
Method Article
Preparação de um gel peptídico definido pelo usuário para modelos de cultura 3D controlados de câncer e doença
Neste Artigo
Resumo
Apresentamos um método para criar um ambiente de cultura de células 3D, que pode ser usado para investigar a importância das interações célula/matriz na progressão do câncer. Usando um octapeptídeo simples de automontagem, a matriz ao redor das células encapsuladas pode ser controlada, com regulação independente de pistas mecânicas e bioquímicas.
Resumo
Há uma consciência crescente de que as células cultivadas em 3D modelam melhor o comportamento in vivo do que aquelas cultivadas em 2D. Neste protocolo, descrevemos um hidrogel 3D simples e ajustável, adequado para cultivar células e tecidos em um ambiente que corresponda ao seu ambiente nativo. Isso é particularmente importante para pesquisadores que investigam a iniciação, crescimento e tratamento do câncer, onde a interação entre as células e sua matriz extracelular local é uma parte fundamental do modelo. Mudar para a cultura 3D pode ser desafiador e é frequentemente associado à falta de reprodutibilidade devido à alta variação de lote para lote em matrizes de cultura 3D derivadas de animais. Da mesma forma, problemas de manuseio podem limitar a utilidade dos hidrogéis sintéticos. Em resposta a essa necessidade, otimizamos um gel peptídico simples de automontagem, para permitir a cultura de modelos de linha celular relevantes de câncer e doença, bem como tecidos/células derivados de pacientes. O gel em si está livre de componentes da matriz, além daqueles adicionados durante o encapsulamento ou depositados no gel pelas células encapsuladas. As propriedades mecânicas dos hidrogéis também podem ser alteradas independentemente da adição da matriz. Portanto, atua como uma 'lousa em branco', permitindo que os pesquisadores construam um ambiente de cultura 3D que reflita o tecido-alvo de interesse e dissequem as influências das forças mecânicas e / ou controle bioquímico do comportamento celular de forma independente.
Introdução
Os muitos papéis desempenhados pelo ambiente extracelular no desenvolvimento e progressão do câncer estão se tornando cada vez mais claros1. Recentemente, análises proteômicas detalhadas se somaram a uma base já convincente da literatura, demonstrando que os componentes da matriz derivados de células estromais associadas ao câncer, ou as próprias células cancerígenas, são fatores-chave em eventos como a promoção da transição mesenquimal epitelial e disseminação metastática 2,3,4. Dada essa reconhecida importância da matriz extracelular (MEC), torna-se crucial avançar para plataformas de cultura de células que permitam o controle sobre o ambiente 3D apresentado às células. Em resposta a essa necessidade, este protocolo apresenta um método para encapsulamento e cultura celular em um hidrogel 3D, com composição de ECM definida pelo usuário e propriedades mecânicas5.
Atualmente, existe uma correlação fraca entre a eficácia terapêutica do câncer em cultura in vitro 2D, o impacto dessas terapêuticas nos modelos in vivo atuais (xenoenxerto derivado do paciente, PDX) e sua eventual atividade em ensaios clínicos 6,7. Isso levou a falhas significativas no pipeline de descoberta de medicamentos, com uma necessidade urgente de modelos in vitro aprimorados que permitam que a terapêutica testada "falhe cedo, falhe barato". Muitos pesquisadores usam o produto derivado de mastocitoma de camundongo, por exemplo, Matrigel (ou produtos similares) para criar ambientes ricos em matriz 3D para crescer e observar o comportamento celular in vitro, incluindo células derivadas de PDX e outras células próximas ao paciente 8,9,10. No entanto, essa abordagem de 'tamanho único' negligencia o papel complexo desempenhado pelas proteínas / glicanos da matriz na iniciação e progressão do câncer.
O reconhecimento do papel da matriz extracelular (MEC) no controle do comportamento celular também encorajou o uso de cultura 3D em ou sobre hidrogéis compostos por componentes específicos da matriz11. Embora isso seja útil para investigar interações específicas, esses sistemas sofrem com a incapacidade de separar instruções mecânicas e bioquímicas entre células e matriz. Eles também podem ser difíceis de manusear e podem fornecer leituras pouco claras do comportamento celular. Os géis de colágeno são um exemplo importante desse problema, uma vez que a contração do gel mediada por células pode reduzir drasticamente a capacidade de visualizar as células dentro do gel5. Existem também alguns sistemas de gel multicomponentes muito elegantes, que os especialistas usaram com grande efeito 12,13,14. Estes podem incorporar ligantes sensíveis a enzimas e motivos bioativos, mas são significativamente mais complexos em sua formulação e aplicação do que o sistema descrito aqui.
Este protocolo descreve um método para criar modelos de cultura 3D totalmente definidos, permitindo que os papéis do ECM no desenvolvimento e na doença sejam modelados in vitro. A base do modelo 3D é um gel peptídico, que descrevemos anteriormente como uma otimização de um hidrogel octapeptídeo de automontagem simples 5,15,16. Ao se afastar de matrizes complexas derivadas de animais, este sistema oferece um benefício significativo de melhor consistência de lote para lote e melhor manuseio. Em seu estado simples, o peptídeo não contém motivos derivados da matriz e efetivamente fornece uma 'lousa em branco' na qual o usuário pode construir funcionalidade.
Demonstramos como as propriedades mecânicas do gel peptídico podem ser reguladas de forma independente, juntamente com a incorporação de proteínas/glicanos da matriz. O sistema é altamente ajustável, permitindo o encapsulamento de uma variedade de tipos de células em vários formatos. Importante para a construção de um modelo de câncer, as células estromais também podem ser incorporadas: em co-cultura direta ou separadas para permitir uma análise específica das interações indiretas entre células cancerígenas e estroma. Mais crucialmente, o protocolo descrito aqui não requer nenhum conhecimento complexo de química e pode ser reproduzido em qualquer laboratório de cultura de células sem a necessidade de conhecimento ou equipamento químico especializado.
Otimizamos métodos para o estudo do comportamento celular nos géis peptídicos, incluindo imagens, análise reológica, extração de material para PCR5 e incorporação para avaliação histológica. Um benefício claro para o sistema de hidrogel simples é a capacidade de visualizar e estudar a matriz depositada pelas células encapsuladas. A importância das matrizes derivadas de células e os benefícios de uma melhor compreensão de como as células reprojetam seu microambiente local foram destacados recentemente17 e refletem uma crescente conscientização da importância de aprisionar os componentes da matriz secretada por células, de maneira semelhante ao que ocorre in vivo. Aproveitar a capacidade de modelar tais processos pode ser um dos fatores fundamentais para melhorar a relevância do paciente dos modelos de doenças baseados em hidrogel.
Protocolo
1. Dissolução do peptídeo
- Em uma capela de cultura de tecidos, adicione 800 μL de água estéril a um tubo de 15 mL usando uma pipeta P1000.
- Pesar o pó de peptídeo num recipiente de pesagem não estático com uma balança fina. Use uma massa (em mg) de 1,25x a concentração final desejada de gel de peptídeo (em mg / mL, Tabela 1).
NOTA: O método descrito aqui produzirá um volume de aproximadamente 1,25 mL de gel peptídico por tubo no ponto de gelificação final. Vários tubos podem ser preparados ao mesmo tempo ou, alternativamente, o volume por tubo pode ser aumentado quando o usuário tiver experiência com o método apresentado.
| Concentração de peptídeos (após gelificação final) | Massa do peptídeo | Adição inicial de NaOH |
| 6 mg/ml | 7,5 mg | 30 μL |
| 10 mg/ml | 12,5 mg | 60 μL |
| 15 mg/ml | 18,75 mg | 100 μL |
Tabela 1: Massa peptídica e adição inicial sugerida de NaOH para concentrações típicas de gel final. As faixas de concentrações de peptídeos listadas podem ser estendidas em qualquer direção, no entanto, é mais provável que concentrações mais baixas de peptídeos não formem géis estáveis, enquanto em altas concentrações o gel resultante pode ser muito denso para permitir troca de nutrientes e viabilidade celular suficientes. A concentração apropriada exigirá otimização para diferentes tipos de células e lotes de peptídeos.
- Adicione o peptídeo pesado no tubo de 15 mL. Agite o recipiente de pesagem para garantir que nenhum pó seja deixado para trás.
NOTA: O pó do peptídeo pode ser muito estático, tome cuidado para minimizar a perda de pó. Não é necessário manter condições estéreis durante as etapas 1.2 e 1.3 (ou para quaisquer medições de pH), pois a esterilização ocorre durante a incubação a 80 °C posteriormente no processo. - Vortex por 3 min, depois centrifugue a 200 x g por 3 min.
- Incubar a solução de peptídeos numa estufa regulada para 80 °C durante um mínimo de 2 h. Se o peptídeo não dissolvido estiver presente após a incubação, repita a etapa 1.4.
NOTA: Como o aquecimento uniforme é essencial, um bloco de calor não é adequado para esta etapa. No entanto, descobrimos que os fornos de hibridização funcionam bem, assim como um banho-maria, desde que a solução peptídica esteja totalmente submersa.
2. Formação de precursores de gel
- Prepare uma solução estéril de hidróxido de sódio (NaOH) 0,5 M usando água estéril.
NOTA: A solução de NaOH recém-diluída é recomendada para resultados mais consistentes. - Em uma capa de cultura de tecidos, adicione NaOH 0,5 M ao centro do peptídeo dissolvido e misture mexendo lentamente com a ponta da pipeta (Tabela 1).
- Vortex o tubo por 10 s e centrifugue a 200 x g por 10 s para remover as bolhas.
- Se o precursor do gel estiver turvo, repita as etapas 2.2 e 2.3, adicionando incrementos de 5 μL de NaOH.
NOTA: A medição do pH pode ajudar a determinar se NaOH suficiente foi adicionado: o pH ideal está entre 9–10,5 e é preferível não exceder isso, pois o uso de ácidos para reduzir o pH pode resultar em heterogeneidade do gel. O volume preciso de NaOH necessário pode variar de acordo com a fonte do peptídeo. - Uma vez que o precursor do gel esteja opticamente claro e autossustentável (ou apenas fluindo) na inversão do tubo de 15 mL, adicione 100 μL de PBS estéril 10x em uma capa de cultura de tecidos. Vórtice por 10 s e centrifugação a 200 x g por 10 s.
NOTA: Se o precursor do gel estiver turvo, repita a etapa 2.4 até que fique claro e semi-sólido. Se o precursor do gel for líquido (acima do pH ~ 10,5), ele não formará um gel estável e deve ser descartado. - Incubar durante a noite em estufa a 80 °C.
- Verifique visualmente o precursor do gel para garantir que esteja totalmente líquido a 80 °C.
- Se o precursor do gel não for totalmente líquido a 80 °C, não tiver sido suficientemente neutralizado, adicionar NaOH seguindo o passo 2.4.
- Se o precursor do gel for líquido, mas houver bolhas de ar ou pequenos precipitados, agite o tubo bruscamente para dispersá-los. Se persistirem bolhas de ar ou precipitados após sacudir o tubo, agitar o precursor do gel e centrifugar a 200 x g durante 10 s cada.
- Depois de seguir um dos passos 2.7.1 ou 2.7.2, incubar géis precursores durante mais 2 h a 80 °C antes de proceder à gelificação final.
- Manter os precursores de gel a 80 °C até serem necessários (máximo 48 h).
NOTA: O protocolo pode ser pausado aqui, armazene os precursores do gel a 4 ° C por até 4 semanas. Se reiniciar a partir deste ponto de pausa, incubar precursores de gel a 80 °C durante pelo menos 2 h e reiniciar a partir do passo 2.7. É altamente recomendável preparar precursores de gel bem antes de serem necessários para a gelificação final, para garantir que haja tempo suficiente para as modificações acima, se necessário.
3. Preparação dos componentes da matriz para semeadura
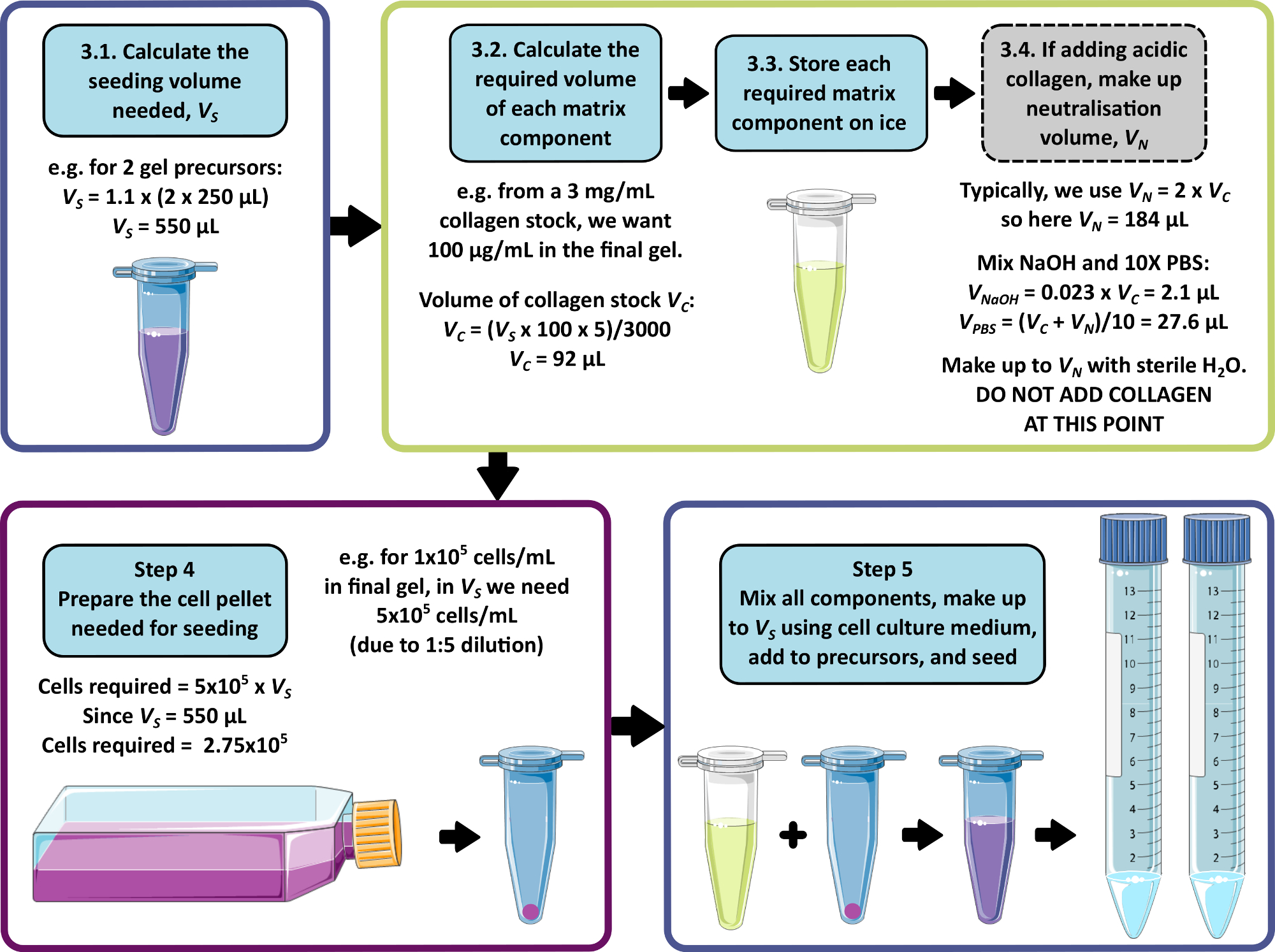
NOTA: Um exemplo de cálculo para as etapas 3 a 5 é mostrado na Figura 1. As etapas 3 e 4 podem ser omitidas para produzir um gel livre de matriz e/ou um gel livre de células, respectivamente.
- Calcule o volume total combinado de células, matriz e meio necessário para a semeadura, permitindo que 250 μL sejam adicionados a cada gel precursor de 1 mL. Multiplique esse volume por 1,1 para permitir o erro de pipetagem. Este é o volume de semeadura, VS.
- Para cada componente da matriz, calcule o volume de solução de reserva a ser adicionado ao volume de semeadura usando a Equação 1:
 (1)
(1)
NOTA: VS corresponde ao volume total de meio de semeadura necessário para semear todos os géis (etapa 3.1). Certifique-se de que a soma de todos os volumes dos componentes da matriz não exceda VS. - Descongele bem todos os componentes da matriz (consulte as instruções do fabricante) e guarde no gelo até que seja necessário.
NOTA: Alguns componentes da matriz serão muito propensos à gelificação. Consulte as instruções do fabricante para evitar que isso ocorra. - Se uma solução de estoque de colágeno ácido que requer neutralização for adicionada (consulte as instruções do fabricante), prepare a solução de neutralização da seguinte forma:
- Calcule o volume de solução estoque de colágeno, VC, necessário usando a Equação 1.
- Determinar um volume adequado para a solução de neutralização, VN. Isso deve ser escolhido de forma que o volume combinado de todas as adições de matriz não exceda o volume de semeadura, VS.
NOTA: Geralmente, um valor sensato para VN = 2 x Vc, mas isso pode ser ajustado de acordo com o número e o volume de outros componentes da matriz a serem adicionados. - Calcule o volume de NaOH 1 M necessário para neutralização usando a Equação 2.
 (2)
(2) - Calcule o volume de 10x PBS necessário na solução de neutralização usando a Equação 3.
 (3)
(3) - Combinar os volumes calculados de NaOH 1 M e 10x PBS e, em seguida, completar o volume até VN com água estéril. Misture bem e guarde no gelo até que seja necessário.
NOTA: Não adicione o colágeno ácido neste momento. A mistura prematura do colágeno com a solução de neutralização levará ao início da fibrilogênese do colágeno, o que pode causar inconsistência nas propriedades do gel peptídico final.
4. Preparação de células para semeadura
- Se ainda não estiver concluído, calcule o volume de semeadura, VS , de acordo com o passo 3.1.
- Calcule a densidade celular necessária nesta suspensão celular, tomando a densidade celular desejada no gel peptídico final e multiplicando por 5.
NOTA: A suspensão celular é 5x a concentração final desejada para contabilizar a diluição após a mistura com o precursor do gel. As densidades celulares devem ser otimizadas para cada nova linhagem celular em consideração. Dependendo do tipo de célula, densidades entre 1 x 104 e 1 x 106 células/mL no gel peptídico final podem ser apropriadas. - Calcular o número total de células necessárias para a propagação (multiplicar a densidade celular pelo volume de propagação VS calculado no passo 4.1.).
- Utilizando métodos padrão de cultura/passagem para as células em utilização, preparar um sedimento de células contendo a quantidade necessária de células, conforme calculado no passo 4.3.
5. Gelificação final/encapsulamento celular
- Quando estiver pronto para iniciar a gelificação final, transfira os precursores de gel do forno a 80 °C para um banho-maria a 37 °C.
NOTA: Os precursores de gel devem ser autoportantes a 37 °C. Se forem líquidos, é improvável que ocorra gelificação completa e os precursores do gel devem ser descartados. - Prepare uma placa de 96 poços ou, alternativamente, uma placa de 24 poços (ou similar) com inserções de cultura de células.
NOTA: Certifique-se de que haja uma lacuna entre a base da inserção e a placa do poço ao revestir géis peptídicos em inserções de 24 poços. Isso garante que o gel esteja em contato com o meio. - Se a matriz for adicionada, combine todos os componentes da matriz e soluções de neutralização (etapa 3). Completar o volume até VS (etapa 3.1) com o meio de cultura celular e homogeneizar. Se for necessário adicionar células, ressuspenda o pellet de células preparado na etapa 4 usando o volume de semeadura VS.
NOTA: Se nenhum componente da matriz tiver sido preparado na etapa 3, use o meio de cultura de células para o volume de semeadura. Este é geralmente o meio de cultura padrão para o tipo de célula em uso, embora possa ser necessário validar se uma mistura de tipos de células estiver sendo usada. - Usando uma pipeta P1000, adicione 250 μL da mistura célula / matriz suavemente em cima do precursor de gel.
NOTA: Os componentes da matriz, particularmente o extrato da membrana basal, podem começar a polimerizar uma vez adicionados ao precursor do gel. É, portanto, importante passar para a próxima etapa o mais rápido possível. - Misture delicadamente pela ação combinada de pipetagem e agitação. O gel é diluído por cisalhamento, então será mais fácil de misturar com pipetagem/agitação suave. Quando bem misturado, adicione 100 μL a cada alvéolo de uma placa de 96 poços ou 200 μL a cada inserção de cultura de células.
NOTA: Ao misturar, a pipetagem reversa usando um P1000 ajustado para 200 μL pode ser benéfica para evitar a introdução de bolhas de ar. O gel pode inicialmente ser difícil de misturar, mas na mistura contínua deve se tornar mais fácil - isso indica que a mistura é eficiente. - Incubar durante 10 min a 37 °C em 5% de CO2 e atmosfera humidificada.
NOTA: Esta etapa não é necessária se o gel peptídico não contiver adições de matriz. - Adicione 200 μL de meio a cada poço da placa de 96 poços ou 1 mL na parte externa do inserto de cultura de células com algumas gotas na parte superior do gel. Incubar a 37 °C em 5% de CO2 e atmosfera umidificada.
- Mude de mídia duas vezes na próxima hora e novamente após várias horas (ou no dia seguinte).
NOTA: Tome cuidado aqui, pois os géis ficarão instáveis por várias horas. - Troque o meio a cada 2 a 3 dias (ou seguindo o protocolo de cultura padrão para as células em uso).
6. Cocultura indireta
NOTA: Este método só é aplicável quando os géis peptídicos são semeados em inserções de placa de 24 poços ou formatos semelhantes nos quais o gel pode ser suportado acima de uma monocamada celular. A co-cultura indireta pode ser introduzida neste caso, preparando uma camada alimentadora 2D de células no fundo da placa do poço.
- Prepare uma placa de 24 poços para semeadura. Esta deve ser uma placa separada dos géis peptídicos, mas deve ser da mesma marca para garantir a compatibilidade com as inserções usadas (consulte a etapa 5.2).
- Calcular a densidade celular necessária para a semeadura da cocultura indireta, de acordo com o tipo de célula considerado.
NOTA: A densidade de semeadura celular deve ser otimizada para cada nova linhagem celular em consideração. As células devem ser plaqueadas para dar aproximadamente 30-50% de confluência. Por exemplo, a linha celular de fibroblastos mamários humanos HMFU19 é tipicamente plaqueada a uma densidade de 1-5 x 104 células/poço. - Usando métodos padrão de cultura/passagem para as células em uso, prepare uma suspensão celular adequada para semeadura a 1 mL/poço, usando o meio de crescimento típico para essas células.
- Semeie a suspensão celular na placa do poço, 1 mL por poço.
- Incubar a 37 °C em 5% de CO2 e uma atmosfera umidificada para fixar por várias horas ou durante a noite. Em seguida, remova o meio dos poços.
- Usando uma pinça estéril, transfira as inserções de placa de 24 poços contendo os géis peptídicos para os novos poços contendo as células pré-semeadas em 2D. Adicione 1 mL de meio gota a gota no exterior da inserção e algumas gotas na superfície do gel.
NOTA: Normalmente, o meio usado neste ponto é o adequado para as células encapsuladas no gel, mas a adequação deste meio para as células em 2D pode precisar de verificação ou otimização para o experimento específico em consideração. - Prepare regularmente novas camadas de alimentação para evitar a confluência excessiva e transfira géis peptídicos para esses novos poços seguindo o mesmo método acima.
NOTA: Normalmente, as células para co-cultura indireta são preparadas ao mesmo tempo que a semeadura do gel peptídico. Os géis peptídicos podem então ser transferidos para a co-cultura no ponto de mudança de meio após algumas horas ou incubação durante a noite, consulte a etapa 5.8.
7. Reologia oscilatória em massa de géis peptídicos
NOTA: Como padrão, a caracterização reológica é realizada 24 h após a semeadura do gel, que deve ocorrer em insertos de placa de 24 poços.
- Configure e calibre o reômetro de acordo com as instruções do fabricante. Use uma geometria de placa paralela com o diâmetro da placa o mais próximo possível do diâmetro do inserto de cultura de células.
NOTA: Os testes podem ser realizados a 37 °C, se desejado, replicando o ambiente durante a cultura. - Remova a primeira amostra de gel de peptídeo a ser testada do inserto de cultura de células, invertendo o inserto e cortando a membrana plástica usando um bisturi.
NOTA: Certifique-se de que a placa que contém os géis peptídicos esteja fora da incubadora de cultura de células pelo menor tempo possível antes do teste. Meios com sistema tamponante de bicarbonato dependem da presença de CO2 para manter o pH. Os géis que ficaram fora da incubadora por muito tempo ficarão à deriva no pH, o que pode afetar a avaliação reológica. Pode ser benéfico manter os géis com HEPES 10 mM adicionados ao meio para evitar esse efeito. - Transfira cuidadosamente o gel para a placa do reômetro. Em seguida, usando um bisturi, apare a altura do gel para aproximadamente 1 mm para minimizar a deformação do gel quando carregado sob a placa do reômetro.
NOTA: Tenha cuidado para não tocar ou danificar a placa do reômetro ao usar o bisturi. - Defina o espaçamento da placa paralela para 1 mm. Apare qualquer excesso de gel que não esteja coberto pelas placas do reômetro.
- Execute as configurações de teste desejadas no reômetro, de acordo com as instruções do fabricante.
NOTA: Para cada nova condição de amostra, é aconselhável executar uma varredura de amplitude de 0,1 a 100% de deformação para garantir que todos os testes sejam realizados em um nível de deformação dentro da região viscoelástica linear da amostra.
8. Coloração viva/morta de células encapsuladas
- Remova o meio dos poços e lave os géis peptídicos duas vezes com 1x PBS, usando a mesma técnica de uma troca de meio (etapa 5.7).
- Remova os géis peptídicos que foram cultivados em inserções de placas de 24 poços seguindo a etapa 7.2. Mantenha os géis em 1x PBS na placa original do poço até que estejam prontos para colorir.
NOTA: Tome cuidado, pois os géis podem ser frágeis neste momento, especialmente após uma cultura prolongada. - Prepare uma solução de coloração viva/morta, permitindo uma coloração de 500 μL por inserto de placa de 24 poços ou 50 μL por poço de uma placa de 96 poços. Uma coloração típica é homodímero de etídio de 4 μM e 2 μM de calceína AM em 1x PBS. Proteja a solução resultante da luz.
NOTA: As concentrações de reagentes vivos/mortos podem exigir otimização adicional, dependendo do tipo de célula usada e do fornecedor do reagente. - Remova cuidadosamente o PBS de cada gel e substitua por algumas gotas da solução de coloração, certificando-se de que cada gel esteja bem coberto.
- Incube géis na solução de coloração no escuro por 10 a 15 minutos e, em seguida, visualize usando um microscópio confocal/fluorescente.
NOTA: Para imagens de alta qualidade, pode ser benéfico transferir os géis para pratos com fundo de vidro com espessura de lamínula.
9. Fixação de géis peptídicos para imagens de endpoint
- Lave os géis peptídicos seguindo o passo 8.1.
- Adicione 4% de paraformaldeído (PFA) em 1x PBS: 100 μL para cada poço de uma placa de 96 poços e 1 mL para cada gel em um inserto de placa de 24 poços (algumas gotas devem ser adicionadas em cima do gel dentro do inserto).
CUIDADO: O paraformaldeído (PFA) é altamente tóxico e é facilmente absorvido pela pele. É extremamente destrutivo para a pele, olhos, membranas mucosas e trato respiratório superior. O PFA deve ser manuseado em um exaustor e os usuários devem usar roupas e luvas de proteção. Fixadores químicos alternativos podem ser usados da mesma maneira de acordo com a aplicação final. - Incube géis peptídicos em fixador PFA por 1 h em temperatura ambiente.
- Remova o fixador PFA e lave os géis peptídicos duas vezes com 1x PBS.
NOTA: O protocolo pode ser pausado aqui, armazene géis peptídicos fixos a 4 °C em 1x PBS por até 4 semanas, certificando-se de que a placa esteja bem selada com filme de parafina.
10. Géis peptídicos de incorporação para seccionamento
NOTA: A incorporação de géis peptídicos em ágar 4% é uma etapa crucial antes da inclusão em parafina para imuno-histoquímica. Alternativamente, os géis podem ser embebidos em ágar 2% e seccionados usando um vibratomo (normalmente seções de 500 μm dão bons resultados). Esta é uma etapa opcional, produzindo seções de gel hidratadas que podem ser benéficas para a coloração da localização da matriz extracelular no gel, usando os métodos da seção 11.
- Prepare a solução de ágar derretida a 2% ou 4% em 1x PBS (veja a nota acima), fervendo no micro-ondas. Deixe esfriar por alguns minutos antes de usar.
NOTA: O ágar derretido apresenta um risco de calor - manuseie com cuidado usando proteção para as mãos e o rosto. Uma vez preparada, a solução de ágar-ágar pode ser armazenada a 4 °C até que seja necessária. - Remova o gel peptídico do inserto da cultura de células seguindo a etapa 7.2.
- Usando uma pipeta de plástico Pasteur, cubra a base de um molde de incorporação histológico com uma fina camada de solução de ágar. Deixar arrefecer a 20 °C durante alguns segundos.
- Usando uma espátula, coloque o gel peptídico no centro do ágar. Em seguida, cubra completamente o gel peptídico em ágar.
NOTA: O gel não deve afundar no ágar. Se isso acontecer, remova o gel e espere mais alguns segundos até que o ágar-ágar solidifique mais e tente novamente. Tente não deixar ocorrer muita solidificação ou haverá uma junção fraca entre as duas camadas. - Deixar arrefecer o gel embebido durante 1 h a 4 °C antes de o retirar do molde histológico.
NOTA: O protocolo pode ser pausado aqui, armazene géis embebidos a 4 °C em 1x PBS por até 4 semanas. - Se a imuno-histoquímica for realizada, coloque géis embebidos em um processador de tecidos e prossiga usando métodos laboratoriais padrão.
NOTA: Alternativamente, os géis embutidos podem ser cortados em seções hidratadas usando um vibratome. As seções hidratadas devem ser armazenadas em placas seladas a 4 ° C em 1x PBS por até 4 semanas.
11. Coloração de células em géis usando imunocitoquímica
- Remova o 1x PBS que cobre as seções de géis/gel peptídicos. Remova todos os géis ainda em inserções de placa de 24 poços seguindo a etapa 7.2.
- Cubra os géis/seções de gel em um tampão de bloqueio e incube por 30 min a 20 °C.
NOTA: Um tampão de bloqueio típico consiste em 0,5% de albumina de soro bovino (BSA) em 1x PBS com 0,1% de Triton X-100. O Triton X-100 é tóxico e causa sérios danos aos olhos, irritação na pele e é muito tóxico para a vida aquática. Os usuários devem usar roupas de proteção, proteção para os olhos e luvas. - Prepare anticorpos primários no tampão de bloqueio em concentrações de trabalho otimizadas. Deixe 200 μL por placa de gel de 24 poços, 100 μL por seção de gel e 50 μL por poço de placa de 96 poços.
NOTA: Normalmente, as concentrações de anticorpos usadas para coloração 3D em géis devem ser o dobro da concentração usada em 2D. - Remova o tampão de bloqueio e adicione a solução de anticorpos aos géis gota a gota.
- Selar a placa com película de parafina e incubar durante a noite a 4 °C.
- Remova a solução de anticorpos e lave duas vezes com tampão de bloqueio.
- Adicionar anticorpo secundário seguindo os mesmos procedimentos descritos nos passos 11.3 e 11.4.
- Incubar no escuro durante a noite a 4 °C ou durante 3 horas a 20 °C.
- Remova a solução de anticorpos e lave duas vezes com 1x PBS.
- Cobrir as amostras com solução de DAPI a 1:1 000 e incubar a 4 °C na obscuridade durante 1 h.
- Transfira o gel para uma lamínula de vidro e obtenha imagens por microscopia fluorescente/confocal.
12. Extração de RNA
NOTA: Os volumes usados neste método são aplicáveis onde os géis peptídicos são semeados em inserções de placa de 24 poços. Outros formatos de gel podem ser usados e os volumes ajustados de acordo.
- Remova o meio dos poços e lave os géis peptídicos duas vezes com 1x PBS, usando a mesma técnica de uma troca de meio (etapa 5.7).
- Remova os géis peptídicos que foram cultivados em inserções de placas de 24 poços seguindo a etapa 7.2. Coloque cada gel em um tubo de centrífuga separado de 15 mL.
NOTA: Tome cuidado, pois os géis podem ser frágeis neste momento, especialmente após uma cultura prolongada. - Usando um P1000, adicione 500 μL de tripsina-EDTA (0,25%) a cada tubo e pipete para cima e para baixo para misturar e romper o gel.
- Incube géis no Tripsina-EDTA a 37 °C por 3-5 min.
NOTA: Os tempos de incubação podem exigir otimização dependendo do tipo de célula usada. - Adicione 5 mL de 1x PBS para diluir a tripsina-EDTA.
- Centrifugue a 200 x g por 5 min para pellet cells.
- Remova o sobrenadante.
NOTA: Tome cuidado, pois uma camada de gel pode ter se formado entre o pellet celular e o sobrenadante. - Ressuspenda o pellet celular em tampão de lise, de acordo com as instruções do fabricante, e proceda seguindo os protocolos padrão para extração de RNA.
Resultados
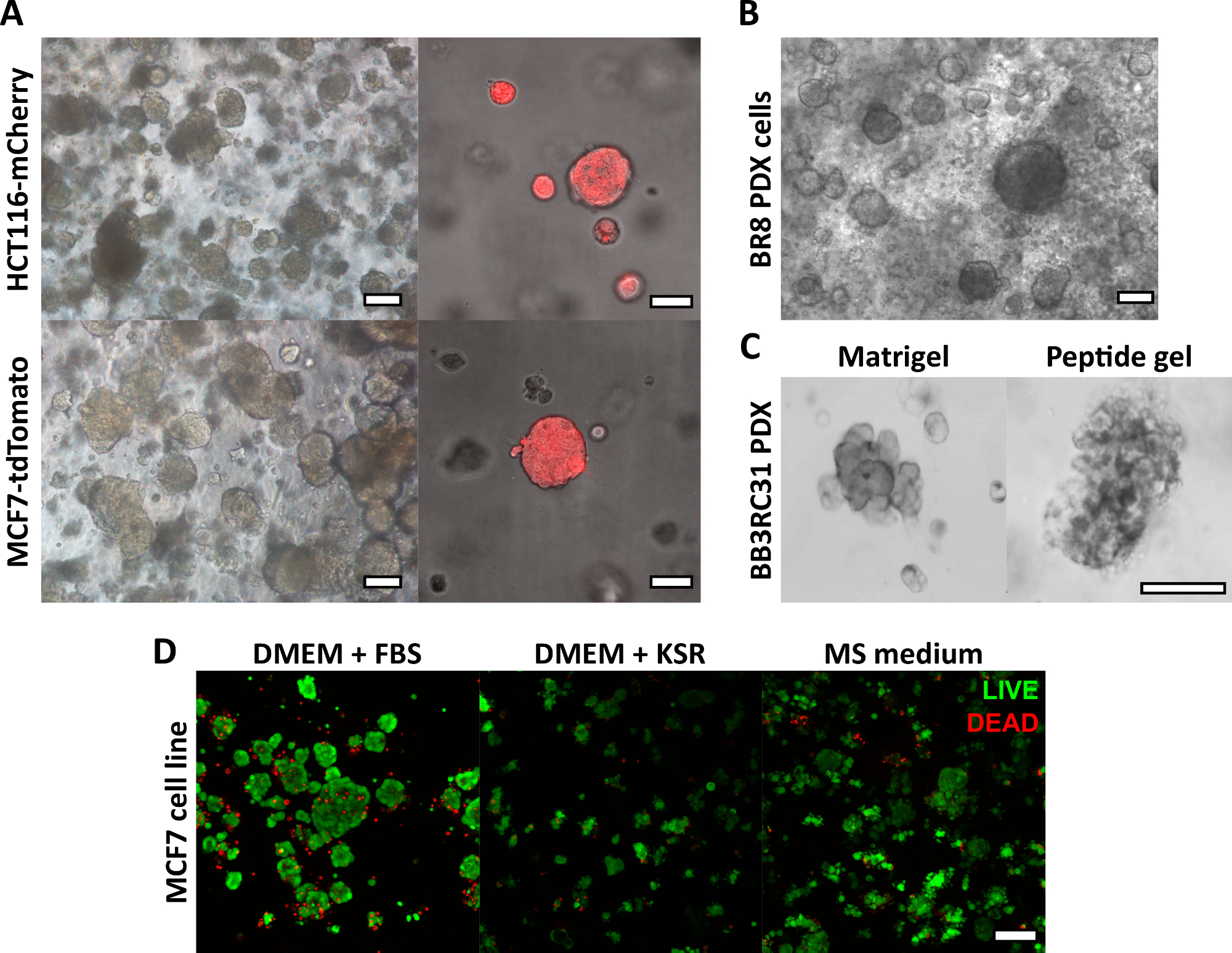
O método de fabricação de gel peptídico descrito aqui permite que o usuário defina e crie um ambiente de cultura 3D sob medida. Embora o ambiente mecânico seja determinado principalmente pela concentração de peptídeos, os componentes da matriz de interesse também podem ser adicionados em densidades controladas, conforme mostrado pelo exemplo de cálculo na Figura 1. Em sua forma mais simples, no entanto, o protocolo de gel peptídico fornece um método para encapsular células em um ambiente 3D livre de matriz. A Figura 2 mostra como essa abordagem pode ser combinada com uma ampla gama de modelos de câncer, incluindo linhas de células cancerígenas marcadas com fluorescência (Figura 2A) e material de xenoenxerto derivado do paciente (PDX) (Figura 2B, C). É importante ressaltar que as linhagens celulares e o material PDX podem ser cultivados dentro dos géis em condições livres de soro (Figura 2C, D), fornecendo um sistema de cultura 3D com composição totalmente definida.
Como o peptídeo em si não contém nenhum motivo de ligação celular, as células encapsuladas normalmente exibem uma morfologia arredondada nos géis peptídicos não modificados. A Figura 3A demonstra isso para fibroblastos mamários humanos em um gel peptídico de 6 mg / mL, em comparação com sua morfologia alongada clássica observada em Matrigel puro e um gel de colágeno puro. É importante, no entanto, que o protocolo de gel peptídico permite a incorporação de componentes da matriz de interesse. A Figura 3A demonstra como a adição de 200 μg/mL de colágeno I pode restaurar a morfologia alongada dos fibroblastos nos géis peptídicos.
As adições de matriz também podem apoiar o crescimento e a organização de outros tipos de células, por exemplo, MCF10A, conforme mostrado na Figura 3B. Nesse caso, a adição de 100 μg/mL de colágeno I a um gel peptídico de 6 mg/mL permite a formação de estruturas acinares no dia 7. Mais complexidade também pode ser introduzida pela incorporação de uma camada de células de suporte em co-cultura indireta. A Figura 3C demonstra como a abordagem combinada de incorporação de matriz e co-cultura indireta com fibroblastos mamários humanos pode aumentar o crescimento e a organização do MCF10A.
Outro parâmetro importante é a concentração de peptídeo usada na fabricação de gel peptídico. A Figura 4A mostra um exemplo de como o controle da concentração de peptídeos, neste caso entre 4 e 10 mg / mL, resulta em uma rigidez que varia entre 100s a 1000s de Pa. Esses géis podem ser fabricados sem matriz ou podem ser criados com adições de matriz para permitir o controle simultâneo da rigidez e da composição. Os géis peptídicos com adições de matriz podem ser seccionados e corados para permitir que a distribuição dessas adições seja visualizada. A Figura 4B, C mostra duas abordagens para fazer isso: incorporação em ágar 4% seguida de processamento de tecido padrão e inclusão em parafina para imuno-histoquímica (Figura 4B) ou incorporação em ágar 2% seguida de seccionamento de vibratomo e coloração fluorescente (Figura 4C).
Ao modificar a composição dos géis peptídicos, é crucial garantir que essas alterações não impactem o ambiente mecânico inicialmente apresentado às células. A Figura 4D demonstra como as modificações na concentração do peptídeo podem ser usadas para compensar quaisquer alterações na rigidez do gel do peptídeo na incorporação da matriz. As medições de reologia oscilatória em massa da rigidez do gel (módulo de armazenamento, G') podem então distinguir entre os efeitos da composição do gel e da rigidez na morfologia celular. Conforme mostrado nas imagens de campo claro, as células MDA MB 231 desenvolvem uma morfologia alongada na adição de colágeno a géis peptídicos de 10 mg / mL ou 15 mg / mL. A Figura 4E mostra que essas células alongadas coram positivo para pFAK, indicando uma interação com a matriz circundante. O ambiente inicialmente livre de matriz dos géis peptídicos também os torna uma plataforma ideal para estudar a síntese celular e a deposição de componentes da matriz de interesse. A Figura 4F mostra a deposição localizada de colágeno I pelas células MCF7 encapsuladas em géis peptídicos de 10 mg/mL.
Uma das principais vantagens dos géis peptídicos é a facilidade com que os métodos laboratoriais padrão podem ser aplicados à sua análise. O material pode ser extraído para qRT-PCR para determinar perfis de expressão gênica (como mostrado em nossa recente publicação5). A imagem por microscopia de campo claro também permite a visualização em tempo real do crescimento celular. A Figura 5 mostra alguns dos problemas típicos de solução de problemas que podem ser encontrados em géis peptídicos malsucedidos: mistura incompleta do precursor do gel (Figura 5A,B); otimização incorreta da concentração de peptídeos (Figura 5C,D) ou densidade de semeadura (Figura 5E,F); e neutralização incorreta do colágeno ácido antes da incorporação nos géis peptídicos (Figura 5G,H). A concentração de peptídeos e a densidade de semeadura, em particular, devem ser otimizadas para cada linhagem celular e fonte de peptídeos, para garantir que o ambiente de cultura seja adequadamente definido e representativo da aplicação de interesse.

Figura 1: Um exemplo de cálculo para composição de matriz e densidade de semeadura. Este exemplo de fluxo de trabalho descreve o procedimento que seria seguido para semear dois precursores de gel de peptídeo com adições de 100 μg/mL de colágeno, a uma densidade celular final de 1 x 105 células/mL. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: Os géis peptídicos livres de matriz fornecem uma plataforma de cultura 3D adequada para modelos de câncer derivados de pacientes e linhagens celulares. (A) As linhagens celulares de câncer colorretal HCT116 e câncer de mama MCF7, expressando constitutivamente os marcadores fluorescentes mCherry e tdTomato, respectivamente, formam aglomerados de células em géis de 6 mg / mL no dia 9 (esquerda) e podem ser visualizadas ao vivo usando microscopia fluorescente (direita, barra de escala de 50 μm); (B) Células de xenoenxerto derivadas de pacientes (PDX) de um paciente com câncer de mama triplo negativo (BR8) formam aglomerados de células no dia 7 em géis peptídicos de 10 mg / mL; (C) As células PDX de tumores de mama positivos para receptores de estrogênio (BB3RC31) podem ser cultivadas em condições livres de soro18, mostradas com controle de matriz de membrana basal (por exemplo, Matrigel) na passagem combinada para comparação; (D) As células de câncer de mama MCF7 são viáveis em géis peptídicos de 6 mg/mL em condições sem matriz e sem soro, conforme avaliado usando um ensaio de células VIVAS/MORTAS no dia 7. KSR = reposição de soro knockout, meio MS = meio mamosfera19. Barra de escala 100 μm, a menos que especificado. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: A complexidade do gel peptídico pode ser aumentada pela introdução de adições de matriz e co-cultura. (A) A linha celular de fibroblastos mamários humanos HMFU19 requer adições de colágeno para restaurar uma morfologia alongada em um gel peptídico de 6 mg / mL, mostrado com matriz de membrana basal pura (por exemplo, Matrigel) e gel de colágeno I de cauda de rato de 1,5 mg / mL para comparação, barra de escala de 50 μm; (B) As células normais da mama MCF10A formam estruturas acinares no dia 7 em géis peptídicos de 6 mg / mL com adição de 100 μg / mL de colágeno humano I, barra de escala de 100 μm; (C) A adição combinada de componentes da matriz fibronectina / HA (ácido hialurônico, peso molecular 804 kDa) e HMFU19 em co-cultura indireta aumenta o tamanho e a organização dos ácinos MCF10A em géis peptídicos de 10 mg / mL, conforme avaliado pela coloração de caspase 3 clivada, barra de escala de 50 μm. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4: Os géis peptídicos permitem o controle independente da rigidez e composição e a avaliação da matriz depositada nas células. (A) Medições de reologia em massa demonstrando uma faixa de rigidez típica (módulo de armazenamento, G ') alcançável pelo controle da concentração de peptídeos, * indica p < 0,05; (B) Imuno-histoquímica mostrando coloração de 150 μg/mL de colágeno I em gel peptídico de 10 mg/mL com MCF7 encapsulado (dia 7, barra de escala de 100 μm); (C) Imunofluorescência da distribuição do colágeno I em um gel peptídico de 6 mg/mL com 200 μg/mL de colágeno I humano, por inclusão em ágar e seccionamento de vibratome, barra de escala de 25 μm; (D) A adição de 200 μg / mL de colágeno I dá uma diminuição modesta no módulo de armazenamento, G ', de géis peptídicos de 10 mg / mL (reologia oscilatória em massa), compensada pelo aumento da concentração de peptídeos para 15 mg / mL. MDA MB 231 células de câncer de mama triplo negativo são mostradas em cada condição (dia 7, barra de escala 50 μm); (E) MDA MB 231 em géis peptídicos de 15 mg/mL com 200 μg/mL de colágeno humano I mostram alongamento e interação com a matriz via coloração pFAK (dia 14, barra de escala 50 μm); (F) Coloração in situ da deposição de colágeno MCF7 I em um gel peptídico de 10 mg/mL inicialmente livre de matriz (dia 10, barra de escala de 100 μm). Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 5: Problemas comuns de solução de problemas de gel peptídico podem ser resolvidos usando microscopia de campo claro. As células mostradas são células epiteliais mamárias normais MCF10A, no dia 7, a menos que especificado. (A) Um precursor de gel corretamente misturado deve ser opticamente claro, sem inconsistências, enquanto (B) mistura / neutralização insuficiente pode causar heterogeneidades / estrias visíveis no gel peptídico (setas brancas); (C) MCF10A forma estruturas acinares em géis peptídicos de 6 mg / mL na adição de co-cultura indireta HMFU19, no entanto (D) a 15 mg / mL a concentração de peptídeos é muito alta para permitir a formação de acinar; (E) MCF10A semeado a 5 x 105 células/mL forma estruturas acinares em géis de 6 mg/mL com adição de 100 μg/mL de colágeno I, no entanto (F) a densidade celular de 2 x 105 células/mL é muito baixa para permitir a formação acinar; (G) As adições de colágeno podem produzir grandes aglomerados de células no dia 14, no entanto, (H) a adição incorreta (neutralização do colágeno muito cedo no processo) pode impedir o crescimento do aglomerado. Barra de escala 100 μm. Clique aqui para ver uma versão ampliada desta figura.
{kind=link}
Discussão
Descobrimos que os géis peptídicos descritos aqui são uma solução simples, econômica e flexível para suportar a cultura 3D de vários tipos de células. Ao fornecer controle total sobre a concentração de peptídeo usada e as adições de proteína ou glicano feitas, esse método permite que os géis peptídicos sejam cuidadosamente adaptados à sua aplicação.
A vantagem crucial dos géis peptídicos sobre os métodos existentes é que a composição da matriz e as propriedades mecânicas podem ser controladas de forma independente, usando um método simples que não requer nenhum procedimento químico complexo. As propriedades mecânicas do gel peptídico são determinadas principalmente pela concentração do peptídeo no precursor inicial do gel. A adição subsequente de células e/ou componentes da matriz permite a criação de um ambiente in vitro totalmente definido pelo usuário. Embora as adições de matriz possam alterar as propriedades mecânicas iniciais do gel, isso pode ser facilmente compensado pela variação independente da concentração de peptídeos5. Isso fornece uma vantagem tangível sobre os sistemas existentes, por exemplo, géis de colágeno, nos quais os parâmetros que controlam a rigidez também geralmente resultam em uma mudança nos motivos de ligação à integrina20,21.
Demonstramos a aplicação do gel peptídico para cultura in vitro de linhagens de células cancerígenas e material derivado de pacientes5. A faixa de rigidez acessível com o gel peptídico (na faixa de 100 a 1000 de Pa) é ideal para replicar ambientes normais e de matriz tumoral em tecidos moles, como a mama. No entanto, reconhecemos que outras aplicações requerem ambientes consideravelmente mais rígidos, por exemplo, na faixa de 10 a 20 kPa para regeneração óssea. Modificações adicionais do protocolo apresentado aqui seriam necessárias para estender a rigidez alcançável para essa faixa, que é mais típica de abordagens alternativas, como géis de alginato22. Da mesma forma, aqui descrevemos um método simples para funcionalização por aprisionamento físico de proteínas / glicanos da matriz dentro do gel peptídico. Para as aplicações descritas aqui, essa abordagem funciona bem e é facilmente adaptada para uso por grupos não especialistas que desejam usar modelos 3D in vitro de doenças. Como muitos outros hidrogéis11, o peptídeo usado aqui pode ser estendido para incluir ligação celular ou outros motivos biológicos e, para algumas aplicações, essa abordagem pode ser preferível.
Identificamos alguns pontos-chave que precisam de atenção cuidadosa para garantir o sucesso. A formação do precursor do gel é uma etapa intermediária crítica que permite ao usuário verificar se as condições usadas estão corretas antes que as células sejam incorporadas. Este precursor pode ser armazenado durante várias semanas (a 4 °C), mas deve ser incubado a 80 °C e, posteriormente, a 37 °C antes da utilização. Um precursor adequado será completamente líquido a 80 °C e autoportante a 37 °C. Essas verificações são essenciais para garantir que a gelificação ocorra corretamente. As células e/ou matriz podem então ser incorporadas em condições fisiológicas.
Os laboratórios que já usam matrizes 3D estarão familiarizados com o manuseio cuidadoso necessário para encapsular as células nos géis peptídicos. Deve-se tomar cuidado para limitar a agitação das células antes e durante as etapas de encapsulamento. Descobrimos que tipos específicos de células são diferencialmente suscetíveis a danos durante esse processo e isso deve ser cuidadosamente avaliado pelo usuário. As concentrações do gel peptídico descritas aqui permitem que a gelificação prossiga em um período de tempo que, para as células mencionadas, permite que as células sejam encapsuladas antes de afundarem no fundo do poço de fundição, mas lentamente o suficiente para que não sejam danificadas por esse processo. No entanto, é importante notar que alguns tipos de células sensíveis podem exigir neutralização mais rápida para evitar a exposição prolongada a pH elevado. Nesse caso, a adição de HEPES 10 mM ao meio ao redor do gel peptídico pode ser benéfica.
Ao adotar o método descrito neste protocolo, é muito importante considerar cuidadosamente a qualidade da fonte do peptídeo. Em vez de ser usado como um motivo ou revestimento funcional, o peptídeo aqui é a totalidade da porção não solúvel do hidrogel. Portanto, quaisquer contaminantes ou variação na estrutura do peptídeo provavelmente terão um impacto significativo na integridade ou capacidade de suportar a viabilidade celular no hidrogel final. Ao mudar para um novo lote de peptídeo, deve-se tomar cuidado para garantir que haja boa consistência lote a lote do fornecedor, bem como verificar o comportamento do peptídeo ao formar o precursor do gel.
Em resumo, este protocolo descreve um sistema de cultura 3D com foco crucial no controle independente de propriedades mecânicas e biológicas. A simplicidade e adaptabilidade do método o tornam adequado para adoção por qualquer laboratório de cultura de células e para uma ampla gama de aplicações5. No futuro, este protocolo pode ser estendido para permitir a modificação covalente da sequência peptídica. Isso pode ser combinado com métodos avançados de microscopia para investigar as forças de tração exercidas pelas células em sua matriz circundante. De importância fundamental, no entanto, é a capacidade de distinguir entre matriz incorporada artificialmente e matriz sintetizada pelas próprias células encapsuladas. Essa capacidade de controlar e monitorar as mudanças da matriz ao longo do tempo permitirá insights sem precedentes sobre os papéis das interações célula-matriz no desenvolvimento do câncer e de outras doenças.
Divulgações
Os autores não têm nada a divulgar.
Agradecimentos
Gostaríamos de agradecer o financiamento do Centro Nacional para a Substituição, Refinamento e Redução de Animais em Pesquisa NC / N0015831 / 1 para JCA, GF e CLRM, NC / T001267 / 1 para RBC, CLRM, JCA, KL-S e KS, NC / T001259 / 1 para JCA, KL-S e CLRM e NC / P002285 / 1 para AMG, SJ e CLRM. Também financiamento do Conselho de Pesquisa em Engenharia e Ciências Físicas EP / R035563 / 1 para KL-S e CLRM e EP / N006615 / 1 para JLT e CLRM. A Figura 1 foi criada usando gráficos adaptados da Servier Medical Art. A Servier Medical Art da Servier está licenciada sob uma Licença Creative Commons Atribuição 3.0 Não Adaptada.
Materiais
| Name | Company | Catalog Number | Comments |
| Gel fabrication - Reagents | |||
| FEFEFKFK | Pepceuticals | n/a | Polypeptide; available from various suppliers. Pepceuticals is our recommended supplier due to the quality of the product. |
| PBS 10X | Gibco | 70011-036 | |
| Sodium hydroxide (1 M) | Sigma-Aldrich | S2770 | NaOH; dilute to 0.5 M prior to use |
| Water | Sigma-Aldrich | W3500 | |
| Gel fabrication - Equipment and Consumables | |||
| 15 mL falcon tubes | Greiner | 188261 | If using different brand ensure the material withstands temperatures of up to 90°C |
| 24 well plate | Corning Costar | 3524 | Alternative brands/suppliers can be used as long as there is a gap between the insert base and the plate surface |
| Centrifuge | Any | 200 x g for 3 minutes | |
| Class II Microbiological Safety Cabinet | Any | ||
| Fine balance | Any | Readability 0.1 mg | |
| Hanging insert for 24 well plate | Millipore | MCRP24H48 | Alternative brands/suppliers can be used as long as there is a gap between the insert base and the plate surface |
| Incubator | Any | 37°C, 5% CO2, humidified environment | |
| Oven | Any | set to 80°C | |
| P1000/200/20/10 pipette | Any | It is essential the pipettes used for the procedure are calibrated | |
| P1000/200/20/10 tips | Any | ||
| pH meter with microprobe | Any | ||
| Spatula | Any | ||
| Vortex | Any | ||
| Matrix addition | |||
| Collagen I (human) | Stem Cell Technologies | 07005 | |
| Collagen I (rat tail) | Gibco | A10483 | |
| Fibronectin | Stem Cell Technologies | 07159 | |
| Hyaluronic Acid | Iduron | HA804 | |
| Matrigel | Corning | 354234 | |
| Cell encapsulation/culture | |||
| B27 Supplement (no retinoic acid) | Gibco | 12587010 | Media additions for serum free cultures (Figure 2D) |
| Cholera toxin | Sigma-Aldrich | C-8052 | Media additions for MCF10A cells (Figure 3, 5) |
| DMEM | Gibco | 21969-035 | |
| DMEM/F12 | Sigma-Aldrich | D8062 | Media additions for MCF10A cells (Figure 3, 5) |
| DMEM/F12 Phenol Red Free | Gibco | 21041-025 | Media additions for serum free cultures (Figure 2D) |
| DPBS | Gibco | 14190-094 | |
| EGF | SourceBiosciences | ABC016 | Media additions for MCF10A cells (Figure 3, 5) |
| Fetal Bovine Serum | Gibco | 10500-064 | |
| Horse serum | Gibco | 26050-070 | Media additions for MCF10A cells (Figure 3, 5) |
| Human cancer/epithelial cell lines | e.g. MCF7/tdTomato MCF7/MCF10a/HCT116-mCherry | ||
| Human mammary fibroblasts | e.g. HMFU19 | ||
| Hydrocortisone | Sigma-Aldrich | H-0888 | Media additions for MCF10A cells (Figure 3, 5) |
| Insulin | Sigma-Aldrich | I9278 | Media additions for MCF10A cells (Figure 3, 5) |
| Knockout serum replacement | Gibco | 10828-028 | Media additions for serum free cultures (Figure 2D) |
| L-glutamine | Gibco | 25030-024 | |
| RPMI | Gibco | 21875-034 | |
| RPMI Phenol Red Free | Sigma-Aldrich | R7509 | |
| Imaging and other assays | |||
| 4% paraformaldehyde | Polysciences | 18814 | |
| Agar | SLS | CHE1070 | |
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | 5482 | |
| Confocal and/or fluorescent microscope | Any | e.g. Leica TCS SPE confocal laser scanning microscope (Figures 2-4) | |
| DAPI solution | Invitrogen | D3571 | 300 uM working solution |
| DPX mounting medium | ThermoFisher Scientific | ||
| Glass cover slips | Any | No1 coverslips 0.13 - 0.17 mm thickness | |
| Glass-bottom dishes | MatTek | ||
| Goat Anti-Rabbit IgG H&L (HRP polymer) | Abcam | ab214880 | |
| Haematoxylin and Eosin | Any | ||
| Histology molds (disposable, plastic) | Any | ||
| Image analysis software | ImageJ | ||
| Live/Dead assay kit | Invitrogen | L3224 | |
| Microtome | Any | ||
| Phalloidin | Life Technologies | F432/R415 | |
| Pierce Peroxidase IHC Detection Kit | ThermoFisher Scientific | 36000 | |
| Primary Ab Caspase 3 | Abcam | ab34710 | Shown in Figure 3C |
| Primary Ab Collagen I | Cell Signalling Technology | 9661 | Shown in Figure 4B, C, F |
| Primary Ab pFAK Tyr 397 | ThermoFisher Scientific | 44-624G | Shown in Figure 4E |
| Prolong gold/diamond anti-fade mountant with DAPI | Molecular Probes | S36939 | |
| Rheometer Physica MCR 301 | Anton Paar | ||
| Scalpel | Any | ||
| Secondary antibody Goat anti Rabbit AF488 | nvitrogen | a11034 | |
| Secondary antibody Goat anti Rabbit AF546 | Invitrogen | a11010 | |
| SuperFrost slides | ThermoFisher Scientific | Coating e.g. APES can help to retain microtome sections on slides. | |
| Triton X 100 | Sigma-Aldrich | X100 | |
| Trypsin-EDTA (0.25%) | Gibco | 25300054 | |
| Vibratome | Leica |
Referências
- Hynes, R. The extracellular matrix: not just pretty fibrils. Science. 326 (5957), 1216-1219 (2009).
- Tian, C., et al. Cancer-cell-derived matrisome proteins promote metastasis in pancreatic ductal adenocarcinoma. Cancer Research. 80 (7), 1461-1474 (2020).
- Hebert, J. D., et al. Proteomic profiling of the ECM of xenograft breast cancer metastases in different organs reveals distinct metastatic niches. Cancer Research. 80 (7), 1475-1485 (2020).
- Vennin, C., et al. CAF hierarchy driven by pancreatic cancer cell p53-status creates a pro-metastatic and chemoresistant environment via perlecan. Nature Communication. 10 (1), 3637 (2019).
- Ashworth, J. C., et al. Peptide gels of fully-defined composition and mechanics for probing cell-cell and cell-matrix interactions in vitro. Matrix Biology. 85, 15-33 (2020).
- Toniatti, C., Jones, P., Graham, H., Pagliara, B., Draetta, G. Oncology drug discovery: Planning a turnaround. Cancer Discovery. 4 (4), 397-404 (2014).
- Mak, I. W., Evaniew, N., Ghert, M. Lost in translation: animal models and clinical trials in cancer treatment. American Journal of Translational Research. 6 (2), 114-118 (2014).
- Aisenbrey, E. A., Murphy, W. L. Synthetic alternatives to Matrigel. Nature Reviews Materials. 5, 539-551 (2020).
- Onion, D., et al. 3-Dimensional patient-derived lung cancer assays reveal resistance to standards-of-care promoted by stromal cells but sensitivity to histone deacetylase inhibitors. Molecular Cancer Therapy. 15 (4), 753-763 (2016).
- Saunders, J. H., et al. Individual patient oesophageal cancer 3D models for tailored treatment. Oncotarget. 8 (15), 24224-24236 (2017).
- Caliari, S. R., Burdick, J. A. A practical guide to hydrogels for cell culture. Nature Methods. 13 (5), 405-414 (2016).
- Kühn, S., et al. Cell-instructive multiphasic gel-in-gel materials. Advanced Functional Materials. 30, 1908857 (2020).
- Gjorevski, N., et al. Designer matrices for intestinal stem cell and organoid culture. Nature. 539 (7630), 560-564 (2016).
- Gjorevski, N., Lutolf, M. P. Synthesis and characterization of well- defined hydrogel matrices and their application to intestinal stem cell and organoid culture. Nature Protocols. 12 (11), 2263-2274 (2017).
- Saiani, A., et al. Self assembly and gelation properties of α-helix versus β-sheet forming peptides. Soft Matter. 5 (1), 193-202 (2008).
- Wan, S., et al. Self-assembling peptide hydrogel for intervertebral disc tissue engineering. Acta Biomaterialia. 46, 29-40 (2016).
- Blache, U., Stevens, M. M., Gentleman, E. Harnessing the secreted extracellular matrix to engineer tissues. Nature Biomedical Engineering. 4 (4), 357-363 (2020).
- Sachs, N., et al. A living biobank of breast cancer organoids captures disease heterogeneity. Cell. 172 (1-2), 373-386 (2018).
- Shaw, F. L., et al. A detailed mammosphere assay protocol for the quantification of breast stem cell activity. Journal of Mammary Gland Biology and Neoplasia. 17 (2), 111-117 (2012).
- Barcus, C. E., Keely, P. J., Eliceiri, K. W., Schule, L. A. Stiff collagen matrices increase tumorigenic prolactin signaling in breast cancer cells. Journal of Biological Chemistry. 288, 12722-12732 (2013).
- Bax, D. V., et al. Impact of UV- and carbodiimide-based crosslinking on the Integrin-binding properties of collagen-based materials. Acta Biomaterialia. 100, 280 (2019).
- Huang, B. P., et al. Multi-peptide presentation and hydrogel mechanics jointly enhance therapeutic duo-potential of entrapped stromal cells. Biomaterials. 245, 119973 (2020).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados