
Method Article
Préparation d’un gel peptidique défini par l’utilisateur pour des modèles de culture 3D contrôlés de cancer et de maladie
Dans cet article
Résumé
Nous présentons une méthode de création d’un environnement de culture cellulaire 3D, qui peut être utilisée pour étudier l’importance des interactions cellule/matrice dans la progression du cancer. À l’aide d’un simple octapeptide auto-assemblé, la matrice entourant les cellules encapsulées peut être contrôlée, avec une régulation indépendante des signaux mécaniques et biochimiques.
Résumé
Il y a une prise de conscience croissante que les cellules cultivées en 3D modélisent mieux le comportement in vivo que celles cultivées en 2D. Dans ce protocole, nous décrivons un hydrogel 3D simple et réglable, adapté à la culture de cellules et de tissus dans un cadre qui correspond à leur environnement d’origine. Ceci est particulièrement important pour les chercheurs qui étudient l’initiation, la croissance et le traitement du cancer, où l’interaction entre les cellules et leur matrice extracellulaire locale est un élément fondamental du modèle. Le passage à la culture 3D peut s’avérer difficile et est souvent associé à un manque de reproductibilité en raison de la forte variation d’un lot à l’autre des matrices de culture 3D d’origine animale. De même, des problèmes de manipulation peuvent limiter l’utilité des hydrogels synthétiques. En réponse à ce besoin, nous avons optimisé un simple gel peptidique auto-assemblé, afin de permettre la culture de modèles de lignées cellulaires pertinents de cancer et de maladie, ainsi que de tissus/cellules dérivés de patients. Le gel lui-même est exempt de composants de la matrice, à l’exception de ceux ajoutés lors de l’encapsulation ou déposés dans le gel par les cellules encapsulées. Les propriétés mécaniques des hydrogels peuvent également être modifiées indépendamment de l’ajout de matrice. Il agit donc comme une « ardoise vierge » permettant aux chercheurs de construire un environnement de culture 3D qui reflète le tissu cible d’intérêt et de disséquer les influences des forces mécaniques et/ou du contrôle biochimique du comportement cellulaire de manière indépendante.
Introduction
Les nombreux rôles joués par l’environnement extracellulaire dans le développement et la progression du cancer deviennent de plus en plus clairs1. Récemment, des analyses protéomiques détaillées ont ajouté à une base de littérature déjà convaincante, démontrant que les composants matriciels dérivés des cellules stromales associées au cancer, ou des cellules cancéreuses elles-mêmes, sont des facteurs clés dans des événements tels que la promotion de la transition mésenchymateuse épithéliale et la propagation métastatique 2,3,4. Compte tenu de l’importance reconnue de la matrice extracellulaire (MEC), il devient crucial de s’orienter vers des plateformes de culture cellulaire permettant de contrôler l’environnement 3D présenté aux cellules. En réponse à ce besoin, ce protocole présente une méthode d’encapsulation et de culture cellulaire dans un hydrogel 3D, avec une composition ECM définie par l’utilisateur et des propriétés mécaniques5.
À l’heure actuelle, il existe une faible corrélation entre l’efficacité thérapeutique du cancer en culture in vitro 2D, l’impact de ces thérapies dans les modèles in vivo actuels (xénogreffe dérivée du patient, PDX) et leur activité éventuelle dans les essais cliniques 6,7. Cela a conduit à des échecs importants dans le pipeline de découverte de médicaments, avec un besoin urgent de modèles in vitro améliorés qui permettent aux traitements testés d'« échouer tôt, échouer à moindre coût ». De nombreux chercheurs utilisent le produit dérivé du mastocytome de souris, par exemple Matrigel (ou des produits similaires) pour créer des environnements riches en matrice 3D afin de croître et d’observer le comportement cellulaire in vitro, y compris des cellules dérivées de PDX et d’autres cellules proches du patient 8,9,10. Cependant, cette approche « taille unique » néglige le rôle complexe joué par les protéines matricielles/glycanes dans l’initiation et la progression du cancer.
La reconnaissance du rôle de la matrice extracellulaire (MEC) dans le contrôle du comportement cellulaire a également encouragé l’utilisation de la culture 3D dans ou sur des hydrogels composés de composants matriciels spécifiques11. Bien que cela soit utile pour étudier des interactions spécifiques, ces systèmes souffrent de l’incapacité à séparer les instructions mécaniques et biochimiques entre les cellules et la matrice. Ils peuvent également être difficiles à manipuler et peuvent donner des lectures peu claires du comportement cellulaire. Les gels de collagène sont un exemple clé de ce problème, car la contraction du gel à médiation cellulaire peut réduire considérablement la capacité de visualiser les cellules à l’intérieur du gel5. Il existe également des systèmes de gel multi-composants très élégants, que les experts ont utilisés à bon escient 12,13,14. Ceux-ci peuvent incorporer des agents de liaison sensibles aux enzymes et des motifs bioactifs, mais sont nettement plus complexes dans leur formulation et leur application que le système décrit ici.
Ce protocole décrit une méthode de création de modèles de culture 3D entièrement définis, permettant de modéliser in vitro les rôles de la MEC dans le développement et la maladie. La base du modèle 3D est un gel peptidique, que nous avons précédemment décrit comme une optimisation d’unhydrogel octapeptide 5,15,16 simple auto-assemblé. En s’éloignant des matrices complexes d’origine animale, ce système offre un avantage significatif d’une meilleure cohérence d’un lot à l’autre et une meilleure manipulation. Dans son état simple, le peptide ne contient aucun motif dérivé de la matrice et fournit effectivement une « ardoise vierge » sur laquelle l’utilisateur peut construire des fonctionnalités.
Nous démontrons comment les propriétés mécaniques du gel peptidique peuvent être régulées indépendamment, parallèlement à l’incorporation de protéines/glycanes de la matrice. Le système est hautement réglable, ce qui permet l’encapsulation d’une gamme de types de cellules dans différents formats. Il est important de noter que pour la construction d’un modèle de cancer, les cellules stromales peuvent également être incorporées : soit en co-culture directe, soit séparées pour permettre une analyse spécifique des interactions indirectes entre les cellules cancéreuses et le stroma. Plus important encore, le protocole décrit ici ne nécessite aucune connaissance complexe de la chimie et peut être reproduit dans n’importe quel laboratoire de culture cellulaire sans avoir besoin de connaissances ou d’équipements chimiques spécialisés.
Nous avons optimisé les méthodes pour l’étude du comportement cellulaire dans les gels peptidiques, y compris l’imagerie, l’analyse rhéologique, l’extraction de matériel pour la PCR5 et l’intégration pour l’évaluation histologique. Un avantage évident du système d’hydrogel simple est la possibilité de visualiser et d’étudier la matrice déposée par les cellules encapsulées. L’importance des matrices dérivées de cellules et les avantages d’une meilleure compréhension de la façon dont les cellules réorganisent leur microenvironnement local ont été récemment soulignées17 et reflètent une prise de conscience croissante de l’importance de piéger les composants de la matrice sécrétée par les cellules, d’une manière similaire à ce qui se produit in vivo. L’exploitation de la capacité de modéliser de tels processus peut être l’un des facteurs fondamentaux de l’amélioration de la pertinence pour les patients des modèles de maladies à base d’hydrogel.
Protocole
1. Dissolution du peptide
- Dans une hotte de culture tissulaire, ajoutez 800 μL d’eau stérile dans un tube de 15 mL à l’aide d’une pipette P1000.
- À l’aide d’une balance fine, pesez la poudre de peptide dans une barque de pesée non statique. Utiliser une masse (en mg) de 1,25 fois la concentration finale souhaitée en gel peptidique (en mg/mL, tableau 1).
REMARQUE : La méthode décrite ici produira un volume d’environ 1,25 mL de gel peptidique par tube au point de gélification finale. Plusieurs tubes peuvent être préparés à la fois, ou bien le volume par tube peut être augmenté lorsque l’utilisateur a de l’expérience avec la méthode présentée.
| Concentration peptidique (après gélification finale) | Masse du peptide | Ajout initial de NaOH |
| 6 mg/mL | 7,5 mg | 30 μL |
| 10 mg/mL | 12,5 mg | 60 μL |
| 15 mg/mL | 18,75 mg | 100 μL |
Tableau 1 : Masse peptidique et ajout initial suggéré de NaOH pour les concentrations finales typiques du gel. Les plages de concentrations peptidiques énumérées peuvent être étendues dans les deux sens, cependant, il est plus probable que des concentrations peptidiques plus faibles ne forment pas de gels stables, tandis qu’à des concentrations élevées, le gel résultant peut être trop dense pour permettre un échange suffisant de nutriments et la viabilité cellulaire. La concentration appropriée nécessitera une optimisation pour différents types de cellules et lots de peptides.
- Ajouter le peptide pesé dans le tube de 15 ml. Secouez le bateau de pesée pour vous assurer qu’aucune poudre n’est laissée derrière.
REMARQUE : La poudre de peptide peut être très statique, veillez à minimiser la perte de poudre. Il n’est pas nécessaire de maintenir des conditions stériles pendant les étapes 1.2 et 1.3 (ou pour toute mesure du pH) car la stérilisation a lieu pendant l’incubation à 80 °C plus tard dans le processus. - Vortex pendant 3 min, puis centrifuger à 200 x g pendant 3 min.
- Incuber la solution peptidique dans un four réglé à 80 °C pendant au moins 2 h. Si un peptide non dissous est présent après l’incubation, répétez l’étape 1.4.
REMARQUE : Comme un chauffage uniforme est essentiel, un bloc chauffant ne convient pas à cette étape. Cependant, nous avons constaté que les fours d’hybridation fonctionnent bien, tout comme un bain d’eau, tant que la solution peptidique est complètement immergée.
2. Formation de précurseurs de gel
- Préparez une solution stérile d’hydroxyde de sodium (NaOH) de 0,5 M avec de l’eau stérile.
REMARQUE : Une solution de NaOH fraîchement diluée est recommandée pour des résultats plus cohérents. - Dans une hotte de culture tissulaire, ajoutez 0,5 M de NaOH au centre du peptide dissous et mélangez en agitant lentement avec la pointe de la pipette (Tableau 1).
- Vortex le tube pendant 10 s et centrifuger à 200 x g pendant 10 s pour éliminer les bulles.
- Si le précurseur du gel est trouble, répétez les étapes 2.2 et 2.3, en ajoutant des incréments de 5 μL de NaOH.
REMARQUE : La mesure du pH peut aider à déterminer si suffisamment de NaOH a été ajouté : le pH optimal se situe entre 9 et 10,5 et il est préférable de ne pas le dépasser, car l’utilisation d’acides pour faire baisser le pH peut entraîner une inhomogénéité du gel. Le volume précis de NaOH requis peut varier selon la source peptidique. - Une fois que le précurseur de gel est optiquement clair et autoportant (ou qu’il s’écoule à peine) lors de l’inversion du tube de 15 ml, ajoutez 100 μL de PBS 10x stérile dans une hotte de culture tissulaire. Vortex pendant 10 s et centrifugeuse à 200 x g pendant 10 s.
REMARQUE : Si le précurseur de gel est trouble, répétez l’étape 2.4 jusqu’à ce qu’il devienne clair et semi-solide. Si le précurseur du gel est liquide (au-dessus du pH ~10,5), il ne formera pas un gel stable et doit être jeté. - Incuber une nuit dans un four à 80 °C.
- Vérifiez visuellement le précurseur du gel pour vous assurer qu’il est entièrement liquide à 80 °C.
- Si le précurseur du gel n’est pas complètement liquide à 80 °C, s’il n’a pas été suffisamment neutralisé, ajoutez du NaOH en suivant l’étape 2.4.
- Si le précurseur du gel est liquide mais qu’il y a des bulles d’air ou de petits précipités, agitez brusquement le tube pour les disperser. Si des bulles d’air ou des précipités persistent après avoir donné un coup de poing sur le tube, agiter le précurseur du gel et centrifuger à 200 x g pendant 10 s chacun.
- Après avoir suivi l’une ou l’autre des étapes 2.7.1 ou 2.7.2, incuber les gels précurseurs pendant 2 h supplémentaires à 80 °C avant de passer à la gélification finale.
- Maintenez les précurseurs de gel à 80 °C jusqu’à ce que vous en ayez besoin (maximum 48 h).
REMARQUE : Le protocole peut être mis en pause ici, stockez les précurseurs de gel à 4 °C jusqu’à 4 semaines. Si vous redémarrez à partir de ce point de pause, incubez les précurseurs du gel à 80 °C pendant au moins 2 h et recommencez à partir de l’étape 2.7. Il est fortement recommandé de préparer les précurseurs de gel bien à l’avance pour la gélification finale, afin de s’assurer qu’il y a suffisamment de temps pour les modifications ci-dessus si nécessaire.
3. Préparation des composants de la matrice pour l’ensemencement
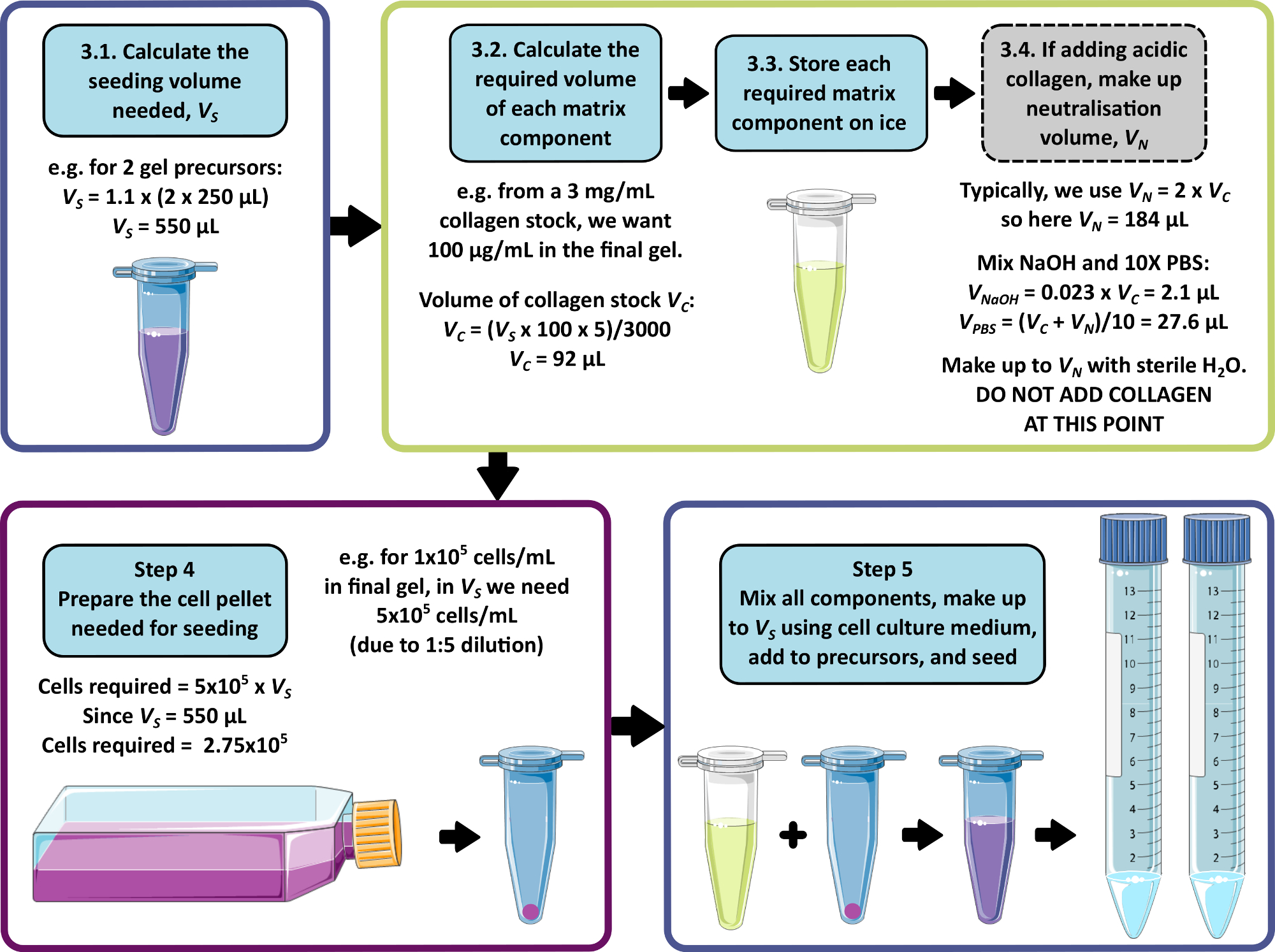
REMARQUE : La Figure 1 présente un exemple de calcul pour les étapes 3 à 5. Les étapes 3 et 4 peuvent être omises pour produire un gel sans matrice et/ou un gel sans cellule, respectivement.
- Calculer le volume total combiné de cellules, de matrice et de milieu requis pour l’ensemencement, en tenant compte de l’ajout de 250 μL à chaque gel de précurseur de 1 mL. Multipliez ce volume par 1,1 pour tenir compte des erreurs de pipetage. Il s’agit du volume d’ensemencement, VS.
- Pour chaque composant de la matrice, calculer le volume de solution mère à ajouter au volume d’ensemencement à l’aide de l’équation 1 :
 (1)
(1)
REMARQUE : VS correspond au volume total de substrat de semis nécessaire pour l’ensemencement de tous les gels (étape 3.1). Assurez-vous que la somme de tous les volumes des composants de la matrice ne dépasse pas VS. - Décongelez soigneusement tous les composants de la matrice (reportez-vous aux instructions du fabricant) et stockez-les sur de la glace jusqu’à ce que vous en ayez besoin.
REMARQUE : Certains composants de la matrice seront très sujets à la gélification. Référez-vous aux instructions du fabricant pour éviter que cela ne se produise. - Si une solution mère de collagène acide nécessitant une neutralisation doit être ajoutée (reportez-vous aux instructions du fabricant), préparez la solution de neutralisation comme suit :
- Calculez le volume de solution mère de collagène, VC, requis à l’aide de l’équation 1.
- Déterminer un volume approprié pour la solution de neutralisation, VN. Celle-ci doit être choisie de telle sorte que le volume combiné de tous les ajouts de matrice ne dépasse pas le volume d’ensemencement, VS.
REMARQUE : En général, une valeur raisonnable pour VN = 2 x Vc, mais elle peut être ajustée en fonction du nombre et du volume des autres composants de la matrice à ajouter. - Calculer le volume de 1 M de NaOH nécessaire à la neutralisation à l’aide de l’équation 2.
 (2)
(2) - Calculez le volume de 10x PBS requis dans la solution de neutralisation à l’aide de l’équation 3.
 (3)
(3) - Combinez les volumes calculés de 1 M de NaOH et de 10x PBS, puis augmentez jusqu’à VN avec de l’eau stérile. Bien mélanger et conserver sur de la glace jusqu’à ce que vous en ayez besoin.
REMARQUE : N’ajoutez pas de collagène acide à ce stade. Le mélange prématuré du collagène avec la solution de neutralisation entraînera l’initiation de la fibrillogenèse du collagène, ce qui peut entraîner une incohérence dans les propriétés du gel peptidique final.
4. Préparation des cellules pour l’ensemencement
- Si ce n’est pas déjà fait, calculez le volume de semis, VS , conformément à l’étape 3.1.
- Calculez la densité cellulaire requise dans cette suspension cellulaire, en prenant la densité cellulaire souhaitée dans le gel peptidique final et en multipliant par 5.
REMARQUE : La suspension cellulaire est 5 fois supérieure à la concentration finale souhaitée pour tenir compte de la dilution lors du mélange avec le précurseur du gel. Les densités cellulaires doivent être optimisées pour chaque nouvelle lignée cellulaire à l’étude. Selon le type de cellule, des densités comprises entre 1 x 104 et 1 x 106 cellules/mL dans le gel peptidique final peuvent être appropriées. - Calculer le nombre total de cellules nécessaires à l’ensemencement (multiplier la densité cellulaire par le volume d’ensemencement VS calculé à l’étape 4.1.).
- En utilisant les méthodes standard de culture/passage des cellules utilisées, préparer une pastille cellulaire contenant la quantité requise de cellules, calculée à l’étape 4.3.
5. Gélification finale/encapsulation cellulaire
- Lorsque vous êtes prêt à commencer la gélification finale, transférez les précurseurs de gel du four à 80 °C dans un bain-marie à 37 °C.
REMARQUE : Les précurseurs de gel doivent être autoportants à 37 °C. S’ils sont liquides, il est peu probable qu’une gélification complète se produise, et les précurseurs du gel doivent être jetés. - Préparez une plaque de 96 puits, ou bien une plaque de 24 puits (ou similaire) avec des inserts de culture cellulaire.
REMARQUE : Assurez-vous qu’il y a un espace entre la base de l’insert et la plaque de puits lors du placage de gels peptidiques dans des inserts à 24 puits. Cela permet de s’assurer que le gel est en contact avec le fluide. - Si une matrice doit être ajoutée, combinez tous les composants de la matrice et les solutions de neutralisation (étape 3). Compléter à VS (étape 3.1) à l’aide d’un milieu de culture cellulaire et bien mélanger. Si des cellules doivent être ajoutées, remettre en suspension la pastille de cellules préparée à l’étape 4 à l’aide du volume d’ensemencement VS.
REMARQUE : Si aucun composant de la matrice n’a été préparé à l’étape 3, utilisez le milieu de culture cellulaire pour le volume d’ensemencement. Il s’agit généralement du milieu de culture standard pour le type de cellule utilisé, bien qu’il puisse être nécessaire de le valider si un mélange de types de cellules est utilisé. - À l’aide d’une pipette P1000, ajoutez doucement 250 μL du mélange cellule/matrice sur le précurseur de gel.
REMARQUE : Les composants de la matrice, en particulier l’extrait de membrane basale, peuvent commencer à polymériser une fois ajoutés au précurseur du gel. Il est donc important de passer à l’étape suivante le plus rapidement possible. - Mélanger délicatement par l’action combinée du pipetage et de l’agitation. Le gel est diluant par cisaillement et deviendra donc plus facile à mélanger avec un pipetage/agitation doux. Lorsqu’il est bien mélangé, ajoutez 100 μL dans chaque puits d’une plaque de 96 puits, ou 200 μL dans chaque insert de culture cellulaire.
REMARQUE : Lors du mélange, le pipetage inversé à l’aide d’un P1000 réglé à 200 μL peut être bénéfique pour éviter l’introduction de bulles d’air. Le gel peut être difficile à mélanger au début, mais en continuant à mélanger, il devrait devenir plus facile - cela indique que le mélange est efficace. - Incuber pendant 10 min à 37 °C dans 5 % de CO2 et une atmosphère humidifiée.
REMARQUE : Cette étape n’est pas nécessaire si le gel peptidique ne contient pas d’ajouts de matrice. - Ajouter 200 μL de milieu dans chaque puits de la plaque à 96 puits, ou 1 mL à l’extérieur de l’insert de culture cellulaire avec quelques gouttes sur le dessus du gel. Incuber à 37 °C dans 5 % de CO2 et une atmosphère humidifiée.
- Changez de support deux fois dans l’heure qui suit, puis de nouveau après plusieurs heures (ou le lendemain).
REMARQUE : Faites attention ici car les gels seront instables pendant plusieurs heures. - Changez de milieu tous les 2 à 3 jours (ou en suivant le protocole de culture standard pour les cellules utilisées).
6. Co-culture indirecte
REMARQUE : Cette méthode n’est applicable que lorsque les gels peptidiques sont ensemencés dans des inserts de plaque à 24 puits, ou dans des formats similaires dans lesquels le gel peut être soutenu au-dessus d’une monocouche cellulaire. La co-culture indirecte peut être introduite dans ce cas en préparant une couche nourricière 2D de cellules au fond de la plaque du puits.
- Préparez une plaque de 24 puits pour l’ensemencement. Il doit s’agir d’une plaque distincte des gels peptidiques, mais elle doit être de la même marque pour assurer la compatibilité avec les inserts utilisés (voir étape 5.2).
- Calculez la densité cellulaire nécessaire à l’ensemencement de la co-culture indirecte, en fonction du type de cellule considéré.
REMARQUE : La densité d’ensemencement cellulaire doit être optimisée pour chaque nouvelle lignée cellulaire à l’étude. Les cellules doivent être plaquées pour donner une confluence d’environ 30 à 50 %. À titre d’exemple, la lignée cellulaire de fibroblastes mammaires humains HMFU19 est généralement plaquée à une densité de 1-5 x 104 cellules/puits. - En utilisant les méthodes standard de culture/passage des cellules utilisées, préparer une suspension cellulaire adaptée à l’ensemencement à 1 mL/puits, en utilisant le milieu de croissance typique de ces cellules.
- Ensemencer la suspension cellulaire dans la plaque du puits, 1 mL par puits.
- Incuber à 37 °C dans 5 % de CO2 et une atmosphère humidifiée pour fixer pendant plusieurs heures, ou toute la nuit. Retirez ensuite le milieu des puits.
- À l’aide d’une pince stérile, transférez les inserts de plaque à 24 puits contenant les gels peptidiques dans les nouveaux puits contenant les cellules pré-ensemencées en 2D. Ajouter 1 mL de goutte à goutte à l’extérieur de l’insert et quelques gouttes à la surface du gel.
REMARQUE : En règle générale, le milieu utilisé à ce stade est celui qui convient aux cellules encapsulées dans le gel, mais l’adéquation de ce milieu pour les cellules en 2D peut nécessiter une vérification ou une optimisation pour l’expérience particulière à l’étude. - Préparez régulièrement de nouvelles couches nourricières pour éviter une confluence excessive, et transférez les gels peptidiques dans ces nouveaux puits en suivant la même méthode que ci-dessus.
REMARQUE : En règle générale, les cellules pour la co-culture indirecte sont préparées en même temps que l’ensemencement du gel peptidique. Les gels peptidiques peuvent ensuite être transférés en co-culture au moment du changement de milieu après quelques heures ou une nuit d’incubation, voir l’étape 5.8.
7. Rhéologie oscillatoire en vrac des gels peptidiques
REMARQUE : En standard, la caractérisation rhéologique est effectuée 24 h après l’ensemencement du gel, qui doit avoir lieu dans des inserts de plaque à 24 puits.
- Installez et calibrez le rhéomètre selon les instructions du fabricant. Utilisez une géométrie de plaque parallèle avec un diamètre de plaque aussi proche que possible du diamètre de l’insert de culture cellulaire.
REMARQUE : Les essais peuvent être effectués à 37 °C si désiré, en reproduisant l’environnement pendant la culture. - Retirez le premier échantillon de gel peptidique à tester de l’insert de culture cellulaire en inversant l’insert et en découpant la membrane plastique à l’aide d’un scalpel.
REMARQUE : Assurez-vous que la plaque contenant les gels peptidiques est à l’extérieur de l’incubateur de culture cellulaire aussi courte que possible avant le test. Les milieux dotés d’un système tampon bicarbonaté dépendent de la présence de CO2 pour maintenir le pH. Les gels qui sont restés trop longtemps à l’extérieur de l’incubateur verront leur pH dériver, ce qui peut affecter l’évaluation rhéologique. Il peut être bénéfique de maintenir les gels avec 10 mM d’HEPES ajoutés au média pour prévenir cet effet. - Transférez délicatement le gel sur la plaque du rhéomètre. Ensuite, à l’aide d’un scalpel, coupez la hauteur du gel à environ 1 mm pour minimiser la déformation du gel lorsqu’il est chargé sous la plaque du rhéomètre.
REMARQUE : Veillez à ne pas toucher ou endommager la plaque du rhéomètre lors de l’utilisation du scalpel. - Réglez l’espacement des plaques parallèles à 1 mm. Coupez tout excès de gel qui n’est pas recouvert par les plaques du rhéomètre.
- Exécutez les paramètres de test souhaités sur le rhéomètre, conformément aux instructions du fabricant.
REMARQUE : Pour chaque nouvelle condition d’échantillon, il est conseillé d’effectuer un balayage d’amplitude de 0,1 à 100 % de déformation pour s’assurer que tous les tests sont effectués à un niveau de déformation dans la région viscoélastique linéaire de l’échantillon.
8. Coloration vivante/morte des cellules encapsulées
- Retirez le milieu des puits et lavez les gels peptidiques deux fois avec 1x PBS, en utilisant la même technique que pour un changement de milieu (étape 5.7).
- Retirez les gels peptidiques qui ont été cultivés dans des inserts de plaque à 24 puits en suivant l’étape 7.2. Conservez les gels dans 1x PBS dans la plaque de puits d’origine jusqu’au moment de teindre.
REMARQUE : Faites attention car les gels peuvent être fragiles à ce stade, surtout après une culture prolongée. - Préparez une solution de coloration vivante/morte, en prévoyant une coloration de 500 μL par insert de plaque à 24 puits, ou de 50 μL par puits d’une plaque à 96 puits. Une coloration typique est un homodimère d’éthidium de 4 μM et une calcéine AM de 2 μM dans 1x PBS. Protégez la solution résultante de la lumière.
REMARQUE : Les concentrations de réactifs vivants/morts peuvent nécessiter une optimisation supplémentaire en fonction du type de cellule utilisé et du fournisseur de réactifs. - Retirez soigneusement le PBS de chaque gel et remplacez-le par quelques gouttes de la solution de coloration, en vous assurant que chaque gel est bien couvert.
- Incuber les gels dans la solution de coloration dans l’obscurité pendant 10 à 15 min, puis visualiser à l’aide d’un microscope confocal/fluorescent.
REMARQUE : Pour des images de meilleure qualité, il peut être avantageux de transférer les gels dans des boîtes à fond de verre de l’épaisseur d’une lamelle.
9. Fixation de gels peptidiques pour l’imagerie finale
- Lavez les gels peptidiques en suivant l’étape 8.1.
- Ajouter 4 % de paraformaldéhyde (PFA) dans 1 PBS : 100 μL pour chaque puits d’une plaque de 96 puits et 1 mL pour chaque gel dans un insert de plaque de 24 puits (quelques gouttes doivent être ajoutées sur le gel à l’intérieur de l’insert).
ATTENTION : Le paraformaldéhyde (PFA) est très toxique et est facilement absorbé par la peau. Il est extrêmement destructeur pour la peau, les yeux, les muqueuses et les voies respiratoires supérieures. Le PFA doit être manipulé dans une hotte et les utilisateurs doivent porter des vêtements de protection et des gants. D’autres fixateurs chimiques peuvent être utilisés de la même manière en fonction de l’application finale. - Incuber des gels peptidiques dans un fixateur PFA pendant 1 h à température ambiante.
- Retirez le fixateur PFA et lavez les gels peptidiques deux fois avec 1x PBS.
REMARQUE : Le protocole peut être interrompu ici, stockez les gels peptidiques fixes à 4 °C dans 1x PBS jusqu’à 4 semaines, en vous assurant que la plaque est bien scellée avec un film de paraffine.
10. Intégration de gels peptidiques pour le sectionnement
REMARQUE : L’intégration de gels peptidiques dans une gélose à 4 % est une étape cruciale avant l’intégration de la paraffine pour l’immunohistochimie. Alternativement, les gels peuvent être incorporés dans de la gélose à 2 % et sectionnés à l’aide d’un vibratome (généralement des sections de 500 μm donnent de bons résultats). Il s’agit d’une étape facultative, produisant des sections de gel hydratées qui peuvent être bénéfiques pour la coloration de la localisation de la matrice extracellulaire dans le gel, en utilisant les méthodes de la section 11.
- Préparez une solution de gélose fondue à 2 % ou 4 % dans 1x PBS (voir note ci-dessus), en la faisant bouillir au micro-ondes. Laisser refroidir quelques minutes avant utilisation.
REMARQUE : La gélose fondue présente un risque de chaleur - manipulez-la avec précaution à l’aide d’une protection pour les mains et le visage. Une fois préparée, la solution de gélose peut être conservée à 4 °C jusqu’à ce que vous en ayez besoin. - Retirez le gel peptidique de l’insert de culture cellulaire en suivant l’étape 7.2.
- À l’aide d’une pipette Pasteur en plastique, recouvrez la base d’un moule d’enrobage histologique d’une fine couche de solution de gélose. Laisser refroidir à 20 °C pendant quelques secondes.
- À l’aide d’une spatule, placez le gel peptidique au centre de l’agar. Ensuite, recouvrez complètement le gel peptidique d’agar.
REMARQUE : Le gel ne doit pas s’enfoncer dans l’agar. Si c’est le cas, retirez le gel et attendez quelques secondes de plus jusqu’à ce que la gélose se soit solidifiée davantage et réessayez. Essayez de ne pas laisser trop de solidification se produire ou il y aura une faible jonction entre les deux couches. - Laisser refroidir le gel noyé pendant 1 h à 4 °C avant de le démouler histologiquement.
REMARQUE : Le protocole peut être mis en pause ici, stocker les gels intégrés à 4 °C dans 1x PBS jusqu’à 4 semaines. - Si l’immunohistochimie doit être effectuée, placez des gels intégrés dans un processeur de tissus et procédez en utilisant des méthodes de laboratoire standard.
REMARQUE : Alternativement, les gels intégrés peuvent être coupés en sections hydratées à l’aide d’un vibratome. Les sections hydratées doivent être stockées dans des plaques scellées à 4 °C dans 1x PBS pendant 4 semaines maximum.
11. Coloration des cellules dans des gels à l’aide de l’immunocytochimie
- Retirez le 1x PBS couvrant les gels peptidiques/sections de gel. Retirez tous les gels encore dans les inserts de plaque à 24 puits en suivant l’étape 7.2.
- Recouvrez les gels/sections de gel d’un tampon de blocage et incubez pendant 30 min à 20 °C.
REMARQUE : Un tampon bloquant typique est constitué de 0,5 % d’albumine sérique bovine (BSA) dans 1x PBS avec 0,1 % de Triton X-100. Triton X-100 est toxique et provoque de graves lésions oculaires, une irritation de la peau et est très toxique pour la vie aquatique. Les utilisateurs doivent porter des vêtements de protection, des lunettes de protection et des gants. - Préparez les anticorps primaires dans le tampon de blocage à des concentrations de travail optimisées. Prévoyez 200 μL par gel de plaque de 24 puits, 100 μL par section de gel et 50 μL par puits de plaque de 96 puits.
REMARQUE : En règle générale, les concentrations d’anticorps utilisées pour la coloration 3D dans les gels doivent être le double de la concentration utilisée dans la 2D. - Retirez le tampon de blocage et ajoutez la solution d’anticorps aux gels goutte à goutte.
- Sceller la plaque avec un film de paraffine et incuber toute la nuit à 4 °C.
- Retirez la solution d’anticorps et lavez-la deux fois avec un tampon de blocage.
- Ajouter l’anticorps secondaire en suivant les mêmes procédures décrites aux étapes 11.3 et 11.4.
- Incuber dans l’obscurité toute la nuit à 4 °C, ou pendant 3 heures à 20 °C.
- Retirez la solution d’anticorps et lavez deux fois avec 1x PBS.
- Couvrir les échantillons dans une solution DAPI 1:1 000 et incuber à 4 °C dans l’obscurité pendant 1 h.
- Transférez le gel sur une lamelle de verre et imagez par microscopie fluorescente/confocale.
12. Extraction de l’ARN
REMARQUE : Les volumes utilisés dans cette méthode s’appliquent lorsque les gels peptidiques sont ensemencés dans des inserts de plaque à 24 puits. D’autres formats de gel peuvent être utilisés, et les volumes ajustés en conséquence.
- Retirez le milieu des puits et lavez les gels peptidiques deux fois avec 1x PBS, en utilisant la même technique que pour un changement de milieu (étape 5.7).
- Retirez les gels peptidiques qui ont été cultivés dans des inserts de plaque à 24 puits en suivant l’étape 7.2. Placez chaque gel dans un tube à centrifuger séparé de 15 ml.
REMARQUE : Faites attention car les gels peuvent être fragiles à ce stade, surtout après une culture prolongée. - À l’aide d’un P1000, ajoutez 500 μL de trypsine-EDTA (0,25 %) dans chaque tube et pipetez de haut en bas pour mélanger et perturber le gel.
- Incuber les gels dans la trypsine-EDTA à 37 °C pendant 3 à 5 min.
REMARQUE : Les temps d’incubation peuvent nécessiter une optimisation en fonction du type de cellule utilisé. - Ajouter 5 ml de 1x PBS pour diluer la trypsine-EDTA.
- Centrifugeuse à 200 x g pendant 5 min pour les cellules de granulés.
- Retirez le surnageant.
REMARQUE : Faites attention car une couche de gel peut s’être formée entre la pastille cellulaire et le surnageant. - Remettre en suspension la pastille cellulaire dans le tampon de lyse, selon les instructions du fabricant, et procéder en suivant les protocoles standard pour l’extraction de l’ARN.
Résultats
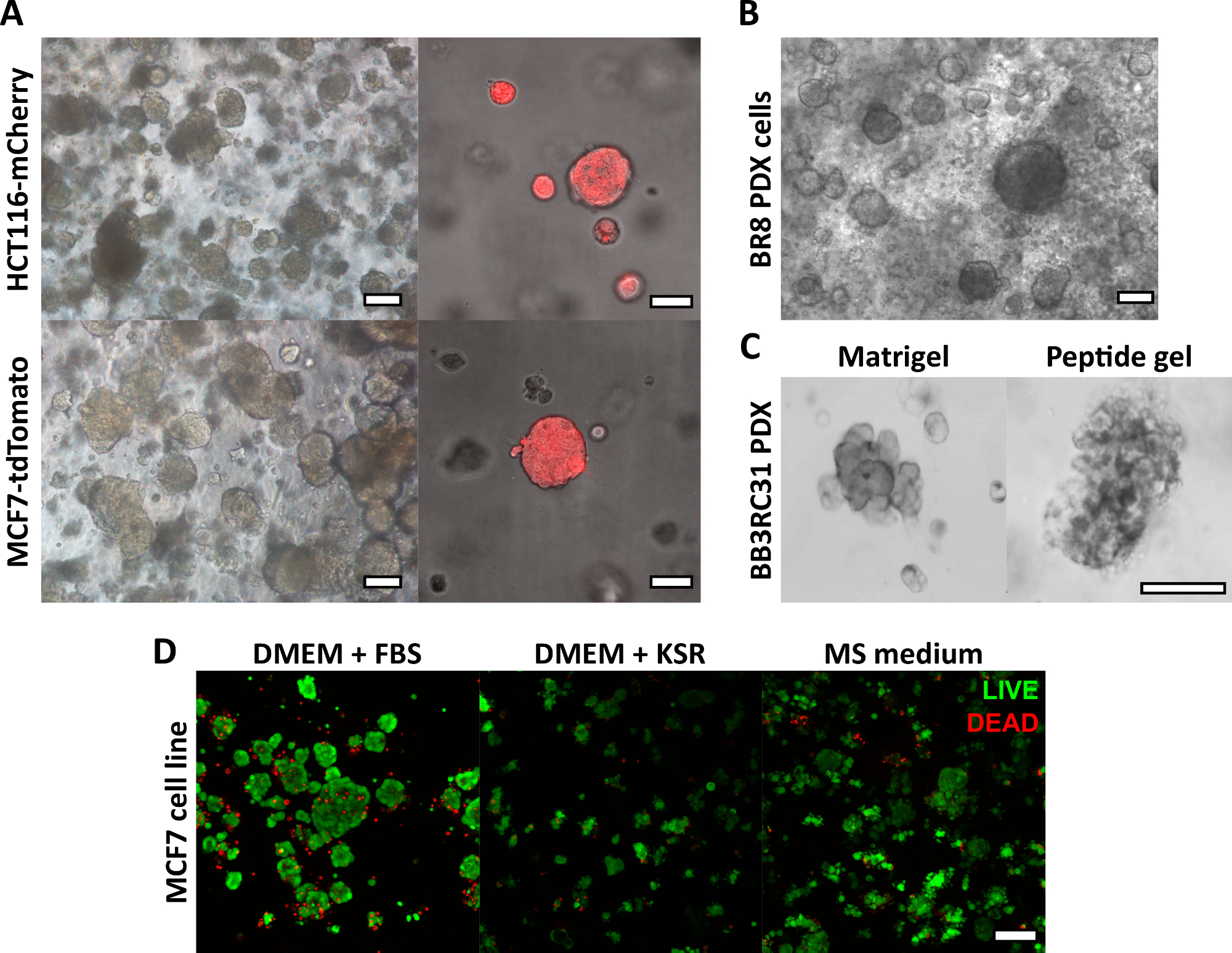
La méthode de fabrication de gel peptidique décrite ici permet à l’utilisateur de définir et de créer un environnement de culture 3D sur mesure. Bien que l’environnement mécanique soit déterminé principalement par la concentration peptidique, des composants matriciels d’intérêt peuvent également être ajoutés à des densités contrôlées, comme le montre l’exemple de calcul de la figure 1. Dans sa forme la plus simple, cependant, le protocole du gel peptidique fournit une méthode pour encapsuler des cellules dans un environnement 3D sans matrice. La figure 2 montre comment cette approche peut être combinée à un large éventail de modèles de cancer, y compris des lignées cellulaires cancéreuses marquées par fluorescence (figure 2A) et du matériel de xénogreffe dérivé du patient (PDX) (figure 2B, C). Il est important de noter que les lignées cellulaires et le matériel PDX peuvent tous deux être cultivés dans les gels dans des conditions sans sérum (Figure 2C,D), fournissant un système de culture 3D avec une composition entièrement définie.
Étant donné que le peptide lui-même ne contient aucun motif de liaison cellulaire, les cellules encapsulées présentent généralement une morphologie arrondie dans les gels peptidiques non modifiés. La figure 3A le démontre pour les fibroblastes mammaires humains dans un gel peptidique de 6 mg/mL, par rapport à leur morphologie allongée classique observée dans le Matrigel pur et un gel de collagène pur. Il est important de noter que le protocole du gel peptidique permet l’incorporation de composants matriciels d’intérêt. La figure 3A montre comment l’ajout de 200 μg/mL de collagène I peut restaurer la morphologie allongée des fibroblastes dans les gels peptidiques.
Les ajouts de matrice peuvent également soutenir la croissance et l’organisation d’autres types de cellules, par exemple MCF10A, comme le montre la figure 3B. Dans ce cas, l’ajout de 100 μg/mL de collagène I à un gel peptidique de 6 mg/mL permet aux structures acineuses de se former au 7e jour. Une complexité supplémentaire peut également être introduite par l’incorporation d’une couche cellulaire de soutien dans la co-culture indirecte. La figure 3C montre comment l’approche combinée de l’incorporation matricielle et de la co-culture indirecte avec des fibroblastes mammaires humains peut améliorer la croissance et l’organisation de MCF10A.
Un autre paramètre important est la concentration de peptide utilisée dans la fabrication de gels peptidiques. La figure 4A montre comment le contrôle de la concentration en peptides, dans ce cas entre 4 et 10 mg/mL, entraîne une rigidité variant entre 100 et 1000 secondes de Pa. Ces gels peuvent être fabriqués sans matrice ou peuvent être créés avec des ajouts de matrice pour permettre un contrôle simultané de la rigidité et de la composition. Les gels peptidiques avec des ajouts matriciels peuvent être sectionnés et colorés pour permettre de visualiser la distribution de ces ajouts. Les figures 4B et C montrent deux approches pour ce faire : l’enrobage dans une gélose à 4 % suivi d’un traitement tissulaire standard et d’un enrobage à la paraffine pour l’immunohistochimie (figure 4B) ou l’enrobage dans une gélose à 2 % suivi d’une section de vibratome et d’une coloration fluorescente (figure 4C).
Lors de la modification de la composition des gels peptidiques, il est crucial de s’assurer que ces changements n’impactent pas l’environnement mécanique initialement présenté aux cellules. La figure 4D montre comment les modifications de la concentration peptidique peuvent être utilisées pour compenser toute modification de la rigidité du gel peptidique lors de l’incorporation de la matrice. Les mesures de rhéologie oscillatoire en vrac de la rigidité du gel (module de stockage, G') permettent alors de distinguer les effets de la composition du gel et de la rigidité sur la morphologie cellulaire. Comme le montrent les images en champ clair, les cellules MDA MB 231 développent une morphologie allongée lors de l’ajout de collagène à des gels peptidiques de 10 mg/mL ou de 15 mg/mL. La figure 4E montre que ces cellules allongées se colorent positivement pour la pFAK, indiquant une interaction avec leur matrice environnante. L’environnement initialement dépourvu de matrice des gels peptidiques en fait également une plate-forme idéale pour l’étude de la synthèse cellulaire et du dépôt des composants matriciels d’intérêt. La figure 4F montre le dépôt localisé de collagène I par des cellules MCF7 encapsulées dans des gels peptidiques de 10 mg/mL.
L’un des principaux avantages des gels peptidiques est la facilité avec laquelle les méthodes de laboratoire standard peuvent être appliquées à leur analyse. Le matériel peut être extrait pour la qRT-PCR afin de déterminer les profils d’expression génique (comme le montre notre récente publication5). L’imagerie par microscopie à fond clair permet en outre de visualiser en temps réel la croissance cellulaire. La figure 5 montre certains des problèmes de dépannage typiques qui peuvent être rencontrés lors de gels peptidiques infructueux : mélange incomplet du précurseur du gel (figure 5A, B) ; une optimisation incorrecte de la concentration peptidique (Figure 5C,D) ou de la densité de semis (Figure 5E,F) ; et neutralisation incorrecte du collagène acide avant incorporation dans les gels peptidiques (Figure 5G,H). La concentration peptidique et la densité d’ensemencement, en particulier, doivent être optimisées pour chaque lignée cellulaire et source peptidique, afin de s’assurer que l’environnement de culture est correctement défini et représentatif de l’application d’intérêt.

Figure 1 : Exemple de calcul de la composition de la matrice et de la densité d’ensemencement. Cet exemple de flux de travail décrit la procédure qui serait suivie pour ensemencer deux précurseurs de gel peptidique avec des ajouts de 100 μg/mL de collagène, à une densité cellulaire finale de 1 x 105 cellules/mL. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 2 : Les gels peptidiques sans matrice fournissent une plate-forme de culture 3D appropriée pour les lignées cellulaires et les modèles de cancer dérivés de patients. (A) les lignées cellulaires du cancer colorectal HCT116 et du cancer du sein MCF7, exprimant constitutivement les marqueurs fluorescents mCherry et tdTomato respectivement, forment des amas de cellules dans des gels de 6 mg/mL au jour 9 (à gauche), et peuvent être imagées en direct à l’aide de la microscopie fluorescente (à droite, barre d’échelle 50 μm) ; (B) Les cellules de xénogreffe dérivée d’une patiente (PDX) d’une patiente atteinte d’un cancer du sein triple négatif (BR8) forment des amas de cellules au jour 7 dans des gels peptidiques de 10 mg/mL ; (C) Les cellules PDX de tumeurs du sein à récepteurs d’œstrogènes positifs (BB3RC31) peuvent être cultivées dans des conditions sans sérum18, montrées avec un contrôle de matrice de membrane basale (par exemple Matrigel) au passage apparié à des fins de comparaison ; (D) Les cellules cancéreuses du sein MCF7 sont viables dans des gels peptidiques de 6 mg/mL dans des conditions sans matrice et sans sérum, comme évalué à l’aide d’un test de cellules LIVE/DEAD au jour 7. KSR = remplacement sérique knock-out, MS medium = mammosphèremedium 19. Barre d’échelle 100 μm sauf indication contraire. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 3 : La complexité du gel peptidique peut être augmentée par l’introduction d’ajouts de matrices et la co-culture. (A) La lignée cellulaire de fibroblastes mammaires humains HMFU19 nécessite des ajouts de collagène pour restaurer une morphologie allongée dans un gel peptidique de 6 mg/mL, montré avec une matrice de membrane basale pure (par exemple Matrigel) et 1,5 mg/mL de gel de collagène de queue de rat I à titre de comparaison, barre d’échelle 50 μm ; (B) Les cellules mammaires normales MCF10A forment des structures acineuses au jour 7 dans des gels peptidiques de 6 mg/mL sur addition de 100 μg/mL de collagène humain I, barre d’échelle de 100 μm ; (C) L’ajout combiné de composants matriciels fibronectine/HA (acide hyaluronique, poids moléculaire 804 kDa) et HMFU19 en co-culture indirecte augmente la taille et l’organisation des acini MCF10A dans les gels peptidiques de 10 mg/mL, tels qu’évalués par coloration à la caspase 3 clivée, barre d’échelle 50 μm. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 4 : Les gels peptidiques permettent un contrôle indépendant de la rigidité et de la composition, ainsi que l’évaluation de la matrice déposée par les cellules. (A) Mesures de rhéologie globale démontrant une plage de rigidité typique (module de stockage, G') réalisable par le contrôle de la concentration peptidique, * indique p < 0,05 ; (B) Immunohistochimie montrant une coloration de 150 μg/mL de collagène I dans un gel peptidique de 10 mg/mL avec MCF7 encapsulé (jour 7, barre d’échelle 100 μm) ; (C) Immunofluorescence de la distribution du collagène I dans un gel peptidique de 6 mg/mL avec 200 μg/mL de collagène humain I, par enrobage de gélose et sectionnement par vibratome, barre d’échelle de 25 μm ; (D) L’ajout de 200 μg/mL de collagène I donne une diminution modeste du module de stockage, G', des gels peptidiques de 10 mg/mL (rhéologie oscillatoire en vrac), compensée par une augmentation de la concentration peptidique à 15 mg/mL. Les cellules cancéreuses du sein triple négatif MDA MB 231 sont représentées dans chaque condition (jour 7, barre d’échelle 50 μm) ; (E) MDA MB 231 dans des gels peptidiques de 15 mg/mL avec 200 μg/mL de collagène humain I montrent un allongement et une interaction avec la matrice via la coloration pFAK (jour 14, barre d’échelle 50 μm) ; (F) Coloration in situ du dépôt de collagène MCF7 I dans un gel peptidique de 10 mg/mL initialement sans matrice (jour 10, barre d’échelle 100 μm). Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 5 : Les problèmes courants de dépannage des gels peptidiques peuvent être résolus à l’aide de la microscopie à fond clair. Les cellules montrées sont des cellules épithéliales mammaires normales MCF10A, au jour 7, sauf indication contraire. (A) Un précurseur de gel correctement mélangé doit être optiquement clair sans incohérences, tandis que (B) un mélange/neutralisation insuffisant peut provoquer des inhomogénéités/stries visibles dans le gel peptidique (flèches blanches) ; (C) MCF10A forme des structures acineuses dans des gels peptidiques de 6 mg/mL lors de l’ajout de co-culture indirecte HMFU19, mais (D) à 15 mg/mL, la concentration peptidique est trop élevée pour permettre la formation d’acineux ; (E) MCF10A ensemencé à 5 x 105 cellules/mL forme des structures acineuses dans des gels de 6 mg/mL sur addition de 100 μg/mL de collagène I, mais (F) à 2 x 105 cellules/mL la densité cellulaire est trop faible pour permettre la formation d’acineux ; (G) Les ajouts de collagène peuvent produire de grands amas de cellules au 14e jour, mais (H) un ajout incorrect (neutralisation du collagène trop tôt dans le processus) peut empêcher la croissance des amas. Barre d’échelle 100 μm. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Discussion
Nous avons constaté que les gels peptidiques décrits ici sont une solution simple, rentable et flexible pour prendre en charge la culture 3D de plusieurs types de cellules. En offrant un contrôle total sur la concentration du peptide utilisé et les ajouts de protéines ou de glycanes effectués, cette méthode permet d’adapter soigneusement les gels peptidiques à leur application.
L’avantage crucial des gels peptidiques par rapport aux méthodes existantes est que la composition de la matrice et les propriétés mécaniques peuvent être contrôlées indépendamment, en utilisant une méthode simple qui ne nécessite aucune procédure chimique complexe. Les propriétés mécaniques du gel peptidique sont principalement déterminées par la concentration peptidique dans le précurseur initial du gel. L’ajout ultérieur de cellules et/ou de composants matriciels permet ensuite de créer un environnement in vitro entièrement défini par l’utilisateur. Bien que les ajouts de matrice puissent modifier les propriétés mécaniques initiales du gel, cela peut facilement être compensé par la variation indépendante de la concentration peptidique5. Cela offre un avantage tangible par rapport aux systèmes existants, par exemple les gels de collagène, dans lesquels les paramètres qui contrôlent la rigidité entraînent également souvent une modification des motifs de liaison de l’intégrine20,21.
Nous avons démontré l’application du gel peptidique pour la culture in vitro de lignées cellulaires cancéreuses et de matériel dérivé de patients5. La plage de rigidité accessible avec le gel peptidique (de l’ordre de 100 à 1000 Pa est parfaitement adaptée pour reproduire des environnements normaux et tumoraux dans les tissus mous tels que le sein. Cependant, nous reconnaissons que d’autres applications nécessitent des environnements considérablement plus rigides, par exemple dans la gamme de 10 à 20 kPa pour la régénération osseuse. D’autres modifications du protocole présenté ici seraient nécessaires pour étendre la rigidité réalisable à cette plage, ce qui est plus typique des approches alternatives telles que les gels d’alginate22. De même, nous avons décrit ici une méthode simple de fonctionnalisation par piégeage physique de protéines/glycanes matriciels dans le gel peptidique. Pour les applications décrites ici, cette approche fonctionne bien et est facilement adaptable à une utilisation par des groupes non spécialistes souhaitant utiliser des modèles 3D in vitro de maladies. Comme beaucoup d’autres hydrogels11, le peptide utilisé ici peut être étendu pour inclure la liaison cellulaire ou d’autres motifs biologiques et, pour certaines applications, cette approche peut être préférable.
Nous avons identifié quelques points clés qui nécessitent une attention particulière pour assurer le succès. La formation du précurseur du gel est une étape intermédiaire critique qui permet à l’utilisateur de vérifier que les conditions utilisées sont correctes avant que les cellules ne soient incorporées. Ce précurseur peut être conservé pendant plusieurs semaines (à 4 °C) mais doit être incubé à 80 °C puis à 37 °C avant d’être utilisé. Un précurseur approprié sera complètement liquide à 80 °C et autoportant à 37 °C. Ces contrôles sont essentiels pour s’assurer que la gélification se déroulera correctement. Les cellules et/ou la matrice peuvent alors être incorporées dans des conditions physiologiques.
Les laboratoires qui utilisent déjà des matrices 3D connaissent la manipulation soigneuse nécessaire pour encapsuler les cellules dans les gels peptidiques. Des précautions doivent être prises pour limiter l’agitation des cellules avant et pendant les étapes d’encapsulation. Nous avons constaté que des types de cellules spécifiques sont différentiellement susceptibles d’être endommagés au cours de ce processus et que cela doit être soigneusement évalué par l’utilisateur. Les concentrations du gel peptidique décrites ici permettent à la gélification de se dérouler dans un laps de temps qui, pour les cellules mentionnées, permet aux cellules d’être encapsulées avant qu’elles ne coulent au fond du puits de coulée, mais suffisamment lentement pour qu’elles ne soient pas endommagées par ce processus. Il convient toutefois de noter que certains types de cellules sensibles peuvent nécessiter une neutralisation plus rapide pour éviter une exposition prolongée à un pH élevé. Dans ce cas, l’ajout de 10 mM de HEPES au milieu entourant le gel peptidique peut être bénéfique.
Lors de l’adoption de la méthode décrite dans ce protocole, il est très important d’examiner attentivement la qualité de la source peptidique. Plutôt que d’être utilisé comme motif fonctionnel ou enrobage, le peptide est ici l’intégralité de la partie non soluble de l’hydrogel. Par conséquent, tout contaminant ou variation dans la structure peptidique est susceptible d’avoir un impact significatif sur l’intégrité ou la capacité à soutenir la viabilité cellulaire dans l’hydrogel final. Lors du passage à un nouveau lot de peptide, il faut veiller à ce qu’il y ait une bonne cohérence d’un lot à l’autre de la part du fournisseur ainsi qu’à vérifier le comportement du peptide lors de la formation du précurseur du gel.
En résumé, ce protocole décrit un système de culture 3D avec un accent crucial sur le contrôle indépendant des propriétés mécaniques et biologiques. La simplicité et l’adaptabilité de la méthode la rendent apte à être adoptée par n’importe quel laboratoire de culture cellulaire et pour un large éventail d’applications5. À l’avenir, ce protocole pourrait être étendu pour permettre la modification covalente de la séquence peptidique. Cela pourrait être combiné à des méthodes de microscopie avancées pour étudier les forces de traction exercées par les cellules sur leur matrice environnante. Cependant, il est essentiel de pouvoir faire la distinction entre la matrice incorporée artificiellement et la matrice synthétisée par les cellules encapsulées elles-mêmes. Cette capacité à contrôler et à surveiller les changements matriciels au fil du temps permettra de mieux comprendre les rôles des interactions cellule-matrice dans le développement du cancer et d’autres maladies.
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Nous tenons à souligner le financement du National Centre for the Replacement, Refinement and Reduction of Animals in Research NC/N0015831/1 à JCA, GF et CLRM, NC/T001267/1 à RBC, CLRM, JCA, KL-S et KS, NC/T001259/1 à JCA, KL-S et CLRM et NC/P002285/1 à AMG, SJ et CLRM. Également financement du Conseil de recherches en ingénierie et en sciences physiques EP/R035563/1 à KL-S et CLRM et EP/N006615/1 à JLT et CLRM. La figure 1 a été réalisée à partir de graphiques adaptés de Servier Medical Art. Servier Medical Art by Servier est sous licence Creative Commons Attribution 3.0 Unported.
matériels
| Name | Company | Catalog Number | Comments |
| Gel fabrication - Reagents | |||
| FEFEFKFK | Pepceuticals | n/a | Polypeptide; available from various suppliers. Pepceuticals is our recommended supplier due to the quality of the product. |
| PBS 10X | Gibco | 70011-036 | |
| Sodium hydroxide (1 M) | Sigma-Aldrich | S2770 | NaOH; dilute to 0.5 M prior to use |
| Water | Sigma-Aldrich | W3500 | |
| Gel fabrication - Equipment and Consumables | |||
| 15 mL falcon tubes | Greiner | 188261 | If using different brand ensure the material withstands temperatures of up to 90°C |
| 24 well plate | Corning Costar | 3524 | Alternative brands/suppliers can be used as long as there is a gap between the insert base and the plate surface |
| Centrifuge | Any | 200 x g for 3 minutes | |
| Class II Microbiological Safety Cabinet | Any | ||
| Fine balance | Any | Readability 0.1 mg | |
| Hanging insert for 24 well plate | Millipore | MCRP24H48 | Alternative brands/suppliers can be used as long as there is a gap between the insert base and the plate surface |
| Incubator | Any | 37°C, 5% CO2, humidified environment | |
| Oven | Any | set to 80°C | |
| P1000/200/20/10 pipette | Any | It is essential the pipettes used for the procedure are calibrated | |
| P1000/200/20/10 tips | Any | ||
| pH meter with microprobe | Any | ||
| Spatula | Any | ||
| Vortex | Any | ||
| Matrix addition | |||
| Collagen I (human) | Stem Cell Technologies | 07005 | |
| Collagen I (rat tail) | Gibco | A10483 | |
| Fibronectin | Stem Cell Technologies | 07159 | |
| Hyaluronic Acid | Iduron | HA804 | |
| Matrigel | Corning | 354234 | |
| Cell encapsulation/culture | |||
| B27 Supplement (no retinoic acid) | Gibco | 12587010 | Media additions for serum free cultures (Figure 2D) |
| Cholera toxin | Sigma-Aldrich | C-8052 | Media additions for MCF10A cells (Figure 3, 5) |
| DMEM | Gibco | 21969-035 | |
| DMEM/F12 | Sigma-Aldrich | D8062 | Media additions for MCF10A cells (Figure 3, 5) |
| DMEM/F12 Phenol Red Free | Gibco | 21041-025 | Media additions for serum free cultures (Figure 2D) |
| DPBS | Gibco | 14190-094 | |
| EGF | SourceBiosciences | ABC016 | Media additions for MCF10A cells (Figure 3, 5) |
| Fetal Bovine Serum | Gibco | 10500-064 | |
| Horse serum | Gibco | 26050-070 | Media additions for MCF10A cells (Figure 3, 5) |
| Human cancer/epithelial cell lines | e.g. MCF7/tdTomato MCF7/MCF10a/HCT116-mCherry | ||
| Human mammary fibroblasts | e.g. HMFU19 | ||
| Hydrocortisone | Sigma-Aldrich | H-0888 | Media additions for MCF10A cells (Figure 3, 5) |
| Insulin | Sigma-Aldrich | I9278 | Media additions for MCF10A cells (Figure 3, 5) |
| Knockout serum replacement | Gibco | 10828-028 | Media additions for serum free cultures (Figure 2D) |
| L-glutamine | Gibco | 25030-024 | |
| RPMI | Gibco | 21875-034 | |
| RPMI Phenol Red Free | Sigma-Aldrich | R7509 | |
| Imaging and other assays | |||
| 4% paraformaldehyde | Polysciences | 18814 | |
| Agar | SLS | CHE1070 | |
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | 5482 | |
| Confocal and/or fluorescent microscope | Any | e.g. Leica TCS SPE confocal laser scanning microscope (Figures 2-4) | |
| DAPI solution | Invitrogen | D3571 | 300 uM working solution |
| DPX mounting medium | ThermoFisher Scientific | ||
| Glass cover slips | Any | No1 coverslips 0.13 - 0.17 mm thickness | |
| Glass-bottom dishes | MatTek | ||
| Goat Anti-Rabbit IgG H&L (HRP polymer) | Abcam | ab214880 | |
| Haematoxylin and Eosin | Any | ||
| Histology molds (disposable, plastic) | Any | ||
| Image analysis software | ImageJ | ||
| Live/Dead assay kit | Invitrogen | L3224 | |
| Microtome | Any | ||
| Phalloidin | Life Technologies | F432/R415 | |
| Pierce Peroxidase IHC Detection Kit | ThermoFisher Scientific | 36000 | |
| Primary Ab Caspase 3 | Abcam | ab34710 | Shown in Figure 3C |
| Primary Ab Collagen I | Cell Signalling Technology | 9661 | Shown in Figure 4B, C, F |
| Primary Ab pFAK Tyr 397 | ThermoFisher Scientific | 44-624G | Shown in Figure 4E |
| Prolong gold/diamond anti-fade mountant with DAPI | Molecular Probes | S36939 | |
| Rheometer Physica MCR 301 | Anton Paar | ||
| Scalpel | Any | ||
| Secondary antibody Goat anti Rabbit AF488 | nvitrogen | a11034 | |
| Secondary antibody Goat anti Rabbit AF546 | Invitrogen | a11010 | |
| SuperFrost slides | ThermoFisher Scientific | Coating e.g. APES can help to retain microtome sections on slides. | |
| Triton X 100 | Sigma-Aldrich | X100 | |
| Trypsin-EDTA (0.25%) | Gibco | 25300054 | |
| Vibratome | Leica |
Références
- Hynes, R. The extracellular matrix: not just pretty fibrils. Science. 326 (5957), 1216-1219 (2009).
- Tian, C., et al. Cancer-cell-derived matrisome proteins promote metastasis in pancreatic ductal adenocarcinoma. Cancer Research. 80 (7), 1461-1474 (2020).
- Hebert, J. D., et al. Proteomic profiling of the ECM of xenograft breast cancer metastases in different organs reveals distinct metastatic niches. Cancer Research. 80 (7), 1475-1485 (2020).
- Vennin, C., et al. CAF hierarchy driven by pancreatic cancer cell p53-status creates a pro-metastatic and chemoresistant environment via perlecan. Nature Communication. 10 (1), 3637 (2019).
- Ashworth, J. C., et al. Peptide gels of fully-defined composition and mechanics for probing cell-cell and cell-matrix interactions in vitro. Matrix Biology. 85, 15-33 (2020).
- Toniatti, C., Jones, P., Graham, H., Pagliara, B., Draetta, G. Oncology drug discovery: Planning a turnaround. Cancer Discovery. 4 (4), 397-404 (2014).
- Mak, I. W., Evaniew, N., Ghert, M. Lost in translation: animal models and clinical trials in cancer treatment. American Journal of Translational Research. 6 (2), 114-118 (2014).
- Aisenbrey, E. A., Murphy, W. L. Synthetic alternatives to Matrigel. Nature Reviews Materials. 5, 539-551 (2020).
- Onion, D., et al. 3-Dimensional patient-derived lung cancer assays reveal resistance to standards-of-care promoted by stromal cells but sensitivity to histone deacetylase inhibitors. Molecular Cancer Therapy. 15 (4), 753-763 (2016).
- Saunders, J. H., et al. Individual patient oesophageal cancer 3D models for tailored treatment. Oncotarget. 8 (15), 24224-24236 (2017).
- Caliari, S. R., Burdick, J. A. A practical guide to hydrogels for cell culture. Nature Methods. 13 (5), 405-414 (2016).
- Kühn, S., et al. Cell-instructive multiphasic gel-in-gel materials. Advanced Functional Materials. 30, 1908857 (2020).
- Gjorevski, N., et al. Designer matrices for intestinal stem cell and organoid culture. Nature. 539 (7630), 560-564 (2016).
- Gjorevski, N., Lutolf, M. P. Synthesis and characterization of well- defined hydrogel matrices and their application to intestinal stem cell and organoid culture. Nature Protocols. 12 (11), 2263-2274 (2017).
- Saiani, A., et al. Self assembly and gelation properties of α-helix versus β-sheet forming peptides. Soft Matter. 5 (1), 193-202 (2008).
- Wan, S., et al. Self-assembling peptide hydrogel for intervertebral disc tissue engineering. Acta Biomaterialia. 46, 29-40 (2016).
- Blache, U., Stevens, M. M., Gentleman, E. Harnessing the secreted extracellular matrix to engineer tissues. Nature Biomedical Engineering. 4 (4), 357-363 (2020).
- Sachs, N., et al. A living biobank of breast cancer organoids captures disease heterogeneity. Cell. 172 (1-2), 373-386 (2018).
- Shaw, F. L., et al. A detailed mammosphere assay protocol for the quantification of breast stem cell activity. Journal of Mammary Gland Biology and Neoplasia. 17 (2), 111-117 (2012).
- Barcus, C. E., Keely, P. J., Eliceiri, K. W., Schule, L. A. Stiff collagen matrices increase tumorigenic prolactin signaling in breast cancer cells. Journal of Biological Chemistry. 288, 12722-12732 (2013).
- Bax, D. V., et al. Impact of UV- and carbodiimide-based crosslinking on the Integrin-binding properties of collagen-based materials. Acta Biomaterialia. 100, 280 (2019).
- Huang, B. P., et al. Multi-peptide presentation and hydrogel mechanics jointly enhance therapeutic duo-potential of entrapped stromal cells. Biomaterials. 245, 119973 (2020).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.