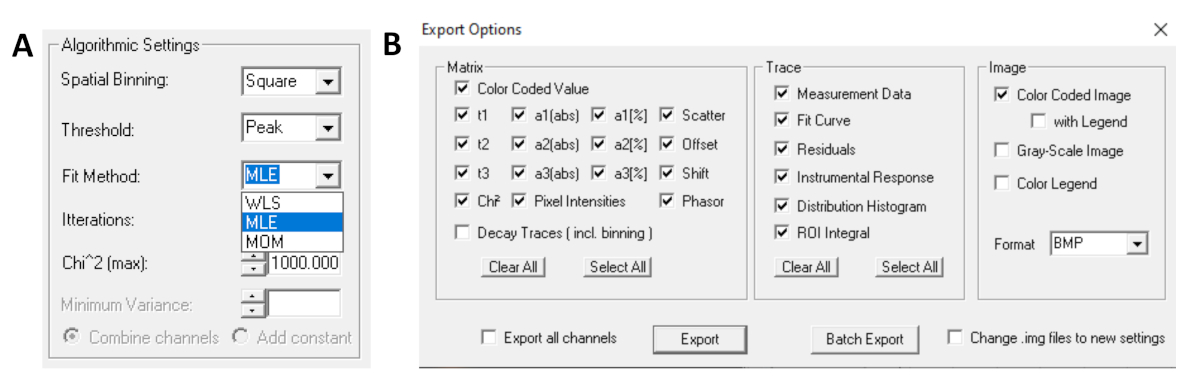
- Wählen Sie vor dem Ausführen der Anpassung den Anpassungsalgorithmus aus. Öffnen Sie das Fenster mit den Algorithmuseinstellungen unter Modelloptionen anzeigen/ausblenden. Wählen Sie den MLE-Algorithmus (Maximum Likelihood Estimation) aus (Abbildung 7A).
- Führen Sie die Anpassung des Bildes aus, indem Sie auf | Zerfallsmatrix berechnen klicken. Nach Abschluss wird das lebenslange CODierte FLIM-Bild im Bildbedienfeld für die Lebensdauer angezeigt(Abbildung 6 violettes Feld).
HINWEIS: Im Fenster der Zerfallskurve(Abbildung 6 grünes Feld) ist es möglich, den Lebensdauerwert zu sehen, der jedem Pixel des Bildes entspricht, indem sie das blaue Kreuz bewegt.
HINWEIS: Um eine große Anzahl ähnlicher FLIM-Datendateien automatisch zu verarbeiten, kann ein Batch-Verarbeitungsmodus verwendet werden. - Überprüfen Sie die Qualität der Passung mit Blick auf die Residuen (idealerweise nach dem Zufallsprinzip um 0 verteilt) und einen Chi-Quadratwert nahe 1.
- Angepasste Daten können in verschiedenen Formaten exportiert werden. Um Dateien in txt-Dateien zu exportieren, wechseln Sie zu Datei-| Export. Wählen Sie im Fenster Exportoptionen (Abbildung 7B) Alle auswählen aus, und klicken Sie dann auf Exportieren.
- Speichern Sie schließlich die Analysedatei. Analysedateien werden als *.img-Dateien gespeichert und können direkt in SPCImage wieder geöffnet werden.
HINWEIS: In bestimmten Fällen unausgewogener Spender-/Akzeptanzmengen kann FLIM-FRET Unterpopulationen in einer Mischung aus interagierenden Proteinkomplexen aufdecken - insbesondere wenn die Konzentrationen der beiden Partner sehr unterschiedlich sind, was zu Mischungen komplexer und freier Arten führt. Nicht interagierende Arten (gekennzeichnet durch einen Zerfall, der dem einzigen Zerfall des Spenders sehr ähnlich ist) können von interagierenden Arten unterschieden werden, die eine räumliche Invarianz der Spenderlebenskomponenten im gesamten Datensatz annehmen. In ähnlicher Weise können sich nicht-stoichiometrische Interaktionskomplexe mit entweder mehr Spendern oder mehr Fluoreszenz-Akzeptoren bilden. Die Fluoreszenzzerzerzenz solcher Komplexe sind in der Regel schwer zu interpretieren. Ein FLIM-Diagrammdiagramm kann verwendet werden, um kritische Informationen über Stoichiometrie und Bindungsmodus von PPI20,23bereitzustellen. Das FLIM-Diagrammdiagramm ist eine grafische Darstellung der kürzesten Lebensdauerkomponente als Funktion ihrer Amplitude. Es kann verwendet werden, um Pixel mit ähnlichen Zerfallssignaturen zu visualisieren. Um solche Darstellungen zu zeichnen, müssen experimentelle Fluoreszenzzerzelle mit einem zwei exponentiellen Modell versehen werden. Die folgenden Schritte können als Leitfaden durch diesen Prozess dienen. - Beginnen Sie mit der Analyse der Daten der Spender nur Konstruktion. Es wird die Bestimmung des Lebenszeitwerts des Spenders ermöglichen. Messen Sie diesen Wert idealerweise über mehrere Bilder, die unter den gleichen Bedingungen wie die Spender-/Akzeptorkonstruktionen aufgenommen wurden, um einen robusten Lebensdauerwert für den Spender abzurufen.
- Einmal bestimmt, passen die Fluoreszenzzerfalle von Spender-/Akzeptorkonstruktionen mit einem zweiexponentiellen Modell. Legen Sie im Parameterfeld Cyan-Zerfall (Abbildung 6) die Anzahl der Komponenten auf 2 fest. Fix t2(ps)-Parameter auf den robusten Lebensdauerwert des Spenders, der in Schritt 1 bestimmt wurde, und aktivieren Sie das Kontrollkästchen, um diesen Parameter zu beheben.
ANMERKUNG: Es ist wichtig, die langlebige Lebensdauer Nr. 2 zu fixieren, um die Überanpassung zu begrenzen, die Anpassungskonvergenz zu verbessern und zuverlässigere zweiexponentielle Anpassungsparameter24,25,26zu erhalten. - Speichern Sie die *img-Datei und exportieren Sie Daten als *.asc-Dateien wie in Schritt 7.1.11.

Abbildung 7: (A) Algorithmuseinstellungen zum Anpassen der Zerfalle mit exponentiellen Modellen. Auswählen von MLE (Maximum-Likelihood-Algorithmus oder Maximum-Likelihood-Schätzung, MLE) als Anpassungsmodell und (B) Exportoptionen Fenster. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
- Erweiterte Analyse der FLIM-Bilder in R
- Installieren Sie bei Bedarf R (https://cran.r-project.org) und RStudio (https://rstudio.com).
- Öffnen Sie RStudio, und erstellen Sie ein neues Projekt.
- Verschieben Sie die gesamte Analysedatei *.asc in einem Ordner namens "Daten" im Hauptordner des Projekts.
- Öffnen Sie eine neue Skriptdatei (oder öffnen Sie das zusätzliche Skript FLIM_analysis. R).
- Installieren Sie das dedizierte paket flimDiagRam für die fim data analysis https://github.com/jgodet/flimDiagRam. Rufen Sie das Paket im Arbeitsbereich auf. (Siehe Hinweis HowTo_FlimDiagRam)
HINWEIS: Die Installation von Paketen muss nur einmal erfolgen. Nach der Installation können Pakete von jeder neuen R-Sitzung aufgerufen werden. Das Herunterladen von R-Paketen von github erfordert die Installation des "devtools"-Pakets. Die Installation von "devtools" kann einige Minuten dauern. Das paketflimDiagRam kann verwendet werden, um die Parameter und Verteilungen der FLIM-Daten darzustellen, FLIM-Daten auf der Ebene einzelner individualisierter Zellen zu extrahieren, FLIM-Ergebnisse über Bedingungen oder Dehnungen hinweg zu vergleichen und FLIM-Daten mithilfe erweiterter Visualisierungswerkzeuge wie des FLIM-Diagrammdiagramms zu untersuchen. - Verwenden Sie den schrittweisen kommentierten Code, und die Daten werden zur unabhängigen Reproduktion aller Unterzahlen zur Verfügung gestellt, die im Abschnitt Repräsentative Ergebnisse unten dargestellt sind. Dieses Tutorial finden Sie in der Mitteilung HowTo_FlimDiagRam unter https://github.com/jgodet/flimDiagRam/blob/master/HowTo.pdf. Der Code kann einfach transponiert werden, um die Daten zu analysieren.
Ergebnisse
Empirische kumulative Verteilungsfunktionen (ecdf) der fluoreszenz-Lebensdauern, die für die verschiedenen Bakterienstämme gemessen wurden, sind in Abbildung 8dargestellt. Tritt FRET auf, werden die Ecdfs in Richtung der kürzeren Lebensdauern verschoben (Abbildung 8A,8B). Beachten Sie, dass, wenn die Wechselwirkung der beiden Proteine zu einem langen Abstand zwischen den beiden Fluorophoren führt, kein FRET auftreten kann (
Diskussion
FLIM-FRET bietet einige wichtige Vorteile gegenüber der intensitätsbasierten FRET-Bildgebung. Die Fluoreszenzlebensdauer ist ein intrinsischer Parameter des Fluorophors. Infolgedessen ist sie weder von lokalen Fluorophorkonzentrationen noch von der Intensität der Lichterregung abhängig. Die Fluoreszenzlebensdauer wird zusätzlich auch durch Photobleichungen schlecht beeinflusst. Besonders interessant ist der Nachweis von PPI in Zellen, in denen die Konzentrationen lokaler Proteine in den subzellulären Kompartimenten...
Offenlegungen
Die Autoren haben nichts zu verraten.
Danksagungen
Wir würdigen Dr. Ludovic Richert für seine wertvolle Unterstützung bei der FLIM-Datenerfassung und bei der technischen Wartung und Entwicklung des FLIM-Setups. Diese Arbeit wurde durch Zuschüsse der Fondation pour la Recherche en Chimie (https://icfrc.fr/) finanziert. VN wird von der Fondation pour la Recherche Médicale (FRM-SPF201809006906) gefördert. YM dankt dem Institut Universitaire de France (IUL) für die Unterstützung und zusätzliche Zeit, um sich der Forschung zu widmen. IJS und JG würdigen das Institut für Arzneimittellieferung in Straßburg für seine finanzielle Unterstützung.
Materialien
| Name | Company | Catalog Number | Comments |
| 525/50 nm band-pass filter | F37-516, AHF, Germany | ||
| 680 nm short pass filter | F75-680, AHF, Germany | ||
| Agarose | Sigma-Aldrich | A9539 | |
| Ammonium Sulfate (NH4)2SO4 | Sigma-Aldrich | A4418 | |
| DreamTaq DNA polymerase 5U/μL | ThermoFisher Scientific | EP0714 | |
| E. coli TOP10 | Invitrogen | C404010 | |
| Fiber-coupled avalanche photo-diode | SPCM-AQR-14- FC, Perkin Elmer | ||
| Glass coverslips (Thickness No. 1.5, 20×20mm | Knitel glass | MS0011 | |
| High-Fidelity DNA polymerase Phusion 2U/μL | ThermoFisher Scientific | F530S | |
| Lysogeny broth (LB) | Millipore | 1.10285 | |
| Magnesium Sulfate Heptahydrate (MgSO4 . 7H2O) | Sigma-Aldrich | 10034-99-8 | |
| Microscope slides (25×75mm) | Knitel glass | MS0057 | |
| NucleoSpin Gel and PCR Clean-up | Macherey-Nagel | 740609.50 | |
| NucleoSpin Plasmid | Macherey-Nagel | 740588.10 | |
| Potassium Phosphate Dibasic (K2HPO4) | Sigma-Aldrich | RES20765 | |
| Potassium Phosphate Monobasic (KH2PO4) | Sigma-Aldrich | P5655 | |
| Sodium Succinate (Disodium) | Sigma-Aldrich | 14160 | |
| SPCImage, SPCM software | Becker & Hickl | ||
| Sterile inoculating loop | Nunc | 7648-1PAK | |
| T4 DNA ligase 1U/μL | ThermoFisher Scientific | 15224017 | |
| TCSPC module | SPC830, Becker & Hickl, Germany | ||
| Ti:Sapphire laser | Insight DeepSee, Spectra Physics | ||
| Tubes 50mL | Falcon | 352070 |
Referenzen
- Braun, P., Gingras, A. C. History of protein-protein interactions: From egg-white to complex networks. Proteomics. 12, 1478-1498 (2012).
- Nooren, I. M. A., Thornton, J. M. Structural characterisation and functional significance of transient protein-protein interactions. Journal of Molecular Biology. 325, 991-1018 (2003).
- Hayes, S., Malacrida, B., Kiely, M., Kiely, P. A. Studying protein-protein interactions: Progress, pitfalls and solutions. Biochemical Society Transactions. 44, 994-1004 (2016).
- Guillon, L., Altenburger, S., Graumann, P. L., Schalk, I. J. Deciphering protein dynamics of the siderophore pyoverdine pathway in Pseudomonas aeruginosa. PLoS ONE. 8, 1-9 (2013).
- Ringel, M. T., Brüser, T. The biosynthesis of pyoverdines. Microbial Cell. 5, 424-437 (2018).
- Schalk, I. J., Rigouin, C., Godet, J. An overview of siderophore biosynthesis among fluorescent Pseudomonads and new insights into their complex cellular organization. Environmental Microbiology. 22, 1447-1466 (2020).
- Cui, Y., et al. Techniques for detecting protein-protein interactions in living cells: principles, limitations, and recent progress. Science China Life Sciences. , (2019).
- Day, R. N., Mazumder, N., Sun, Y., Christopher, K. G. FRET microscopy: Basics, issues and advantages of FLIM-FRET imaging. Springer Series in Chemical Physics. 111, 249-276 (2015).
- Bastiaens, P. I. H., Squire, A. Fluorescence lifetime imaging microscopy: Spatial resolution of biochemical processes in the cell. Trends in Cell Biology. 9, 48-52 (1999).
- Yasuda, R. Imaging spatiotemporal dynamics of neuronal signaling using fluorescence resonance energy transfer and fluorescence lifetime imaging microscopy. Current Opinion in Neurobiology. 16, 551-561 (2006).
- Tramier, M., Zahid, M., Mevel, J. -. C., Masse, M. -. J., Coppey-Moisan, M. Sensitivity of CFP/YFP and GFP/mCherry Pairs to Donor Photobleaching on FRET Determination by Fluorescence Lifetime Imaging Microscopy in Living Cells. Microscopy Research and Technique. 71, 146-157 (2008).
- Bajar, B. T., Wang, E. S., Zhang, S., Lin, M. Z., Chu, J. A guide to fluorescent protein FRET pairs. Sensors (Switzerland). 16, 1-24 (2016).
- Piston, D. W., Kremers, G. J. Fluorescent protein FRET: the good, the bad and the ugly. Trends in Biochemical Sciences. 32, 407-414 (2007).
- Leray, A., et al. Optimized protocol of a frequency domain fluorescence lifetime imaging microscope for fret measurements. Microscopy Research and Technique. 72, 371-379 (2009).
- Elson, D. S., et al. Real-time time-domain fluorescence lifetime imaging including single-shot acquisition with a segmented optical image intensifier. New Journal of Physics. 6, 1-13 (2004).
- Rajoria, S., Zhao, L., Intes, X., Barroso, M. FLIM-FRET for Cancer Applications. Current Molecular Imaging. 3, 144-161 (2014).
- Drobizhev, M., Makarov, N. S., Tillo, S. E., Hughes, T. E., Rebane, A. Two-photon absorption properties of fluorescent proteins. Nature Methods. 8, 393-399 (2011).
- Visca, P., Ciervo, A., Orsi, N. Cloning and nucleotide sequence of the pvdA gene encoding the pyoverdin biosynthetic enzyme L-ornithine N5-oxygenase in Pseudomonas aeruginosa. Journal of Bacteriology. 176, 1128-1140 (1994).
- Imperi, F., Visca, P. Subcellular localization of the pyoverdine biogenesis machinery of Pseudomonas aeruginosa: A membrane-associated 'siderosome'. FEBS Letters. 587, 3387-3391 (2013).
- Gasser, V., et al. In cellulo FRET-FLIM and single molecule tracking reveal the supra-molecular organization of the pyoverdine bio-synthetic enzymes in Pseudomonas aeruginosa. Quarterly Reviews of Biophysics. , 1-11 (2019).
- Rietsch, A., Mekalanos, J. J. Metabolic regulation of type III secretion gene expression in Pseudomonas aeruginosa. Molecular Microbiology. 59, 807-820 (2006).
- Herrero, M., De Lorenzo, V., Timmis, K. N. Transposon vectors containing non-antibiotic resistance selection markers for cloning and stable chromosomal insertion of foreign genes in gram-negative bacteria. Journal of Bacteriology. 172, 6557-6567 (1990).
- Godet, J., Mély, Y. Exploring protein-protein interactions with large differences in protein expression levels using FLIM-FRET. Methods and Applications in Fluorescence. 8, 014007 (2019).
- El Meshri, S. E., et al. Role of the nucleocapsid domain in HIV-1 gag oligomerization and trafficking to the plasma membrane: A fluorescence lifetime imaging microscopy investigation. Journal of Molecular Biology. 427, 1480-1494 (2015).
- Becker, W. The bh TCSPC Handbook. Scanning. , 1 (2010).
- Richert, L., Didier, P., de Rocquigny, H., Mély, Y. Monitoring HIV-1 protein oligomerization by FLIM FRET microscopy. Springer Series in Chemical Physics. , 111 (2015).
- Fereidouni, F., Blab, G. A., Gerritsen, H. C. Phasor based analysis of FRET images recorded using spectrally resolved lifetime imaging. Methods and Applications in Fluorescence. 2, (2014).
- Fereidouni, F., Gorpas, D., Ma, D., Fatakdawala, H., Marcu, L. Rapid fluorescence lifetime estimation with modified phasor approach and Laguerre deconvolution: a comparative study. Methods and Applications in Fluorescence. 5, 035003 (2017).
- Margineanu, A., et al. Screening for protein-protein interactions using Förster resonance energy transfer (FRET) and fluorescence lifetime imaging microscopy (FLIM). Scientific Reports. 6, (2016).
- Guzmán, C., Oetken-Lindholm, C., Abankwa, D. Automated High-Throughput Fluorescence Lifetime Imaging Microscopy to Detect Protein-Protein Interactions. Journal of Laboratory Automation. 21, 238-245 (2016).
- Liu, W., Cui, Y., Ren, W., Irudayaraj, J. Epigenetic biomarker screening by FLIM-FRET for combination therapy in ER+ breast cancer. Clinical Epigenetics. 11, 1-9 (2019).
- Liu, X., et al. Fast fluorescence lifetime imaging techniques: A review on challenge and development. Journal of Innovative Optical Health Sciences. 12, 1-27 (2019).
- Padilla-Parra, S., Auduge, N., Coppey-Moisan, M., Tramier, M. Non fitting based FRET-FLIM analysis approaches applied to quantify protein-protein interactions in live cells. Biophysical Reviews. 3, 63-70 (2011).
- Padilla-Parra, S., Audugé, N., Coppey-Moisan, M., Tramier, M. Quantitative FRET analysis by fast acquisition time domain FLIM at high spatial resolution in living cells. Biophysical Journal. 95, 2976-2988 (2008).
- Stringari, C., et al. Phasor approach to fluorescence lifetime microscopy distinguishes different metabolic states of germ cells in a live tissue. Proceedings of the National Academy of Sciences of the United States of America. 108, 13582-13587 (2011).
- Digman, M. A., Caiolfa, V. R., Zamai, M., Gratton, E. The phasor approach to fluorescence lifetime imaging analysis. Biophysical Journal. 94, 14-16 (2008).
- Liang, Z., Lou, J., Scipioni, L., Gratton, E., Hinde, E. Quantifying nuclear wide chromatin compaction by phasor analysis of histone Förster resonance energy transfer (FRET) in frequency domain fluorescence lifetime imaging microscopy (FLIM) data. Data in Brief. 30, 105401 (2020).
- Grimm, J. B., Heckman, L. M., Lavis, L. D. The chemistry of small-molecule fluorogenic probes. Progress in Molecular Biology and Translational Science. 113, (2013).
- Li, L., Sun, H. Next Generation of Small-Molecule Fluorogenic Probes for Bioimaging. Biochemistry. 59, 216-217 (2020).
- Yao, R., Ochoa, M., Yan, P., Intes, X. Net-FLICS: fast quantitative wide-field fluorescence lifetime imaging with compressed sensing - deep learning approach. Light: Science and Applications. 8, 1-7 (2019).
- Smith, J. T., et al. Fast fit-free analysis of fluorescence lifetime imaging via deep learning. Proceedings of the National Academy of Sciences of the United States of America. 116, 24019-24030 (2019).
- Yao, R., Ochoa, M., Intes, X., Yan, P. Deep compressive macroscopic fluorescence lifetime imaging. Proceedings - International Symposium on Biomedical Imaging. 2018, 908-911 (2018).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten