
Method Article
Preparation of a User-Defined Peptide Gel for Controlled 3D Culture Models of Cancer and Disease
In This Article
Summary
We present a method for creating a 3D cell culture environment, which can be used to investigate the importance of cell/matrix interactions in cancer progression. Using a simple self-assembling octapeptide, the matrix surrounding encapsulated cells can be controlled, with independent regulation of mechanical and biochemical cues.
Abstract
There is a growing awareness that cells grown in 3D better model in vivo behavior than those grown in 2D. In this protocol, we describe a simple and tunable 3D hydrogel, suitable for culturing cells and tissue in a setting that matches their native environment. This is particularly important for researchers investigating the initiation, growth, and treatment of cancer where the interaction between cells and their local extracellular matrix is a fundamental part of the model. Moving to 3D culture can be challenging and is often associated with a lack of reproducibility due to high batch-to-batch variation in animal-derived 3D culture matrices. Similarly, handling issues can limit the usefulness of synthetic hydrogels. In response to this need, we have optimized a simple self-assembling peptide gel, to enable the culture of relevant cell line models of cancer and disease, as well as patient-derived tissue/cells. The gel itself is free from matrix components, apart from those added during encapsulation or deposited into the gel by the encapsulated cells. The mechanical properties of the hydrogels can also be altered independent of matrix addition. It, therefore, acts as a ‘blank slate’ allowing researchers to build a 3D culture environment that reflects the target tissue of interest and to dissect the influences of mechanical forces and/or biochemical control of cell behavior independently.
Introduction
The many roles played by the extracellular environment in cancer development and progression are becoming ever clearer1. Recently, detailed proteomic-based analyses have added to an already convincing literature base, demonstrating that matrix components derived from cancer-associated stromal cells, or the cancer cells themselves, are key factors in events such as the promotion of epithelial mesenchymal transition and metastatic spread2,3,4. Given this recognized importance of the extracellular matrix (ECM), it is becoming crucial to move towards cell culture platforms that allow control over the 3D environment presented to cells. In response to this need, this protocol presents a method for cell encapsulation and culture in a 3D hydrogel, with user-defined ECM composition and mechanical properties5.
Currently, there is a poor correlation between cancer therapeutic efficacy in 2D in vitro culture, the impact of these therapeutics in current in vivo (patient-derived xenograft, PDX) models and their eventual activity in clinical trials6,7. This has led to significant failures in the drug discovery pipeline with an urgent need for improved in vitro models that allow tested therapeutics to ‘fail early, fail cheap’. Many researchers use the mouse mastocytoma-derived product e.g., Matrigel (or similar products) to create 3D matrix-rich environments to grow and observe cell behavior in vitro, including PDX-derived and other close-to-patient cells8,9,10. However, this ‘one size fits all’ approach neglects the complex role played by matrix proteins/glycans in cancer initiation and progression.
Recognition of the role of extracellular matrix (ECM) in the control of cell behavior has also encouraged the use of 3D culture in or on hydrogels composed of specific matrix components11. Whilst this is useful for investigating specific interactions, these systems suffer from the inability to separate mechanical and biochemical instructions between cells and matrix. They may also be difficult to handle and can give unclear read-outs of cell behavior. Collagen gels are a key example of this problem, since cell-mediated gel contraction can dramatically reduce the ability to visualize cells within the gel5. There are also some very elegant, multi-component gel systems, which experts have used to great effect12,13,14. These can incorporate enzyme-sensitive linkers and bio-active motifs but are significantly more complex in their formulation and application than the system described here.
This protocol describes a method for creating fully defined 3D culture models, allowing the roles of the ECM in development and disease to be modeled in vitro. The basis of the 3D model is a peptide gel, which we have previously described as an optimization of a simple self-assembling octapeptide hydrogel5,15,16. By moving away from complex, animal-derived matrices this system offers a significant benefit of improved batch-to-batch consistency and improved handling. In its simple state, the peptide contains no matrix-derived motifs and effectively provides a ‘blank slate’ onto which the user can build functionality.
We demonstrate how the mechanical properties of the peptide gel can be regulated independently, alongside incorporation of matrix proteins/glycans. The system is highly tunable, allowing the encapsulation of a range of cell types in various formats. Importantly for building a cancer model, stromal cells can also be incorporated: either in direct co-culture or separated to allow specific analysis of indirect cancer cell-stroma interactions. Most crucially, the protocol described here requires no complex knowledge of chemistry and can be reproduced in any cell culture laboratory without the need for specialized chemical knowledge or equipment.
We have optimized methods for the study of cell behavior in the peptide gels, including imaging, rheological analysis, extraction of material for PCR5 and embedding for histological assessment. A clear benefit to the simple hydrogel system is the ability to visualize and study the matrix deposited by encapsulated cells. The importance of cell-derived matrices and the benefits of better understanding of how cells re-engineer their local microenvironment was highlighted recently17 and reflects a growing awareness of the importance of trapping cell-secreted matrix components, in a similar way to that which occurs in vivo. Harnessing the ability to model such processes may be one of the fundamental drivers of the improved patient relevance of hydrogel-based disease models.
Protocol
1. Dissolution of peptide
- In a tissue culture hood, add 800 μL of sterile water to a 15 mL tube using a P1000 pipette.
- Using a fine balance, weigh the peptide powder into a non-static weighing-boat. Use a mass (in mg) of 1.25x the desired final peptide gel concentration (in mg/mL, Table 1).
NOTE: The method described here will produce a volume of approximately 1.25 mL of peptide gel per tube at the point of final gelation. Multiple tubes may be prepared at a time, or alternatively the volume per tube can be increased when the user is experienced with the method presented.
| Peptide concentration (after final gelation) | Mass of peptide | Initial NaOH addition |
| 6 mg/mL | 7.5 mg | 30 µL |
| 10 mg/mL | 12.5 mg | 60 µL |
| 15 mg/mL | 18.75 mg | 100 µL |
Table 1: Peptide mass and suggested initial NaOH addition for typical final gel concentrations. The ranges of peptide concentrations listed may be extended in either direction, however, it is more likely that lower peptide concentrations may not form stable gels, whereas at high concentrations the resulting gel may be too dense to allow sufficient nutrient exchange and cell viability. The appropriate concentration will require optimization for different cell types and peptide batches.
- Add the weighed peptide into the 15 mL tube. Flick the weighing boat to ensure no powder is left behind.
NOTE: The peptide powder can be very static, take care to minimize the loss of powder. It is not necessary to maintain sterile conditions during steps 1.2 and 1.3 (or for any pH measurements) as sterilization occurs during the incubation at 80 °C later in the process. - Vortex for 3 min, then centrifuge at 200 x g for 3 min.
- Incubate the peptide solution in an oven set at 80 °C for a minimum of 2 h. If undissolved peptide is present after incubation, repeat step 1.4.
NOTE: As uniform heating is essential, a heatblock is not suitable for this step. However, we have found that hybridization ovens work well as does a water bath as long as the peptide solution is fully submerged.
2. Formation of gel precursors
- Prepare a sterile 0.5 M sodium hydroxide (NaOH) solution using sterile water.
NOTE: Freshly diluted NaOH solution is recommended for most consistent results. - In a tissue culture hood, add 0.5 M NaOH to the center of the dissolved peptide and mix by slowly stirring with the pipette tip (Table 1).
- Vortex the tube for 10 s and centrifuge at 200 x g for 10 s to remove bubbles.
- If the gel precursor is cloudy, repeat steps 2.2 and 2.3, adding 5 µL increments of NaOH.
NOTE: pH measurement can help determine whether sufficient NaOH has been added: the optimal pH is between 9–10.5 and it is preferable not exceed this as using acids to bring the pH back down can result in gel inhomogeneity. The precise volume of NaOH required may vary with peptide source. - Once the gel precursor is optically clear and self-supporting (or only just flowing) on inversion of the 15 mL tube, add 100 μL of sterile 10x PBS in a tissue culture hood. Vortex for 10 s and centrifuge at 200 x g for 10 s.
NOTE: If the gel precursor is cloudy, repeat step 2.4 until it becomes clear and semi-solid. If the gel precursor is liquid (above pH ~10.5), it will not form a stable gel and should be discarded. - Incubate overnight in an oven at 80 °C.
- Visually check the gel precursor to ensure it is fully liquid at 80 °C.
- If the gel precursor is not fully liquid at 80 °C, it has not been sufficiently neutralized, add NaOH following step 2.4.
- If the gel precursor is liquid but air bubbles or small precipitates are present, flick the tube sharply to disperse them. If air bubbles or precipitates persist after flicking the tube, vortex the gel precursor and centrifuge at 200 x g for 10 s each.
- After following either of steps 2.7.1 or 2.7.2, incubate precursor gels for a further 2 h at 80 °C before proceeding to final gelation.
- Keep the gel precursors at 80 °C until needed (maximum 48 h).
NOTE: The protocol can be paused here, store gel precursors at 4 °C for up to 4 weeks. If restarting from this pause point, incubate gel precursors at 80 °C for at least 2 h, and restart from step 2.7. It is strongly recommended to prepare gel precursors well in advance of them being needed for the final gelation, to ensure there is sufficient time for the above modifications if required.
3. Preparation of matrix components for seeding
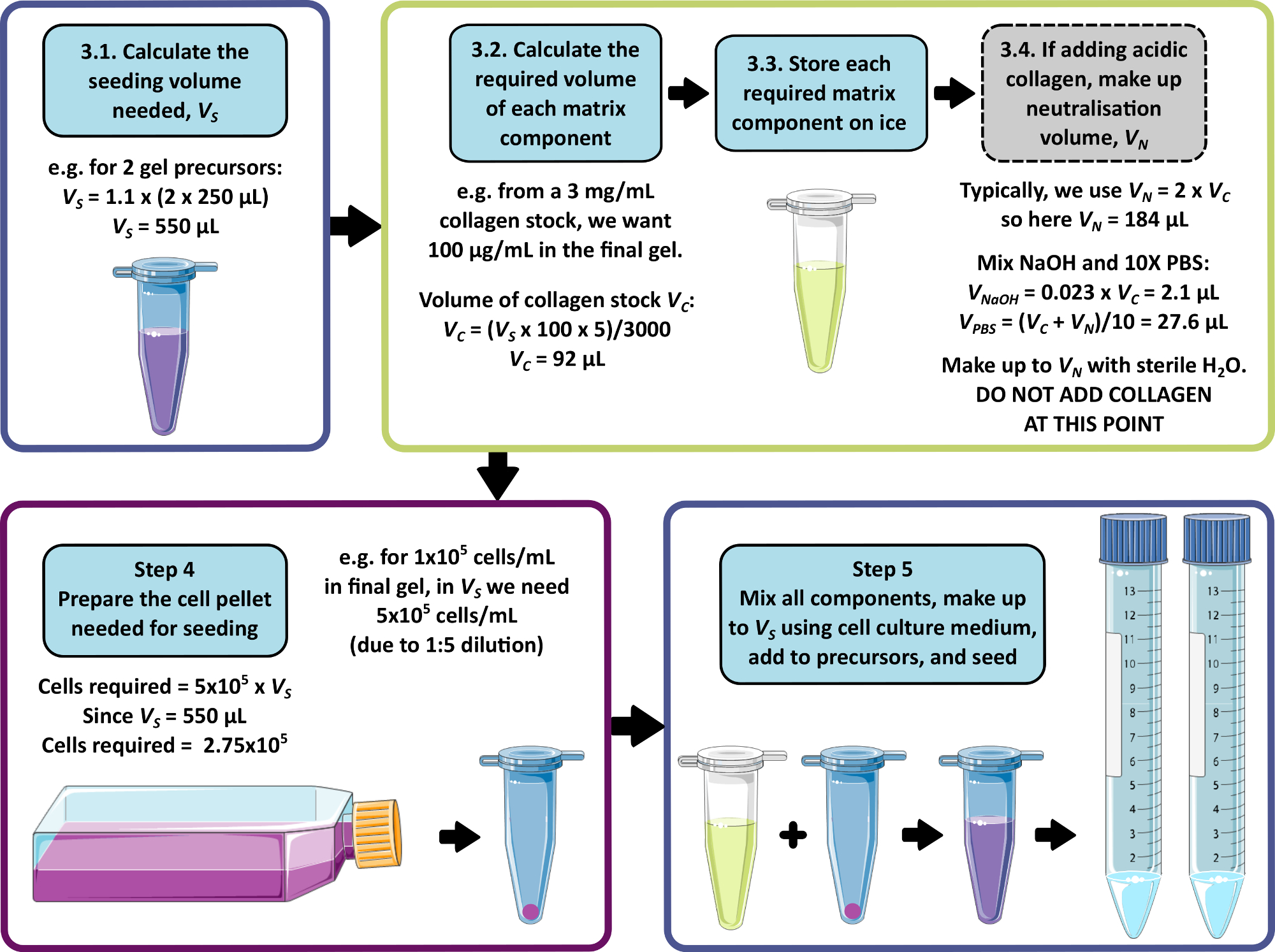
NOTE: An example calculation for steps 3–5 is shown in Figure 1. Step 3 and step 4 may be omitted to produce a matrix-free and/or a cell-free gel respectively.
- Calculate the total combined volume of cells, matrix and media required for seeding, allowing for 250 µL to be added to each 1 mL precursor gel. Multiply this volume by 1.1 to allow for pipetting error. This is the seeding volume, VS.
- For each matrix component, calculate the volume of stock solution to be added to the seeding volume using Equation 1:
 (1)
(1)
NOTE: VS corresponds to the total volume of seeding medium required for seeding all gels (step 3.1). Ensure that the sum of all matrix component volumes does not exceed VS. - Thaw all matrix components thoroughly (refer to manufacturer’s instructions) and store on ice until needed.
NOTE: Some matrix components will be very prone to gelation. Refer to manufacturer’s instructions to avoid this occurring. - If an acidic collagen stock solution requiring neutralization is to be added (refer to manufacturer’s instructions), prepare the neutralization solution as follows:
- Calculate the volume of collagen stock solution, VC, required using Equation 1.
- Determine a suitable volume for the neutralization solution, VN. This should be chosen such that the combined volume of all matrix additions does not exceed the seeding volume, VS.
NOTE: Generally, a sensible value for VN = 2 x Vc, but this may be adjusted according to the number and volume of other matrix components to be added. - Calculate the volume of 1 M NaOH required for neutralization using Equation 2.
 (2)
(2) - Calculate the volume of 10x PBS required in the neutralization solution using Equation 3.
 (3)
(3) - Combine the calculated volumes of 1 M NaOH and 10x PBS, then make up to VN with sterile water. Mix well, and store on ice until needed.
NOTE: Do not add the acidic collagen at this point. Premature mixing of the collagen with the neutralization solution will lead to the initiation of collagen fibrillogenesis, which can cause inconsistency in the properties of the final peptide gel.
4. Preparation of cells for seeding
- If not already completed, calculate the seeding volume, VS according to step 3.1.
- Calculate the cell density required in this cell suspension, by taking the desired cell density in the final peptide gel and multiplying by 5.
NOTE: The cell suspension is 5x the desired end concentration to account for dilution upon mixing with the gel precursor. Cell densities should be optimized for each new cell line under consideration. Depending on the cell type, densities between 1 x 104 and 1 x 106 cells/mL in the final peptide gel may be appropriate. - Calculate the total number of cells required for seeding (multiply the cell density by the seeding volume VS calculated in step 4.1.).
- Using standard culture/passage methods for the cells in use, prepare a cell pellet containing the required quantity of cells as calculated in step 4.3.
5. Final gelation/cell encapsulation
- When ready to begin final gelation, transfer the gel precursors from the 80 °C oven into a 37 °C water bath.
NOTE: The gel precursors should be self-supporting at 37 °C. If they are liquid, it is unlikely that complete gelation will occur, and the gel precursors should be discarded. - Prepare a 96 well plate, or alternatively a 24 well plate (or similar) with cell culture inserts.
NOTE: Ensure there is a gap between the base of the insert and the well plate when plating peptide gels in 24 well inserts. This ensures the gel is in contact with media. - If matrix is to be added, combine all matrix components and neutralization solutions (step 3). Make up to VS (step 3.1) using cell culture medium and mix thoroughly. If cells are to be added, resuspend the cell pellet prepared in step 4 using the seeding volume VS.
NOTE: If no matrix components have been prepared in step 3, use the cell culture medium for the seeding volume. This is generally the standard culture medium for the cell type in use, although this may need to be validated if a mixture of cell types is being used. - Using a P1000 pipette, add 250 μL of the cell/matrix mix gently on top of the gel precursor.
NOTE: Matrix components, particularly basement membrane extract, may begin to polymerize once added to the gel precursor. It is, therefore, important to move to the next step as quickly as possible. - Gently mix by the combined action of pipetting and stirring. The gel is shear thinning so will become easier to mix with gentle pipetting/stirring. When thoroughly mixed, add 100 μL to each well of a 96 well plate, or 200 μL to each cell culture insert.
NOTE: When mixing, reverse pipetting using a P1000 set at 200 μL can be beneficial to avoid introducing air bubbles. The gel may initially be difficult to mix, but on continued mixing should become easier - this indicates that the mixing is efficient. - Incubate for 10 min at 37 °C in 5% CO2 and a humidified atmosphere.
NOTE: This step is not necessary if the peptide gel contains no matrix additions. - Add 200 μL of media to each well of the 96 well plate, or 1 mL to the outside of the cell culture insert with a few drops on the top of the gel. Incubate at 37 °C in 5% CO2 and a humidified atmosphere.
- Change media twice within the next hour, and again after several hours (or the following day).
NOTE: Take care here as the gels will be unstable for several hours. - Change media every 2–3 days (or following standard culture protocol for the cells in use).
6. Indirect co-culture
NOTE: This method is only applicable where the peptide gels are seeded into 24 well plate inserts, or similar formats in which the gel can be supported above a cell monolayer. Indirect co-culture can be introduced in this case by preparing a 2D feeder layer of cells on the bottom of the well plate.
- Prepare a 24 well plate for seeding. This should be a separate plate to the peptide gels but should be of the same brand to ensure compatibility with the inserts used (see step 5.2).
- Calculate the cell density needed for seeding the indirect co-culture, according to the cell type under consideration.
NOTE: The cell seeding density should be optimized for each new cell line under consideration. Cells should be plated to give approximately 30–50% confluence. As an example, the human mammary fibroblast cell line HMFU19 is typically plated at a density of 1-5 x 104 cells/well. - Using standard culture/passage methods for the cells in use, prepare a cell suspension suitable for seeding at 1 mL/well, using the typical growth medium for these cells.
- Seed the cell suspension into the well plate, 1 mL per well.
- Incubate at 37 °C in 5% CO2 and a humidified atmosphere to attach for several hours, or overnight. Then remove the medium from the wells.
- Using sterile forceps, transfer the 24 well plate inserts containing the peptide gels into the new wells containing the pre-seeded cells in 2D. Add 1 mL of medium dropwise to the exterior of the insert and a few drops on the surface of the gel.
NOTE: Typically, the medium used at this point is the one suitable for the cells encapsulated in the gel, but the suitability of this medium for the cells in 2D may need verification or optimization for the particular experiment under consideration. - Regularly prepare fresh feeder layers to avoid over-confluence, and transfer peptide gels to these new wells following the same method as above.
NOTE: Typically, cells for indirect co-culture are prepared at the same time as the peptide gel seeding. Peptide gels can then be transferred to co-culture at the point of media change after a few hours or overnight incubation, see step 5.8.
7. Bulk oscillatory rheology of peptide gels
NOTE: As standard, rheological characterization is carried out 24 h after gel seeding, which should take place in 24 well plate inserts.
- Set up and calibrate the rheometer according to manufacturer’s instructions. Use a parallel plate geometry with plate diameter as close as possible to the diameter of the cell culture insert.
NOTE: Tests may be carried out at 37 °C if desired, replicating the environment during culture. - Remove the first peptide gel sample to be tested from the cell culture insert by inverting the insert and cutting out the plastic membrane using a scalpel.
NOTE: Ensure the plate containing the peptide gels is outside the cell culture incubator for as short a time as possible prior to testing. Media with a bicarbonate buffering system relies on the presence of CO2 to maintain pH. Gels that have been outside the incubator too long will drift in pH, which can affect rheological assessment. It can be beneficial to maintain the gels with 10 mM HEPES added to the media to prevent this effect. - Carefully transfer the gel to the rheometer plate. Then, using a scalpel, trim the height of the gel to approximately 1 mm to minimize gel deformation when loaded under the rheometer plate.
NOTE: Be careful not to touch or damage the rheometer plate when using the scalpel. - Set the parallel plate spacing to 1 mm. Trim any excess gel that is not covered by the rheometer plates.
- Run the desired test settings on the rheometer, according to manufacturer’s instructions.
NOTE: For each new sample condition, it is advised to run an amplitude sweep from 0.1–100% strain to ensure that all tests are carried out at a strain level within the sample’s linear viscoelastic region.
8. Live/dead staining of encapsulated cells
- Remove the medium from the wells, and wash peptide gels twice with 1x PBS, using the same technique as for a media change (step 5.7).
- Remove peptide gels that have been cultured in 24 well plate inserts following step 7.2. Keep gels in 1x PBS in the original well plate until ready to stain.
NOTE: Take care as the gels can be fragile at this point, especially after extended culture. - Prepare a live/dead staining solution, allowing for 500 μL stain per 24 well plate insert, or 50 μL per well of a 96 well plate. A typical stain is 4 μM ethidium homodimer and 2 μM calcein AM in 1x PBS. Protect the resulting solution from light.
NOTE: Concentrations of live/dead reagents may require further optimization depending on the cell type used and reagent supplier. - Carefully remove PBS from each gel and replace with a few drops of the staining solution, making sure each gel is well-covered.
- Incubate gels in the staining solution in the dark for 10–15 min, then visualize using a confocal/fluorescent microscope.
NOTE: For higher quality images, it can be beneficial to transfer the gels to glass-bottom dishes of coverslip thickness.
9. Fixing peptide gels for end-point imaging
- Wash peptide gels following step 8.1.
- Add 4% paraformaldehyde (PFA) in 1x PBS: 100 μL for each well of a 96 well plate and 1 mL for each gel in a 24 well plate insert (a few drops should be added on top of the gel inside the insert).
CAUTION: Paraformaldehyde (PFA) is highly toxic and is readily absorbed through the skin. It is extremely destructive to the skin, eyes, mucous membranes, and upper respiratory tract. PFA should be handled in a fume hood and users should wear protective clothing and gloves. Alternative chemical fixatives may be used in the same way according to the end application. - Incubate peptide gels in PFA fixative for 1 h at room temperature.
- Remove PFA fixative and wash peptide gels twice with 1x PBS.
NOTE: The protocol can be paused here, store fixed peptide gels at 4 °C in 1x PBS for up to 4 weeks, making sure the plate is well sealed with paraffin film.
10. Embedding peptide gels for sectioning
NOTE: Embedding peptide gels in 4% agar is a crucial step prior to paraffin-embedding for immunohistochemistry. Alternatively, gels may be embedded in 2% agar and sectioned using a vibratome (typically 500 μm sections give good results). This is an optional step, producing hydrated gel sections that can be beneficial for staining extracellular matrix localization in the gel, using the methods in section 11.
- Prepare molten 2% or 4% agar solution in 1x PBS (see note above), by boiling in a microwave. Allow to cool for a few minutes before using.
NOTE: Molten agar presents a heat hazard – handle with care using hand and face protection. Once prepared, agar solution may be stored at 4 °C until needed. - Remove peptide gel from the cell culture insert following step 7.2.
- Using a plastic Pasteur pipette, cover the base of a histological embedding mold with a thin layer of agar solution. Leave to cool at 20 °C for a few seconds.
- Using a spatula, place the peptide gel into the center of the agar. Then completely cover the peptide gel in agar.
NOTE: The gel should not sink into the agar. If it does, remove the gel and wait a few more seconds until the agar has solidified more and try again. Try not to let too much solidification take place or there will be a weak join between the two layers. - Allow the embedded gel to cool for 1 h at 4 °C before removing from the histological mold.
NOTE: The protocol can be paused here, store embedded gels at 4 °C in 1x PBS for up to 4 weeks. - If immunohistochemistry is to be carried out, put embedded gels into a tissue processor, and proceed using standard lab methods.
NOTE: Alternatively, embedded gels may be cut into hydrated sections using a vibratome. Hydrated sections should be stored in sealed plates at 4 °C in 1x PBS for up to 4 weeks.
11. Staining cells in gels using immunocytochemistry
- Remove the 1x PBS covering the peptide gels/gel sections. Remove any gels still in 24 well plate inserts following step 7.2.
- Cover gels/gel sections in a blocking buffer and incubate for 30 min at 20 °C.
NOTE: A typical blocking buffer consists of 0.5% bovine serum albumin (BSA) in 1x PBS with 0.1% Triton X-100. Triton X-100 is toxic and causes serious eye damage, skin irritation and is very toxic to aquatic life. Users should wear protective clothing, eye protection and gloves. - Prepare primary antibodies in blocking buffer at optimized working concentrations. Allow 200 μL per 24 well plate gel, 100 μL per gel section, and 50 μL per 96 well plate well.
NOTE: Typically, the antibody concentrations used for 3D staining in gels should be double the concentration used in 2D. - Remove blocking buffer and add antibody solution to the gels dropwise.
- Seal plate with paraffin film and incubate overnight at 4 °C.
- Remove antibody solution and wash twice with blocking buffer.
- Add secondary antibody following the same procedures described in steps 11.3 and 11.4.
- Incubate in the dark overnight at 4 °C, or for 3 hours at 20 °C.
- Remove antibody solution and wash twice with 1x PBS.
- Cover samples in 1:1,000 DAPI solution and incubate at 4 °C in the dark for 1 h.
- Transfer the gel to a glass coverslip and image by fluorescent/confocal microscopy.
12. RNA extraction
NOTE: The volumes used in this method are applicable where the peptide gels are seeded into 24-well plate inserts. Other gel formats can be used, and volumes adjusted accordingly.
- Remove the medium from the wells, and wash peptide gels twice with 1x PBS, using the same technique as for a media change (step 5.7).
- Remove peptide gels that have been cultured in 24 well plate inserts following step 7.2. Place each gel into a separate 15 mL centrifuge tube.
NOTE: Take care as the gels can be fragile at this point, especially after extended culture. - Using a P1000, add 500 μL Trypsin-EDTA (0.25%) to each tube and pipette up and down to mix, and disrupt the gel.
- Incubate gels in the Trypsin-EDTA at 37 °C for 3-5 min.
NOTE: Incubation times may require optimization depending on the cell type used. - Add 5 mL of 1x PBS to dilute the Trypsin-EDTA.
- Centrifuge at 200 x g for 5 min to pellet cells.
- Remove the supernatant.
NOTE: Take care as a gel layer may have formed in between the cell pellet and the supernatant. - Resuspend the cell pellet in lysis buffer, according to the manufacturer’s instructions, and proceed following standard protocols for RNA extraction.
Results
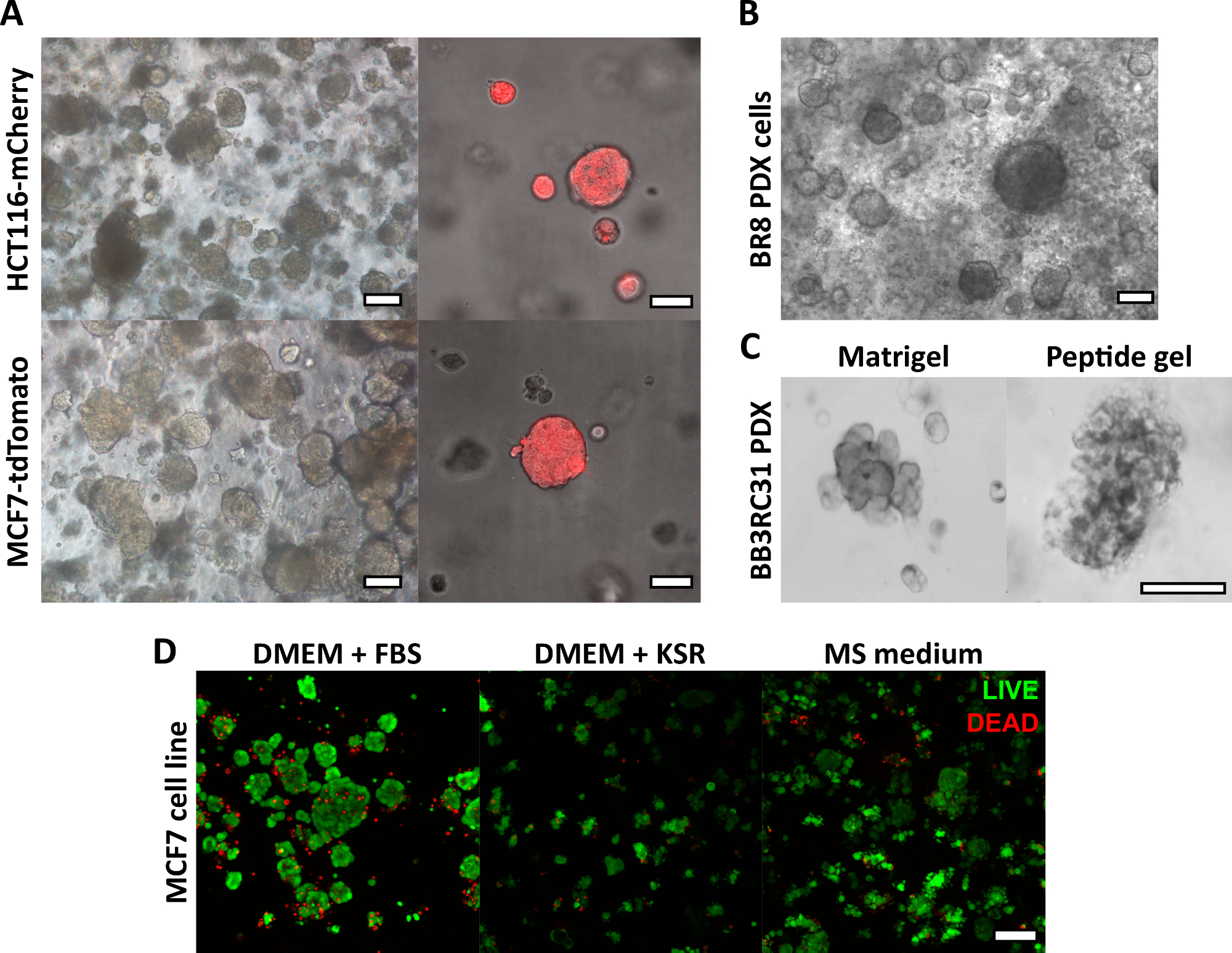
The peptide gel fabrication method described here allows the user to define and create a bespoke 3D culture environment. While the mechanical environment is determined primarily by peptide concentration, matrix components of interest may also be added at controlled densities, as shown by the example calculation in Figure 1. In its simplest form, however, the peptide gel protocol provides a method for encapsulating cells in a matrix-free 3D environment. Figure 2 shows how this approach may be combined with a wide range of cancer models, including fluorescently labeled cancer cell lines (Figure 2A) and patient-derived xenograft (PDX) material (Figure 2B,C). Importantly, cell lines and PDX material may both be cultured within the gels in serum-free conditions (Figure 2C,D), providing a 3D culture system with fully defined composition.
Since the peptide itself does not contain any cell-binding motifs, encapsulated cells typically display a rounded morphology in the unmodified peptide gels. Figure 3A demonstrates this for human mammary fibroblasts in a 6 mg/mL peptide gel, compared with their classic elongate morphology seen in pure Matrigel and a pure collagen gel. Importantly however, the peptide gel protocol allows incorporation of matrix components of interest. Figure 3A demonstrates how addition of 200 μg/mL collagen I can restore the elongate fibroblast morphology in the peptide gels.
Matrix additions can also support the growth and organization of other cell types, for instance MCF10A, as shown in Figure 3B. In this case, addition of 100 μg/mL collagen I to a 6 mg/mL peptide gel allows acinar structures to form by day 7. Further complexity may also be introduced by incorporation of a supportive cell layer in indirect co-culture. Figure 3C demonstrates how the combined approach of matrix incorporation and indirect co-culture with human mammary fibroblasts can enhance MCF10A growth and organization.
Another important parameter is the concentration of peptide used in peptide gel fabrication. Figure 4A shows an example of how controlling peptide concentration, in this case between 4 and 10 mg/mL, results in a stiffness ranging between 100s to 1000s of Pa. These gels may be fabricated matrix free or can be created with matrix additions to allow simultaneous control of both stiffness and composition. Peptide gels with matrix additions may be sectioned and stained to allow the distribution of these additions to be visualized. Figure 4B, C show two approaches for doing this: embedding in 4% agar followed by standard tissue processing and paraffin embedding for immunohistochemistry (Figure 4B) or embedding in 2% agar followed by vibratome sectioning and fluorescent staining (Figure 4C).
When modifying the composition of the peptide gels, it is crucial to ensure that these changes do not impact the mechanical environment initially presented to the cells. Figure 4D demonstrates how modifications to the peptide concentration can be used to offset any changes to the peptide gel stiffness on matrix incorporation. Bulk oscillatory rheology measurements of gel stiffness (storage modulus, G’) can then distinguish between the effects of gel composition and stiffness on cell morphology. As shown in the bright field images, MDA MB 231 cells develop an elongate morphology on collagen addition to either 10 mg/mL or 15 mg/mL peptide gels. Figure 4E shows that these elongated cells stain positive for pFAK, indicating an interaction with their surrounding matrix. The initially matrix-free environment of the peptide gels also makes them an ideal platform for studying cellular synthesis and deposition of matrix components of interest. Figure 4F shows the localized deposition of collagen I by MCF7 cells encapsulated in 10 mg/mL peptide gels.
One of the key advantages of the peptide gels is the ease with which standard laboratory methods can be applied to their analysis. Material can be extracted for qRT-PCR to determine gene expression profiles (as shown in our recent publication5). Imaging by bright-field microscopy additionally allows real-time visualization of cell growth. Figure 5 shows some of the typical troubleshooting issues that may be encountered in unsuccessful peptide gels: incomplete mixing of the gel precursor (Figure 5A,B); incorrect optimization of peptide concentration (Figure 5C,D) or seeding density (Figure 5E,F); and incorrect neutralization of acidic collagen prior to incorporation in the peptide gels (Figure 5G,H). Peptide concentration and seeding density, in particular, must be optimized for each cell line and peptide source, to ensure that the culture environment is appropriately defined, and representative of the application of interest.

Figure 1: An example calculation for matrix composition and seeding density. This example workflow describes the procedure that would be followed to seed two peptide gel precursors with additions of 100 µg/mL collagen, at a final cell density of 1 x 105 cells/mL. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Matrix-free peptide gels provide a suitable 3D culture platform for cell line and patient-derived cancer models. (A) HCT116 colorectal cancer and MCF7 breast cancer cell lines, constitutively expressing the fluorescent markers mCherry and tdTomato respectively, form cell clusters in 6 mg/mL gels by day 9 (left), and may be imaged live using fluorescent microscopy (right, scale bar 50 µm); (B) Patient-derived xenograft (PDX) cells from a triple negative breast cancer patient (BR8) form cell clusters by day 7 in 10 mg/mL peptide gels; (C) PDX cells from estrogen receptor positive breast tumours (BB3RC31) may be grown in serum-free conditions18, shown with basement membrane matrix (e.g. Matrigel) control at matched passage for comparison; (D) MCF7 breast cancer cells are viable in 6 mg/mL peptide gels in matrix-free and serum-free conditions, as assessed using a LIVE/DEAD cell assay at day 7. KSR = knockout serum replacement, MS medium = mammosphere medium19. Scale bar 100 µm unless specified. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Peptide gel complexity may be increased by introduction of matrix additions and co-culture. (A) Human mammary fibroblast cell line HMFU19 requires collagen additions to restore an elongate morphology in a 6 mg/mL peptide gel, shown with pure basement membrane matrix (e.g. Matrigel) and 1.5 mg/mL rat tail collagen I gel for comparison, scale bar 50 µm; (B) MCF10A normal breast cells form acinar structures by day 7 in 6 mg/mL peptide gels on addition of 100 µg/mL human collagen I, scale bar 100 µm; (C) Combined addition of matrix components fibronectin/HA (hyaluronic acid, molecular weight 804 kDa) and HMFU19 in indirect co-culture increase the size and organization of MCF10A acini in 10 mg/mL peptide gels as assessed by cleaved caspase 3 staining, scale bar 50 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Peptide gels allow independent control of stiffness and composition, and assessment of cell-deposited matrix. (A) Bulk rheology measurements demonstrating a typical stiffness range (storage modulus, G’) achievable by control of peptide concentration, * indicates p < 0.05; (B) Immunohistochemistry showing staining of 150 µg/mL collagen I in a 10 mg/mL peptide gel with encapsulated MCF7 (day 7, scale bar 100 µm); (C) Immunofluorescence of collagen I distribution in a 6 mg/mL peptide gel with 200 µg/mL human collagen I, by agar embedding and vibratome sectioning, scale bar 25 µm; (D) Addition of 200 µg/mL collagen I gives a modest decrease in the storage modulus, G’, of 10 mg/mL peptide gels (bulk oscillatory rheology), offset by increasing peptide concentration to 15 mg/mL. MDA MB 231 triple negative breast cancer cells are shown in each condition (day 7, scale bar 50 µm); (E) MDA MB 231 in 15 mg/mL peptide gels with 200 µg/mL human collagen I show elongation and interaction with the matrix via pFAK staining (day 14, scale bar 50 µm); (F) In situ staining of MCF7 collagen I deposition in an initially matrix-free 10 mg/mL peptide gel (day 10, scale bar 100 µm). Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Common peptide gel troubleshooting issues may be resolved using bright-field microscopy. Cells shown are MCF10A normal breast epithelial cells, at day 7 unless specified. (A) A correctly mixed gel precursor should be optically clear with no inconsistencies, whereas (B) insufficient mixing/neutralization can cause visible inhomogeneities/streaks in the peptide gel (white arrows); (C) MCF10A form acinar structures in 6 mg/mL peptide gels on addition of HMFU19 indirect co-culture, however (D) at 15 mg/mL the peptide concentration is too high to allow acinar formation; (E) MCF10A seeded at 5 x 105 cells/mL form acinar structures in 6 mg/mL gels on addition of 100 μg/mL collagen I, however (F) at 2 x 105 cells/mL cell density is too low to allow acinar formation; (G) Collagen additions can produce large cell clusters by day 14, however (H) incorrect addition (collagen neutralization too early in the process) can prevent cluster growth. Scale bar 100 µm. Please click here to view a larger version of this figure.
{kind=link}
Discussion
We have found the peptide gels described here to be a simple, cost-effective, and flexible solution to support 3D culture of multiple cell types. By providing full control over the concentration of peptide used and the protein or glycan additions made, this method allows the peptide gels to be carefully tailored to their application.
The crucial advantage of the peptide gels over existing methods is that matrix composition and mechanical properties may be controlled independently, using a simple method that does not require any complex chemical procedures. The mechanical properties of the peptide gel are primarily determined by the peptide concentration in the initial gel precursor. Subsequent addition of cells and/or matrix components then allows creation of a fully user-defined in vitro environment. Although matrix additions may alter the initial mechanical properties of the gel, this may readily be offset by the independent variation of peptide concentration5. This provides a tangible advantage over existing systems, for instance collagen gels, in which parameters that control stiffness also commonly result in a change in integrin binding motifs20,21.
We have demonstrated the application of the peptide gel for in vitro culture of cancer cell lines and patient-derived material5. The range of stiffness accessible with the peptide gel (in the range of 100s to 1000s of Pa) is ideally suited to replicate normal and tumor matrix environments in soft tissues such as breast. However, we recognize that other applications require considerably stiffer environments, e.g. in the 10–20 kPa range for osseous regeneration. Further modification of the protocol presented here would be required to extend the achievable stiffness into this range, which is more typical of alternative approaches such as alginate gels22. Similarly, here we have described a simple method for functionalization by physical entrapment of matrix proteins/glycans within the peptide gel. For the applications described here, this approach works well and is easily adapted for use by non-specialist groups wishing to use 3D in vitro models of disease. Like many other hydrogels11, the peptide used here can be extended to include cell-binding or other biological motifs and for some applications this approach may be preferable.
We have identified a few key points that need careful attention to ensure success. Formation of the gel precursor is a critical intermediate step that enables the user to check that the conditions used are correct before cells are incorporated. This precursor can be stored for several weeks (at 4°C) but must be incubated at 80 °C and subsequently at 37 °C prior to use. A suitable precursor will be completely liquid at 80 °C, and self-supporting at 37 °C. These checks are essential to ensure that gelation will occur correctly. Cells and/or matrix may then be incorporated under physiological conditions.
Labs already using 3D matrices will be familiar with the careful handling needed to encapsulate cells in the peptide gels. Care must be taken to limit the agitation of cells before and during the encapsulation steps. We have found that specific cell types are differentially susceptible to damage during this process and this must be carefully evaluated by the user. The concentrations of the peptide gel described here allow gelation to proceed in a time frame that, for the cells mentioned, allows cells to be encapsulated before they sink to the bottom of the casting well but slowly enough that they are not damaged by this process. It is, however, of note that some sensitive cell types may require more rapid neutralization to avoid prolonged exposure to raised pH. In this case, the addition of 10 mM HEPES to the medium surrounding the peptide gel can be beneficial.
When adopting the method described in this protocol, it is very important to carefully consider the quality of the peptide source. Rather than being used as a functional motif or coating, the peptide here is the entirety of the non-soluble portion of the hydrogel. Therefore, any contaminants or variation in the peptide structure are likely to have a significant impact on the integrity or capacity to support cell viability in the final hydrogel. When moving to a new batch of peptide, care must be taken to ensure that there is good batch-to-batch consistency from the supplier as well as checking the behavior of the peptide when forming the gel precursor.
In summary, this protocol describes a 3D culture system with a crucial focus on the independent control of mechanical and biological properties. The simplicity and adaptability of the method makes it suitable for adoption by any cell culture laboratory, and for a wide range of applications5. In the future, this protocol may be extended to allow the covalent modification of the peptide sequence. This could be combined with advanced microscopy methods to investigate the tensile forces exerted by cells on their surrounding matrix. Of key importance, however, is the ability to distinguish between artificially incorporated matrix, and matrix synthesized by the encapsulated cells themselves. This ability to control and monitor matrix changes over time will allow unprecedented insights into the roles of cell-matrix interactions in the development of cancer and other diseases.
Disclosures
The authors have nothing to disclose.
Acknowledgements
We would like to acknowledge funding from the National Centre for the Replacement, Refinement and Reduction of Animals in Research NC/N0015831/1 to JCA, GF and CLRM, NC/T001267/1 to RBC, CLRM, JCA, KL-S, and KS, NC/T001259/1 to JCA, KL-S and CLRM and NC/P002285/1 to AMG, SJ and CLRM. Also funding from the Engineering and Physical Sciences Research Council EP/R035563/1 to KL-S and CLRM and EP/N006615/1 to JLT and CLRM. Figure 1 was created using adapted graphics from Servier Medical Art. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License.
Materials
| Name | Company | Catalog Number | Comments |
| Gel fabrication - Reagents | |||
| FEFEFKFK | Pepceuticals | n/a | Polypeptide; available from various suppliers. Pepceuticals is our recommended supplier due to the quality of the product. |
| PBS 10X | Gibco | 70011-036 | |
| Sodium hydroxide (1 M) | Sigma-Aldrich | S2770 | NaOH; dilute to 0.5 M prior to use |
| Water | Sigma-Aldrich | W3500 | |
| Gel fabrication - Equipment and Consumables | |||
| 15 mL falcon tubes | Greiner | 188261 | If using different brand ensure the material withstands temperatures of up to 90°C |
| 24 well plate | Corning Costar | 3524 | Alternative brands/suppliers can be used as long as there is a gap between the insert base and the plate surface |
| Centrifuge | Any | 200 x g for 3 minutes | |
| Class II Microbiological Safety Cabinet | Any | ||
| Fine balance | Any | Readability 0.1 mg | |
| Hanging insert for 24 well plate | Millipore | MCRP24H48 | Alternative brands/suppliers can be used as long as there is a gap between the insert base and the plate surface |
| Incubator | Any | 37°C, 5% CO2, humidified environment | |
| Oven | Any | set to 80°C | |
| P1000/200/20/10 pipette | Any | It is essential the pipettes used for the procedure are calibrated | |
| P1000/200/20/10 tips | Any | ||
| pH meter with microprobe | Any | ||
| Spatula | Any | ||
| Vortex | Any | ||
| Matrix addition | |||
| Collagen I (human) | Stem Cell Technologies | 07005 | |
| Collagen I (rat tail) | Gibco | A10483 | |
| Fibronectin | Stem Cell Technologies | 07159 | |
| Hyaluronic Acid | Iduron | HA804 | |
| Matrigel | Corning | 354234 | |
| Cell encapsulation/culture | |||
| B27 Supplement (no retinoic acid) | Gibco | 12587010 | Media additions for serum free cultures (Figure 2D) |
| Cholera toxin | Sigma-Aldrich | C-8052 | Media additions for MCF10A cells (Figure 3, 5) |
| DMEM | Gibco | 21969-035 | |
| DMEM/F12 | Sigma-Aldrich | D8062 | Media additions for MCF10A cells (Figure 3, 5) |
| DMEM/F12 Phenol Red Free | Gibco | 21041-025 | Media additions for serum free cultures (Figure 2D) |
| DPBS | Gibco | 14190-094 | |
| EGF | SourceBiosciences | ABC016 | Media additions for MCF10A cells (Figure 3, 5) |
| Fetal Bovine Serum | Gibco | 10500-064 | |
| Horse serum | Gibco | 26050-070 | Media additions for MCF10A cells (Figure 3, 5) |
| Human cancer/epithelial cell lines | e.g. MCF7/tdTomato MCF7/MCF10a/HCT116-mCherry | ||
| Human mammary fibroblasts | e.g. HMFU19 | ||
| Hydrocortisone | Sigma-Aldrich | H-0888 | Media additions for MCF10A cells (Figure 3, 5) |
| Insulin | Sigma-Aldrich | I9278 | Media additions for MCF10A cells (Figure 3, 5) |
| Knockout serum replacement | Gibco | 10828-028 | Media additions for serum free cultures (Figure 2D) |
| L-glutamine | Gibco | 25030-024 | |
| RPMI | Gibco | 21875-034 | |
| RPMI Phenol Red Free | Sigma-Aldrich | R7509 | |
| Imaging and other assays | |||
| 4% paraformaldehyde | Polysciences | 18814 | |
| Agar | SLS | CHE1070 | |
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | 5482 | |
| Confocal and/or fluorescent microscope | Any | e.g. Leica TCS SPE confocal laser scanning microscope (Figures 2-4) | |
| DAPI solution | Invitrogen | D3571 | 300 uM working solution |
| DPX mounting medium | ThermoFisher Scientific | ||
| Glass cover slips | Any | No1 coverslips 0.13 - 0.17 mm thickness | |
| Glass-bottom dishes | MatTek | ||
| Goat Anti-Rabbit IgG H&L (HRP polymer) | Abcam | ab214880 | |
| Haematoxylin and Eosin | Any | ||
| Histology molds (disposable, plastic) | Any | ||
| Image analysis software | ImageJ | ||
| Live/Dead assay kit | Invitrogen | L3224 | |
| Microtome | Any | ||
| Phalloidin | Life Technologies | F432/R415 | |
| Pierce Peroxidase IHC Detection Kit | ThermoFisher Scientific | 36000 | |
| Primary Ab Caspase 3 | Abcam | ab34710 | Shown in Figure 3C |
| Primary Ab Collagen I | Cell Signalling Technology | 9661 | Shown in Figure 4B, C, F |
| Primary Ab pFAK Tyr 397 | ThermoFisher Scientific | 44-624G | Shown in Figure 4E |
| Prolong gold/diamond anti-fade mountant with DAPI | Molecular Probes | S36939 | |
| Rheometer Physica MCR 301 | Anton Paar | ||
| Scalpel | Any | ||
| Secondary antibody Goat anti Rabbit AF488 | nvitrogen | a11034 | |
| Secondary antibody Goat anti Rabbit AF546 | Invitrogen | a11010 | |
| SuperFrost slides | ThermoFisher Scientific | Coating e.g. APES can help to retain microtome sections on slides. | |
| Triton X 100 | Sigma-Aldrich | X100 | |
| Trypsin-EDTA (0.25%) | Gibco | 25300054 | |
| Vibratome | Leica |
References
- Hynes, R. The extracellular matrix: not just pretty fibrils. Science. 326 (5957), 1216-1219 (2009).
- Tian, C., et al. Cancer-cell-derived matrisome proteins promote metastasis in pancreatic ductal adenocarcinoma. Cancer Research. 80 (7), 1461-1474 (2020).
- Hebert, J. D., et al. Proteomic profiling of the ECM of xenograft breast cancer metastases in different organs reveals distinct metastatic niches. Cancer Research. 80 (7), 1475-1485 (2020).
- Vennin, C., et al. CAF hierarchy driven by pancreatic cancer cell p53-status creates a pro-metastatic and chemoresistant environment via perlecan. Nature Communication. 10 (1), 3637 (2019).
- Ashworth, J. C., et al. Peptide gels of fully-defined composition and mechanics for probing cell-cell and cell-matrix interactions in vitro. Matrix Biology. 85, 15-33 (2020).
- Toniatti, C., Jones, P., Graham, H., Pagliara, B., Draetta, G. Oncology drug discovery: Planning a turnaround. Cancer Discovery. 4 (4), 397-404 (2014).
- Mak, I. W., Evaniew, N., Ghert, M. Lost in translation: animal models and clinical trials in cancer treatment. American Journal of Translational Research. 6 (2), 114-118 (2014).
- Aisenbrey, E. A., Murphy, W. L. Synthetic alternatives to Matrigel. Nature Reviews Materials. 5, 539-551 (2020).
- Onion, D., et al. 3-Dimensional patient-derived lung cancer assays reveal resistance to standards-of-care promoted by stromal cells but sensitivity to histone deacetylase inhibitors. Molecular Cancer Therapy. 15 (4), 753-763 (2016).
- Saunders, J. H., et al. Individual patient oesophageal cancer 3D models for tailored treatment. Oncotarget. 8 (15), 24224-24236 (2017).
- Caliari, S. R., Burdick, J. A. A practical guide to hydrogels for cell culture. Nature Methods. 13 (5), 405-414 (2016).
- Kühn, S., et al. Cell-instructive multiphasic gel-in-gel materials. Advanced Functional Materials. 30, 1908857 (2020).
- Gjorevski, N., et al. Designer matrices for intestinal stem cell and organoid culture. Nature. 539 (7630), 560-564 (2016).
- Gjorevski, N., Lutolf, M. P. Synthesis and characterization of well- defined hydrogel matrices and their application to intestinal stem cell and organoid culture. Nature Protocols. 12 (11), 2263-2274 (2017).
- Saiani, A., et al. Self assembly and gelation properties of α-helix versus β-sheet forming peptides. Soft Matter. 5 (1), 193-202 (2008).
- Wan, S., et al. Self-assembling peptide hydrogel for intervertebral disc tissue engineering. Acta Biomaterialia. 46, 29-40 (2016).
- Blache, U., Stevens, M. M., Gentleman, E. Harnessing the secreted extracellular matrix to engineer tissues. Nature Biomedical Engineering. 4 (4), 357-363 (2020).
- Sachs, N., et al. A living biobank of breast cancer organoids captures disease heterogeneity. Cell. 172 (1-2), 373-386 (2018).
- Shaw, F. L., et al. A detailed mammosphere assay protocol for the quantification of breast stem cell activity. Journal of Mammary Gland Biology and Neoplasia. 17 (2), 111-117 (2012).
- Barcus, C. E., Keely, P. J., Eliceiri, K. W., Schule, L. A. Stiff collagen matrices increase tumorigenic prolactin signaling in breast cancer cells. Journal of Biological Chemistry. 288, 12722-12732 (2013).
- Bax, D. V., et al. Impact of UV- and carbodiimide-based crosslinking on the Integrin-binding properties of collagen-based materials. Acta Biomaterialia. 100, 280 (2019).
- Huang, B. P., et al. Multi-peptide presentation and hydrogel mechanics jointly enhance therapeutic duo-potential of entrapped stromal cells. Biomaterials. 245, 119973 (2020).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved