
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Medições FLIM-FRET de Interações proteína-proteína em bactérias vivas.
Neste Artigo
Resumo
Descrevemos aqui um protocolo para caracterizar interações proteína-proteína entre duas proteínas altamente diferentes expressas em Pseudomonas aeruginosa viva usando medidas FLIM-FRET. O protocolo inclui construções de cepas de bactérias, imobilização de bactérias, rotinas de análise de dados de imagem e pós-imagem.
Resumo
As interações proteína-proteína (PPIs) controlam vários processos-chave nas células. A microscopia de imagem por imagem de fluorescência (FLIM) combinada com a transferência de energia de ressonância de Förster (FRET) fornece informações precisas sobre PPIs em células vivas. A FLIM-FRET baseia-se na medição da decadência ao longo da vida de um doador de FRET em cada pixel da imagem FLIM, fornecendo informações quantitativas e precisas sobre PPIs e suas organizações celulares espaciais. Propomos aqui um protocolo detalhado para as medições FLIM-FRET que aplicamos para monitorar PPIs em Pseudomonas aeruginosa ao vivo no caso particular de duas proteínas interativas expressas com números de cópias altamente diferentes para demonstrar a qualidade e robustez da técnica em revelar características críticas dos PPIs. Este protocolo descreve detalhadamente todas as etapas necessárias para a caracterização do PPI - desde construções mutantes bacterianas até a análise final usando ferramentas recentemente desenvolvidas fornecendo possibilidades avançadas de visualização para uma interpretação direta de dados complexos FLIM-FRET.
Introdução
Interações proteína-proteína (PPIs) controlam vários processos-chave nas células1. Os papéis dos PPIs diferem com base na composição proteica, as afinidades funcionam e locais nas células2. Os PPIs podem ser investigados por meio de diferentes técnicas3. Por exemplo, a co-imunoprecipitação é uma técnica relativamente simples, robusta e barata comumente usada para identificar ou confirmar PPIs. No entanto, estudar PPIs pode ser desafiador quando as proteínas interativas têm baixos níveis de expressão ou quando as interações são transitórias ou relevantes apenas em ambientes específicos. Estudar os PPIs que ocorrem entre as diferentes enzimas da via pyoverdine em P. aeruginosa requer que a repressão do repressor geral de ferro-co-fatorado Fur seja aliviada para permitir que a expressão de todas as proteínas da via de pyoverdine seja expressa na célula4,5,6. Esta regulação comum para todas as proteínas da via resulta em expressões oportunas na célula esperadas para promover suas interações. A diversidade em termos de tamanho, natureza, níveis de expressão e o número de proteínas dessa via metabólica dificultam o estudo em sistemas reconstituídos6. Explorar PPIs em seu ambiente celular é, portanto, fundamental para entender melhor as funções biológicas das proteínas em seu contexto nativo.
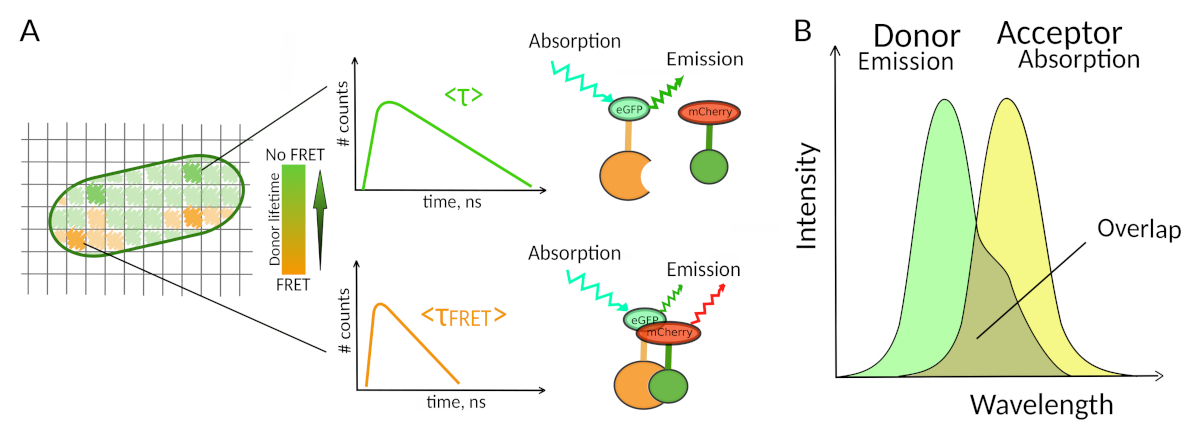
Apenas poucos métodos, incluindo a fluorescência, permitem explorar PPIs em células vivas7. Entre os diferentes parâmetros de fluorescência que podem ser medidos, a vida útil da fluorescência (ou seja, o tempo médio que um fluoróforo permanece em seu estado animado antes de emitir um fóton) é provavelmente um dos parâmetros mais interessantes para explorar em células vivas. A vida fluorescência de um fluoróforo é altamente sensível ao seu ambiente e a FLIM pode, portanto, fornecer informações químicas ou físicas sobre o entorno do fluorohore8. Isso inclui a presença de transferência de energia de ressonância Förster (FRET) que pode ocorrer na presença de um "aceitador" de fluorescência localizado a uma curta distância de um "doador" de fluorescência. A transferência de energia resulta em encurtamento significativo da vida útil da fluorescência do doador(Figura 1A),tornando a Fluorescence Lifetime Imaging Microscopy (FLIM) uma abordagem poderosa para explorar interações proteína-proteína diretamente em células vivas. A FLIM pode fornecer ainda informações espaciais sobre onde as interações ocorrem nas células7,8. Essa abordagem é extremamente poderosa para investigar PPIs em situações em que a rotulagem com fluoroforos dos dois parceiros interagindo é possível.
Para que o FRET ocorra - são necessárias condições críticas na distância entre dois fluoroforos8,9. Os dois fluoroforos não devem estar distantes um do outro por mais de 10 nm. Portanto, devem ser tomadas precauções ao projetar experimentos FLIM-FRET para garantir que o doador e o tomador da fluorescência tenham a chance de estarem próximos um do outro no complexo de interação. Embora isso possa parecer constrangedora, é de fato uma verdadeira vantagem, pois a dependência da distância do FRET garante que duas proteínas rotuladas submetidas ao FRET tenham que interagir fisicamente(Figura 1A). As dificuldades em obter respostas claras sobre o PPI em experimentos de colocalização (duas proteínas colocalized podem não necessariamente interagir) não são, portanto, um problema usando FLIM-FRET.

Figura 1: Princípio de análise FLIM-FRET. Cada pixel da imagem multidimensional FLIM-FRET contém informações sobre a decadência da fluorescência registrada neste local específico (#counts = número de fótons detectados no canal t). (A) A representação clássica da imagem FLIM é geralmente uma imagem 2D codificada por cores falsas (esquerda). Uma diminuição na vida média de fluorescência do doador - como visto por uma mudança na escala de cor - pode ser observada na presença de FRET e é informativa sobre a presença de PPIs nesta área espacial. (B) Sobreposição entre o espectro de emissão de doadores e o espectro de absorção aceitador é necessária para que o FRET ocorra. Clique aqui para ver uma versão maior desta figura.
{kind=link}
Um segundo requisito para fret é que o espectro de emissão do doador e os espectros de absorção do aceitador devem se sobrepor8 (Figura 1B). A excitação da fluorescência do doador deve estar em comprimentos de onda que contribuem muito pouco para a excitação da fluorescência direta do aceitador. Nem todas as combinações de fluoroforos são possíveis e também recomendamos usar preferencialmente doadores com decaimentos monoexponenciais de fluorescência para facilitar as interpretações FLIM-FRET10. Vários casais de proteínas de fluorescência atendem a esses requisitos, incluindo o popular casal eGFP-mCherry11 (para uma revisão na paleta de pares FRET de proteína fluorescente disponíveis ver12,13).
FLIM-FRET permite medir a fluorescência de um doador de FRET em cada pixel de uma imagem FLIM(Figura 1A). Existem duas técnicas principais para determinar a vida útil da fluorescência que diferem na aquisição e análise: domínio de frequência (FD)14 e domínio de tempo (TD). O TD FLIM é mais difundido e é realizado usando uma iluminação pulsada combinada com diferentes configurações possíveis de detecção, incluindo os métodos de gating15, câmera de raia16 ou técnicas de contagem de fótons único (TCSPC)8. Para as técnicas de FD e TD, a vida útil da fluorescência não é diretamente medida, mas requer uma análise dos dados medidos para estimar a vida útil ou a presença de interações. Para as técnicas de TCSPC, a análise mais utilizada baseia-se na adequação das decaimentos com funções únicas ou multinenciais utilizando re-convoluções iterativas menos quadradas que minimizam a soma ponderada dos resíduos.
Finalmente, flim-FRET pode ser realizado tanto usando excitações de fótons únicos ou multifotos. As últimas têm várias vantagens como reduzir a autofluorescência e fotodamage fora do plano focal. Excitações multifoton permitem também uma maior profundidade de excitação se trabalhar em amostras 3D grossas8. Pelo contrário, a excitação de fótons único é geralmente mais eficiente, pois as seções transversais de absorção de dois fótons de proteínas fluorescentes são limitadasa 17.
Aqui, propomos um protocolo para medições FLIM-FRET de PPIs em P. aeruginosa ao vivo no caso particular de duas proteínas interativas (PvdA e PvdL) expressas com números altamente diferentes de cópias para demonstrar a qualidade e robustez da técnica em revelar características críticas dos PPIs. As proteínas PvdA e PvdL estão envolvidas na biossíntese de pyoverdine. PvdA é um L-ornithine N5-oxygenase e sintetiza o L-N5-formyl-N5-hydroxyornithine de L-ornithine por hidroxication (PvdA) e formylation (PvdF)18. PvdL é uma enzima de síntese de peptídeos não ribossômicos (NRPS) composta por quatro módulos. O primeiro módulo catalisa a aciilação do ácido mirístico. O segundo módulo catalisa a ativação do L-Glu e sua condensação para o myristic-coA. Em seguida, o terceiro módulo condensa um aminoácido L-Tyr que é então isomerizado em D-Tyr. Finalmente, o quarto módulo une um aminoácido L-Dab (ácido diaminobutírico) para formar o tripeptídeo aciclado L-Glu/D-Tyr/L-Dab6. O PvdL é, portanto, responsável pela síntese dos três primeiros aminoácidos do precursor de pyoverdine. A interação da proteína PvdA com PvdL é surpreendente, pois o PvdL, ao contrário do PvdI e pvdJ, não carrega um módulo específico para o L-N5-formyl-N5-hidroxiornithine. Essa interação sugere que todas as enzimas responsáveis pela biossíntese precursora de pyoverdine estão dispostas em grandes complexos multienzimáticos transitórios e dinâmicos19,20.
Neste relatório explicamos em detalhes como construir as cepas bacterianas expressando nativamente as duas proteínas interajadas e SFP e mCherry. Descrevemos também a preparação da amostra e as condições para imagens eficientes das células FLIM-FRET. Finalmente, propomos um tutorial passo a passo para análise de imagens, incluindo uma ferramenta recentemente desenvolvida que fornece possibilidades avançadas de visualização para interpretação direta de dados complexos FLIM-FRET. Com este relatório, gostaríamos de convencer não apenas os aventureiros, mas a maioria dos biólogos de que o FRET-FLIM é uma técnica acessível e poderosa capaz de responder às suas perguntas sobre os PPIs diretamente no ambiente celular nativo.
Protocolo
1. Construção plasmid
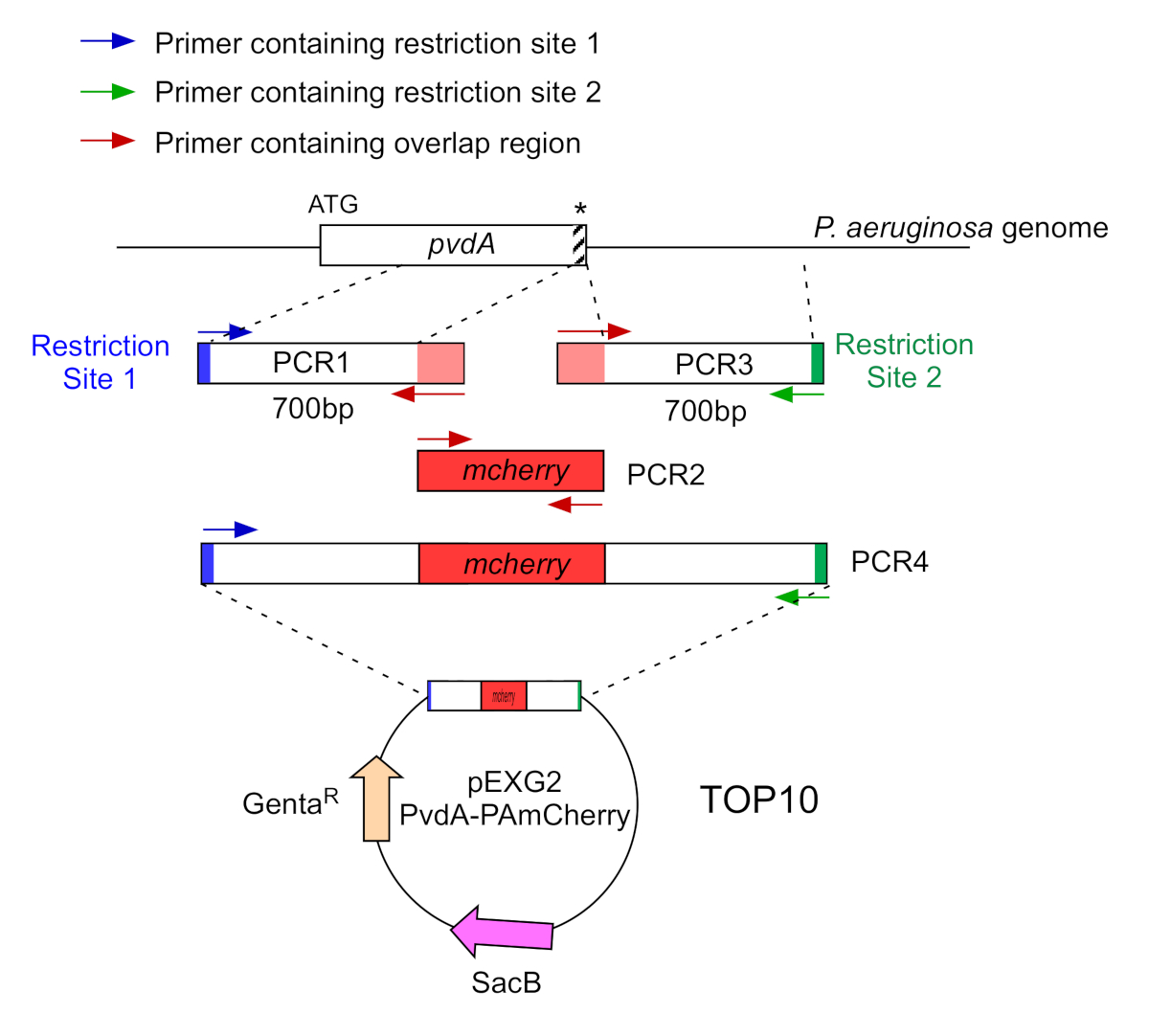
- Amplificar por dois PCR (PCR1 e 3) as sequências de DNA (usar DNA genômico de P. aeruginosa PAO1) dos 700 pares de base rio acima e rio abaixo das regiões correspondentes ao local de inserção em P. aeruginosa genoma com polimerase de DNA de alta fidelidade. Adicione sites de restrição aos primers em azul e verde e adicione uma sequência sobreposta com mCherry aos primers em vermelho(Figura 2).
- Para PvdA rotulado no C-terminus com eGFP, amplie a região de 700 bp upstream em relação ao códon stop pelos primers em azul, e amplie a região a jusante de 700 bp contendo o códon stop com os primers em verde.
- Para PvdL rotulado no N-terminus com mCherry, amplie a região de 700 bp upstream para o gene PvdL, incluindo o códon inicial, pelos primers em azul, e amplifique a região de 700 bp downstream com os primers em verde.

Figura 2: Visão geral da estratégia pcr e construção de plasmídeos utilizados para a construção de PvdA-mCherry. Consulte texto para detalhes - pvdA codifica uma enzima envolvida na biossíntese da pioverdina siderophore, um metabólito secundário envolvido na aquisição de ferro. Clique aqui para ver uma versão maior desta figura.
{kind=link}
- Amplie o DNA de codificação eGFP (sem os códons de partida e parada) com primers em vermelho com polimerase de DNA de alta fidelidade.
- Purifique os produtos PCR em uma coluna de limpeza PCR(Tabela de Materiais).
- Misture produtos PCR sobrepostos na relação equimolar e realize um segundo PCR usando primers com site de restrição usado para PCR 1 e 3 (verde e azul na Figura 2).
- Migrem o produto PCR em gel tae agarose-1x (Tris-Base Acetato EDTA pH 8.0), corte a banda correspondente e extraia a amplicon com um kit de limpeza PCR(Tabela de Materiais).
NOTA: O protocolo pode ser pausado aqui. - Digestão PCR amplicon e plasmid pEXG2 utilizando as enzimas de restriçãocorrespondentes 21.
- Ligate plasmídeo e insira com liga ligase de DNA T4 usando 90 ng de plasmídeo e razão molecular 1:1 (plasmid:insert).
- Transforme a construção de plasmídeos em células quimicamente competentes células E. coli TOP10 misturando produto de ligadura e 100 μL de TOP10. Incubar a competente mistura bactérias/plasmídeos no gelo por 30 minutos antes de prosseguir com um choque térmico de 42 °C para 60 s. Então, coloque o tubo no gelo por 10 minutos.
- Adicione 1 mL de caldo de lise (LB) às bactérias e incubar a 37 °C por 1h.
- Placa 100 μL bactérias em ágar LB contendo 15 μg/mL gentamicina.
- Incubar durante a noite a 37 °C.
- Tela a presença da inserção pela colônia PCR: de uma colônia transformadora isolada, pegue uma quantidade minuciosa de bactérias a serem adicionadas a uma mistura pcr contendo primers hibridizador no plasmídeo de tal forma que a presença do amplicon poderia ser detectada executando o produto em um gel de agarose (DNA polimerase). Da mesma colônia usada para PCR, transfira uma pequena quantidade de bactérias em uma placa fresca contendo 15 μg/mL de gentamicina para ser isolada e usada para extração plasmida. Por fim, isole e purifique o plasmídeo(Tabela de Materiais)e verifique a inserção por sequenciamento.
- Armazene as bactérias TOP10 contendo o plasmídeo em LB com 20 % de glicerol em microtubo de 1,5 mL a -80 °C e o plasmídeo purificado a -20 °C em tubo de 1,5 mL.
NOTA: O protocolo pode ser pausado aqui
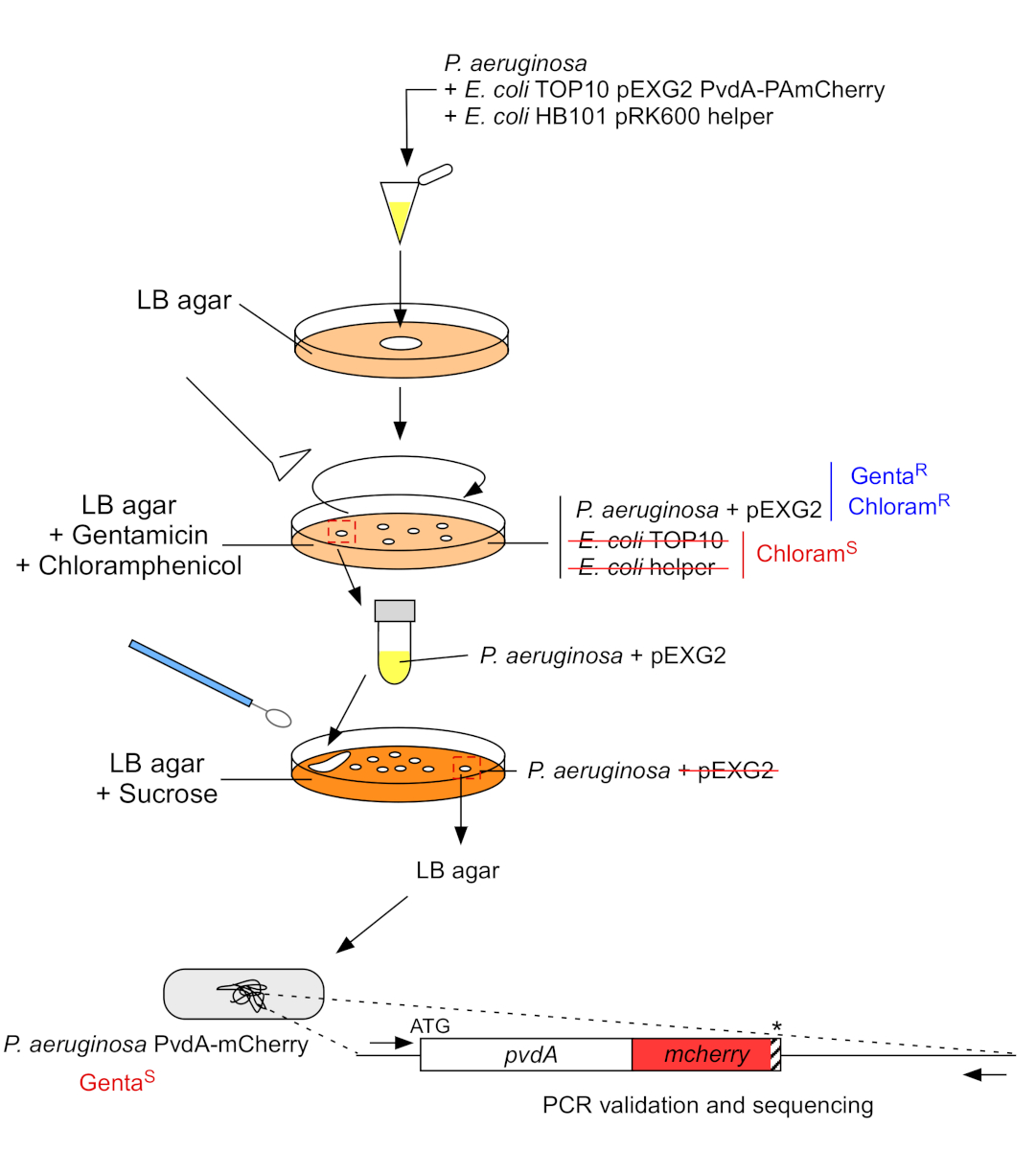
2. Inserção de etiqueta fluorescente no genoma cromossômico de P. aeruginosa (Figura 3)
- Cresça as bactérias P. aeruginosa,TOP10 e E. coli, cada uma em 5 mL de LB sem antibiótico a 30 °C sob agitação orbital durante a noite22. Gerar inserção de etiqueta fluorescente no genoma de P. aeruginosa transferindo o plasmídeo de E. coli TOP10 para a cepa PAO1 e integrando o plasmídeo no genoma por recombinação homologous. Um segundo evento de travessia extirquido do vetor gera o mutante correspondente.
- Meça a densidade óptica a 600 nm (OD600 nm) da cultura bacteriana e misture uma quantidade igual de P. aeruginosa (500 μL, OD600 nm = 1,0) com ajudante E. coli TOP10 pEXG2 (500 μL, OD600 nm = 1,0) e E. coli HB101 pRK600 (500 μL, OD600 nm = 1,0) em microtubo de 1,5 mL.
- Centrifugar 5 min a 9.300 x g para pelotar as bactérias.
NOTA: Ferramentas online podem ser usadas para converter força g centrífuga em rotação por minuto (rpm) para ajustar a velocidade da centrífuga. - Mantenha a pelota bacteriana e descarte o supernasce.
- Resuspend a pelota contendo bactérias em 50 μL de LB.
- Emplaque uma mancha (~50 μL) da mistura no meio do ágar LB (pré-aqueça a 37 °C) e incubar 5h a 37 °C.
- Raspe o local com um laço de inoculação estéril e resuspenque em 1 mL de LB.
- Placa 100 μL desta suspensão bacteriana em ágar LB contendo 10 μg/mL clororamfenicol para eliminar E. coli (E. coli TOP10 pEXG2 e E. coli HB101 pRK600 auxiliar são sensíveis ao clorofenicol, mas P. aeruginosa é naturalmente resistente) e 30 μg/mL gentamicina e incubar 2 dias a 37 °C.
- Resuspend uma colônia em 1 mL LB e incubar a 37 °C sob agitação orbital 4h.
- Centrifugar 3 min a 9.300 x g e descartar 950 μL de supernaspeuta. Resuspenha a pelota em 50 μL de LB e isole a mistura em ágar LB contendo sacarose e sem NaCl.
- Incubar durante a noite a 30 °C.
- Localmente colônias isoladas em ágar LB e ágar LB contendo 15 mg/mL gentamicina, a fim de verificar a sensibilidade da gentamicina.
- Verifique a inserção eGFP ou mCherry por colônias PCR (DNA polymerase) e sequenciamento usando primers específicos.

Figura 3: Protocolo de construção de cepas de P. aeruginosa por inserção de etiqueta fluorescente. Consulte o texto para obter detalhes. Clique aqui para ver uma versão maior desta figura.
{kind=link}
3. Medição de Pyoverdine
- Cresça bactérias em 5 mL de LB a 30 °C sob agitação orbital durante a noite.
- Bactérias de pelotas por centrifugação, lavem e plantem em 5 mL de SM (Succinate Medium, composição: 6 g∙L−1 K2HPO4, 3 g∙L−1 KH2PO4, 1 g∙L−1 (NH4)2 SO4, 0.2 g∙L−1 MgSO4, 7 H2O e 4 g∙L−1 succinato de sódio com o pH ajustado para 7,0 adicionando NaOH) a 30 °C sob agitação orbital durante a noite. SM é um meio privado de ferro - a ausência de ferro ativará a expressão das proteínas da via pyoverdine normalmente reprimidas na presença de ferro.
- Medir OD600 nm e diluir bactérias novamente em meio SM fresco em OD600 nm = 0,1 e cultivá-los a 30 °C sob agitação orbital durante a noite.
- Meça600 nm para determinar a quantidade de bactérias em cada amostra.
- Prepare um cuvette de quartzo contendo 100 μL de cultura P. aeruginosa e complete até 1 mL de SM (900 μL). Prepare uma cuvette de quartzo contendo 1 mL de meio SM (em branco).
- Usando um espectrofotômetro visível UV, meça a absorvência no máximo do pico de absorção. No pH 7.0, o máximo de absorção de pyoverdine ocorrerá em ~400 nm. Determine a concentração de pyoverdina (forma apo) na amostra usando a lei Beer-Lambert usando um coeficiente de extinção molar a 400 nm de e = 19 000 M-1∙cm-1.
NOTA: A pyoverdina pode ser quantificada na faixa de absorvância de ~0,1 a ~1 (dependendo do espectrofotômetro UV-Visível) em que a absorvância aumenta linearmente com concentração.
4. Cultura de bactérias e condições para as células expressarem PvdA, PvdL e PvdJ
- No primeiro dia, inocular um tubo com 5 mL de LB do estoque glicerol apropriado de bactérias e cultivar bactérias durante a noite a 30 °C a 200 rpm em uma incubadora de shaker orbital.
- No dia 2, células de pelotas por centrifugação a 3.000 x g por 3 min e descartam o supernasce.
- Resuspense as células em 10 mL de SM.
- Repita as etapas 4.2-4.3 uma vez e cresça bactérias em SM durante a noite a 30 °C 200 rpm.
- No dia 3, diluir 1/10 a cultura das bactérias em SM fresco.
- Cresça bactérias diluídas novamente durante a noite nas mesmas condições.
NOTA: A presença de pyoverdine pode ser detectada visualmente, pois colore em verde-amarelo a mídia em crescimento. Mostra que a expressão das proteínas da via pyoverdine foi ativada e que enzimas de interesse estão sendo expressas nas células.
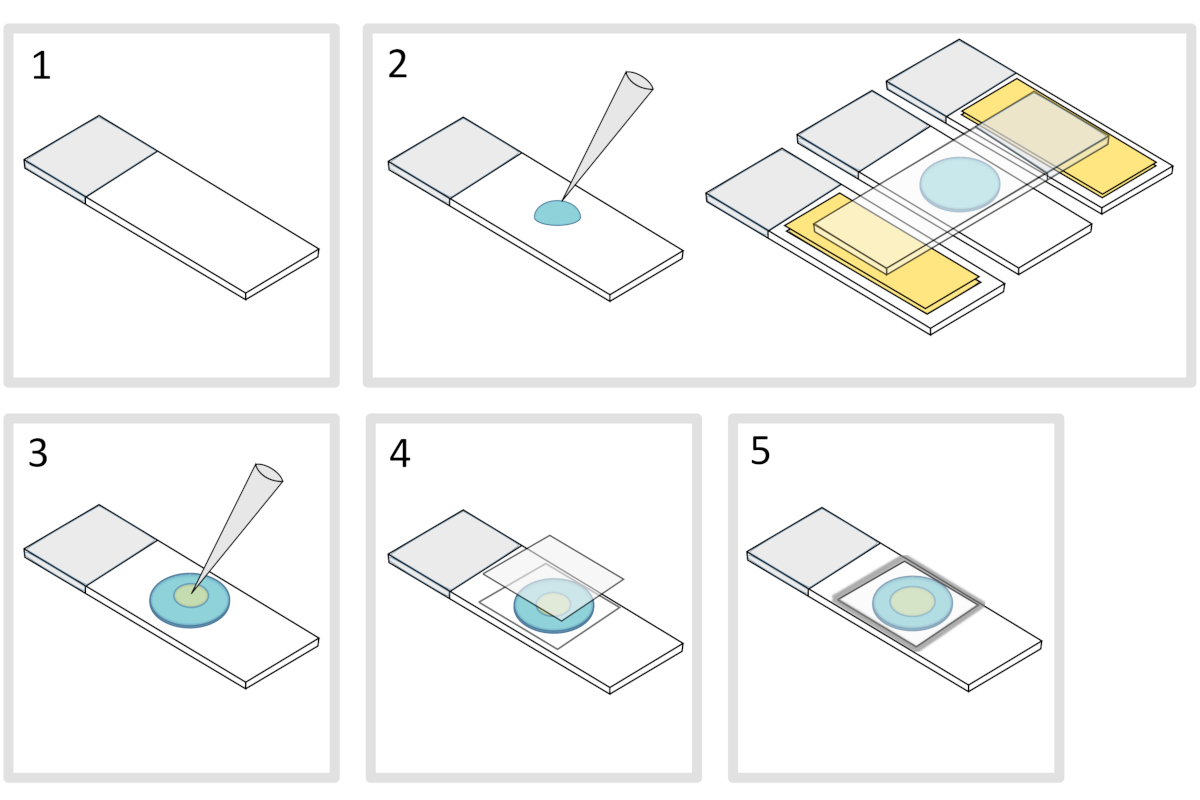
5. Preparação de almofada de agarose (Figura 4)
- Coloque um microscópio de vidro deslizante em uma superfície horizontal plana. Disponha dois slides de vidro cobertos com duas camadas de fita adesiva em cada lado do slide inicial.
NOTA: Mantenha um espaço de 1-2 mm entre os três slides alinhados para evitar que a agarose meleted se espalhe nos slides com fita adesiva. - Pipeta e despeje uma gota de 70 μL de 1% de derretido agarose no deslizamento de vidro. Adicione um quarto slide na parte superior para achatar a gota de agarose e pressione suavemente para baixo. Espere cerca de um minuto.
- Retire o slide superior e solte com uma pipeta de cerca de 3 μL de bactérias em 3 a 4 pontos em diferentes locais para a almofada de agarose.
- Cubra com uma mancha de vidro microscópico (por exemplo, uma espessura de 22x22 mm #1,5).
NOTA: O achatamento e a espessura das tampas são importantes para trabalhar com excitações de dois fótons. Tampas de precisão com achatamento uniforme controlado e baixa autofluorescência são geralmente uma boa escolha. - Fixar a mancha de cobertura com parafina derretida para selar a mancha de cobertura na lâmina de vidro. Comece consertando os quatro cantos da tampa.

Figura 4: Preparação do bloco de agarose. Clique aqui para ver uma versão maior desta figura.
{kind=link}
6. Imagem com configuração de microscopia de dois fótons
NOTA: Estamos usando um microscópio de varredura de dois fótons caseiros com um objetivo de imersão de água 60x 1.2NA operando no modo de coleta de fluorescência despersada. O comprimento de onda de excitação de dois fótons é definido em 930 nm. É fornecido por um laser Ti:Sapphire (taxa de repetição de 80 MHz, ≈ 70 fs de largura de pulso) trabalhando a 10-20 mW. Os fótons de fluorescência foram coletados através de um filtro de passagem curta de 680 nm e um filtro de passe de banda de 525/50 nm antes de serem direcionados para um foto-diodo de avalanche acoplado a fibras conectado a um módulo de contagem de fótons único correlacionado por tempo (TCSPC). O microscópio também é equipado com uma lâmpada de fluorescência de transmissão. Vários microscópios FLIM-FRET estão agora disponíveis comercialmente e muitas instalações de imagem estão equipadas com configurações capazes de realizar medições FLIM-FRET.
- Use a lâmpada de fluorescência para focar o objetivo na monocamada de bactérias na amostra e selecionar regiões de interesse.
- Verifique se o obturador a laser de excitação está fechado e que a luz infravermelha proveniente do laser está bloqueada e não entra no microscópio.
Atenção: Atenção cuidadosa e vigilância constante devem ser dadas trabalhando com lasers pulsados de RI, pois a luz laser não pode ser vista pelos olhos, mas qualquer exposição direta transitória ou reflexão a laser pode ser extremamente prejudicial e criar danos irreversíveis nos olhos. Consulte os procedimentos e treinamento locais de segurança a laser antes de usar as configurações de microscopia. - Coloque o slide de microscopia no palco com as tampas voltadas para o objetivo.
- Verifique se a lâmpada de fluorescência está acesa.
- Gire a torre do cubo do filtro para selecionar o cubo eGFP e abra o obturador da lâmpada de fluorescência.
- Envie a luz da fluorescência para a ocular do microscópio.
Atenção: Certifique-se de que os filtros apropriados sejam descartados no caminho da luz para descartar a luz de excitação direta proveniente da lâmpada de fluorescência que pode danificar os olhos. - Concentre o objetivo em bactérias usando o botão do microscópio.
- Selecione uma região de interesse na amostra traduzindo-a usando o joystick que controla o estágio motorizado
NOTA: O foco é mais fácil com uma amostra altamente fluorescente permitindo que a fluorescência seja vista diretamente com os olhos. - Altere a excitação para o laser de 2PE para medições FLIM-FRET.
- Envie o caminho de emissão de fluorescência de volta para o detector.
- Volte a torre do cubo do filtro para selecionar para o cubo dicroico para o laser de 930 nm.
- Defina a potência laser para 20 mW.
- Defina o tamanho da região de interesse para 30 μm. Esta operação ajusta a tensão que opera os espelhos-galvo e define o alcance de seus movimentos(Figura 5).
- Ligue o detector e comece a digitalizar a amostra - os botões de partida e parada que controlam a varredura também controlam a abertura e o fechamento do obturador a laser tanto por razões de segurança quanto para limitar o fotobleaching da amostra(Figura 5).

Figura 5: Representação esquemática da interface do software de controle de microscópio. Clique aqui para ver uma versão maior desta figura.
{kind=link}
- Se necessário, ajuste o foco movendo ligeiramente o botão de foco fino do microscópio.
- Escolha o campo de visão para imagens movendo-se finamente o estágio da interface do computador. Isso pode ser feito na configuração movendo a cruz na imagem no software de controle de microscópio(Figura 5)que definirá o novo centro da imagem e pressionará o Move Stage. Um bom campo de visão para aquisição corresponde a uma imagem com 10-30 bactérias imóveis, todas corretamente focadas (todas as bactérias estão no mesmo plano). Se estiver interessado em extrair dados FLIM-FRET de células únicas, certifique-se de que as bactérias sejam bem individualizadas (a segmentação de imagens será muito mais fácil).
- Abra o software SPCM (software comercial para aquisição de dados) e verifique se a taxa de contagem de fótons não é muito alta para evitar efeitos de acúmulo que podem afetar as medições ao longo da vida. Se necessário, diminua a intensidade do laser para manter a taxa de contagem de fótons baixa (cerca de 1% da taxa de repetição do laser).
NOTA: O efeito pile-up descreve os efeitos dos fótons perdidos em altas taxas de contagem de fótons devido ao tempo morto dos dispositivos TCSPC (Time Correlated Single Photon Counting). Se o Pile-Up ocorrer, a vida média medida torna-se artificialmente mais curta com possivelmente um componente adicional mais curto que pode aparecer na decadência devido à sobresamplagem dos fótons emissores rápidos. - Ajuste os parâmetros de aquisição, incluindo o tempo de coleta de aquisição (normalmente de 60 a 180 s são necessários para coletar fótons suficientes).
- Pressione o botão Iniciar e aguarde a conclusão da aquisição.
- Guarde os dados.
- Pare de digitalizar a amostra e desligue o detector.
- Selecione outro campo de visão na amostra e repita as etapas 6.14-6.22 ou imagem um novo slide de microscopia repetindo as etapas 6.1-6.22.
NOTA: P. aeruginosa pode viver e dividir por até 6-8 horas em temperatura ambiente na almofada de agarose (correspondente ao último ~4 tempo de duplicação a 20°C). O ideal é não esperar muito tempo para realizar a medição FLIM-FRET para evitar observar uma almofada completamente coberta com bactérias.
7. Análise de dados

Figura 6: Painel principal da janela de análise de dados do software SPCImage. Imagem de intensidade (caixa azul), imagem vitalícia (caixa roxa), histograma vitalício (superior direito), curva de decaimento na posição selecionada (caixa verde) e parâmetros de decaimento na posição selecionada (caixa de ciano) de uma decadência pvdA-eGFP representativa registrada em P. aeruginosa ao vivo usando um cartão de aquisição BH SPC830 em uma configuração de Excitação de Dois FLIM-FRET feita em casa. A curva experimental de decaimento do pixel apontada na imagem acima, seu ajuste mono-exponencial (curva vermelha) desconvolundo a decadência de sua função de resposta instrumental calculada (curva verde) pode ser vista no painel verde. Clique aqui para ver uma versão maior desta figura.
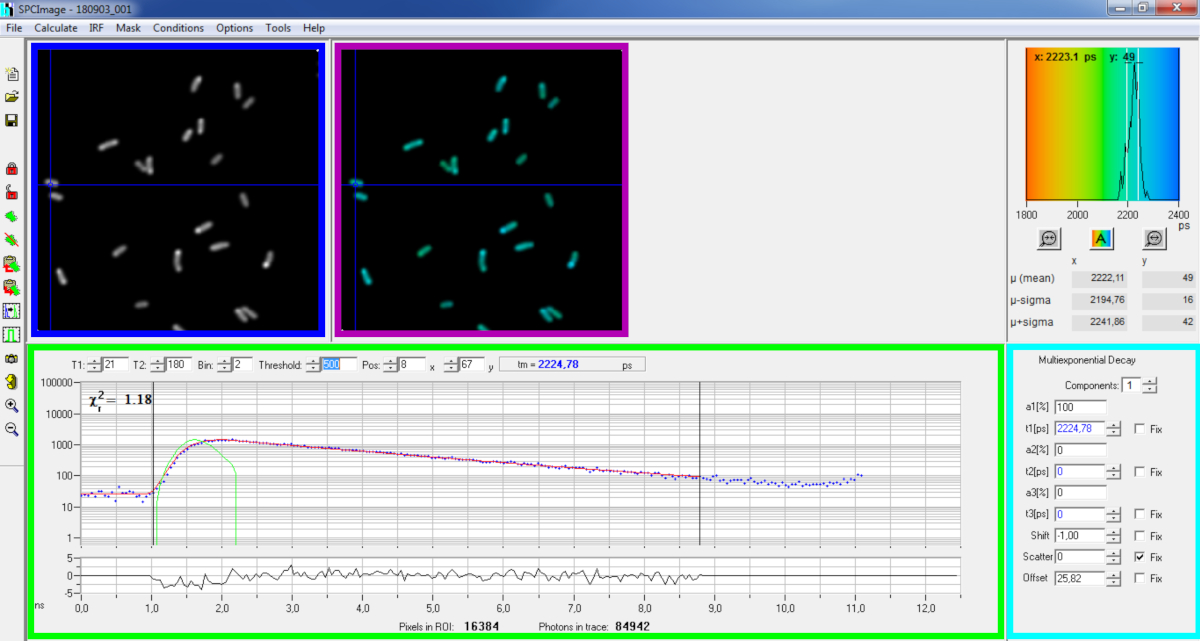{kind=link}
- Análise básica
- Execute o software SPCImage.
- Importe o arquivo SPCM salvo. A imagem de intensidade é exibida no painel superior esquerdo do software(figura 6 caixa azul).
- Examine a janela da curva de decomposição(Figura 6 caixa verde) que exibe os dados de decaimento correspondentes ao pixel selecionado na imagem de intensidade(Figura 6 caixa azul). Os números de fótons de cada canal de tempo são mostrados como pontos azuis e o ajuste da decadência é desenhado como uma linha vermelha. Observe que após o carregamento dos dados o software exibe o pixel mais brilhante da imagem. Mova a cruz azul através da imagem para examinar pixels com menor intensidade. A janela de decomposição será atualizada automaticamente a cada nova posição de pixel.
NOTA: Medir a Função de Resposta Instrumental (IRF) de um sistema de varredura a laser é muito difícil. Um IRF calculado a partir da borda ascendente das curvas de decaimento da fluorescência nos dados FLIM pode ser usado para desconvolução de decaimento. Esta é a opção feita por padrão na SPCimage ( Curva verdeFigura 6). - Ajuste o intervalo de encaixe movendo os canais de partida e término da caixa de montagem (T1 e T2 na caixa verde). T1 deve começar nos primeiros canais da decadência crescente e T2 definir o último canal no final da decadência e pode ser escolhido como um dos últimos canais da decadência com um número de fótons acima do deslocamento de contagem de fótons (ou seja, os níveis de fótons contados antes da decadência sobe).
- Escolha o binning alterando o valor da lixeira. A decadência da curva integra a contagem de fótons do pixel selecionado juntamente com uma área de i pixels ao redor da posição do cursor definida pelo parâmetro de bin (aumentar o binning aumentará o número de fótons na decadência e pode ser útil para alcançar a contagem de fótons necessária para modelos multinenciais).
- Ajuste o valor limiar. Pixels que não possuem pelo menos um canal com um número de fótons acima do valor limiar não serão incluídos no procedimento de encaixe. Claro que quanto maior o número de pixels para caber, maior a análise.
NOTA: Os dados FLIM podem conter um enorme número de pixels e canais de tempo. As últimas versões do software permitem o uso de GPU (Graphics Processor Unit) para processar um grande número de pixels em paralelo, o que reduz maciçamente os tempos de processamento. Pode ser interessante ajustar os parâmetros de binning e limiar usando imagens correspondentes a construções de bactérias que exibem a menor intensidade de fluorescência (por exemplo, com cepas bacterianas com os menores níveis de expressão). Isso garantirá que as decaimentos relevantes observadas nessas amostras atendam aos critérios de filtragem e sejam incluídas na análise. Esses parâmetros podem então ser usados para todas as imagens. - Ajuste, se necessário, os parâmetros de decomposição (caixa de ciano). Deixe a mudança variar, a maior parte do tempo de dispersão e deslocamento pode ser fixado a zero se um olhar para as funções de decadência mostrar que sua contribuição é insignificante. O deslocamento pode ser estimado olhando para os primeiros canais da decadência - observe que a imagem por um longo tempo devido à baixa fluorescência na amostra geralmente resulta em deslocamento não-zero. A dispersão ocorre principalmente em amostras grossas e pode ser considerada insignificante de outra forma.
- Antes de executar o ajuste, selecione o algoritmo de montagem. Abra a janela de configurações do algoritmo em Opções de Modelo Show/Hide. Selecione o algoritmo de estimativa máxima de probabilidade (MLE)(Figura 7A).
- Execute o encaixe da imagem clicando em Calcular | matriz Decay. Uma vez concluída, a imagem FLIM codificada por toda a vida aparece no painel de imagem vitalício (caixa roxafigura 6).
NOTA: Na janela curva de decomposição(Figura 6 caixa verde) é possível ver o valor de vida que corresponde a cada pixel da imagem movendo a cruz azul.
NOTA: Para processar um grande número de arquivos de dados FLIM semelhantes automaticamente, um modo de processamento em lote pode ser usado. - Verifique a qualidade do ajuste olhando para os resíduos (idealmente distribuídos aleatoriamente em torno de 0) e um valor quadrado chi perto de 1.
- Os dados instalados podem ser exportados em diferentes formatos. Para exportar arquivos em arquivos txt, vá para File | Exportação. Na janela Opções de exportação (Figura 7B),escolha Selecionar Tudo e, em seguida, clique em Exportar.
- Finalmente salve o arquivo de análise. Os arquivos de análise são salvos como arquivos *.img e podem ser reabertos diretamente no SPCImage.
NOTA: Em casos particulares de quantidades de doadores/aceitadores desequilibrados, a FLIM-FRET pode revelar sub-populações em uma mistura de complexos proteicos interagindo - em particular quando as concentrações dos dois parceiros são muito diferentes, resultando assim em misturas de espécies complexas e livres. Espécies não interativas (caracterizadas por uma decadência muito semelhante à decadência apenas do doador) podem ser discriminadas por interagirem assumindo uma invariância espacial dos componentes de vida do doador em todo o conjunto de dados. Da mesma forma, complexos de interação não-estequiométricos com mais doadores ou mais aceitadores de fluorescência podem se formar. As decaimentos de fluorescência desses complexos são geralmente difíceis de interpretar. Um gráfico de diagrama flim pode ser usado para fornecer informações críticas sobre estequiometria e modo de vinculação de PPIs20,23. O enredo do diagrama FLIM é uma representação gráfica do componente de vida mais curto em função de sua amplitude. Pode ser usado para visualizar pixels com assinaturas semelhantes de decadência. Para desenhar tais representações, as decaimentos experimentais de fluorescência devem ser equipadas com um modelo de dois modelos exponenciais. As etapas a seguir podem ser um guia através deste processo. - Comece analisando os dados da construção apenas do doador. Permitirá determinar o valor vitalício do doador. O ideal é medir esse valor em várias imagens registradas nas mesmas condições das construções doador/aceitadora para recuperar um valor de vida robusto para o doador.
- Uma vez determinada, ajuste as decaimentos de fluorescência de construções doador/aceitadora com um modelo bi-exponencial. Na caixa de parâmetro de decomposição do ciano(Figura 6),defina o número de componentes para 2. Corrija o parâmetro t2(ps) para o valor de vida robusto do doador determinado na etapa 1 e verifique a caixa para corrigir este parâmetro.
NOTA: É importante corrigir a vida útil de longa duração τ2, a fim de limitar o excesso de montagem, melhorar a convergência de montagem e obter parâmetros de ajuste mais confiáveis24,25,26. - Salve o arquivo *img e os dados de exportação como arquivos *.asc como na etapa 7.1.11.

Figura 7: (A) Configurações de algoritmo para encaixar as decaimentos com modelos exponenciais. Selecionando mle (algoritmo de maior probabilidade ou estimativa de máxima probabilidade, MLE) como o modelo de ajuste e (B) janela de opções de exportação. Clique aqui para ver uma versão maior desta figura.
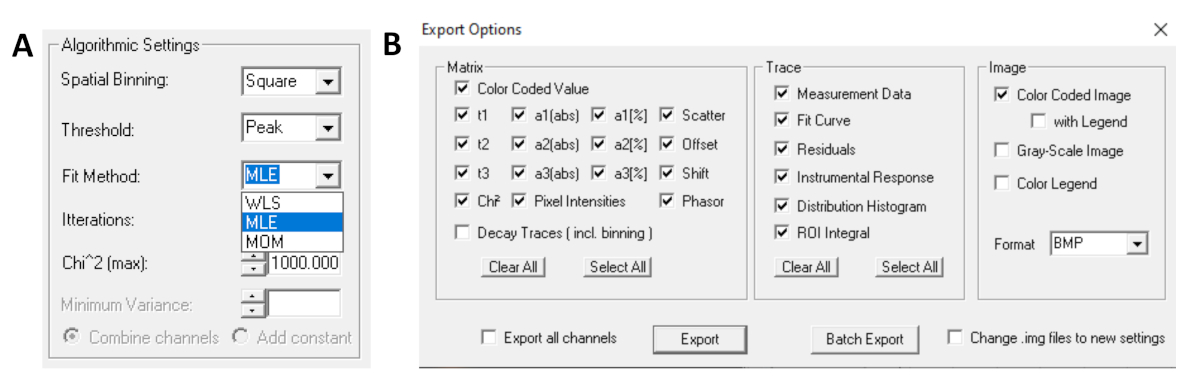{kind=link}
- Análise avançada das imagens FLIM em R
- Instale R (https://cran.r-project.org) e RStudio (https://rstudio.com) se necessário.
- Abra o RStudio e crie um novo projeto.
- Mova todas as análises *.asc arquivo em uma pasta chamada "dados" na pasta principal do projeto.
- Abra um novo arquivo de script (ou abra o script suplementar FLIM_analysis. R).
- Instale o pacote flimDiagRam dedicado para análise de dados frágeis https://github.com/jgodet/flimDiagRam. Ligue para o pacote no espaço de trabalho. (Veja o aviso HowTo_FlimDiagRam)
NOTA: A instalação dos pacotes deve ser feita apenas uma vez. Uma vez instalados, os pacotes podem ser chamados a partir de qualquer nova sessão R. Baixar pacotes R do github requer a instalação de pacotes 'devtools'. A instalação de 'devtools' pode levar vários minutos. O pacote FlimDiagRam pode ser usado para representar os parâmetros e distribuições dos dados FLIM, para extrair dados FLIM ao nível de células individualizadas únicas, para comparar os resultados de FLIM em condições ou cepas e para explorar dados FLIM usando ferramentas avançadas de visualização como o gráfico de diagrama flim. - Use o código de comentários passo a passo e os dados são disponibilizados para reproduzir independentemente todas as subprezas apresentadas na seção Resultados Representativos abaixo. Este tutorial pode ser encontrado no HowTo_FlimDiagRam de avisos em https://github.com/jgodet/flimDiagRam/blob/master/HowTo.pdf. O código pode ser facilmente transposto para analisar os dados.
Resultados
Funções de distribuição cumulativa empírica (ecdf) das vidas de fluorescência medidas para as diferentes cepas bacterianas são mostradas na Figura 8. Se ocorrer o FRET, os ecdfs são deslocados para as vidas de menor duração(Figura 8A,8B). Note-se que quando a interação das duas proteínas resulta em uma longa distância entre os dois fluoroforos, não pode ocorrer nenhum TRAF(Figura 8C). Essa situação ...
Discussão
FLIM-FRET oferece algumas vantagens importantes sobre a imagem FRET baseada em intensidade. Fluorescência vitalícia é um parâmetro intrínseco do fluoróforo. Como consequência, não depende das concentrações locais de fluoroforos nem da intensidade da excitação da luz. A vida útil da fluorescência também é também mal afetada pelo branqueamento fotográfico. É particularmente interessante evidenciar PPIs em células onde concentrações de proteínas locais podem ser altamente heterogêneas em todos os com...
Divulgações
Os autores não têm nada a revelar.
Agradecimentos
Reconhecemos o Dr. Ludovic Richert por sua valiosa assistência na aquisição de dados da FLIM e pela manutenção técnica e desenvolvimento da configuração da FLIM. Este trabalho foi financiado por subsídios da Fondation pour la Recherche en Chimie (https://icfrc.fr/). A VN é financiada pela Fondation pour la Recherche Médicale (FRM‐SPF201809006906). A YM agradece ao Institut Universitaire de France (IUF) pelo apoio e pelo tempo adicional para se dedicar à pesquisa. O IJS e a JG reconhecem o Instituto de Entrega de Drogas de Estrasburgo pelo seu apoio financeiro.
Materiais
| Name | Company | Catalog Number | Comments |
| 525/50 nm band-pass filter | F37-516, AHF, Germany | ||
| 680 nm short pass filter | F75-680, AHF, Germany | ||
| Agarose | Sigma-Aldrich | A9539 | |
| Ammonium Sulfate (NH4)2SO4 | Sigma-Aldrich | A4418 | |
| DreamTaq DNA polymerase 5U/μL | ThermoFisher Scientific | EP0714 | |
| E. coli TOP10 | Invitrogen | C404010 | |
| Fiber-coupled avalanche photo-diode | SPCM-AQR-14- FC, Perkin Elmer | ||
| Glass coverslips (Thickness No. 1.5, 20×20mm | Knitel glass | MS0011 | |
| High-Fidelity DNA polymerase Phusion 2U/μL | ThermoFisher Scientific | F530S | |
| Lysogeny broth (LB) | Millipore | 1.10285 | |
| Magnesium Sulfate Heptahydrate (MgSO4 . 7H2O) | Sigma-Aldrich | 10034-99-8 | |
| Microscope slides (25×75mm) | Knitel glass | MS0057 | |
| NucleoSpin Gel and PCR Clean-up | Macherey-Nagel | 740609.50 | |
| NucleoSpin Plasmid | Macherey-Nagel | 740588.10 | |
| Potassium Phosphate Dibasic (K2HPO4) | Sigma-Aldrich | RES20765 | |
| Potassium Phosphate Monobasic (KH2PO4) | Sigma-Aldrich | P5655 | |
| Sodium Succinate (Disodium) | Sigma-Aldrich | 14160 | |
| SPCImage, SPCM software | Becker & Hickl | ||
| Sterile inoculating loop | Nunc | 7648-1PAK | |
| T4 DNA ligase 1U/μL | ThermoFisher Scientific | 15224017 | |
| TCSPC module | SPC830, Becker & Hickl, Germany | ||
| Ti:Sapphire laser | Insight DeepSee, Spectra Physics | ||
| Tubes 50mL | Falcon | 352070 |
Referências
- Braun, P., Gingras, A. C. History of protein-protein interactions: From egg-white to complex networks. Proteomics. 12, 1478-1498 (2012).
- Nooren, I. M. A., Thornton, J. M. Structural characterisation and functional significance of transient protein-protein interactions. Journal of Molecular Biology. 325, 991-1018 (2003).
- Hayes, S., Malacrida, B., Kiely, M., Kiely, P. A. Studying protein-protein interactions: Progress, pitfalls and solutions. Biochemical Society Transactions. 44, 994-1004 (2016).
- Guillon, L., Altenburger, S., Graumann, P. L., Schalk, I. J. Deciphering protein dynamics of the siderophore pyoverdine pathway in Pseudomonas aeruginosa. PLoS ONE. 8, 1-9 (2013).
- Ringel, M. T., Brüser, T. The biosynthesis of pyoverdines. Microbial Cell. 5, 424-437 (2018).
- Schalk, I. J., Rigouin, C., Godet, J. An overview of siderophore biosynthesis among fluorescent Pseudomonads and new insights into their complex cellular organization. Environmental Microbiology. 22, 1447-1466 (2020).
- Cui, Y., et al. Techniques for detecting protein-protein interactions in living cells: principles, limitations, and recent progress. Science China Life Sciences. , (2019).
- Day, R. N., Mazumder, N., Sun, Y., Christopher, K. G. FRET microscopy: Basics, issues and advantages of FLIM-FRET imaging. Springer Series in Chemical Physics. 111, 249-276 (2015).
- Bastiaens, P. I. H., Squire, A. Fluorescence lifetime imaging microscopy: Spatial resolution of biochemical processes in the cell. Trends in Cell Biology. 9, 48-52 (1999).
- Yasuda, R. Imaging spatiotemporal dynamics of neuronal signaling using fluorescence resonance energy transfer and fluorescence lifetime imaging microscopy. Current Opinion in Neurobiology. 16, 551-561 (2006).
- Tramier, M., Zahid, M., Mevel, J. -. C., Masse, M. -. J., Coppey-Moisan, M. Sensitivity of CFP/YFP and GFP/mCherry Pairs to Donor Photobleaching on FRET Determination by Fluorescence Lifetime Imaging Microscopy in Living Cells. Microscopy Research and Technique. 71, 146-157 (2008).
- Bajar, B. T., Wang, E. S., Zhang, S., Lin, M. Z., Chu, J. A guide to fluorescent protein FRET pairs. Sensors (Switzerland). 16, 1-24 (2016).
- Piston, D. W., Kremers, G. J. Fluorescent protein FRET: the good, the bad and the ugly. Trends in Biochemical Sciences. 32, 407-414 (2007).
- Leray, A., et al. Optimized protocol of a frequency domain fluorescence lifetime imaging microscope for fret measurements. Microscopy Research and Technique. 72, 371-379 (2009).
- Elson, D. S., et al. Real-time time-domain fluorescence lifetime imaging including single-shot acquisition with a segmented optical image intensifier. New Journal of Physics. 6, 1-13 (2004).
- Rajoria, S., Zhao, L., Intes, X., Barroso, M. FLIM-FRET for Cancer Applications. Current Molecular Imaging. 3, 144-161 (2014).
- Drobizhev, M., Makarov, N. S., Tillo, S. E., Hughes, T. E., Rebane, A. Two-photon absorption properties of fluorescent proteins. Nature Methods. 8, 393-399 (2011).
- Visca, P., Ciervo, A., Orsi, N. Cloning and nucleotide sequence of the pvdA gene encoding the pyoverdin biosynthetic enzyme L-ornithine N5-oxygenase in Pseudomonas aeruginosa. Journal of Bacteriology. 176, 1128-1140 (1994).
- Imperi, F., Visca, P. Subcellular localization of the pyoverdine biogenesis machinery of Pseudomonas aeruginosa: A membrane-associated 'siderosome'. FEBS Letters. 587, 3387-3391 (2013).
- Gasser, V., et al. In cellulo FRET-FLIM and single molecule tracking reveal the supra-molecular organization of the pyoverdine bio-synthetic enzymes in Pseudomonas aeruginosa. Quarterly Reviews of Biophysics. , 1-11 (2019).
- Rietsch, A., Mekalanos, J. J. Metabolic regulation of type III secretion gene expression in Pseudomonas aeruginosa. Molecular Microbiology. 59, 807-820 (2006).
- Herrero, M., De Lorenzo, V., Timmis, K. N. Transposon vectors containing non-antibiotic resistance selection markers for cloning and stable chromosomal insertion of foreign genes in gram-negative bacteria. Journal of Bacteriology. 172, 6557-6567 (1990).
- Godet, J., Mély, Y. Exploring protein-protein interactions with large differences in protein expression levels using FLIM-FRET. Methods and Applications in Fluorescence. 8, 014007 (2019).
- El Meshri, S. E., et al. Role of the nucleocapsid domain in HIV-1 gag oligomerization and trafficking to the plasma membrane: A fluorescence lifetime imaging microscopy investigation. Journal of Molecular Biology. 427, 1480-1494 (2015).
- Becker, W. The bh TCSPC Handbook. Scanning. , 1 (2010).
- Richert, L., Didier, P., de Rocquigny, H., Mély, Y. Monitoring HIV-1 protein oligomerization by FLIM FRET microscopy. Springer Series in Chemical Physics. , 111 (2015).
- Fereidouni, F., Blab, G. A., Gerritsen, H. C. Phasor based analysis of FRET images recorded using spectrally resolved lifetime imaging. Methods and Applications in Fluorescence. 2, (2014).
- Fereidouni, F., Gorpas, D., Ma, D., Fatakdawala, H., Marcu, L. Rapid fluorescence lifetime estimation with modified phasor approach and Laguerre deconvolution: a comparative study. Methods and Applications in Fluorescence. 5, 035003 (2017).
- Margineanu, A., et al. Screening for protein-protein interactions using Förster resonance energy transfer (FRET) and fluorescence lifetime imaging microscopy (FLIM). Scientific Reports. 6, (2016).
- Guzmán, C., Oetken-Lindholm, C., Abankwa, D. Automated High-Throughput Fluorescence Lifetime Imaging Microscopy to Detect Protein-Protein Interactions. Journal of Laboratory Automation. 21, 238-245 (2016).
- Liu, W., Cui, Y., Ren, W., Irudayaraj, J. Epigenetic biomarker screening by FLIM-FRET for combination therapy in ER+ breast cancer. Clinical Epigenetics. 11, 1-9 (2019).
- Liu, X., et al. Fast fluorescence lifetime imaging techniques: A review on challenge and development. Journal of Innovative Optical Health Sciences. 12, 1-27 (2019).
- Padilla-Parra, S., Auduge, N., Coppey-Moisan, M., Tramier, M. Non fitting based FRET-FLIM analysis approaches applied to quantify protein-protein interactions in live cells. Biophysical Reviews. 3, 63-70 (2011).
- Padilla-Parra, S., Audugé, N., Coppey-Moisan, M., Tramier, M. Quantitative FRET analysis by fast acquisition time domain FLIM at high spatial resolution in living cells. Biophysical Journal. 95, 2976-2988 (2008).
- Stringari, C., et al. Phasor approach to fluorescence lifetime microscopy distinguishes different metabolic states of germ cells in a live tissue. Proceedings of the National Academy of Sciences of the United States of America. 108, 13582-13587 (2011).
- Digman, M. A., Caiolfa, V. R., Zamai, M., Gratton, E. The phasor approach to fluorescence lifetime imaging analysis. Biophysical Journal. 94, 14-16 (2008).
- Liang, Z., Lou, J., Scipioni, L., Gratton, E., Hinde, E. Quantifying nuclear wide chromatin compaction by phasor analysis of histone Förster resonance energy transfer (FRET) in frequency domain fluorescence lifetime imaging microscopy (FLIM) data. Data in Brief. 30, 105401 (2020).
- Grimm, J. B., Heckman, L. M., Lavis, L. D. The chemistry of small-molecule fluorogenic probes. Progress in Molecular Biology and Translational Science. 113, (2013).
- Li, L., Sun, H. Next Generation of Small-Molecule Fluorogenic Probes for Bioimaging. Biochemistry. 59, 216-217 (2020).
- Yao, R., Ochoa, M., Yan, P., Intes, X. Net-FLICS: fast quantitative wide-field fluorescence lifetime imaging with compressed sensing - deep learning approach. Light: Science and Applications. 8, 1-7 (2019).
- Smith, J. T., et al. Fast fit-free analysis of fluorescence lifetime imaging via deep learning. Proceedings of the National Academy of Sciences of the United States of America. 116, 24019-24030 (2019).
- Yao, R., Ochoa, M., Intes, X., Yan, P. Deep compressive macroscopic fluorescence lifetime imaging. Proceedings - International Symposium on Biomedical Imaging. 2018, 908-911 (2018).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados