
Method Article
Preparación de un gel peptídico definido por el usuario para modelos de cultivo 3D controlados de cáncer y enfermedades
En este artículo
Resumen
Presentamos un método para crear un entorno de cultivo celular en 3D, que se puede utilizar para investigar la importancia de las interacciones célula/matriz en la progresión del cáncer. Usando un simple octapéptido autoensamblable, se puede controlar la matriz que rodea a las células encapsuladas, con regulación independiente de señales mecánicas y bioquímicas.
Resumen
Cada vez hay más conciencia de que las células cultivadas en 3D modelan mejor el comportamiento in vivo que las cultivadas en 2D. En este protocolo, describimos un hidrogel 3D simple y ajustable, adecuado para cultivar células y tejidos en un entorno que coincida con su entorno nativo. Esto es particularmente importante para los investigadores que investigan el inicio, el crecimiento y el tratamiento del cáncer, donde la interacción entre las células y su matriz extracelular local es una parte fundamental del modelo. El paso a la cultura 3D puede ser un reto y, a menudo, se asocia a una falta de reproducibilidad debido a la gran variación de un lote a otro en las matrices de cultivo 3D derivadas de animales. Del mismo modo, los problemas de manipulación pueden limitar la utilidad de los hidrogeles sintéticos. En respuesta a esta necesidad, hemos optimizado un gel peptídico simple y autoensamblable para permitir el cultivo de modelos de líneas celulares relevantes de cáncer y enfermedades, así como tejidos/células derivadas de pacientes. El gel en sí está libre de componentes de la matriz, aparte de los añadidos durante la encapsulación o depositados en el gel por las células encapsuladas. Las propiedades mecánicas de los hidrogeles también pueden modificarse independientemente de la adición de la matriz. Por lo tanto, actúa como una "pizarra en blanco" que permite a los investigadores construir un entorno de cultivo en 3D que refleje el tejido objetivo de interés y diseccionar las influencias de las fuerzas mecánicas y/o el control bioquímico del comportamiento celular de forma independiente.
Introducción
Las múltiples funciones que desempeña el entorno extracelular en el desarrollo y la progresión del cáncer son cada vez más claras1. Recientemente, análisis detallados basados en la proteómica se han sumado a una base bibliográfica ya convincente, demostrando que los componentes de la matriz derivados de las células estromales asociadas al cáncer, o las propias células cancerosas, son factores clave en eventos como la promoción de la transición mesenquimal epitelial y la diseminación metastásica 2,3,4. Dada esta reconocida importancia de la matriz extracelular (MEC), se está volviendo crucial avanzar hacia plataformas de cultivo celular que permitan el control sobre el entorno 3D que se presenta a las células. En respuesta a esta necesidad, este protocolo presenta un método para la encapsulación y cultivo celular en un hidrogel 3D, con composición de ECM y propiedades mecánicas definidas por el usuario5.
En la actualidad, existe una escasa correlación entre la eficacia terapéutica del cáncer en cultivo in vitro 2D, el impacto de estas terapias en los modelos in vivo actuales (xenoinjerto derivado del paciente, PDX) y su eventual actividad en ensayos clínicos 6,7. Esto ha llevado a fracasos significativos en el proceso de descubrimiento de fármacos, con una necesidad urgente de mejorar los modelos in vitro que permitan que las terapias probadas "fracasen temprano, fracasen baratas". Muchos investigadores utilizan el producto derivado del mastocitoma de ratón, por ejemplo, Matrigel (o productos similares) para crear entornos ricos en matrices 3D para crecer y observar el comportamiento celular in vitro, incluidas las células derivadas de PDX y otras células cercanas al paciente 8,9,10. Sin embargo, este enfoque de "talla única" pasa por alto el complejo papel que desempeñan las proteínas/glicanos de la matriz en el inicio y la progresión del cáncer.
El reconocimiento del papel de la matriz extracelular (MEC) en el control del comportamiento celular también ha fomentado el uso del cultivo 3D en o sobre hidrogeles compuestos por componentes específicos de la matriz11. Si bien esto es útil para investigar interacciones específicas, estos sistemas sufren de la incapacidad de separar las instrucciones mecánicas y bioquímicas entre las células y la matriz. También pueden ser difíciles de manejar y pueden dar lecturas poco claras del comportamiento de las células. Los geles de colágeno son un ejemplo clave de este problema, ya que la contracción delgel mediada por células puede reducir drásticamente la capacidad de visualizar las células dentro del gel. También hay algunos sistemas de gel multicomponente muy elegantes, que los expertos han utilizado con gran efecto 12,13,14. Estos pueden incorporar enlazadores sensibles a las enzimas y motivos bioactivos, pero son significativamente más complejos en su formulación y aplicación que el sistema descrito aquí.
Este protocolo describe un método para crear modelos de cultivo 3D completamente definidos, lo que permite modelar in vitro las funciones de la MEC en el desarrollo y la enfermedad. La base del modelo 3D es un gel peptídico, que hemos descrito anteriormente como una optimización de un hidrogel octapeptídico autoensamblablesimple 5,15,16. Al alejarse de las matrices complejas de origen animal, este sistema ofrece un beneficio significativo de una mayor consistencia entre lotes y un mejor manejo. En su estado simple, el péptido no contiene motivos derivados de la matriz y proporciona efectivamente una "pizarra en blanco" sobre la que el usuario puede construir la funcionalidad.
Demostramos cómo las propiedades mecánicas del gel peptídico pueden regularse de forma independiente, junto con la incorporación de proteínas de la matriz/glicanos. El sistema es altamente ajustable, lo que permite la encapsulación de una variedad de tipos de células en varios formatos. Es importante destacar que para la construcción de un modelo de cáncer, las células estromales también se pueden incorporar: ya sea en cocultivo directo o separadas para permitir un análisis específico de las interacciones indirectas entre las células cancerosas y el estroma. Lo más importante es que el protocolo descrito aquí no requiere conocimientos complejos de química y puede reproducirse en cualquier laboratorio de cultivo celular sin necesidad de conocimientos o equipos químicos especializados.
Hemos optimizado los métodos para el estudio del comportamiento celular en los geles peptídicos, incluyendo la obtención de imágenes, el análisis reológico, la extracción de material para PCR5 y la inclusión para la evaluación histológica. Un claro beneficio del sistema de hidrogel simple es la capacidad de visualizar y estudiar la matriz depositada por las células encapsuladas. La importancia de las matrices derivadas de células y los beneficios de una mejor comprensión de cómo las células rediseñan su microambiente local se ha puesto de relieve recientemente17 y refleja una creciente conciencia de la importancia de atrapar los componentes de la matriz secretados por las células, de forma similar a lo que ocurre in vivo. El aprovechamiento de la capacidad de modelar estos procesos puede ser uno de los impulsores fundamentales de la mayor relevancia para los pacientes de los modelos de enfermedades basados en hidrogel.
Protocolo
1. Disolución del péptido
- En una campana de cultivo de tejidos, agregue 800 μL de agua estéril a un tubo de 15 mL con una pipeta P1000.
- Usando una balanza fina, pese el polvo peptídico en un bote de pesaje no estático. Utilice una masa (en mg) de 1,25 veces la concentración final deseada de gel peptídico (en mg/mL, Tabla 1).
NOTA: El método descrito aquí producirá un volumen de aproximadamente 1,25 mL de gel peptídico por tubo en el punto de gelificación final. Se pueden preparar varios tubos a la vez, o alternativamente se puede aumentar el volumen por tubo cuando el usuario tiene experiencia con el método presentado.
| Concentración de péptidos (después de la gelificación final) | Masa de péptido | Adición inicial de NaOH |
| 6 mg/mL | 7,5 mg | 30 μL |
| 10 mg/mL | 12,5 mg | 60 μL |
| 15 mg/mL | 18,75 mg | 100 μL |
Tabla 1: Masa peptídica y adición inicial sugerida de NaOH para concentraciones finales típicas de gel. Los rangos de concentraciones de péptidos enumerados pueden extenderse en cualquier dirección, sin embargo, es más probable que las concentraciones más bajas de péptidos no formen geles estables, mientras que a altas concentraciones el gel resultante puede ser demasiado denso para permitir un intercambio suficiente de nutrientes y viabilidad celular. La concentración adecuada requerirá optimización para diferentes tipos de células y lotes de péptidos.
- Agregue el péptido pesado en el tubo de 15 ml. Agite el bote de pesaje para asegurarse de que no quede polvo.
NOTA: El polvo peptídico puede ser muy estático, tenga cuidado de minimizar la pérdida de polvo. No es necesario mantener condiciones estériles durante los pasos 1.2 y 1.3 (o para cualquier medición de pH) ya que la esterilización se produce durante la incubación a 80 °C más adelante en el proceso. - Vórtice durante 3 minutos, luego centrifugar a 200 x g durante 3 minutos.
- Incubar la solución peptídica en un horno a 80 °C durante un mínimo de 2 h. Si hay péptido no disuelto después de la incubación, repita el paso 1.4.
NOTA: Dado que el calentamiento uniforme es esencial, un bloque de calor no es adecuado para este paso. Sin embargo, hemos descubierto que los hornos de hibridación funcionan bien, al igual que un baño de agua, siempre que la solución peptídica esté completamente sumergida.
2. Formación de precursores de gel
- Prepare una solución estéril de hidróxido de sodio (NaOH) de 0,5 M con agua estéril.
NOTA: Se recomienda una solución de NaOH recién diluida para obtener los resultados más consistentes. - En una campana de cultivo de tejidos, agregue 0,5 M de NaOH al centro del péptido disuelto y mezcle agitando lentamente con la punta de la pipeta (Tabla 1).
- Agite el tubo durante 10 s y centrifugue a 200 x g durante 10 s para eliminar las burbujas.
- Si el precursor del gel está turbio, repita los pasos 2.2 y 2.3, añadiendo incrementos de 5 μL de NaOH.
NOTA: La medición del pH puede ayudar a determinar si se ha añadido suficiente NaOH: el pH óptimo está entre 9 y 10,5 y es preferible no superarlo, ya que el uso de ácidos para reducir el pH puede dar lugar a una falta de homogeneidad del gel. El volumen preciso de NaOH requerido puede variar según la fuente del péptido. - Una vez que el precursor de gel sea ópticamente transparente y autoportante (o solo fluya) al invertir el tubo de 15 mL, agregue 100 μL de PBS 10x estéril en una campana de cultivo de tejidos. Vórtice durante 10 s y centrífuga a 200 x g durante 10 s.
NOTA: Si el precursor de gel está turbio, repita el paso 2.4 hasta que se vuelva transparente y semisólido. Si el precursor del gel es líquido (por encima del pH ~10.5), no formará un gel estable y debe desecharse. - Incubar durante la noche en un horno a 80 °C.
- Compruebe visualmente el precursor de gel para asegurarse de que está completamente líquido a 80 °C.
- Si el precursor del gel no es completamente líquido a 80 °C, no se ha neutralizado suficientemente, agregue NaOH siguiendo el paso 2.4.
- Si el precursor del gel es líquido pero hay burbujas de aire o pequeños precipitados, agite el tubo bruscamente para dispersarlos. Si persisten las burbujas de aire o los precipitados después de agitar el tubo, haga un vórtice con el precursor de gel y centrifugue a 200 x g durante 10 s cada uno.
- Después de seguir cualquiera de los pasos 2.7.1 o 2.7.2, incubar los geles precursores durante 2 h más a 80 °C antes de proceder a la gelificación final.
- Mantener los precursores de gel a 80 °C hasta que se necesiten (máximo 48 h).
NOTA: El protocolo se puede pausar aquí, almacene los precursores de gel a 4 °C hasta por 4 semanas. Si se reinicia desde este punto de pausa, incube los precursores de gel a 80 °C durante al menos 2 h y reinicie desde el paso 2.7. Se recomienda encarecidamente preparar los precursores de gel con mucha antelación a que se necesiten para la gelificación final, a fin de garantizar que haya tiempo suficiente para las modificaciones anteriores si es necesario.
3. Preparación de los componentes de la matriz para la siembra
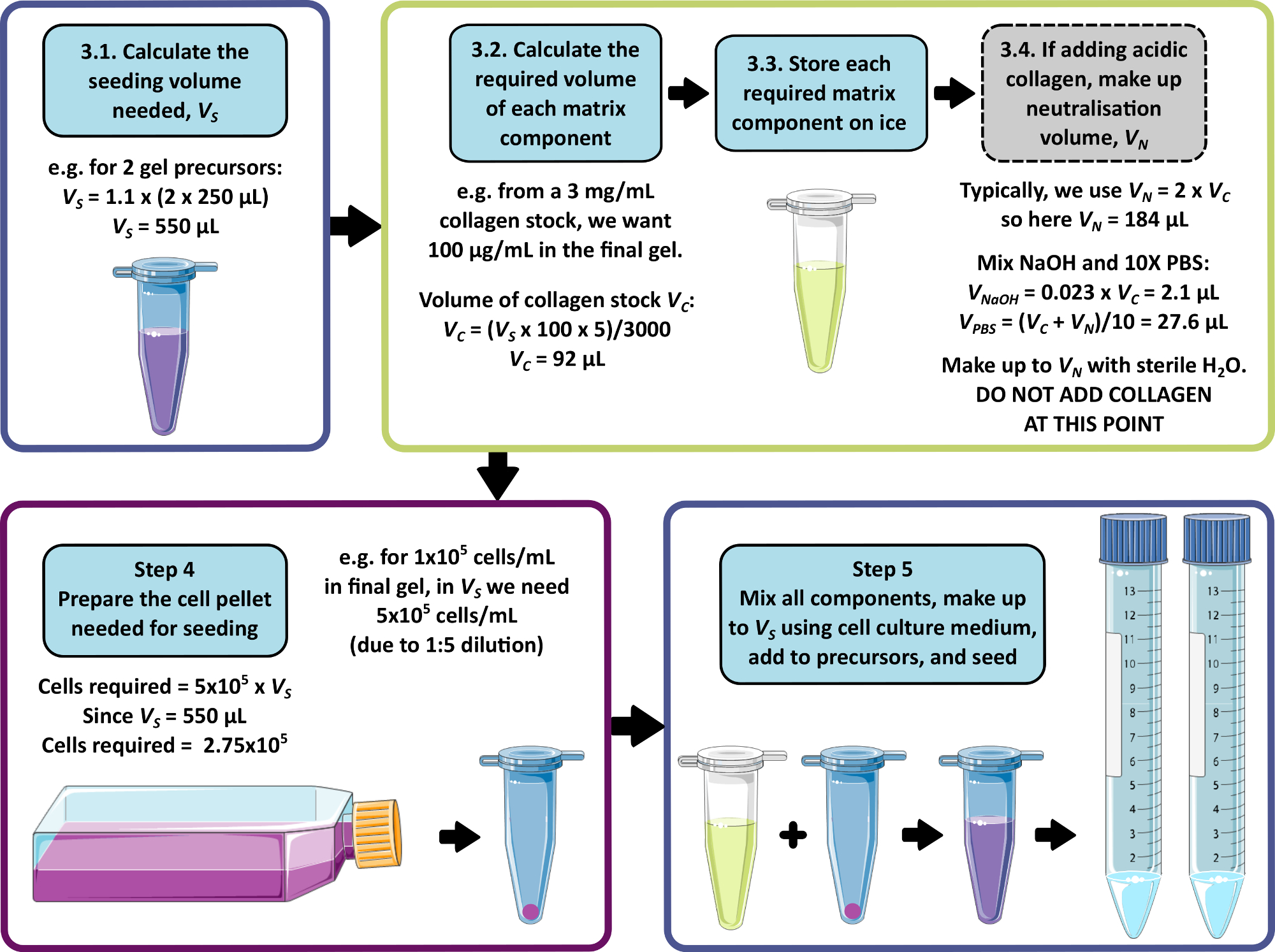
NOTA: En la Figura 1 se muestra un ejemplo de cálculo para los pasos 3 a 5. Los pasos 3 y 4 pueden omitirse para producir un gel sin matriz y/o sin células, respectivamente.
- Calcule el volumen total combinado de células, matriz y medios necesarios para la siembra, permitiendo añadir 250 μL a cada 1 mL de gel precursor. Multiplique este volumen por 1,1 para permitir el error de pipeteo. Este es el volumen de siembra, VS.
- Para cada componente de la matriz, calcule el volumen de solución madre que se agregará al volumen de siembra utilizando la Ecuación 1:
 (1)
(1)
NOTA: VS corresponde al volumen total de medio de siembra requerido para sembrar todos los geles (paso 3.1). Asegúrese de que la suma de todos los volúmenes de los componentes de la matriz no supere VS. - Descongele completamente todos los componentes de la matriz (consulte las instrucciones del fabricante) y guárdelos en hielo hasta que los necesite.
NOTA: Algunos componentes de la matriz serán muy propensos a la gelificación. Consulte las instrucciones del fabricante para evitar que esto ocurra. - Si se va a agregar una solución madre de colágeno ácido que requiere neutralización (consulte las instrucciones del fabricante), prepare la solución de neutralización de la siguiente manera:
- Calcule el volumen de solución madre de colágeno, VC, requerido usando la Ecuación 1.
- Determine un volumen adecuado para la solución de neutralización, VN. Debe elegirse de manera que el volumen combinado de todas las adiciones de la matriz no exceda el volumen de siembra, VS.
NOTA: En general, un valor razonable para VN = 2 x Vc, pero esto puede ajustarse de acuerdo con el número y el volumen de otros componentes de la matriz que se agregarán. - Calcule el volumen de NaOH de 1 M requerido para la neutralización utilizando la Ecuación 2.
 (2)
(2) - Calcule el volumen de 10x PBS requerido en la solución de neutralización usando la Ecuación 3.
 (3)
(3) - Combine los volúmenes calculados de 1 M de NaOH y 10x PBS, luego haga hasta VN con agua estéril. Mezcle bien y almacene en hielo hasta que lo necesite.
NOTA: No agregue el colágeno ácido en este punto. La mezcla prematura del colágeno con la solución de neutralización conducirá al inicio de la fibrilogénesis del colágeno, lo que puede causar inconsistencia en las propiedades del gel peptídico final.
4. Preparación de las células para la siembra
- Si aún no se ha completado, calcule el volumen de siembra, VS de acuerdo con el paso 3.1.
- Calcule la densidad celular requerida en esta suspensión celular, tomando la densidad celular deseada en el gel peptídico final y multiplicándola por 5.
NOTA: La suspensión celular es 5 veces la concentración final deseada para tener en cuenta la dilución al mezclarse con el precursor en gel. Las densidades celulares deben optimizarse para cada nueva línea celular que se esté considerando. Dependiendo del tipo de célula, pueden ser apropiadas densidades entre 1 x 104 y 1 x 106 células/ml en el gel peptídico final. - Calcule el número total de celdas necesarias para la siembra (multiplique la densidad de celdas por el volumen de siembra VS calculado en el paso 4.1.).
- Utilizando métodos estándar de cultivo/paso para las células en uso, prepare una pastilla de celda que contenga la cantidad requerida de células calculada en el paso 4.3.
5. Gelificación final/encapsulación celular
- Cuando esté listo para comenzar la gelificación final, transfiera los precursores de gel del horno a 80 °C a un baño de agua a 37 °C.
NOTA: Los precursores de gel deben ser autoportantes a 37 °C. Si son líquidos, es poco probable que ocurra una gelificación completa y los precursores del gel deben desecharse. - Prepare una placa de 96 pocillos, o alternativamente una placa de 24 pocillos (o similar) con insertos de cultivo celular.
NOTA: Asegúrese de que haya un espacio entre la base del inserto y la placa de pocillos cuando siembra geles de péptidos en insertos de 24 pocillos. Esto asegura que el gel esté en contacto con los medios. - Si se va a agregar una matriz, combine todos los componentes de la matriz y las soluciones de neutralización (paso 3). Hacer hasta VS (paso 3.1) con medio de cultivo celular y mezclar bien. Si se van a añadir células, vuelva a suspender el pellet de células preparado en el paso 4 utilizando el volumen de siembra VS.
NOTA: Si no se han preparado componentes de la matriz en el paso 3, utilice el medio de cultivo celular para el volumen de siembra. Por lo general, este es el medio de cultivo estándar para el tipo de célula que se utiliza, aunque puede ser necesario validarlo si se utiliza una mezcla de tipos de células. - Con una pipeta P1000, añada suavemente 250 μL de la mezcla de células/matrices sobre el precursor de gel.
NOTA: Los componentes de la matriz, particularmente el extracto de membrana basal, pueden comenzar a polimerizarse una vez que se agregan al precursor del gel. Por lo tanto, es importante pasar al siguiente paso lo más rápido posible. - Mezclar suavemente mediante la acción combinada de pipetear y agitar. El gel es adelgazante por cizallamiento, por lo que será más fácil de mezclar con un pipeteo / agitación suave. Cuando esté completamente mezclado, agregue 100 μL a cada pocillo de una placa de 96 pocillos, o 200 μL a cada inserto de cultivo celular.
NOTA: Al mezclar, el pipeteo inverso con un P1000 ajustado a 200 μL puede ser beneficioso para evitar la introducción de burbujas de aire. Al principio, el gel puede ser difícil de mezclar, pero si se continúa mezclando, debería ser más fácil, lo que indica que la mezcla es eficiente. - Incubar durante 10 min a 37 °C en 5% de CO2 y en atmósfera humidificada.
NOTA: Este paso no es necesario si el gel peptídico no contiene adiciones a la matriz. - Agregue 200 μL de medio a cada pocillo de la placa de 96 pocillos, o 1 mL al exterior del inserto de cultivo celular con unas gotas en la parte superior del gel. Incubar a 37 °C en 5% de CO2 y en atmósfera humidificada.
- Cambie el medio dos veces en la próxima hora y nuevamente después de varias horas (o al día siguiente).
NOTA: Tenga cuidado aquí ya que los geles estarán inestables durante varias horas. - Cambie el medio cada 2 o 3 días (o siga el protocolo de cultivo estándar para las células en uso).
6. Cocultura indirecta
NOTA: Este método solo es aplicable cuando los geles peptídicos se siembran en insertos de placa de 24 pocillos o formatos similares en los que el gel se puede apoyar sobre una monocapa celular. En este caso, se puede introducir el cocultivo indirecto mediante la preparación de una capa alimentadora 2D de células en el fondo de la placa de pocillo.
- Prepare un plato de 24 pocillos para sembrar. Esta debe ser una placa separada de los geles peptídicos, pero debe ser de la misma marca para garantizar la compatibilidad con los insertos utilizados (consulte el paso 5.2).
- Calcule la densidad de celdas necesaria para sembrar el cocultivo indirecto, de acuerdo con el tipo de celda que se esté considerando.
NOTA: La densidad de siembra de células debe optimizarse para cada nueva línea celular que se esté considerando. Las células deben estar cubiertas para dar aproximadamente un 30-50% de confluencia. A modo de ejemplo, la línea celular de fibroblastos mamarios humanos HMFU19 suele estar sembrada con una densidad de 1-5 x 104 células/pocillo. - Utilizando métodos estándar de cultivo/paso para las células en uso, prepare una suspensión celular adecuada para la siembra a 1 mL/pocillo, utilizando el medio de crecimiento típico para estas células.
- Siembre la suspensión celular en la placa de pocillo, 1 mL por pocillo.
- Incubar a 37 °C en 5% de CO2 y en una atmósfera humidificada para adherir durante varias horas o toda la noche. Luego retire el medio de los pozos.
- Con pinzas estériles, transfiera los insertos de placa de 24 pocillos que contienen los geles peptídicos a los nuevos pocillos que contienen las células presembrados en 2D. Añadir 1 mL de gota a gota media en el exterior del inserto y unas gotas en la superficie del gel.
NOTA: Por lo general, el medio utilizado en este punto es el adecuado para las células encapsuladas en el gel, pero la idoneidad de este medio para las células en 2D puede necesitar verificación u optimización para el experimento particular que se está considerando. - Prepare regularmente las ponedoras frescas del comedero para evitar la confluencia excesiva y transfiera los geles de péptidos a estos nuevos pocillos siguiendo el mismo método que el anterior.
NOTA: Por lo general, las células para el cocultivo indirecto se preparan al mismo tiempo que la siembra del gel de péptidos. A continuación, los geles peptídicos pueden transferirse al cocultivo en el punto de cambio de medio después de unas horas o durante la incubación nocturna, véase el paso 5.8.
7. Reología oscilatoria a granel de geles peptídicos
NOTA: Como estándar, la caracterización reológica se lleva a cabo 24 h después de la siembra en gel, que debe realizarse en insertos de placa de 24 pocillos.
- Configure y calibre el reómetro de acuerdo con las instrucciones del fabricante. Utilice una geometría de placa paralela con un diámetro de placa lo más cercano posible al diámetro del inserto de cultivo celular.
NOTA: Las pruebas pueden realizarse a 37 °C si se desea, replicando el entorno durante el cultivo. - Extraiga la primera muestra de gel peptídico que se vaya a analizar del inserto de cultivo celular invirtiendo el inserto y cortando la membrana de plástico con un bisturí.
NOTA: Asegúrese de que la placa que contiene los geles peptídicos esté fuera de la incubadora de cultivos celulares durante el menor tiempo posible antes de la prueba. Los medios con un sistema de amortiguación de bicarbonato dependen de la presencia de CO2 para mantener el pH. Los geles que han estado fuera de la incubadora demasiado tiempo tendrán un pH a la deriva, lo que puede afectar la evaluación reológica. Puede ser beneficioso mantener los geles con 10 mM de HEPES añadidos al medio para evitar este efecto. - Transfiera con cuidado el gel a la placa del reómetro. Luego, con un bisturí, recorte la altura del gel a aproximadamente 1 mm para minimizar la deformación del gel cuando se cargue debajo de la placa del reómetro.
NOTA: Tenga cuidado de no tocar ni dañar la placa del reómetro cuando utilice el bisturí. - Ajuste la distancia entre placas paralelas a 1 mm. Quite el exceso de gel que no esté cubierto por las placas del reómetro.
- Ejecute la configuración de prueba deseada en el reómetro, de acuerdo con las instrucciones del fabricante.
NOTA: Para cada nueva condición de la muestra, se recomienda ejecutar un barrido de amplitud de 0,1 a 100% de deformación para garantizar que todas las pruebas se lleven a cabo a un nivel de deformación dentro de la región viscoelástica lineal de la muestra.
8. Tinción viva/muerta de células encapsuladas
- Retire el medio de los pocillos y lave los geles peptídicos dos veces con 1x PBS, utilizando la misma técnica que para un cambio de medio (paso 5.7).
- Retire los geles peptídicos que se hayan cultivado en insertos de placa de 24 pocillos siguiendo el paso 7.2. Mantenga los geles en 1x PBS en la placa de pocillo original hasta que estén listos para teñir.
NOTA: Tenga cuidado ya que los geles pueden ser frágiles en este punto, especialmente después de un cultivo prolongado. - Prepare una solución de tinción viva/muerta, permitiendo una tinción de 500 μL por inserto de placa de 24 pocillos, o 50 μL por pocillo de una placa de 96 pocillos. Una tinción típica es 4 μM de homodímero de etidio y 2 μM de calceína AM en 1x PBS. Proteja la solución resultante de la luz.
NOTA: Las concentraciones de reactivos vivos/muertos pueden requerir una mayor optimización en función del tipo de célula utilizada y del proveedor de reactivos. - Retire con cuidado el PBS de cada gel y reemplácelo con unas gotas de la solución para manchar, asegurándose de que cada gel esté bien cubierto.
- Incubar los geles en la solución de tinción en la oscuridad durante 10 a 15 minutos, luego visualizarlos con un microscopio confocal/fluorescente.
NOTA: Para obtener imágenes de mayor calidad, puede ser beneficioso transferir los geles a platos con fondo de vidrio del grosor del cubreobjetos.
9. Fijación de geles peptídicos para la obtención de imágenes de punto final
- Lave los geles peptídicos siguiendo el paso 8.1.
- Agregue un 4% de paraformaldehído (PFA) en 1x PBS: 100 μL por cada pocillo de una placa de 96 pocillos y 1 mL por cada gel en un inserto de placa de 24 pocillos (se deben agregar unas gotas encima del gel dentro del inserto).
PRECAUCIÓN: El paraformaldehído (PFA) es altamente tóxico y se absorbe fácilmente a través de la piel. Es extremadamente destructivo para la piel, los ojos, las membranas mucosas y el tracto respiratorio superior. El PFA debe manipularse en una campana extractora y los usuarios deben usar ropa protectora y guantes. Los fijadores químicos alternativos se pueden usar de la misma manera según la aplicación final. - Incubar geles de péptidos en fijador de PFA durante 1 h a temperatura ambiente.
- Retire el fijador de PFA y lave los geles de péptidos dos veces con 1x PBS.
NOTA: El protocolo se puede pausar aquí, almacene los geles de péptidos fijos a 4 °C en 1x PBS hasta por 4 semanas, asegurándose de que la placa esté bien sellada con una película de parafina.
10. Inclusión de geles peptídicos para el corte
NOTA: La inclusión de geles de péptidos en agar al 4% es un paso crucial antes de la inclusión en parafina para la inmunohistoquímica. Alternativamente, los geles pueden incrustarse en agar al 2% y seccionarse con un vibratomo (por lo general, las secciones de 500 μm dan buenos resultados). Este es un paso opcional, que produce secciones de gel hidratadas que pueden ser beneficiosas para teñir la localización de la matriz extracelular en el gel, utilizando los métodos de la sección 11.
- Prepare una solución de agar fundida al 2% o al 4% en 1x PBS (ver nota anterior), hirviendo en un microondas. Deje enfriar durante unos minutos antes de usar.
NOTA: El agar fundido presenta un peligro de calor: manéjelo con cuidado con protección para las manos y la cara. Una vez preparada, la solución de agar puede almacenarse a 4 °C hasta que se necesite. - Retire el gel peptídico del inserto de cultivo celular siguiendo el paso 7.2.
- Con una pipeta Pasteur de plástico, cubra la base de un molde de inclusión histológica con una capa fina de solución de agar. Dejar enfriar a 20 °C durante unos segundos.
- Con una espátula, coloque el gel de péptidos en el centro del agar. A continuación, cubra completamente el gel peptídico en agar.
NOTA: El gel no debe hundirse en el agar. Si es así, retira el gel y espera unos segundos más hasta que el agar se haya solidificado más y vuelve a intentarlo. Trate de no dejar que se produzca demasiada solidificación o habrá una unión débil entre las dos capas. - Deje que el gel incrustado se enfríe durante 1 h a 4 °C antes de retirarlo del molde histológico.
NOTA: El protocolo se puede pausar aquí, almacene geles incorporados a 4 °C en 1x PBS hasta por 4 semanas. - Si se va a llevar a cabo la inmunohistoquímica, coloque geles incrustados en un procesador de tejidos y proceda utilizando métodos de laboratorio estándar.
NOTA: Alternativamente, los geles incrustados se pueden cortar en secciones hidratadas usando un vibrátomo. Las secciones hidratadas deben almacenarse en placas selladas a 4 °C en 1x PBS durante un máximo de 4 semanas.
11. Tinción de células en geles mediante inmunocitoquímica
- Retire el 1x PBS que cubre las secciones de geles de péptidos/gel. Retire los geles que aún estén en los insertos de placa de 24 pocillos siguiendo el paso 7.2.
- Cubrir las secciones de gel/gel con un tampón de bloqueo e incubar durante 30 min a 20 °C.
NOTA: Un tampón de bloqueo típico consiste en 0,5% de albúmina sérica bovina (BSA) en 1x PBS con 0,1% de Triton X-100. Triton X-100 es tóxico y causa graves daños oculares, irritación de la piel y es muy tóxico para la vida acuática. Los usuarios deben usar ropa protectora, protección para los ojos y guantes. - Prepare los anticuerpos primarios en el tampón de bloqueo a concentraciones de trabajo optimizadas. Permita 200 μL por plato de gel de 24 pocillos, 100 μL por sección de gel y 50 μL por pocillo de placa de 96 pocillos.
NOTA: Por lo general, las concentraciones de anticuerpos utilizadas para la tinción 3D en geles deben ser el doble de la concentración utilizada en 2D. - Retire el tampón de bloqueo y agregue la solución de anticuerpos a los geles gota a gota.
- Sellar la placa con una película de parafina e incubar durante la noche a 4 °C.
- Retire la solución de anticuerpos y lávese dos veces con tampón de bloqueo.
- Agregue el anticuerpo secundario siguiendo los mismos procedimientos descritos en los pasos 11.3 y 11.4.
- Incubar en la oscuridad durante la noche a 4 °C, o durante 3 horas a 20 °C.
- Retire la solución de anticuerpos y lávese dos veces con 1x PBS.
- Cubra las muestras con una solución DAPI 1:1.000 e incube a 4 °C en la oscuridad durante 1 h.
- Transfiera el gel a un cubreobjetos de vidrio y obtenga una imagen mediante microscopía fluorescente/confocal.
12. Extracción de ARN
NOTA: Los volúmenes utilizados en este método son aplicables cuando los geles peptídicos se siembran en insertos de placa de 24 pocillos. Se pueden utilizar otros formatos de gel y ajustar los volúmenes en consecuencia.
- Retire el medio de los pocillos y lave los geles peptídicos dos veces con 1x PBS, utilizando la misma técnica que para un cambio de medio (paso 5.7).
- Retire los geles peptídicos que se hayan cultivado en insertos de placa de 24 pocillos siguiendo el paso 7.2. Coloque cada gel en un tubo de centrífuga de 15 ml por separado.
NOTA: Tenga cuidado ya que los geles pueden ser frágiles en este punto, especialmente después de un cultivo prolongado. - Con un P1000, agregue 500 μL de tripsina-EDTA (0,25%) a cada tubo y pipetee hacia arriba y hacia abajo para mezclar y romper el gel.
- Incubar los geles en la tripsina-EDTA a 37 °C durante 3-5 min.
NOTA: Los tiempos de incubación pueden requerir optimización según el tipo de celda utilizado. - Añadir 5 mL de 1x PBS para diluir la tripsina-EDTA.
- Centrifugar a 200 x g durante 5 min para pellets celdas.
- Retire el sobrenadante.
NOTA: Tenga cuidado ya que puede haberse formado una capa de gel entre el pellet de la célula y el sobrenadante. - Vuelva a suspender el pellet celular en el tampón de lisis, de acuerdo con las instrucciones del fabricante, y proceda siguiendo los protocolos estándar para la extracción de ARN.
Resultados
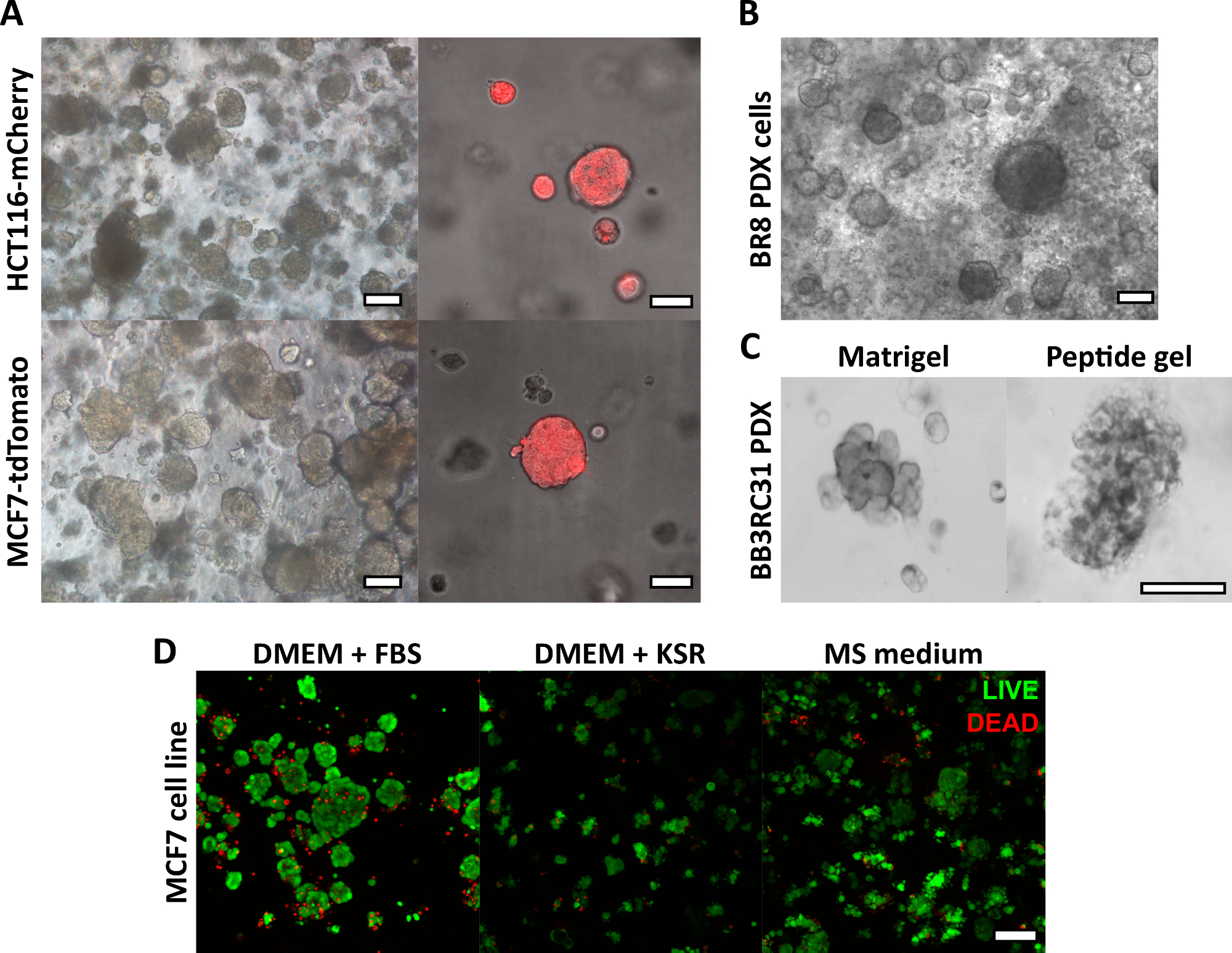
El método de fabricación de gel peptídico descrito aquí permite al usuario definir y crear un entorno de cultivo 3D a medida. Si bien el entorno mecánico está determinado principalmente por la concentración de péptidos, los componentes de la matriz de interés también se pueden agregar a densidades controladas, como se muestra en el cálculo de ejemplo en la Figura 1. Sin embargo, en su forma más simple, el protocolo de gel de péptidos proporciona un método para encapsular células en un entorno 3D sin matriz. La Figura 2 muestra cómo este enfoque puede combinarse con una amplia gama de modelos de cáncer, incluidas las líneas celulares cancerosas marcadas con fluorescencia (Figura 2A) y el material de xenoinjerto derivado del paciente (PDX) (Figura 2B,C). Es importante destacar que las líneas celulares y el material PDX pueden cultivarse dentro de los geles en condiciones libres de suero (Figura 2C,D), lo que proporciona un sistema de cultivo 3D con una composición completamente definida.
Dado que el péptido en sí no contiene ningún motivo de unión celular, las células encapsuladas suelen mostrar una morfología redondeada en los geles de péptidos no modificados. La Figura 3A demuestra esto para los fibroblastos mamarios humanos en un gel de péptidos de 6 mg/mL, en comparación con su morfología alargada clásica observada en Matrigel puro y un gel de colágeno puro. Sin embargo, es importante destacar que el protocolo de gel peptídico permite la incorporación de componentes de la matriz de interés. La Figura 3A demuestra cómo la adición de 200 μg/mL de colágeno I puede restaurar la morfología alargada de los fibroblastos en los geles peptídicos.
Las adiciones de matrices también pueden apoyar el crecimiento y la organización de otros tipos de células, por ejemplo, MCF10A, como se muestra en la Figura 3B. En este caso, la adición de 100 μg/mL de colágeno I a un gel de péptidos de 6 mg/mL permite que se formen estructuras acinares para el día 7. También se puede introducir una mayor complejidad mediante la incorporación de una capa celular de soporte en el cocultivo indirecto. La Figura 3C demuestra cómo el enfoque combinado de incorporación de la matriz y el cocultivo indirecto con fibroblastos mamarios humanos puede mejorar el crecimiento y la organización de MCF10A.
Otro parámetro importante es la concentración de péptido utilizado en la fabricación de gel de péptido. La figura 4A muestra un ejemplo de cómo el control de la concentración de péptidos, en este caso entre 4 y 10 mg/mL, da como resultado una rigidez que oscila entre 100 y 1000 s de Pa. Estos geles pueden fabricarse sin matriz o se pueden crear con adiciones de matriz para permitir el control simultáneo de la rigidez y la composición. Los geles peptídicos con adiciones a la matriz se pueden seccionar y teñir para permitir que se visualice la distribución de estas adiciones. Las Figuras 4B y C muestran dos enfoques para hacer esto: inclusión en agar al 4% seguido de procesamiento de tejidos estándar e inclusión en parafina para inmunohistoquímica (Figura 4B) o inclusión en agar al 2% seguido de corte de vibratomo y tinción fluorescente (Figura 4C).
Al modificar la composición de los geles peptídicos, es crucial asegurarse de que estos cambios no afecten el entorno mecánico presentado inicialmente a las células. La Figura 4D demuestra cómo se pueden utilizar las modificaciones en la concentración de péptidos para compensar cualquier cambio en la rigidez del gel peptídico en la incorporación de la matriz. Las mediciones reológicas oscilatorias masivas de la rigidez del gel (módulo de almacenamiento, G') pueden distinguir entre los efectos de la composición del gel y la rigidez en la morfología celular. Como se muestra en las imágenes de campo claro, las células MDA MB 231 desarrollan una morfología alargada en la adición de colágeno a geles de péptidos de 10 mg/mL o 15 mg/mL. La Figura 4E muestra que estas células alargadas dan positivo para pFAK, lo que indica una interacción con su matriz circundante. El entorno inicialmente libre de matriz de los geles peptídicos también los convierte en una plataforma ideal para estudiar la síntesis celular y la deposición de componentes de la matriz de interés. La figura 4F muestra la deposición localizada de colágeno I por células MCF7 encapsuladas en geles de péptidos de 10 mg/mL.
Una de las principales ventajas de los geles peptídicos es la facilidad con la que se pueden aplicar los métodos estándar de laboratorio a su análisis. Se puede extraer material para qRT-PCR para determinar los perfiles de expresión génica (como se muestra en nuestra reciente publicación5). Las imágenes por microscopía de campo claro permiten, además, la visualización en tiempo real del crecimiento celular. La Figura 5 muestra algunos de los problemas típicos de solución de problemas que se pueden encontrar en los geles peptídicos fallidos: mezcla incompleta del precursor del gel (Figura 5A,B); optimización incorrecta de la concentración de péptidos (Figura 5C,D) o de la densidad de siembra (Figura 5E,F); y neutralización incorrecta del colágeno ácido antes de su incorporación en los geles peptídicos (Figura 5G,H). La concentración de péptidos y la densidad de siembra, en particular, deben optimizarse para cada línea celular y fuente de péptidos, para garantizar que el entorno de cultivo esté adecuadamente definido y sea representativo de la aplicación de interés.

Figura 1: Un ejemplo de cálculo para la composición de la matriz y la densidad de siembra. Este ejemplo de flujo de trabajo describe el procedimiento que se seguiría para sembrar dos precursores de gel peptídico con adiciones de 100 μg/mL de colágeno, a una densidad celular final de 1 x 105 células/mL. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Los geles peptídicos sin matriz proporcionan una plataforma de cultivo 3D adecuada para líneas celulares y modelos de cáncer derivados de pacientes. (A) Las líneas celulares de cáncer colorrectal HCT116 y cáncer de mama MCF7, que expresan constitutivamente los marcadores fluorescentes mCherry y tdTomato respectivamente, forman grupos de células en geles de 6 mg/mL en el día 9 (izquierda), y pueden ser visualizadas en vivo usando microscopía fluorescente (derecha, barra de escala de 50 μm); (B) Las células de xenoinjerto (PDX) derivadas de pacientes de una paciente con cáncer de mama triple negativo (BR8) forman grupos de células en el día 7 en geles de péptidos de 10 mg/mL; (C) Las células PDX de tumores de mama con receptor de estrógeno positivo (BB3RC31) pueden cultivarse en condiciones sin suero18, mostradas con control de la matriz de la membrana basal (por ejemplo, Matrigel) en el paso emparejado para la comparación; (D) Las células de cáncer de mama MCF7 son viables en geles peptídicos de 6 mg/mL en condiciones sin matriz y sin suero, según se evaluó mediante un ensayo de células vivas/muertas en el día 7. KSR = reemplazo de suero knockout, medio MS = medio mamosférico19. Barra de escala de 100 μm a menos que se especifique. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: La complejidad del gel peptídico puede incrementarse mediante la introducción de adiciones de matriz y cocultivo. (A) La línea celular de fibroblastos mamarios humanos HMFU19 requiere adiciones de colágeno para restaurar una morfología alargada en un gel de péptidos de 6 mg/mL, que se muestra con matriz pura de membrana basal (por ejemplo, Matrigel) y 1,5 mg/mL de gel de colágeno I de cola de rata para comparar, barra de escala de 50 μm; (B) Las células mamarias normales MCF10A forman estructuras acinares en el día 7 en geles de péptidos de 6 mg/mL con la adición de 100 μg/mL de colágeno humano I, barra de escala de 100 μm; (C) La adición combinada de los componentes de la matriz fibronectina/HA (ácido hialurónico, peso molecular 804 kDa) y HMFU19 en cocultivo indirecto aumenta el tamaño y la organización de los acini MCF10A en geles peptídicos de 10 mg/mL, según lo evaluado mediante tinción con caspasa 3 escindida, barra de escala de 50 μm. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4: Los geles peptídicos permiten el control independiente de la rigidez y la composición, y la evaluación de la matriz depositada en las células. (A) Las mediciones reológicas a granel que demuestren un rango de rigidez típico (módulo de almacenamiento, G') alcanzable mediante el control de la concentración de péptidos, * indica p < 0,05; (B) Inmunohistoquímica que muestra tinción de 150 μg/mL de colágeno I en un gel de péptido de 10 mg/mL con MCF7 encapsulado (día 7, barra de escala de 100 μm); (C) Inmunofluorescencia de la distribución de colágeno I en un gel de péptidos de 6 mg/mL con 200 μg/mL de colágeno humano I, mediante inclusión en agar y corte de vibratome, barra de escala de 25 μm; (D) La adición de 200 μg/mL de colágeno I proporciona una modesta disminución en el módulo de almacenamiento, G', de 10 mg/mL de geles peptídicos (reología oscilatoria a granel), compensada por el aumento de la concentración de péptidos a 15 mg/mL. Se muestran células de cáncer de mama triple negativo MDA MB 231 en cada condición (día 7, barra de escala 50 μm); (E) MDA MB 231 en geles peptídicos de 15 mg/mL con 200 μg/mL de colágeno humano I muestra elongación e interacción con la matriz a través de la tinción con pFAK (día 14, barra de escala 50 μm); (F) Tinción in situ de la deposición de colágeno I MCF7 en un gel de péptidos de 10 mg/mL inicialmente libre de matriz (día 10, barra de escala de 100 μm). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5: Los problemas comunes de solución de problemas de gel peptídico pueden resolverse mediante microscopía de campo claro. Las células que se muestran son células epiteliales de mama normales MCF10A, en el día 7 a menos que se especifique. (A) Un precursor de gel correctamente mezclado debe ser ópticamente transparente sin inconsistencias, mientras que (B) una mezcla/neutralización insuficiente puede causar inhomogeneidades/rayas visibles en el gel peptídico (flechas blancas); (C) MCF10A forma estructuras acinares en geles peptídicos de 6 mg/mL tras la adición de cocultivo indirecto HMFU19, sin embargo, (D) a 15 mg/mL la concentración de péptidos es demasiado alta para permitir la formación de acinares; (E) MCF10A sembrado a 5 x 105 células/mL forma estructuras acinares en geles de 6 mg/mL con la adición de 100 μg/mL de colágeno I, sin embargo, (F) a 2 x 105 células/mL la densidad celular es demasiado baja para permitir la formación de acinares; (G) Las adiciones de colágeno pueden producir grandes grupos de células para el día 14, sin embargo, (H) la adición incorrecta (neutralización del colágeno demasiado temprano en el proceso) puede prevenir el crecimiento del grupo. Barra de escala de 100 μm. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Discusión
Hemos descubierto que los geles peptídicos descritos aquí son una solución sencilla, rentable y flexible para soportar el cultivo en 3D de múltiples tipos de células. Al proporcionar un control total sobre la concentración de péptido utilizado y las adiciones de proteínas o glicanos realizadas, este método permite que los geles de péptidos se adapten cuidadosamente a su aplicación.
La ventaja crucial de los geles peptídicos sobre los métodos existentes es que la composición de la matriz y las propiedades mecánicas se pueden controlar de forma independiente, utilizando un método simple que no requiere ningún procedimiento químico complejo. Las propiedades mecánicas del gel peptídico están determinadas principalmente por la concentración de péptidos en el precursor inicial del gel. La adición posterior de células y/o componentes de la matriz permite la creación de un entorno in vitro totalmente definido por el usuario. Aunque las adiciones de matriz pueden alterar las propiedades mecánicas iniciales del gel, esto puede compensarse fácilmente mediante la variación independiente de la concentración de péptidos5. Esto proporciona una ventaja tangible sobre los sistemas existentes, por ejemplo, los geles de colágeno, en los que los parámetros que controlan la rigidez también suelen dar lugar a un cambio en los motivos de unión de las integrinas20,21.
Hemos demostrado la aplicación del gel peptídico para el cultivo in vitro de líneas celulares cancerosas y material derivado del paciente5. El rango de rigidez accesible con el gel peptídico (en el rango de 100 a 1000 de Pa) es ideal para replicar entornos de matriz normal y tumoral en tejidos blandos como la mama. Sin embargo, reconocemos que otras aplicaciones requieren entornos considerablemente más rígidos, por ejemplo, en el rango de 10-20 kPa para la regeneración ósea. Sería necesaria una modificación adicional del protocolo presentado aquí para ampliar la rigidez alcanzable a este rango, que es más típico de enfoques alternativos como los geles de alginato22. De manera similar, aquí hemos descrito un método simple para la funcionalización por atrapamiento físico de proteínas de la matriz/glicanos dentro del gel peptídico. Para las aplicaciones aquí descritas, este enfoque funciona bien y se adapta fácilmente para su uso por parte de grupos no especializados que deseen utilizar modelos 3D in vitro de enfermedades. Al igual que muchos otros hidrogeles11, el péptido utilizado aquí puede ampliarse para incluir la unión celular u otros motivos biológicos y, para algunas aplicaciones, este enfoque puede ser preferible.
Hemos identificado algunos puntos clave que requieren una atención cuidadosa para garantizar el éxito. La formación del precursor del gel es un paso intermedio crítico que permite al usuario comprobar que las condiciones utilizadas son correctas antes de incorporar las células. Este precursor puede almacenarse durante varias semanas (a 4 °C), pero debe incubarse a 80 °C y, posteriormente, a 37 °C antes de su uso. Un precursor adecuado será completamente líquido a 80 °C y autoportante a 37 °C. Estos controles son esenciales para garantizar que la gelificación se produzca correctamente. A continuación, se pueden incorporar células y/o matrices en condiciones fisiológicas.
Los laboratorios que ya utilizan matrices 3D estarán familiarizados con el manejo cuidadoso necesario para encapsular las células en los geles peptídicos. Se debe tener cuidado de limitar la agitación de las células antes y durante los pasos de encapsulación. Hemos descubierto que tipos específicos de células son diferencialmente susceptibles al daño durante este proceso y esto debe ser evaluado cuidadosamente por el usuario. Las concentraciones del gel peptídico descrito aquí permiten que la gelificación proceda en un marco de tiempo que, para las células mencionadas, permite encapsular las células antes de que se hundan hasta el fondo del pozo de fundición, pero lo suficientemente lento como para que no se dañen por este proceso. Sin embargo, hay que tener en cuenta que algunos tipos de células sensibles pueden requerir una neutralización más rápida para evitar la exposición prolongada a un pH elevado. En este caso, la adición de 10 mM HEPES al medio que rodea el gel peptídico puede ser beneficiosa.
Al adoptar el método descrito en este protocolo, es muy importante considerar cuidadosamente la calidad de la fuente de péptidos. En lugar de utilizarse como motivo funcional o recubrimiento, el péptido aquí es la totalidad de la parte no soluble del hidrogel. Por lo tanto, es probable que cualquier contaminante o variación en la estructura del péptido tenga un impacto significativo en la integridad o la capacidad de respaldar la viabilidad celular en el hidrogel final. Al pasar a un nuevo lote de péptido, se debe tener cuidado para asegurarse de que haya una buena consistencia entre lotes por parte del proveedor, así como para verificar el comportamiento del péptido al formar el precursor del gel.
En resumen, este protocolo describe un sistema de cultivo 3D con un enfoque crucial en el control independiente de las propiedades mecánicas y biológicas. La simplicidad y adaptabilidad del método lo hace adecuado para su adopción por cualquier laboratorio de cultivo celular y para una amplia gama de aplicaciones5. En el futuro, este protocolo puede extenderse para permitir la modificación covalente de la secuencia peptídica. Esto podría combinarse con métodos avanzados de microscopía para investigar las fuerzas de tracción ejercidas por las células en su matriz circundante. De importancia clave, sin embargo, es la capacidad de distinguir entre la matriz incorporada artificialmente y la matriz sintetizada por las propias células encapsuladas. Esta capacidad de controlar y monitorear los cambios en la matriz a lo largo del tiempo permitirá obtener información sin precedentes sobre el papel de las interacciones entre células y matrices en el desarrollo del cáncer y otras enfermedades.
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Nos gustaría agradecer la financiación del Centro Nacional para el Reemplazo, Refinamiento y Reducción de Animales en Investigación NC/N0015831/1 a JCA, GF y CLRM, NC/T001267/1 a RBC, CLRM, JCA, KL-S y KS, NC/T001259/1 a JCA, KL-S y CLRM y NC/P002285/1 a AMG, SJ y CLRM. También financiación del Consejo de Investigación de Ingeniería y Ciencias Físicas EP/R035563/1 a KL-S y CLRM y EP/N006615/1 a JLT y CLRM. La Figura 1 se ha creado utilizando gráficos adaptados de Servier Medical Art. Servier Medical Art by Servier está bajo una Licencia Creative Commons Atribución 3.0 Unported.
Materiales
| Name | Company | Catalog Number | Comments |
| Gel fabrication - Reagents | |||
| FEFEFKFK | Pepceuticals | n/a | Polypeptide; available from various suppliers. Pepceuticals is our recommended supplier due to the quality of the product. |
| PBS 10X | Gibco | 70011-036 | |
| Sodium hydroxide (1 M) | Sigma-Aldrich | S2770 | NaOH; dilute to 0.5 M prior to use |
| Water | Sigma-Aldrich | W3500 | |
| Gel fabrication - Equipment and Consumables | |||
| 15 mL falcon tubes | Greiner | 188261 | If using different brand ensure the material withstands temperatures of up to 90°C |
| 24 well plate | Corning Costar | 3524 | Alternative brands/suppliers can be used as long as there is a gap between the insert base and the plate surface |
| Centrifuge | Any | 200 x g for 3 minutes | |
| Class II Microbiological Safety Cabinet | Any | ||
| Fine balance | Any | Readability 0.1 mg | |
| Hanging insert for 24 well plate | Millipore | MCRP24H48 | Alternative brands/suppliers can be used as long as there is a gap between the insert base and the plate surface |
| Incubator | Any | 37°C, 5% CO2, humidified environment | |
| Oven | Any | set to 80°C | |
| P1000/200/20/10 pipette | Any | It is essential the pipettes used for the procedure are calibrated | |
| P1000/200/20/10 tips | Any | ||
| pH meter with microprobe | Any | ||
| Spatula | Any | ||
| Vortex | Any | ||
| Matrix addition | |||
| Collagen I (human) | Stem Cell Technologies | 07005 | |
| Collagen I (rat tail) | Gibco | A10483 | |
| Fibronectin | Stem Cell Technologies | 07159 | |
| Hyaluronic Acid | Iduron | HA804 | |
| Matrigel | Corning | 354234 | |
| Cell encapsulation/culture | |||
| B27 Supplement (no retinoic acid) | Gibco | 12587010 | Media additions for serum free cultures (Figure 2D) |
| Cholera toxin | Sigma-Aldrich | C-8052 | Media additions for MCF10A cells (Figure 3, 5) |
| DMEM | Gibco | 21969-035 | |
| DMEM/F12 | Sigma-Aldrich | D8062 | Media additions for MCF10A cells (Figure 3, 5) |
| DMEM/F12 Phenol Red Free | Gibco | 21041-025 | Media additions for serum free cultures (Figure 2D) |
| DPBS | Gibco | 14190-094 | |
| EGF | SourceBiosciences | ABC016 | Media additions for MCF10A cells (Figure 3, 5) |
| Fetal Bovine Serum | Gibco | 10500-064 | |
| Horse serum | Gibco | 26050-070 | Media additions for MCF10A cells (Figure 3, 5) |
| Human cancer/epithelial cell lines | e.g. MCF7/tdTomato MCF7/MCF10a/HCT116-mCherry | ||
| Human mammary fibroblasts | e.g. HMFU19 | ||
| Hydrocortisone | Sigma-Aldrich | H-0888 | Media additions for MCF10A cells (Figure 3, 5) |
| Insulin | Sigma-Aldrich | I9278 | Media additions for MCF10A cells (Figure 3, 5) |
| Knockout serum replacement | Gibco | 10828-028 | Media additions for serum free cultures (Figure 2D) |
| L-glutamine | Gibco | 25030-024 | |
| RPMI | Gibco | 21875-034 | |
| RPMI Phenol Red Free | Sigma-Aldrich | R7509 | |
| Imaging and other assays | |||
| 4% paraformaldehyde | Polysciences | 18814 | |
| Agar | SLS | CHE1070 | |
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | 5482 | |
| Confocal and/or fluorescent microscope | Any | e.g. Leica TCS SPE confocal laser scanning microscope (Figures 2-4) | |
| DAPI solution | Invitrogen | D3571 | 300 uM working solution |
| DPX mounting medium | ThermoFisher Scientific | ||
| Glass cover slips | Any | No1 coverslips 0.13 - 0.17 mm thickness | |
| Glass-bottom dishes | MatTek | ||
| Goat Anti-Rabbit IgG H&L (HRP polymer) | Abcam | ab214880 | |
| Haematoxylin and Eosin | Any | ||
| Histology molds (disposable, plastic) | Any | ||
| Image analysis software | ImageJ | ||
| Live/Dead assay kit | Invitrogen | L3224 | |
| Microtome | Any | ||
| Phalloidin | Life Technologies | F432/R415 | |
| Pierce Peroxidase IHC Detection Kit | ThermoFisher Scientific | 36000 | |
| Primary Ab Caspase 3 | Abcam | ab34710 | Shown in Figure 3C |
| Primary Ab Collagen I | Cell Signalling Technology | 9661 | Shown in Figure 4B, C, F |
| Primary Ab pFAK Tyr 397 | ThermoFisher Scientific | 44-624G | Shown in Figure 4E |
| Prolong gold/diamond anti-fade mountant with DAPI | Molecular Probes | S36939 | |
| Rheometer Physica MCR 301 | Anton Paar | ||
| Scalpel | Any | ||
| Secondary antibody Goat anti Rabbit AF488 | nvitrogen | a11034 | |
| Secondary antibody Goat anti Rabbit AF546 | Invitrogen | a11010 | |
| SuperFrost slides | ThermoFisher Scientific | Coating e.g. APES can help to retain microtome sections on slides. | |
| Triton X 100 | Sigma-Aldrich | X100 | |
| Trypsin-EDTA (0.25%) | Gibco | 25300054 | |
| Vibratome | Leica |
Referencias
- Hynes, R. The extracellular matrix: not just pretty fibrils. Science. 326 (5957), 1216-1219 (2009).
- Tian, C., et al. Cancer-cell-derived matrisome proteins promote metastasis in pancreatic ductal adenocarcinoma. Cancer Research. 80 (7), 1461-1474 (2020).
- Hebert, J. D., et al. Proteomic profiling of the ECM of xenograft breast cancer metastases in different organs reveals distinct metastatic niches. Cancer Research. 80 (7), 1475-1485 (2020).
- Vennin, C., et al. CAF hierarchy driven by pancreatic cancer cell p53-status creates a pro-metastatic and chemoresistant environment via perlecan. Nature Communication. 10 (1), 3637(2019).
- Ashworth, J. C., et al. Peptide gels of fully-defined composition and mechanics for probing cell-cell and cell-matrix interactions in vitro. Matrix Biology. 85, 15-33 (2020).
- Toniatti, C., Jones, P., Graham, H., Pagliara, B., Draetta, G. Oncology drug discovery: Planning a turnaround. Cancer Discovery. 4 (4), 397-404 (2014).
- Mak, I. W., Evaniew, N., Ghert, M. Lost in translation: animal models and clinical trials in cancer treatment. American Journal of Translational Research. 6 (2), 114-118 (2014).
- Aisenbrey, E. A., Murphy, W. L. Synthetic alternatives to Matrigel. Nature Reviews Materials. 5, 539-551 (2020).
- Onion, D., et al. 3-Dimensional patient-derived lung cancer assays reveal resistance to standards-of-care promoted by stromal cells but sensitivity to histone deacetylase inhibitors. Molecular Cancer Therapy. 15 (4), 753-763 (2016).
- Saunders, J. H., et al. Individual patient oesophageal cancer 3D models for tailored treatment. Oncotarget. 8 (15), 24224-24236 (2017).
- Caliari, S. R., Burdick, J. A. A practical guide to hydrogels for cell culture. Nature Methods. 13 (5), 405-414 (2016).
- Kühn, S., et al. Cell-instructive multiphasic gel-in-gel materials. Advanced Functional Materials. 30, 1908857(2020).
- Gjorevski, N., et al. Designer matrices for intestinal stem cell and organoid culture. Nature. 539 (7630), 560-564 (2016).
- Gjorevski, N., Lutolf, M. P. Synthesis and characterization of well- defined hydrogel matrices and their application to intestinal stem cell and organoid culture. Nature Protocols. 12 (11), 2263-2274 (2017).
- Saiani, A., et al. Self assembly and gelation properties of α-helix versus β-sheet forming peptides. Soft Matter. 5 (1), 193-202 (2008).
- Wan, S., et al. Self-assembling peptide hydrogel for intervertebral disc tissue engineering. Acta Biomaterialia. 46, 29-40 (2016).
- Blache, U., Stevens, M. M., Gentleman, E. Harnessing the secreted extracellular matrix to engineer tissues. Nature Biomedical Engineering. 4 (4), 357-363 (2020).
- Sachs, N., et al. A living biobank of breast cancer organoids captures disease heterogeneity. Cell. 172 (1-2), 373-386 (2018).
- Shaw, F. L., et al. A detailed mammosphere assay protocol for the quantification of breast stem cell activity. Journal of Mammary Gland Biology and Neoplasia. 17 (2), 111-117 (2012).
- Barcus, C. E., Keely, P. J., Eliceiri, K. W., Schule, L. A. Stiff collagen matrices increase tumorigenic prolactin signaling in breast cancer cells. Journal of Biological Chemistry. 288, 12722-12732 (2013).
- Bax, D. V., et al. Impact of UV- and carbodiimide-based crosslinking on the Integrin-binding properties of collagen-based materials. Acta Biomaterialia. 100, 280(2019).
- Huang, B. P., et al. Multi-peptide presentation and hydrogel mechanics jointly enhance therapeutic duo-potential of entrapped stromal cells. Biomaterials. 245, 119973(2020).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados