
Method Article
がんおよび疾患の制御された3D培養モデルのためのユーザー定義ペプチドゲルの調製
要約
私たちは、がんの進行における細胞/マトリックス相互作用の重要性を調査するために使用できる3D細胞培養環境を作り出す方法を提示します。単純な自己組織化オクタペプチドを使用して、カプセル化された細胞を囲むマトリックスを制御し、機械的および生化学的手がかりを独立して制御することができます。
要約
3Dで増殖した細胞は、2Dで増殖した細胞よりもin vivoでの挙動をよりモデル化するという認識が高まっています。このプロトコールでは、細胞や組織を本来の環境に適した環境で培養するのに適した、シンプルで調整可能な3Dハイドロゲルについて説明します。これは、細胞とその局所的な細胞外マトリックスとの間の相互作用がモデルの基本的な部分であるがんの開始、増殖、および治療を調査する研究者にとって特に重要です。3D培養への移行は困難な場合があり、動物由来の3D培養マトリックスのバッチ間のばらつきが大きいため、再現性の欠如と関連していることがよくあります。同様に、取り扱いの問題により、合成ハイドロゲルの有用性が制限される可能性があります。このニーズに応えるため、私たちは、がんや疾患の関連細胞株モデル、および患者由来の組織/細胞の培養を可能にするために、シンプルな自己組織化ペプチドゲルを最適化しました。ゲル自体には、カプセル化中に添加されたものや、カプセル化された細胞によってゲルに堆積されたものを除いて、マトリックス成分は含まれていません。ハイドロゲルの機械的特性は、マトリックスの添加とは無関係に変化させることもできます。したがって、研究者は「白紙の状態」として機能し、研究者は目的の組織を反映した3D培養環境を構築し、機械的な力の影響や細胞挙動の生化学的制御を独立して分析することができます。
概要
がんの発生と進行において細胞外環境が果たす多くの役割は、ますます明らかになっています1。最近、詳細なプロテオミクスベースの分析が、すでに説得力のある文献ベースに追加され、がん関連間質細胞またはがん細胞自体に由来するマトリックス成分が、上皮間葉転換や転移拡大の促進などのイベントにおける重要な要因であることが示されています2,3,4.このように認識されている細胞外マトリックス(ECM)の重要性を考えると、細胞に提示される3D環境を制御できる細胞培養プラットフォームへの移行が重要になってきています。このニーズに応えて、このプロトコルは、ユーザー定義のECM組成と機械的特性5を備えた3Dハイドロゲルでの細胞カプセル化および培養の方法を提示する。
現在、2D in vitro培養におけるがん治療の有効性と、現在のin vivo(患者由来異種移植片、PDX)モデルにおけるこれらの治療薬の影響、および臨床試験におけるそれらの最終的な活性との間には、相関関係が乏しい6,7。これにより、創薬パイプラインに重大な失敗が生じており、試験済みの治療薬を「早期に、安価に失敗する」ことを可能にする改良されたin vitroモデルが緊急に必要とされています。多くの研究者は、マウス肥満細胞腫由来の製品、例えば、マトリゲル(または類似の製品)を使用して、PDX由来の細胞や他の患者に近い細胞8,9,10を含む細胞の挙動をin vitroで増殖および観察するための3Dマトリックスリッチな環境を作り出している。しかし、この「万能」のアプローチでは、がんの発生と進行においてマトリックスタンパク質/糖鎖が果たす複雑な役割が無視されています。
細胞挙動の制御における細胞外マトリックス(ECM)の役割の認識は、特定のマトリックス成分11で構成されるハイドロゲル内またはハイドロゲル上での3D培養の使用も奨励している。これは特定の相互作用を調べるのに有用ですが、これらのシステムは、細胞とマトリックスの間で機械的および生化学的指示を分離できないという問題を抱えています。また、取り扱いが難しく、細胞の挙動が不明瞭な読み出しになることもあります。コラーゲンゲルはこの問題の重要な例であり、細胞媒介性ゲルの収縮はゲル内の細胞を視覚化する能力を劇的に低下させる可能性があるためです5。また、非常にエレガントな多成分ゲルシステムもあり、専門家はこれらを使用して大きな効果を上げています12,13,14。これらは、酵素感受性リンカーや生理活性モチーフを組み込むことができますが、ここで説明するシステムよりも製剤化と適用が大幅に複雑です。
このプロトコルは、完全に定義された3D培養モデルを作成する方法を説明しており、開発および疾患におけるECMの役割をin vitroでモデル化することができます。3Dモデルの基礎はペプチドゲルであり、これは以前に単純な自己組織化オクタペプチドヒドロゲルの最適化として説明しました5,15,16。このシステムは、複雑な動物由来のマトリックスから離れることで、バッチ間の一貫性の向上とハンドリングの改善という大きなメリットを提供します。単純な状態では、ペプチドはマトリックス由来のモチーフを含まず、ユーザーが機能を構築するための「白紙の状態」を効果的に提供します。
私たちは、ペプチドゲルの機械的特性を、マトリックスタンパク質/糖鎖の取り込みと並行して、独立して制御できることを示しています。このシステムは高度に調整可能で、さまざまな細胞タイプをさまざまな形式でカプセル化できます。がんモデルの構築にとって重要なことは、間質細胞を直接共培養するか、間接的ながん細胞と間質の相互作用を特異的に解析するために分離して組み込むこともできることです。最も重要なことは、ここで説明するプロトコルは化学の複雑な知識を必要とせず、専門的な化学知識や機器を必要とせずに、どの細胞培養実験室でも再現できることです。
私たちは、イメージング、レオロジー分析、PCR5 用の材料の抽出、組織学的評価のための埋め込みなど、ペプチドゲル内の細胞挙動の研究に最適化された方法を提供しています。シンプルなハイドロゲルシステムの明らかな利点は、カプセル化された細胞によって沈着したマトリックスを視覚化し、研究する能力です。細胞由来マトリックスの重要性と、細胞が局所微小環境をどのように再設計するかについての理解を深めることの利点は、最近強調され17 、in vivoで発生するのと同様の方法で細胞分泌マトリックス成分をトラップすることの重要性に対する認識の高まりを反映しています。このようなプロセスをモデル化する能力を活用することは、ハイドロゲルベースの疾患モデルの患者関連性を向上させる基本的な推進力の1つである可能性があります。
プロトコル
1. ペプチドの溶解
- 組織培養フードで、P1000ピペットを使用して800 μLの滅菌水を15 mLチューブに加えます。
- ファインバランスを使用して、ペプチド粉末を静電気防止の計量ボートに計量します。目的の最終ペプチドゲル濃度(mg / mL、 表1)の1.25倍の質量(mg)を使用してください。
注:ここで説明する方法では、最終ゲル化の時点でチューブあたり約1.25mLのペプチドゲルが生成されます。一度に複数のチューブを調製することも、あるいは、ユーザーが提示された方法に慣れたときにチューブあたりの容量を増やすこともできます。
| ペプチド濃度(最終ゲル化後) | ペプチドの質量 | 初期NaOH付加 |
| 6 mg / mL | 7.5ミリグラム | 30μL |
| 10 mg / mLの | 12.5ミリグラム | 60μL |
| 15 mg / mLの | 18.75ミリグラム | 100μL |
表1:典型的な最終ゲル濃度に対するペプチド質量と推奨される初期NaOH添加。 リストされているペプチド濃度の範囲は、どちらの方向にも拡張できますが、低濃度のペプチドでは安定したゲルが形成されない可能性が高く、高濃度では得られるゲルが密度が高すぎて十分な栄養交換と細胞生存率が得られない可能性があります。適切な濃度を得るには、さまざまな細胞タイプやペプチドバッチの最適化が必要です。
- 秤量したペプチドを15 mLチューブに加えます。計量ボートをフリックして、粉体が残らないようにします。
注:ペプチド粉末は非常に静的である可能性があるため、粉末の損失を最小限に抑えるように注意してください。ステップ1.2および1.3(またはpH測定)では、プロセスの後半で80°Cでのインキュベーション中に滅菌が行われるため、滅菌状態を維持する必要はありません。 - ボルテックスで3分間渦巻いた後、200 x g で3分間遠心分離します。
- ペプチド溶液を80°Cに設定したオーブンで最低2時間インキュベートします。インキュベーション後に未溶解のペプチドが存在する場合は、手順1.4を繰り返します。
注意: 均一な加熱が不可欠であるため、ヒートブロックはこの手順には適していません。しかし、ハイブリダイゼーションオーブンは、ペプチド溶液が完全に沈んでいる限り、ウォーターバスと同様にうまく機能することがわかりました。
2. ゲル前駆体の形成
- 滅菌水を使用して滅菌0.5 M水酸化ナトリウム(NaOH)溶液を調製します。
注:最も一貫した結果を得るには、希釈したばかりのNaOH溶液が推奨されます。 - 組織培養フードで、溶解したペプチドの中心に0.5 M NaOHを加え、ピペットチップでゆっくりと攪拌しながら混合します(表1)。
- チューブを10秒間ボルテックスし、200 x g で10秒間遠心分離して気泡を除去します。
- ゲル前駆体が曇っている場合は、ステップ2.2と2.3を繰り返し、NaOHを5μLずつ追加します。
注:pH測定は、十分なNaOHが添加されているかどうかを判断するのに役立ちます:最適なpHは9〜10.5であり、酸を使用してpHを下げるとゲルの不均一性が生じる可能性があるため、これを超えないことが望ましいです。必要なNaOHの正確な量は、ペプチド源によって異なる場合があります。 - 15 mLチューブを反転させてゲル前駆体が光学的に透明で自立する(または単に流れるだけ)になったら、組織培養フードに100 μLの滅菌10x PBSを加えます。ボルテックスで10秒間、200 x g で10秒間遠心分離します。
注:ゲル前駆体が曇っている場合は、透明で半固体になるまで手順2.4を繰り返します。ゲル前駆体が液体(pH~10.5以上)の場合、安定したゲルを形成しないため、廃棄する必要があります。 - 80°Cのオーブンで一晩インキュベートします。
- ゲル前駆体を目視で確認し、80°Cで完全に液体であることを確認します。
- ゲル前駆体が80°Cで完全に液体でない場合、十分に中和されていない場合は、ステップ2.4に従ってNaOHを添加します。
- ゲル前駆体が液体であるが、気泡や小さな沈殿物が存在する場合は、チューブを鋭くフリックしてそれらを分散させます。チューブをフリックしても気泡や沈殿物が持続する場合は、ゲル前駆体をボルテックスし、200 x g でそれぞれ10秒間遠心分離します。
- ステップ2.7.1または2.7.2のいずれかを行った後、前駆体ゲルを80°Cでさらに2時間インキュベートしてから、最終ゲル化に進みます。
- 必要になるまでゲル前駆体を80°Cに保ちます(最大48時間)。
注:プロトコールはここで一時停止でき、ゲル前駆体を4°Cで最大4週間保存します。この一時停止ポイントから再開する場合は、ゲル前駆体を80°Cで少なくとも2時間インキュベートし、ステップ2.7から再開します。ゲル前駆体は、最終的なゲル化に必要になる前に十分に調製し、必要に応じて上記の修飾に十分な時間を確保することを強くお勧めします。
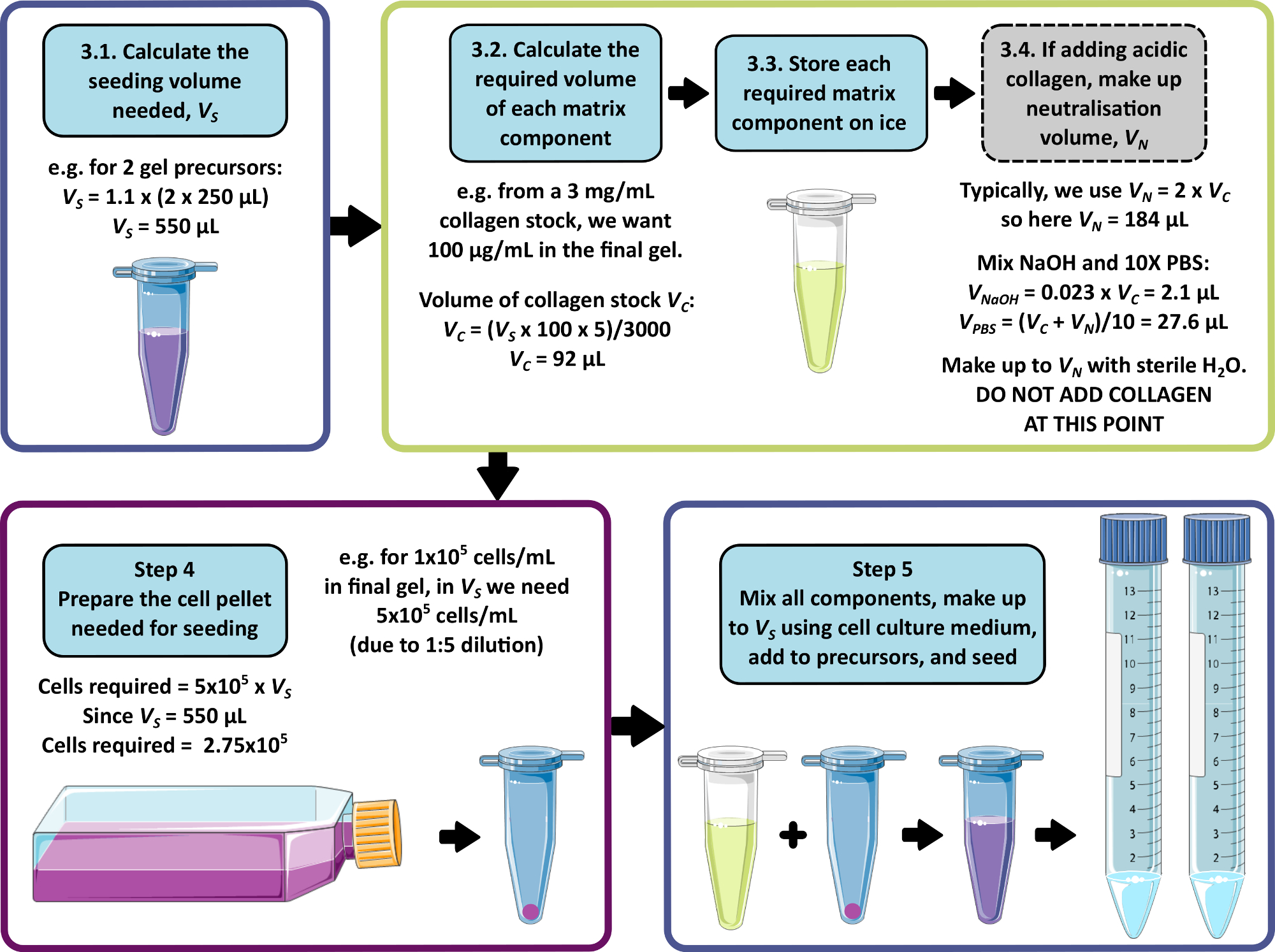
3. 播種用マトリックス成分の調製
注 : 手順 3 から 5 の計算例を 図 1 に示します。ステップ3およびステップ4は、それぞれマトリックスフリーゲルおよび/または無細胞ゲルを作製するために省略されてもよい。
- 播種に必要な細胞、マトリックス、培地の合計容量を計算し、各1 mL前駆体ゲルに250 μLを添加できます。この容量に1.1を掛けると、ピペッティングエラーが生じます。これが播種量VSです。
- 各マトリックス成分について、 式 1 を使用して、播種量に追加するストック溶液の体積を計算します。
 (1)
(1)
注:VSは、すべてのゲルを播種するために必要な播種培地の総量に対応します(ステップ3.1)。すべてのマトリックス成分の体積の合計がVSを超えないようにしてください。 - すべてのマトリックスコンポーネントを完全に解凍し(製造元の指示を参照)、必要になるまで氷上に保管します。
注:一部のマトリックス成分は、ゲル化しやすい傾向があります。これを回避するには、製造元の指示を参照してください。 - 中和が必要な酸性コラーゲン原液を追加する場合(製造元の指示を参照)、次のように中和溶液を準備します。
- 式1を使用して、必要なコラーゲンストック溶液VCの量を計算します。
- 中和溶液の適切な容量、VNを決定します。これは、すべてのマトリックス加算の合計体積が播種量VSを超えないように選択する必要があります。
注:一般的に、VN = 2 x VCの適切な値ですが、これは追加する他のマトリックスコンポーネントの数と体積に応じて調整できます。 - 式2を使用して、中和に必要な1 M NaOHの体積を計算します。
 (2)
(2) - 式3を使用して、中和溶液に必要な10x PBSの体積を計算します。
 (3)
(3) - 1 M NaOHと10x PBSの計算容量を組み合わせ、滅菌水で最大VNを調製します。よく混ぜて、必要になるまで氷の上に保存します。
注:この時点では酸性コラーゲンを追加しないでください。コラーゲンと中和溶液との早期混合は、コラーゲン線維形成の開始につながり、最終的なペプチドゲルの特性に不整合を引き起こす可能性があります。
4.播種用細胞の調製
- まだ完了していない場合は、手順3.1に従って播種量VSを計算します。
- この細胞懸濁液に必要な細胞密度を計算するには、最終ペプチドゲル中の所望の細胞密度を取り、5を掛けます。
注:細胞懸濁液は、ゲル前駆体と混合した際の希釈を考慮して、所望の最終濃度の5倍です。細胞密度は、検討中の新しい細胞株ごとに最適化する必要があります。細胞の種類によっては、最終ペプチドゲル中の1 x 104 から1 x 106 細胞/mLの密度が適切かもしれません。 - 播種に必要なセルの総数を計算します(セル密度にステップ4.1で計算した播種量VSを掛けます)。
- 使用する細胞の標準的な培養/継代法を使用して、ステップ4.3で計算した必要量の細胞を含む細胞ペレットを調製します。
5. 最終ゲル化/細胞カプセル化
- 最終的なゲル化を開始する準備ができたら、ゲル前駆体を80°Cのオーブンから37°Cのウォーターバスに移します。
注:ゲル前駆体は37°Cで自立している必要があります。 それらが液体である場合、完全なゲル化が起こる可能性は低いため、ゲル前駆体は廃棄する必要があります。 - 96ウェルプレート、または細胞培養インサートを備えた24ウェルプレート(または類似のもの)を調製します。
注: ペプチドゲルを24ウェルインサートにプレーティングする場合は、インサートの基部とウェルプレートとの間に隙間があることを確認してください。これにより、ゲルが媒体と接触することが保証されます。 - マトリックスを追加する場合は、すべてのマトリックス成分と中和溶液を組み合わせます(ステップ3)。細胞培養培地を用いてV Sまで調製し(ステップ3.1)、十分に混合します。細胞を添加する場合は、ステップ4で調製した細胞ペレットを播種量VSを使用して再懸濁します。
注:ステップ3でマトリックス成分が調製されていない場合は、播種量に細胞培養培地を使用してください。これは通常、使用する細胞タイプの標準的な培地ですが、複数の細胞タイプの混合物を使用している場合は、バリデーションが必要になる場合があります。 - P1000ピペットを使用して、ゲル前駆体の上に250 μLの細胞/マトリックス混合物を穏やかに加えます。
注:マトリックス成分、特に基底膜抽出物は、ゲル前駆体に添加されると重合し始めることがあります。したがって、できるだけ早く次のステップに進むことが重要です。 - ピペッティングと攪拌の組み合わせで穏やかに混合します。ゲルはせん断が薄くなるため、穏やかなピペッティング/攪拌で混合しやすくなります。十分に混合したら、96ウェルプレートの各ウェルに100 μLを、または各細胞培養インサートに200 μLを加えます。
注:混合時には、200 μLにセットしたP1000を使用したリバースピペッティングが、気泡の発生を防ぐのに有益です。ゲルは最初は混合が難しいかもしれませんが、継続的な混合はより容易になるはずです - これは混合が効率的であることを示しています。 - 5% CO2 および加湿雰囲気中で37°Cで10分間インキュベートします。
注:ペプチドゲルにマトリックス添加物が含まれていない場合、このステップは必要ありません。 - 96ウェルプレートの各ウェルに200 μLの培地を、または細胞培養インサートの外側に1 mLをゲルの上部に数滴加えます。37°Cで5%CO2 と加湿雰囲気中でインキュベートします。
- メディアは、次の 1 時間以内に 2 回交換し、数時間後 (または翌日) に再度交換します。
注:ゲルは数時間不安定になるので、ここでは注意してください。 - 培地は2〜3日ごとに交換してください(または、使用中の細胞の標準的な培養プロトコルに従って)。
6. 間接的な共培養
注:この方法は、ペプチドゲルが24ウェルプレートインサートに播種されている場合、またはゲルを細胞単分子膜の上に支持できる同様の形式にのみ適用できます。この場合、ウェルプレートの底部に細胞の2Dフィーダー層を調製することにより、間接共培養を導入することができる。
- 播種用の24ウェルプレートを準備します。これはペプチドゲルとは別のプレートである必要がありますが、使用するインサートとの互換性を確保するために同じブランドである必要があります(ステップ5.2を参照)。
- 検討中の細胞タイプに応じて、間接共培養の播種に必要な細胞密度を計算します。
注:細胞播種密度は、検討中の新しい細胞株ごとに最適化する必要があります。細胞は、約30〜50%のコンフルエントが得られるようにプレーティングする必要があります。一例として、ヒト乳腺線維芽細胞株HMFU19は、典型的には1〜5×104 細胞/ウェルの密度でプレーティングされる。 - 使用中の細胞の標準的な培養/継代法を使用して、これらの細胞の典型的な増殖培地を使用して、1 mL/ウェルでの播種に適した細胞懸濁液を調製します。
- 細胞懸濁液をウェルプレートに播種し、ウェルあたり1mLです。
- 5% CO2 と加湿雰囲気中で 37 °C でインキュベートし、数時間または一晩接着します。次に、培地をウェルから取り出します。
- 滅菌鉗子を使用して、ペプチドゲルを含む24ウェルプレートインサートを、事前に播種した細胞を2Dで含む新しいウェルに移します。インサートの外側に1mLの培地を滴下し、ゲルの表面に数滴加えます。
注:通常、この時点で使用される培地は、ゲルにカプセル化された細胞に適したものですが、この培地の2D細胞への適合性については、検討中の特定の実験について検証または最適化が必要な場合があります。 - 過剰なコンフルエントを避けるために、新しいフィーダー層を定期的に調製し、上記と同じ方法に従ってペプチドゲルをこれらの新しいウェルに移します。
注:通常、間接共培養用の細胞は、ペプチドゲル播種と同時に調製されます。その後、ペプチドゲルは、数時間後または一晩のインキュベーション後に培地交換の時点で共培養に移すことができます(ステップ5.8を参照)。
7. ペプチドゲルのバルク振動レオロジー
注:標準として、レオロジー特性評価はゲル播種から24時間後に実施され、これは24ウェルプレートインサートで行われるべきです。
- レオメータのセットアップと校正は、メーカーの指示に従って行ってください。プレートの直径が細胞培養インサートの直径にできるだけ近い平行プレート形状を使用してください。
注:必要に応じて、培養中の環境を再現して、37°Cで試験を実施できます。 - 試験する最初のペプチドゲルサンプルを細胞培養インサートから取り出します。これには、インサートを反転させ、メスを使用してプラスチックメンブレンを切り取ります。
注:試験前に、ペプチドゲルを含むプレートが細胞培養インキュベーターの外側にある時間をできるだけ短くしてください。重炭酸塩緩衝システムを備えた培地は、pHを維持するためにCO2 の存在に依存しています。インキュベーターの外に長時間置かれたゲルはpHがドリフトし、レオロジー評価に影響を与える可能性があります。この影響を防ぐために、10 mM HEPESを培地に添加したゲルを維持することは有益です。 - ゲルをレオメータプレートに慎重に移します。次に、メスを使用して、レオメータプレートの下にロードしたときのゲルの変形を最小限に抑えるために、ゲルの高さを約1mmにトリミングします。
注意: メスを使用するときは、レオメータプレートに触れたり損傷したりしないように注意してください。 - 平行プレートの間隔を1mmに設定します。レオメータプレートで覆われていない余分なゲルをトリミングします。
- メーカーの指示に従って、レオメータで必要なテスト設定を実行します。
注:新しいサンプル条件ごとに、0.1〜100%のひずみで振幅スイープを実行して、すべてのテストがサンプルの線形粘弾性領域内のひずみレベルで実行されるようにすることをお勧めします。
8. カプセル化細胞の生死染色
- 培地をウェルから取り出し、培地交換と同じ手法でペプチドゲルを1x PBSで2回洗浄します(ステップ5.7)。
- ステップ7.2に従って、24ウェルプレートインサートで培養したペプチドゲルを除去します。ゲルを元のウェルプレートの1x PBSに保持し、染色する準備ができるまで保管します。
注:この時点では、特に長期培養後、ゲルが壊れやすくなる可能性があるため、注意してください。 - 生/死染色液を調製し、24ウェルプレートインサートあたり500 μL、または96ウェルプレートのウェルあたり50 μLの染色を可能にします。典型的な染色は、1x PBS中の4 μMエチジウムホモダイマーと2 μMカルセインAMです。得られた溶液を光から保護します。
注:生/死試薬の濃度は、使用する細胞タイプと試薬の供給者によっては、さらに最適化が必要な場合があります。 - 各ゲルからPBSを慎重に取り除き、染色液を数滴入れて、各ゲルがしっかりと覆われていることを確認します。
- 暗所で染色溶液中でゲルを10〜15分間インキュベートし、その後、共焦点顕微鏡/蛍光顕微鏡を使用して視覚化します。
注:より高品質の画像を得るには、ゲルをカバーガラスの厚さのガラス底皿に移すことが有益な場合があります。
9. エンドポイントイメージングのためのペプチドゲルの固定
- ステップ8.1に従ってペプチドゲルを洗浄します。
- 4% パラホルムアルデヒド(PFA)を 1x PBS に添加します: 96 ウェルプレートの各ウェルに 100 μL、24 ウェルプレートインサートの各ゲルに 1 mL (インサート内のゲルの上に数滴追加する必要があります)。
注意:パラホルムアルデヒド(PFA)は非常に毒性が高く、皮膚から容易に吸収されます。皮膚、目、粘膜、上気道に非常に破壊的です。PFAはドラフトで取り扱っており、使用者は防護服と手袋を着用する必要があります。代替の化学固定剤は、最終用途に応じて同様に使用できます。 - ペプチドゲルをPFA固定液で室温で1時間インキュベートします。
- PFA固定液を取り出し、ペプチドゲルを1x PBSで2回洗浄します。
注:プロトコールはここで一時停止し、固定ペプチドゲルを1x PBSで4°Cで最大4週間保存し、プレートがパラフィンフィルムで十分に密封されていることを確認します。
10. 切片化のためのペプチドゲルの埋め込み
注:ペプチドゲルを4%寒天に包埋することは、免疫組織化学のためのパラフィン包埋前の重要なステップです。あるいは、ゲルを2%寒天に包埋し、ビブラトームを使用して切片化することもできます(通常、500μm切片で良好な結果が得られます)。これは任意のステップであり、セクション11の方法を使用して、ゲル中の細胞外マトリックス局在を染色するのに有益な水和ゲル切片を作製します。
- 1x PBS(上記注を参照)で溶融した2%または4%寒天溶液を電子レンジで煮沸して調製します。使用する前に数分間冷ましてください。
注意: 溶けた寒天は熱の危険をもたらします–手と顔の保護具を使用して注意して取り扱ってください。調製した寒天溶液は、必要になるまで4°Cで保存できます。 - ステップ7.2に従って、ペプチドゲルを細胞培養インサートから取り出します。
- プラスチック製のパスツールピペットを使用して、組織学的包埋型の基部を寒天溶液の薄層で覆います。20°Cで数秒間冷ましておきます。
- へらを使用して、ペプチドゲルを寒天の中央に置きます。次に、ペプチドゲルを寒天で完全に覆います。
注:ゲルが寒天に沈まないようにしてください。その場合は、ゲルを取り出し、寒天がさらに固まるまでさらに数秒待ってから、再試行してください。固化が起こりすぎると、2つのレイヤー間に弱い結合が生じないようにしてください。 - 包埋したゲルを4°Cで1時間冷ましてから、組織型から取り出します。
注:プロトコールはここで一時停止でき、埋め込みゲルを1x PBSで4°Cで最大4週間保存します。 - 免疫組織化学を実施する場合は、ゲルを組織プロセッサーに入れ、標準的なラボ法で進めます。
注:あるいは、埋め込まれたゲルをビブラトームを使用して水和セクションにカットすることもできます。水和切片は、1x PBS中4°Cの密閉プレートに最大4週間保存する必要があります。
11. 免疫細胞化学を用いたゲル中での細胞染色
- ペプチドゲル/ゲル切片を覆っている1x PBSを取り外してください。ステップ7.2に従って、24ウェルプレートインサートに残っているゲルをすべて取り除きます。
- ゲル/ゲル切片をブロッキングバッファーで覆い、20°Cで30分間インキュベートします。
注:一般的なブロッキングバッファーは、0.5%ウシ血清アルブミン(BSA)と1x PBSと0.1%Triton X-100で構成されています。Triton X-100は毒性があり、深刻な眼の損傷、皮膚の炎症を引き起こし、水生生物に非常に有毒です。ユーザーは、防護服、目の保護具、手袋を着用する必要があります。 - ブロッキングバッファーで一次抗体を最適な使用濃度で調製します。24ウェルプレートゲルあたり200 μL、ゲル切片あたり100 μL、96ウェルプレートウェルあたり50 μLを許容します。
注:通常、ゲルでの3D染色に使用する抗体濃度は、2Dで使用する濃度の2倍にする必要があります。 - ブロッキングバッファーを取り外し、抗体溶液をゲルに滴下します。
- プレートをパラフィンフィルムでシールし、4°Cで一晩インキュベートします。
- 抗体溶液を取り出し、ブロッキングバッファーで2回洗浄します。
- ステップ11.3および11.4で説明したのと同じ手順に従って、二次抗体を添加します。
- 暗所で4°Cで一晩、または20°Cで3時間インキュベートします。
- 抗体溶液を取り出し、1x PBSで2回洗浄します。
- サンプルを1:1,000 DAPI溶液に覆い、暗所で4°Cで1時間インキュベートします。
- ゲルをガラスカバースリップに移し、蛍光/共焦点顕微鏡で画像化します。
12. RNA抽出
注:この方法で使用される容量は、ペプチドゲルが24ウェルプレートインサートに播種される場合に適用できます。他のゲルフォーマットも使用でき、それに応じて容量を調整できます。
- 培地をウェルから取り出し、培地交換と同じ手法でペプチドゲルを1x PBSで2回洗浄します(ステップ5.7)。
- ステップ7.2に従って、24ウェルプレートインサートで培養したペプチドゲルを除去します。各ゲルを別々の15 mL遠心分離チューブに入れます。
注:この時点では、特に長期培養後、ゲルが壊れやすくなる可能性があるため、注意してください。 - P1000を使用して、各チューブに500μLのトリプシン-EDTA(0.25%)を加え、ピペットで上下に動かして混合し、ゲルを破壊します。
- トリプシン-EDTAでゲルを37°Cで3〜5分間インキュベートします。
注:インキュベーション時間は、使用する細胞の種類によっては最適化が必要な場合があります。 - 1x PBSを5 mL加えて、トリプシン-EDTAを希釈します。
- 200 x g で5分間遠心分離し、細胞をペレット化します。
- 上清を取り除きます。
注:細胞ペレットと上清との間にゲル層が形成されている可能性があるため、注意してください。 - 製造元の指示に従って、細胞ペレットを溶解バッファーに再懸濁し、RNA抽出の標準プロトコルに従って進めます。
結果
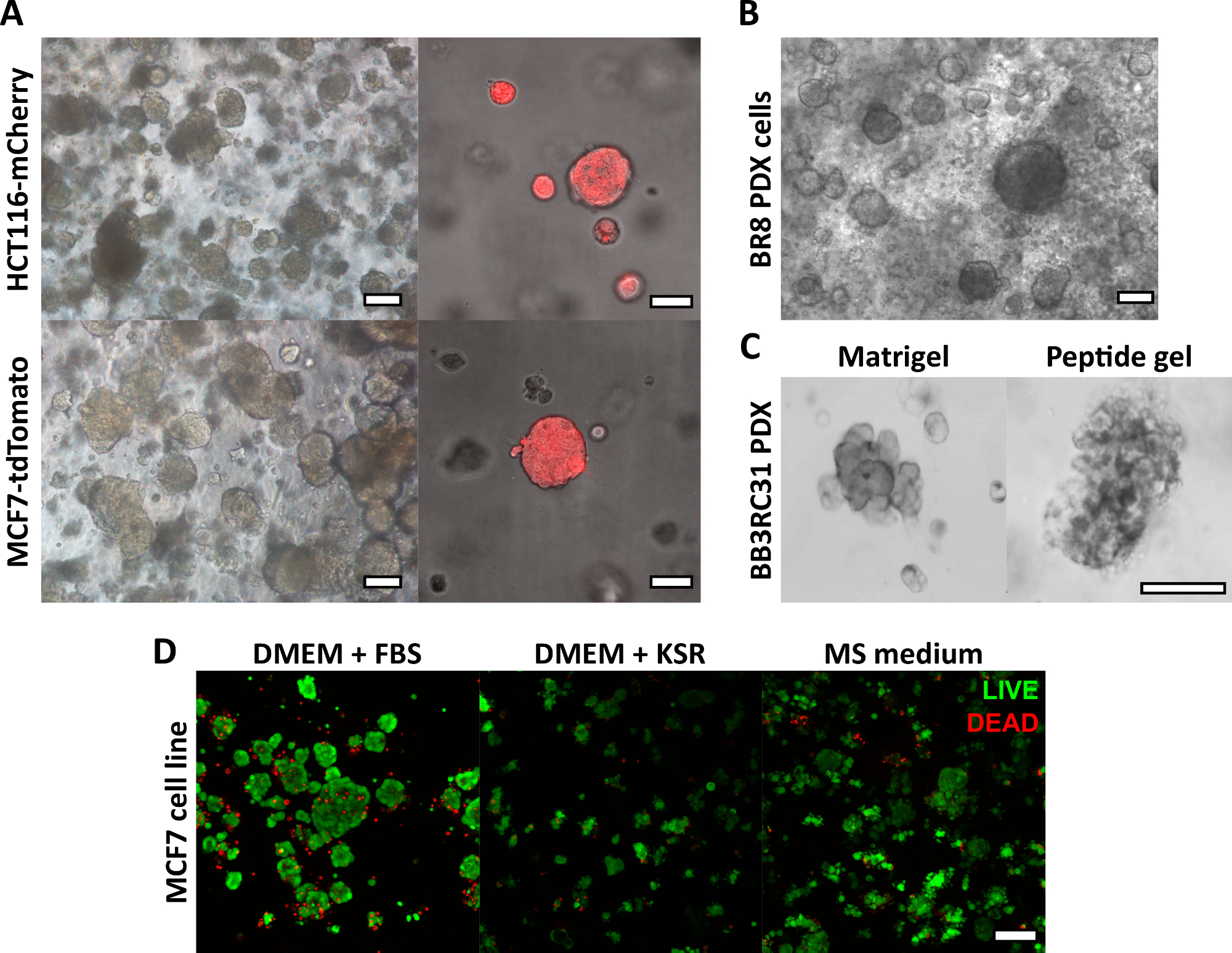
ここで説明するペプチドゲル作製法により、ユーザーはオーダーメイドの3D培養環境を定義し、作成することができます。機械的環境は主にペプチド濃度によって決定されますが、 図1の計算例に示すように、目的のマトリックス成分を制御された密度で添加することもできます。しかし、最も単純な形では、ペプチドゲルプロトコールは、マトリックスフリーの3D環境で細胞をカプセル化する方法を提供します。 図2 は、このアプローチを、蛍光標識されたがん細胞株(図2A)や患者由来異種移植片(PDX)材料(図2B、C)など、さまざまながんモデルと組み合わせる方法を示しています。重要なことは、細胞株とPDX材料の両方をゲル内で無血清条件で培養できること(図2C、D)であり、完全に定義された組成の3D培養システムを提供することです。
ペプチド自体には細胞結合モチーフが含まれていないため、カプセル化された細胞は通常、未修飾のペプチドゲル内で丸みを帯びた形態を示します。 図3A は、6 mg/mLペプチドゲル中のヒト乳腺線維芽細胞について、純粋なマトリゲルゲルおよび純粋なコラーゲンゲルで見られる古典的な細長い形態と比較して、これを示しています。しかし、重要なことに、ペプチドゲルプロトコールは、目的のマトリックス成分の組み込みを可能にする。 図3A は、200 μg/mLコラーゲンIを添加することで、ペプチドゲルの細長い線維芽細胞の形態を回復させる方法を示しています。
マトリックス添加は、 図3Bに示すように、MCF10Aなどの他の細胞タイプの増殖と組織化もサポートできます。この場合、100 μg/mL のコラーゲン I を 6 mg/mL ペプチドゲルに添加すると、7 日目までに腺房構造が形成されます。さらなる複雑さは、間接共培養における支持細胞層の組み込みによってももたらされ得る。 図3C は、マトリックスの取り込みとヒト乳腺線維芽細胞との間接的な共培養を組み合わせたアプローチが、MCF10Aの成長と組織化をどのように促進できるかを示しています。
もう1つの重要なパラメータは、ペプチドゲル製造に使用されるペプチドの濃度です。 図4A は、ペプチド濃度(この場合は4〜10 mg/mL)を制御すると、100〜1000 Paの範囲の剛性が得られる例を示しています。これらのゲルは、マトリックスフリーで製造することも、マトリックス添加で作成して剛性と組成の両方を同時に制御することもできます。マトリックス添加物を含むペプチドゲルは、これらの添加物の分布を可視化するために切片化および染色することができます。 図4B、C は、これを行うための2つのアプローチを示しています:4%寒天培地に包埋した後、標準的な組織プロセシングとパラフィン包埋による免疫組織化学(図4B)、または2%寒天培地への包埋とその後のビブラトーム切片化と蛍光染色(図4C)です。
ペプチドゲルの組成を改変する場合、これらの変化が細胞に最初に提示される機械的環境に影響を与えないようにすることが重要です。 Figure 4D は、ペプチド濃度の修飾を使用して、マトリックスインクルート時のペプチドゲルの硬さの変化を相殺する方法を示しています。ゲルの硬さ(貯蔵弾性率、 G')のバルク振動レオロジー測定により、ゲルの組成と硬さが細胞形態に及ぼす影響を区別することができます。明視野画像に示されているように、MDA MB 231細胞は、10 mg/mLまたは15 mg/mLペプチドゲルにコラーゲンを添加すると、細長い形態を発達させます。 図4E は、これらの細長い細胞がpFAKに対して陽性染色されることを示しており、周囲のマトリックスとの相互作用を示しています。ペプチドゲルは、最初はマトリックスフリーの環境であるため、目的のマトリックス成分の細胞合成および沈着を研究するための理想的なプラットフォームにもなります。 図4F は、10 mg/mLペプチドゲルにカプセル化されたMCF7細胞によるコラーゲンIの局在化を示しています。
ペプチドゲルの主な利点の1つは、標準的なラボ法を分析に容易に適用できることです。qRT-PCR用の材料を抽出して、遺伝子発現プロファイルを決定できます(最近の論文5を参照)。明視野顕微鏡によるイメージングにより、細胞増殖のリアルタイムな可視化がさらに可能になります。図5は、失敗したペプチドゲルで遭遇する可能性のある一般的なトラブルシューティングの問題の一部を示しています:ゲル前駆体の混合が不完全である(図5A、B)。ペプチド濃度(図5、C、D)または播種密度(図5、E、F)の不適切な最適化。ペプチドゲルに取り込まれる前の酸性コラーゲンの誤った中和(図5G、H)。特に、ペプチド濃度と播種密度は、培養環境が適切に定義され、目的のアプリケーションを代表するように、各細胞株とペプチド供給源に対して最適化する必要があります。

図1:マトリックスの組成と播種密度の計算例。 このワークフロー例では、100 μg/mL のコラーゲンを添加した 2 つのペプチドゲル前駆体を、最終細胞密度 1 x 105 cells/mL でシードする手順を説明しています。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図2:マトリックスフリーペプチドゲルは、細胞株および患者由来のがんモデルに適した3D培養プラットフォームを提供します。 (A)HCT116結腸直腸癌およびMCF7乳癌細胞株は、それぞれ蛍光マーカーmCherryおよびtdTomatoを構成的に発現し、9日目までに6 mg / mLゲル中に細胞クラスターを形成し(左)、蛍光顕微鏡法を使用してライブイメージングできます(右、スケールバー50μm)。(B)トリプルネガティブ乳がん患者(BR8)由来の患者由来異種移植片(PDX)細胞は、10mg / mLペプチドゲルで7日目までに細胞クラスターを形成します。(C)エストロゲン受容体陽性乳房腫瘍(BB3RC31)由来のPDX細胞は、比較のために一致した継代で基底膜マトリックス(例えば、マトリゲル)コントロールで示される無血清条件18で増殖させることができる。(D)MCF7乳がん細胞は、7日目にLIVE/DEAD細胞アッセイを用いて評価したように、マトリックスフリーおよび無血清の条件で6 mg/mLペプチドゲル中で生存可能である。KSR = ノックアウト血清補充、MS medium = マンモスフィア培地19。スケールバー 100μm (指定のない限り) この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図3:ペプチドゲルの複雑さは、マトリックス添加物と共培養の導入により増加する可能性があります。 (A)ヒト乳腺線維芽細胞株HMFU19は、純粋な基底膜マトリックス(例:Matrigel)および比較のために1.5 mg/mLラットテールコラーゲンIゲルで示される6 mg/mLペプチドゲルで細長い形態を回復するためにコラーゲン添加を必要とします。スケールバー50 μm;(B)MCF10A正常な乳房細胞は、100μg/mLのヒトコラーゲンI、スケールバー100μmを添加した6mg/mLペプチドゲル中で7日目までに腺房構造を形成する。(C)マトリックス成分フィブロネクチン/HA(ヒアルロン酸、分子量804kDa)とHMFU19を間接共培養に併用すると、10 mg/mLペプチドゲル中のMCF10A aciniのサイズと組織化が増加します。切断されたカスパーゼ3染色、スケールバー50μmによって評価されます 。この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図4:ペプチドゲルは、硬さと組成を独立して制御し、細胞に堆積したマトリックスの評価を可能にします。 (A)ペプチド濃度の制御によって達成可能な典型的な剛性範囲(貯蔵弾性率、 G')を示すバルクレオロジー測定、*は p < 0.05を示します。(B)カプセル化されたMCF7を含む10 mg / mLペプチドゲル中の150 μg / mLコラーゲンIの染色を示す免疫組織化学(7日目、スケールバー100 μm);(C)200 μg/mL ヒトコラーゲン I を含む 6 mg/mL ペプチドゲル中のコラーゲン I 分布の免疫蛍光、寒天包埋およびビブラトーム切片化、スケールバー 25 μm による。(D)200 μg/mL コラーゲン I を添加すると、10 mg/mL ペプチドゲルの保存弾性率 G' がわずかに減少し (バルク振動レオロジー)、ペプチド濃度を 15 mg/mL に増加させることで相殺されます。MDA MB 231 トリプルネガティブ乳がん細胞を各条件に示します (7 日目、スケール バー 50 μm)。(E)MDA MB 231、15 mg/mLペプチドゲル、200 μg/mLヒトコラーゲンIは、pFAK染色によるマトリックスとの伸長および相互作用を示します(14日目、スケールバー50 μm)。(F)最初にマトリックスフリーの10 mg/mLペプチドゲルに沈着したMCF7コラーゲンIのin situ染色(10日目、スケールバー100 μm)。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図5:一般的なペプチドゲルのトラブルシューティングの問題は、明視野顕微鏡を使用して解決できる場合があります。 示されている細胞は、指定がない限り、7日目のMCF10A正常乳房上皮細胞です。(A)正しく混合されたゲル前駆体は、不整合がなく光学的に透明であるべきですが、(B)不十分な混合/中和は、ペプチドゲルに目に見える不均一性/縞を引き起こす可能性があります(白い矢印)。(C)MCF10Aは、HMFU19間接共培養を添加すると、6 mg/mLペプチドゲル中で腺房構造を形成しますが、(D)15 mg/mLではペプチド濃度が高すぎて腺房形成ができません。(E)5 x 105 細胞/mLで播種したMCF10Aは、100 μg/mLコラーゲンIを添加すると、6 mg/mLゲルで腺房構造を形成しますが、(F)2 x 105 細胞/mLの細胞密度は低すぎて腺房形成ができません。(G)コラーゲンの添加は14日目までに大きな細胞クラスターを生成する可能性がありますが、(H)誤った添加(プロセスのコラーゲン中和が早すぎる)はクラスターの成長を妨げる可能性があります。スケールバー 100 μm. この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
ディスカッション
ここで説明するペプチドゲルは、複数の細胞タイプの3D培養をサポートするためのシンプルで費用対効果が高く、柔軟性のあるソリューションであることがわかっています。この方法では、使用するペプチドの濃度とタンパク質または糖鎖の添加を完全に制御できるため、ペプチドゲルをその用途に合わせて慎重に調整できます。
既存の方法に対するペプチドゲルの決定的な利点は、複雑な化学的手順を必要としない単純な方法を使用して、マトリックスの組成と機械的特性を独立して制御できることです。ペプチドゲルの機械的特性は、主に初期ゲル前駆体中のペプチド濃度によって決定されます。その後、細胞および/またはマトリックス成分を添加することで、完全にユーザー定義のin vitro環境を作り出すことができます。マトリックスの添加はゲルの初期の機械的特性を変化させるかもしれないが、これはペプチド濃度の独立した変化によって容易に相殺され得る5。これは、既存のシステム、例えばコラーゲンゲルに比べて明らかな利点を提供し、そこでは、剛性を制御するパラメータもまた、一般的にインテグリン結合モチーフの変化をもたらす20,21。
私たちは、がん細胞株および患者由来材料のin vitro培養へのペプチドゲルの適用を実証しました5。ペプチドゲルでアクセス可能な硬さの範囲(Paの数百から数千の範囲)は、乳房などの軟組織で正常および腫瘍マトリックス環境を再現するのに理想的です。しかし、他のアプリケーションでは、骨再生のために10〜20 kPaの範囲など、かなり硬い環境が必要であることを認識しています。ここで提示されたプロトコルのさらなる修正は、達成可能な剛性をこの範囲に拡張するために必要であり、これはアルギン酸塩ゲル22のような代替アプローチのより典型的である。同様に、ここでは、ペプチドゲル内にマトリックスタンパク質/糖鎖を物理的に閉じ込めることによる官能基化の簡単な方法について説明しました。ここで説明するアプリケーションでは、このアプローチはうまく機能し、疾患の3Din vitroモデルを使用したい非専門家グループによる使用に容易に適応できます。他の多くのハイドロゲル11と同様に、ここで使用されるペプチドは、細胞結合または他の生物学的モチーフを含むように拡張することができ、いくつかのアプリケーションでは、このアプローチが好ましい場合がある。
私たちは、成功を確実にするために細心の注意を払う必要があるいくつかの重要なポイントを特定しました。ゲル前駆体の形成は、細胞を組み込む前に、使用する条件が正しいことをユーザーが確認できるようにする重要な中間ステップです。この前駆体は数週間(4°C)保存できますが、使用前に80°Cでインキュベートし、その後37°Cでインキュベートする必要があります。適切な前駆体は、80°Cで完全に液体で、37°Cで自立します。 これらのチェックは、ゲル化が正しく行われることを確認するために不可欠です。次いで、細胞および/またはマトリックスを生理学的条件下で組み込んでもよい。
すでに3Dマトリックスを使用しているラボでは、ペプチドゲルに細胞をカプセル化するために必要な慎重な取り扱いに精通しているでしょう。カプセル化ステップ前およびカプセル化ステップ中の細胞の攪拌を制限するように注意する必要があります。このプロセスでは、特定の細胞タイプが損傷を受けやすいことが分かっており、これはユーザーが慎重に評価する必要があります。ここで説明するペプチドゲルの濃度は、言及された細胞について、細胞がキャスティングウェルの底に沈む前に細胞をカプセル化することを可能にする時間枠でゲル化を進行させるが、このプロセスによって細胞が損傷を受けない程度に十分にゆっくりと進行する。ただし、一部の敏感な細胞タイプでは、pHの上昇に長時間さらされるのを避けるために、より迅速な中和が必要な場合があることは注目に値します。この場合、ペプチドゲルを周囲の培地に10 mM HEPESを添加することが有益です。
このプロトコールに記載されている方法を採用する際には、ペプチド源の品質を慎重に検討することが非常に重要です。ここでのペプチドは、機能的なモチーフやコーティングとして使用されるのではなく、ハイドロゲルの非可溶性部分の全体です。したがって、ペプチド構造の汚染物質や変動は、最終的なハイドロゲルの細胞生存率をサポートする完全性または能力に大きな影響を与える可能性があります。ペプチドの新しいバッチに移行する際には、サプライヤーからのバッチ間の一貫性が良好であることを確認するとともに、ゲル前駆体を形成する際のペプチドの挙動を確認するように注意する必要があります。
要約すると、このプロトコルは、機械的および生物学的特性の独立した制御に重要な焦点を当てた3D培養システムについて説明しています。この方法のシンプルさと適応性により、あらゆる細胞培養ラボでの採用や、幅広いアプリケーションに適しています5。将来的には、このプロトコールは、ペプチド配列の共有結合的修飾を可能にするために拡張され得る。これを高度な顕微鏡法と組み合わせることで、細胞が周囲のマトリックスに加える引張力を調べることができます。しかし、重要なのは、人工的に組み込まれたマトリックスと、カプセル化された細胞自体によって合成されたマトリックスを区別する能力です。マトリックスの変化を経時的に制御および監視するこの能力により、がんやその他の疾患の発症における細胞とマトリックスの相互作用の役割について、これまでにない洞察が可能になります。
開示事項
著者は何も開示していません。
謝辞
National Centre for the Replacement, Refinement and Reduction of Animals in Research NC/N0015831/1からJCA、GFおよびCLRM、NC/T001267/1からRBC、CLRM、JCA、KL-S、KS、NC/T001259/1からJCA、KL-S、CLRM、NC/P002285/1からAMG、SJ、CLRMに、NC//1からの資金提供に感謝いたします。また、Engineering and Physical Sciences Research Council EP/R035563/1 から KL-S と CLRM に、EP/N006615/1 から JLT と CLRM に資金提供を受けています。 図 1 は、Servier Medical Art のグラフィックを使用して作成されました。セルヴィエ メディカルアート by セルヴィエ is licensed under a Creative Commons Attribution 3.0 Unported License.
資料
| Name | Company | Catalog Number | Comments |
| Gel fabrication - Reagents | |||
| FEFEFKFK | Pepceuticals | n/a | Polypeptide; available from various suppliers. Pepceuticals is our recommended supplier due to the quality of the product. |
| PBS 10X | Gibco | 70011-036 | |
| Sodium hydroxide (1 M) | Sigma-Aldrich | S2770 | NaOH; dilute to 0.5 M prior to use |
| Water | Sigma-Aldrich | W3500 | |
| Gel fabrication - Equipment and Consumables | |||
| 15 mL falcon tubes | Greiner | 188261 | If using different brand ensure the material withstands temperatures of up to 90°C |
| 24 well plate | Corning Costar | 3524 | Alternative brands/suppliers can be used as long as there is a gap between the insert base and the plate surface |
| Centrifuge | Any | 200 x g for 3 minutes | |
| Class II Microbiological Safety Cabinet | Any | ||
| Fine balance | Any | Readability 0.1 mg | |
| Hanging insert for 24 well plate | Millipore | MCRP24H48 | Alternative brands/suppliers can be used as long as there is a gap between the insert base and the plate surface |
| Incubator | Any | 37°C, 5% CO2, humidified environment | |
| Oven | Any | set to 80°C | |
| P1000/200/20/10 pipette | Any | It is essential the pipettes used for the procedure are calibrated | |
| P1000/200/20/10 tips | Any | ||
| pH meter with microprobe | Any | ||
| Spatula | Any | ||
| Vortex | Any | ||
| Matrix addition | |||
| Collagen I (human) | Stem Cell Technologies | 07005 | |
| Collagen I (rat tail) | Gibco | A10483 | |
| Fibronectin | Stem Cell Technologies | 07159 | |
| Hyaluronic Acid | Iduron | HA804 | |
| Matrigel | Corning | 354234 | |
| Cell encapsulation/culture | |||
| B27 Supplement (no retinoic acid) | Gibco | 12587010 | Media additions for serum free cultures (Figure 2D) |
| Cholera toxin | Sigma-Aldrich | C-8052 | Media additions for MCF10A cells (Figure 3, 5) |
| DMEM | Gibco | 21969-035 | |
| DMEM/F12 | Sigma-Aldrich | D8062 | Media additions for MCF10A cells (Figure 3, 5) |
| DMEM/F12 Phenol Red Free | Gibco | 21041-025 | Media additions for serum free cultures (Figure 2D) |
| DPBS | Gibco | 14190-094 | |
| EGF | SourceBiosciences | ABC016 | Media additions for MCF10A cells (Figure 3, 5) |
| Fetal Bovine Serum | Gibco | 10500-064 | |
| Horse serum | Gibco | 26050-070 | Media additions for MCF10A cells (Figure 3, 5) |
| Human cancer/epithelial cell lines | e.g. MCF7/tdTomato MCF7/MCF10a/HCT116-mCherry | ||
| Human mammary fibroblasts | e.g. HMFU19 | ||
| Hydrocortisone | Sigma-Aldrich | H-0888 | Media additions for MCF10A cells (Figure 3, 5) |
| Insulin | Sigma-Aldrich | I9278 | Media additions for MCF10A cells (Figure 3, 5) |
| Knockout serum replacement | Gibco | 10828-028 | Media additions for serum free cultures (Figure 2D) |
| L-glutamine | Gibco | 25030-024 | |
| RPMI | Gibco | 21875-034 | |
| RPMI Phenol Red Free | Sigma-Aldrich | R7509 | |
| Imaging and other assays | |||
| 4% paraformaldehyde | Polysciences | 18814 | |
| Agar | SLS | CHE1070 | |
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | 5482 | |
| Confocal and/or fluorescent microscope | Any | e.g. Leica TCS SPE confocal laser scanning microscope (Figures 2-4) | |
| DAPI solution | Invitrogen | D3571 | 300 uM working solution |
| DPX mounting medium | ThermoFisher Scientific | ||
| Glass cover slips | Any | No1 coverslips 0.13 - 0.17 mm thickness | |
| Glass-bottom dishes | MatTek | ||
| Goat Anti-Rabbit IgG H&L (HRP polymer) | Abcam | ab214880 | |
| Haematoxylin and Eosin | Any | ||
| Histology molds (disposable, plastic) | Any | ||
| Image analysis software | ImageJ | ||
| Live/Dead assay kit | Invitrogen | L3224 | |
| Microtome | Any | ||
| Phalloidin | Life Technologies | F432/R415 | |
| Pierce Peroxidase IHC Detection Kit | ThermoFisher Scientific | 36000 | |
| Primary Ab Caspase 3 | Abcam | ab34710 | Shown in Figure 3C |
| Primary Ab Collagen I | Cell Signalling Technology | 9661 | Shown in Figure 4B, C, F |
| Primary Ab pFAK Tyr 397 | ThermoFisher Scientific | 44-624G | Shown in Figure 4E |
| Prolong gold/diamond anti-fade mountant with DAPI | Molecular Probes | S36939 | |
| Rheometer Physica MCR 301 | Anton Paar | ||
| Scalpel | Any | ||
| Secondary antibody Goat anti Rabbit AF488 | nvitrogen | a11034 | |
| Secondary antibody Goat anti Rabbit AF546 | Invitrogen | a11010 | |
| SuperFrost slides | ThermoFisher Scientific | Coating e.g. APES can help to retain microtome sections on slides. | |
| Triton X 100 | Sigma-Aldrich | X100 | |
| Trypsin-EDTA (0.25%) | Gibco | 25300054 | |
| Vibratome | Leica |
参考文献
- Hynes, R. The extracellular matrix: not just pretty fibrils. Science. 326 (5957), 1216-1219 (2009).
- Tian, C., et al. Cancer-cell-derived matrisome proteins promote metastasis in pancreatic ductal adenocarcinoma. Cancer Research. 80 (7), 1461-1474 (2020).
- Hebert, J. D., et al. Proteomic profiling of the ECM of xenograft breast cancer metastases in different organs reveals distinct metastatic niches. Cancer Research. 80 (7), 1475-1485 (2020).
- Vennin, C., et al. CAF hierarchy driven by pancreatic cancer cell p53-status creates a pro-metastatic and chemoresistant environment via perlecan. Nature Communication. 10 (1), 3637(2019).
- Ashworth, J. C., et al. Peptide gels of fully-defined composition and mechanics for probing cell-cell and cell-matrix interactions in vitro. Matrix Biology. 85, 15-33 (2020).
- Toniatti, C., Jones, P., Graham, H., Pagliara, B., Draetta, G. Oncology drug discovery: Planning a turnaround. Cancer Discovery. 4 (4), 397-404 (2014).
- Mak, I. W., Evaniew, N., Ghert, M. Lost in translation: animal models and clinical trials in cancer treatment. American Journal of Translational Research. 6 (2), 114-118 (2014).
- Aisenbrey, E. A., Murphy, W. L. Synthetic alternatives to Matrigel. Nature Reviews Materials. 5, 539-551 (2020).
- Onion, D., et al. 3-Dimensional patient-derived lung cancer assays reveal resistance to standards-of-care promoted by stromal cells but sensitivity to histone deacetylase inhibitors. Molecular Cancer Therapy. 15 (4), 753-763 (2016).
- Saunders, J. H., et al. Individual patient oesophageal cancer 3D models for tailored treatment. Oncotarget. 8 (15), 24224-24236 (2017).
- Caliari, S. R., Burdick, J. A. A practical guide to hydrogels for cell culture. Nature Methods. 13 (5), 405-414 (2016).
- Kühn, S., et al. Cell-instructive multiphasic gel-in-gel materials. Advanced Functional Materials. 30, 1908857(2020).
- Gjorevski, N., et al. Designer matrices for intestinal stem cell and organoid culture. Nature. 539 (7630), 560-564 (2016).
- Gjorevski, N., Lutolf, M. P. Synthesis and characterization of well- defined hydrogel matrices and their application to intestinal stem cell and organoid culture. Nature Protocols. 12 (11), 2263-2274 (2017).
- Saiani, A., et al. Self assembly and gelation properties of α-helix versus β-sheet forming peptides. Soft Matter. 5 (1), 193-202 (2008).
- Wan, S., et al. Self-assembling peptide hydrogel for intervertebral disc tissue engineering. Acta Biomaterialia. 46, 29-40 (2016).
- Blache, U., Stevens, M. M., Gentleman, E. Harnessing the secreted extracellular matrix to engineer tissues. Nature Biomedical Engineering. 4 (4), 357-363 (2020).
- Sachs, N., et al. A living biobank of breast cancer organoids captures disease heterogeneity. Cell. 172 (1-2), 373-386 (2018).
- Shaw, F. L., et al. A detailed mammosphere assay protocol for the quantification of breast stem cell activity. Journal of Mammary Gland Biology and Neoplasia. 17 (2), 111-117 (2012).
- Barcus, C. E., Keely, P. J., Eliceiri, K. W., Schule, L. A. Stiff collagen matrices increase tumorigenic prolactin signaling in breast cancer cells. Journal of Biological Chemistry. 288, 12722-12732 (2013).
- Bax, D. V., et al. Impact of UV- and carbodiimide-based crosslinking on the Integrin-binding properties of collagen-based materials. Acta Biomaterialia. 100, 280(2019).
- Huang, B. P., et al. Multi-peptide presentation and hydrogel mechanics jointly enhance therapeutic duo-potential of entrapped stromal cells. Biomaterials. 245, 119973(2020).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved