
Method Article
Vorbereitung eines benutzerdefinierten Peptidgels für kontrollierte 3D-Kulturmodelle von Krebs und Krankheit
In diesem Artikel
Zusammenfassung
Wir stellen eine Methode zur Schaffung einer 3D-Zellkulturumgebung vor, mit der die Bedeutung von Zell-Matrix-Interaktionen bei der Krebsprogression untersucht werden kann. Mit einem einfachen, selbstorganisierenden Octapeptid kann die Matrix, die verkapselte Zellen umgibt, gesteuert werden, mit unabhängiger Regulierung mechanischer und biochemischer Signale.
Zusammenfassung
Es gibt ein wachsendes Bewusstsein dafür, dass Zellen, die in 3D gezüchtet wurden, das Verhalten in vivo besser modellieren als solche, die in 2D gezüchtet wurden. In diesem Protokoll beschreiben wir ein einfaches und abstimmbares 3D-Hydrogel, das für die Kultivierung von Zellen und Gewebe in einer Umgebung geeignet ist, die ihrer natürlichen Umgebung entspricht. Dies ist besonders wichtig für Forscher, die die Entstehung, das Wachstum und die Behandlung von Krebs untersuchen, bei denen die Interaktion zwischen Zellen und ihrer lokalen extrazellulären Matrix ein grundlegender Bestandteil des Modells ist. Die Umstellung auf 3D-Kultur kann eine Herausforderung darstellen und ist oft mit einem Mangel an Reproduzierbarkeit verbunden, da die Variation von Charge zu Charge in tierischen 3D-Kulturmatrizen hoch ist. In ähnlicher Weise können Handhabungsprobleme die Nützlichkeit synthetischer Hydrogele einschränken. Als Reaktion auf diesen Bedarf haben wir ein einfaches, selbstorganisierendes Peptidgel optimiert, um die Kultivierung relevanter Zelllinienmodelle für Krebs und Krankheiten sowie von patientengewonnenen Geweben/Zellen zu ermöglichen. Das Gel selbst ist frei von Matrixbestandteilen, abgesehen von denen, die während der Verkapselung hinzugefügt oder von den verkapselten Zellen in das Gel eingebracht werden. Die mechanischen Eigenschaften der Hydrogele können auch unabhängig von der Matrixzugabe verändert werden. Es fungiert daher als "unbeschriebenes Blatt", das es den Forschern ermöglicht, eine 3D-Kulturumgebung zu erstellen, die das interessierende Zielgewebe widerspiegelt und die Einflüsse mechanischer Kräfte und/oder biochemischer Kontrolle des Zellverhaltens unabhängig voneinander zu analysieren.
Einleitung
Die vielen Rollen, die die extrazelluläre Umgebung bei der Entstehung und dem Fortschreiten von Krebs spielt, werden immer deutlicher1. In jüngster Zeit haben detaillierte proteombasierte Analysen eine bereits überzeugende Literaturbasis ergänzt und gezeigt, dass Matrixkomponenten, die von krebsassoziierten Stromazellen abgeleitet werden, oder die Krebszellen selbst, Schlüsselfaktoren für Ereignisse wie die Förderung des epithelialen mesenchymalen Übergangs und der Metastasierung sind 2,3,4. Angesichts dieser anerkannten Bedeutung der extrazellulären Matrix (EZM) wird es immer wichtiger, sich in Richtung Zellkulturplattformen zu bewegen, die die Kontrolle über die 3D-Umgebung ermöglichen, die den Zellen präsentiert wird. Als Antwort auf diesen Bedarf stellt dieses Protokoll eine Methode zur Zellverkapselung und -kultur in einem 3D-Hydrogel mit benutzerdefinierter ECM-Zusammensetzung und mechanischen Eigenschaftenvor 5.
Derzeit besteht eine geringe Korrelation zwischen der Wirksamkeit von Krebstherapeutika in 2D-In-vitro-Kulturen, der Wirkung dieser Therapeutika in aktuellen In-vivo-Modellen (patientenabgeleitetes Xenotransplantat, PDX) und ihrer letztendlichen Aktivität in klinischen Studien 6,7. Dies hat zu erheblichen Misserfolgen in der Pipeline der Wirkstoffforschung geführt, mit einem dringenden Bedarf an verbesserten In-vitro-Modellen, die es ermöglichen, dass getestete Therapeutika "früh versagen, billig scheitern". Viele Forscher verwenden das aus Maus-Mastozytomen gewonnene Produkt, z. B. Matrigel (oder ähnliche Produkte), um 3D-matrixreiche Umgebungen für das Wachstum und die Beobachtung des Zellverhaltens in vitro zu schaffen, einschließlich PDX-abgeleiteter und anderer patientennaher Zellen 8,9,10. Dieser "one size fits all"-Ansatz vernachlässigt jedoch die komplexe Rolle, die Matrixproteine/Glykane bei der Entstehung und Progression von Krebs spielen.
Die Anerkennung der Rolle der extrazellulären Matrix (EZM) bei der Kontrolle des Zellverhaltens hat auch die Verwendung von 3D-Kulturen in oder auf Hydrogelen gefördert, die aus spezifischen Matrixkomponenten bestehen11. Dies ist zwar nützlich, um spezifische Wechselwirkungen zu untersuchen, aber diese Systeme leiden unter der Unfähigkeit, mechanische und biochemische Anweisungen zwischen Zellen und Matrix zu unterscheiden. Sie können auch schwierig zu handhaben sein und unklare Auslesungen des Zellverhaltens liefern. Kollagengele sind ein Schlüsselbeispiel für dieses Problem, da die zellvermittelte Gelkontraktion die Fähigkeit, Zellen innerhalb des Gels sichtbar zu machen, drastisch reduzierenkann 5. Es gibt auch einige sehr elegante, mehrkomponentige Gelsysteme, die Experten mit großem Erfolg eingesetzt haben 12,13,14. Diese können enzymsensitive Linker und bioaktive Motive enthalten, sind aber in ihrer Formulierung und Anwendung deutlich komplexer als das hier beschriebene System.
Dieses Protokoll beschreibt ein Verfahren zur Erstellung vollständig definierter 3D-Kulturmodelle, mit dem die Rolle der EZM in Entwicklung und Krankheit in vitro modelliert werden kann. Die Basis des 3D-Modells ist ein Peptidgel, das wir zuvor als Optimierung eines einfachen selbstorganisierenden Octapeptid-Hydrogels 5,15,16 beschrieben haben. Durch die Abkehr von komplexen, tierischen Matrizen bietet dieses System einen erheblichen Vorteil in Form einer verbesserten Konsistenz von Charge zu Charge und einer verbesserten Handhabung. In seinem einfachen Zustand enthält das Peptid keine von der Matrix abgeleiteten Motive und bietet effektiv ein "unbeschriebenes Blatt", auf dem der Benutzer Funktionalität aufbauen kann.
Wir zeigen, wie die mechanischen Eigenschaften des Peptidgels unabhängig voneinander reguliert werden können, neben dem Einbau von Matrixproteinen/Glykanen. Das System ist hochgradig abstimmbar und ermöglicht die Verkapselung einer Reihe von Zelltypen in verschiedenen Formaten. Wichtig für den Aufbau eines Krebsmodells ist, dass Stromazellen auch eingebaut werden können: entweder in direkter Co-Kultur oder getrennt, um eine spezifische Analyse der indirekten Wechselwirkungen zwischen Krebszellen und Stroma zu ermöglichen. Am wichtigsten ist, dass das hier beschriebene Protokoll keine komplexen Kenntnisse der Chemie erfordert und in jedem Zellkulturlabor reproduziert werden kann, ohne dass spezielle chemische Kenntnisse oder Geräte erforderlich sind.
Wir haben die Methoden für die Untersuchung des Zellverhaltens in den Peptidgelen optimiert, einschließlich Bildgebung, rheologischer Analyse, Extraktion von Material für PCR5 und Einbettung für die histologische Beurteilung. Ein klarer Vorteil des einfachen Hydrogel-Systems ist die Möglichkeit, die von verkapselten Zellen abgelagerte Matrix zu visualisieren und zu untersuchen. Die Bedeutung zellbezogener Matrizen und die Vorteile eines besseren Verständnisses der Art und Weise, wie Zellen ihre lokale Mikroumgebung umgestalten, wurden kürzlich hervorgehoben17 und spiegeln ein wachsendes Bewusstsein für die Bedeutung des Einfangens von zellsekretierten Matrixkomponenten wider, ähnlich wie dies in vivo der Fall ist. Die Möglichkeit, solche Prozesse zu modellieren, könnte einer der grundlegenden Treiber für die verbesserte Patientenrelevanz von Hydrogel-basierten Krankheitsmodellen sein.
Protokoll
1. Auflösung des Peptids
- Geben Sie in einer Gewebekulturhaube 800 μl steriles Wasser mit einer P1000-Pipette in ein 15-ml-Röhrchen.
- Wiegen Sie das Peptidpulver mit einer feinen Waage in ein nicht-statisches Waagschiffchen. Verwenden Sie eine Masse (in mg) von 1,25x der gewünschten endgültigen Peptidgelkonzentration (in mg/ml, Tabelle 1).
HINWEIS: Das hier beschriebene Verfahren erzeugt ein Volumen von ca. 1,25 ml Peptidgel pro Röhrchen zum Zeitpunkt der endgültigen Gelierung. Es können mehrere Röhrchen gleichzeitig vorbereitet werden, oder alternativ kann das Volumen pro Röhrchen erhöht werden, wenn der Benutzer mit dem vorgestellten Verfahren vertraut ist.
| Peptidkonzentration (nach abschließender Gelierung) | Masse des Peptids | Anfängliche NaOH-Zugabe |
| 6 mg/ml | 7,5 mg | 30 μL |
| 10 mg/ml | 12,5 mg | 60 μL |
| 15 mg/ml | 18,75 mg | 100 μL |
Tabelle 1: Peptidmasse und empfohlene anfängliche NaOH-Zugabe für typische endgültige Gelkonzentrationen. Die aufgeführten Bereiche der Peptidkonzentrationen können in beide Richtungen erweitert werden, es ist jedoch wahrscheinlicher, dass niedrigere Peptidkonzentrationen keine stabilen Gele bilden, während bei hohen Konzentrationen das resultierende Gel zu dicht sein kann, um einen ausreichenden Nährstoffaustausch und die Lebensfähigkeit der Zellen zu ermöglichen. Die geeignete Konzentration erfordert eine Optimierung für verschiedene Zelltypen und Peptidchargen.
- Geben Sie das gewogene Peptid in das 15-ml-Röhrchen. Bewegen Sie das Wägeschiffchen, um sicherzustellen, dass kein Pulver zurückbleibt.
HINWEIS: Das Peptidpulver kann sehr statisch sein, achten Sie darauf, den Pulververlust zu minimieren. Es ist nicht erforderlich, während der Schritte 1.2 und 1.3 (oder für pH-Messungen) sterile Bedingungen aufrechtzuerhalten, da die Sterilisation während der Inkubation bei 80 °C zu einem späteren Zeitpunkt des Prozesses erfolgt. - 3 min vortexen, dann 3 min bei 200 x g zentrifugieren.
- Inkubieren Sie die Peptidlösung in einem Ofen bei 80 °C für mindestens 2 h. Wenn nach der Inkubation ein ungelöstes Peptid vorhanden ist, wiederholen Sie Schritt 1.4.
HINWEIS: Da eine gleichmäßige Erwärmung unerlässlich ist, ist ein Heizblock für diesen Schritt nicht geeignet. Wir haben jedoch festgestellt, dass Hybridisierungsöfen gut funktionieren, ebenso wie ein Wasserbad, solange die Peptidlösung vollständig untergetaucht ist.
2. Bildung von Gelvorläufern
- Bereiten Sie eine sterile 0,5 M Natriumhydroxid (NaOH)-Lösung mit sterilem Wasser vor.
HINWEIS: Für möglichst konsistente Ergebnisse wird frisch verdünnte NaOH-Lösung empfohlen. - In einer Gewebekulturhaube 0,5 M NaOH in die Mitte des gelösten Peptids geben und durch langsames Rühren mit der Pipettenspitze mischen (Tabelle 1).
- Das Röhrchen 10 s lang vortexen und 10 s bei 200 x g zentrifugieren, um Blasen zu entfernen.
- Wenn der Gelvorläufer trüb ist, wiederholen Sie die Schritte 2.2 und 2.3 und fügen Sie 5 μl NaOH hinzu.
HINWEIS: Die pH-Messung kann helfen festzustellen, ob ausreichend NaOH zugesetzt wurde: Der optimale pH-Wert liegt zwischen 9 und 10,5 und es ist vorzuziehen, diesen nicht zu überschreiten, da die Verwendung von Säuren, um den pH-Wert wieder zu senken, zu einer Inhomogenität des Gels führen kann. Das genaue benötigte NaOH-Volumen kann je nach Peptidquelle variieren. - Sobald der Gelvorläufer auf der Inversion des 15-ml-Röhrchens optisch klar und selbsttragend (oder nur noch fließend) ist, geben Sie 100 μl steriles 10x PBS in eine Gewebekulturhaube. 10 s vortexen und 10 s bei 200 x g zentrifugieren.
HINWEIS: Wenn der Gelvorläufer trüb ist, wiederholen Sie Schritt 2.4, bis er klar und halbfest ist. Wenn der Gelvorläufer flüssig ist (über pH ~10,5), bildet er kein stabiles Gel und sollte verworfen werden. - Über Nacht im Ofen bei 80 °C inkubieren.
- Überprüfen Sie den Gelvorläufer visuell, um sicherzustellen, dass er bei 80 °C vollständig flüssig ist.
- Wenn der Gelvorläufer bei 80 °C nicht vollständig flüssig ist und nicht ausreichend neutralisiert wurde, ist NaOH gemäß Schritt 2.4 hinzuzufügen.
- Wenn der Gelvorläufer flüssig ist, aber Luftblasen oder kleine Ausfällungen vorhanden sind, schnippen Sie die Tube scharf, um sie zu verteilen. Wenn nach dem Schnippen des Röhrchens Luftblasen oder Ausfällungen bestehen bleiben, den Gelvorläufer vortexen und bei 200 x g für jeweils 10 s zentrifugieren.
- Nachdem Sie einen der Schritte 2.7.1 oder 2.7.2 befolgt haben, inkubieren Sie die Vorläufergele für weitere 2 Stunden bei 80 °C, bevor Sie mit der endgültigen Gelierung fortfahren.
- Bewahren Sie die Gelvorläufer bis zur Verwendung (maximal 48 h) bei 80 °C auf.
HINWEIS: Das Protokoll kann hier pausiert werden, lagern Sie die Gelvorläufer bis zu 4 Wochen bei 4 °C. Wenn Sie von diesem Pausenpunkt aus neu starten, inkubieren Sie die Gelvorläufer mindestens 2 Stunden lang bei 80 °C und beginnen Sie mit Schritt 2.7 neu. Es wird dringend empfohlen, die Gelvorläufer rechtzeitig vorzubereiten, bevor sie für die endgültige Gelierung benötigt werden, um sicherzustellen, dass genügend Zeit für die oben genannten Modifikationen zur Verfügung steht, falls erforderlich.
3. Vorbereitung der Matrixkomponenten für die Aussaat
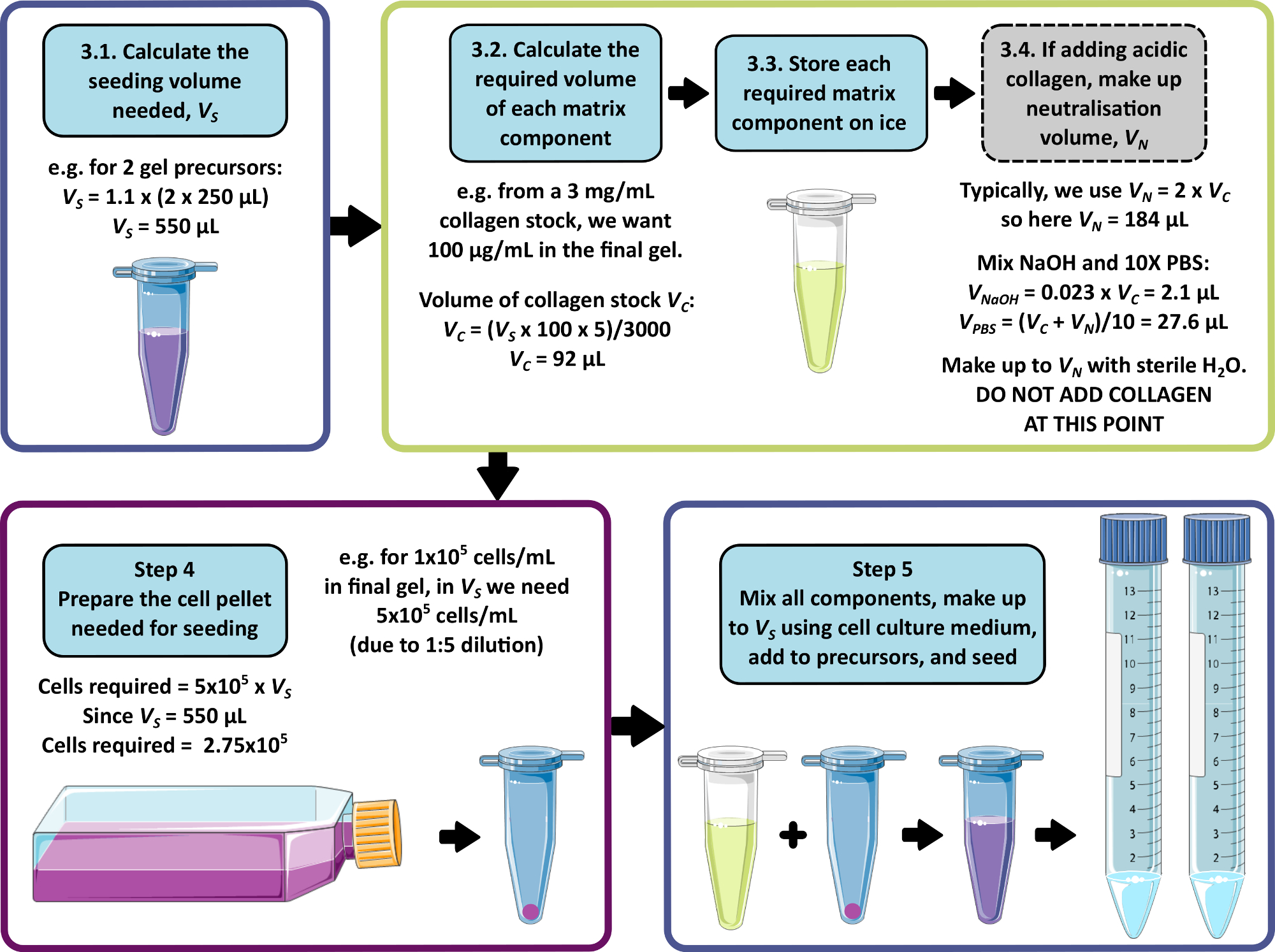
HINWEIS: Eine Beispielrechnung für die Schritte 3 bis 5 ist in Abbildung 1 dargestellt. Schritt 3 und Schritt 4 können weggelassen werden, um ein matrixfreies bzw. ein zellfreies Gel herzustellen.
- Berechnen Sie das kombinierte Gesamtvolumen von Zellen, Matrix und Medien, das für die Aussaat erforderlich ist, wobei 250 μl zu jedem 1 ml-Vorläufergel hinzugefügt werden können. Multiplizieren Sie dieses Volumen mit 1,1, um Pipettierfehler auszuschließen. Dies ist das Aussaatvolumen, VS.
- Berechnen Sie für jede Matrixkomponente das Volumen der Stammlösung, das dem Aussaatvolumen zugesetzt werden soll, indem Sie Gleichung 1 verwenden:
 (1)
(1)
HINWEIS: VS entspricht dem Gesamtvolumen des Aussaatmediums, das für die Aussaat aller Gele erforderlich ist (Schritt 3.1). Stellen Sie sicher, dass die Summe aller Volumina der Matrixkomponenten V S nicht überschreitet. - Tauen Sie alle Matrixkomponenten gründlich auf (siehe Herstellerangaben) und lagern Sie sie bis zur Verwendung auf Eis.
HINWEIS: Einige Matrixkomponenten sind sehr anfällig für Gelierung. Beachten Sie die Anweisungen des Herstellers, um dies zu vermeiden. - Wenn eine saure Kollagen-Stammlösung zugesetzt werden soll, die eine Neutralisation erfordert (siehe Herstellerangaben), bereiten Sie die Neutralisationslösung wie folgt vor:
- Berechnen Sie das erforderliche Volumen der Kollagen-Stammlösung, VC, anhand von Gleichung 1.
- Bestimmen Sie ein geeignetes Volumen für die Neutralisationslösung, VN. Dies sollte so gewählt werden, dass das kombinierte Volumen aller Matrixadditionen das Seeding-Volumen VS nicht überschreitet.
HINWEIS: Im Allgemeinen ein sinnvoller Wert für VN = 2 x Vc, dies kann jedoch entsprechend der Anzahl und des Volumens der anderen hinzuzufügenden Matrixkomponenten angepasst werden. - Berechnen Sie das Volumen von 1 M NaOH, das für die Neutralisation erforderlich ist, unter Verwendung von Gleichung 2.
 (2)
(2) - Berechnen Sie das Volumen von 10x PBS, das in der Neutralisationslösung erforderlich ist , mit Hilfe von Gleichung 3.
 (3)
(3) - Kombinieren Sie die berechneten Volumina von 1 M NaOH und 10x PBS und füllen Sie sie dann mit sterilem Wasser auf VN auf. Gut mischen und bis zur Verwendung auf Eis lagern.
HINWEIS: Fügen Sie das saure Kollagen zu diesem Zeitpunkt nicht hinzu. Ein vorzeitiges Mischen des Kollagens mit der Neutralisationslösung führt zur Einleitung der Kollagenfibrillogenese, was zu Inkonsistenzen in den Eigenschaften des endgültigen Peptidgels führen kann.
4. Vorbereitung der Zellen für die Aussaat
- Falls noch nicht abgeschlossen, berechnen Sie das Aussaatvolumen VS gemäß Schritt 3.1.
- Berechnen Sie die in dieser Zellsuspension erforderliche Zelldichte, indem Sie die gewünschte Zelldichte im endgültigen Peptidgel nehmen und mit 5 multiplizieren.
HINWEIS: Die Zellsuspension hat das 5-fache der gewünschten Endkonzentration, um eine Verdünnung beim Mischen mit dem Gelvorläufer zu berücksichtigen. Die Zelldichten sollten für jede neue Zelllinie, die in Betracht gezogen wird, optimiert werden. Je nach Zelltyp können Dichten zwischen 1 x 104 und 1 x 106 Zellen/ml im endgültigen Peptidgel angemessen sein. - Berechnen Sie die Gesamtzahl der Zellen, die für die Aussaat benötigt werden (multiplizieren Sie die Zelldichte mit dem in Schritt 4.1 berechneten Aussaatvolumen VS ).
- Unter Verwendung von Standardkultur-/Passagemethoden für die verwendeten Zellen ist ein Zellpellet herzustellen, das die erforderliche Zellmenge enthält, wie in Schritt 4.3 berechnet.
5. Endgültige Gelierung/Zellverkapselung
- Wenn Sie bereit sind, mit der endgültigen Gelierung zu beginnen, geben Sie die Gelvorläufer aus dem 80 °C Ofen in ein 37 °C heißes Wasserbad.
HINWEIS: Die Gelvorläufer sollten bei 37 °C selbsttragend sein. Wenn sie flüssig sind, ist es unwahrscheinlich, dass eine vollständige Gelierung auftritt, und die Gelvorläufer sollten verworfen werden. - Bereiten Sie eine 96-Well-Platte oder alternativ eine 24-Well-Platte (oder ähnliches) mit Zellkultureinsätzen vor.
HINWEIS: Stellen Sie sicher, dass zwischen der Basis des Einsatzes und der Well-Platte ein Spalt vorhanden ist, wenn Sie Peptidgele in 24-Well-Einsätzen plattieren. Dadurch wird sichergestellt, dass das Gel mit den Medien in Kontakt kommt. - Wenn eine Matrix hinzugefügt werden soll, kombinieren Sie alle Matrixkomponenten und Neutralisationslösungen (Schritt 3). Mit Zellkulturmedium auf VS auffüllen (Schritt 3.1) und gründlich mischen. Wenn Zellen hinzugefügt werden sollen, wird das in Schritt 4 hergestellte Zellpellet mit dem Aussaatvolumen VS resuspendiert.
HINWEIS: Wenn in Schritt 3 keine Matrixkomponenten hergestellt wurden, verwenden Sie das Zellkulturmedium für das Aussaatvolumen. Dies ist in der Regel das Standardkulturmedium für den verwendeten Zelltyp, obwohl dies möglicherweise validiert werden muss, wenn eine Mischung von Zelltypen verwendet wird. - Geben Sie mit einer P1000-Pipette 250 μl der Zell-/Matrixmischung vorsichtig auf den Gelvorläufer.
HINWEIS: Matrixkomponenten, insbesondere Basalmembranextrakt, können nach Zugabe zum Gelvorläufer zu polymerisieren beginnen. Es ist daher wichtig, so schnell wie möglich zum nächsten Schritt überzugehen. - Sanft mischen durch die kombinierte Wirkung von Pipettieren und Rühren. Das Gel ist scherverdünnend, so dass es durch sanftes Pipettieren/Rühren leichter zu mischen ist. Wenn es gründlich gemischt ist, fügen Sie 100 μl in jede Vertiefung einer 96-Well-Platte oder 200 μl in jeden Zellkultureinsatz hinzu.
HINWEIS: Beim Mischen kann das Rückwärtspipettieren mit einem P1000 mit 200 μl von Vorteil sein, um das Einbringen von Luftblasen zu vermeiden. Das Gel mag anfangs schwer zu mischen sein, aber bei weiterem Mischen sollte es einfacher werden - dies deutet darauf hin, dass das Mischen effizient ist. - 10 min bei 37 °C in 5 % CO2 und befeuchteter Atmosphäre inkubieren.
HINWEIS: Dieser Schritt ist nicht erforderlich, wenn das Peptidgel keine Matrixzusätze enthält. - Geben Sie 200 μl Medium in jede Vertiefung der 96-Well-Platte oder 1 ml auf die Außenseite des Zellkultureinsatzes mit einigen Tropfen auf die Oberseite des Gels. Inkubieren bei 37 °C in 5 % CO2 und befeuchteter Atmosphäre.
- Wechseln Sie das Medium zweimal innerhalb der nächsten Stunde und erneut nach mehreren Stunden (oder am folgenden Tag).
HINWEIS: Seien Sie hier vorsichtig, da die Gele mehrere Stunden lang instabil sind. - Wechseln Sie das Medium alle 2–3 Tage (oder befolgen Sie das Standardkulturprotokoll für die verwendeten Zellen).
6. Indirekte Co-Kultur
HINWEIS: Diese Methode ist nur anwendbar, wenn die Peptidgele in 24-Well-Platteneinsätze oder ähnliche Formate ausgesät werden, in denen das Gel über einer Zellmonoschicht gestützt werden kann. In diesem Fall kann eine indirekte Co-Kultur eingeführt werden, indem eine 2D-Feeder-Schicht aus Zellen auf dem Boden der Well-Platte vorbereitet wird.
- Bereiten Sie eine 24-Well-Platte für die Aussaat vor. Dabei sollte es sich um eine separate Platte zu den Peptidgelen handeln, die jedoch von der gleichen Marke sein sollte, um die Kompatibilität mit den verwendeten Einsätzen zu gewährleisten (siehe Schritt 5.2).
- Berechnen Sie die Zelldichte, die für die Aussaat der indirekten Co-Kultur benötigt wird, entsprechend dem betrachteten Zelltyp.
HINWEIS: Die Zellaussaatdichte sollte für jede neue Zelllinie, die in Betracht gezogen wird, optimiert werden. Die Zellen sollten so plattiert werden, dass sie eine Konfluenz von etwa 30–50 % ergeben. Als Beispiel wird die humane Mamma-Fibroblasten-Zelllinie HMFU19 typischerweise mit einer Dichte von 1-5 x 104 Zellen/Well plattiert. - Unter Verwendung von Standardkultur-/Passagemethoden für die verwendeten Zellen ist eine Zellsuspension herzustellen, die für die Aussaat bei 1 ml/Vertiefung geeignet ist, wobei das für diese Zellen typische Wachstumsmedium verwendet wird.
- Säen Sie die Zellsuspension in die Well-Platte, 1 ml pro Well.
- Bei 37 °C in 5 % CO2 und einer befeuchteten Atmosphäre für mehrere Stunden oder über Nacht inkubieren. Entfernen Sie dann das Medium aus den Vertiefungen.
- Übertragen Sie mit einer sterilen Pinzette die 24-Well-Platteneinsätze mit den Peptidgelen in die neuen Wells mit den vorgesäten Zellen in 2D. Geben Sie 1 ml Medium tropfenweise auf die Außenseite des Einsatzes und einige Tropfen auf die Oberfläche des Gels.
HINWEIS: In der Regel ist das zu diesem Zeitpunkt verwendete Medium dasjenige, das für die im Gel eingekapselten Zellen geeignet ist, aber die Eignung dieses Mediums für die Zellen in 2D erfordert möglicherweise eine Überprüfung oder Optimierung für das jeweilige betrachtete Experiment. - Bereiten Sie regelmäßig frische Feeder-Schichten vor, um eine Überkonfluenz zu vermeiden, und übertragen Sie die Peptidgele nach der gleichen Methode wie oben in diese neuen Wells.
HINWEIS: In der Regel werden Zellen für die indirekte Co-Kultur gleichzeitig mit der Peptid-Gel-Aussaat vorbereitet. Die Peptidgele können dann zum Zeitpunkt des Medienwechsels nach einigen Stunden oder über Nacht in die Co-Kultur überführt werden, siehe Schritt 5.8.
7. Oszillatorische Rheologie von Peptidgelen
HINWEIS: Standardmäßig wird die rheologische Charakterisierung 24 Stunden nach der Gelaussaat durchgeführt, die in 24-Well-Platteneinsätzen erfolgen sollte.
- Richten Sie das Rheometer gemäß den Anweisungen des Herstellers ein und kalibrieren Sie es. Verwenden Sie eine parallele Plattengeometrie mit einem Plattendurchmesser, der so nah wie möglich am Durchmesser des Zellkultureinsatzes liegt.
HINWEIS: Auf Wunsch können die Tests bei 37 °C durchgeführt werden, um die Umgebung während der Kultur zu replizieren. - Entnehmen Sie die erste zu testende Peptidgelprobe aus dem Zellkultureinsatz, indem Sie den Einsatz umdrehen und die Kunststoffmembran mit einem Skalpell herausschneiden.
HINWEIS: Stellen Sie sicher, dass sich die Platte mit den Peptidgelen vor dem Test so kurz wie möglich außerhalb des Zellkulturinkubators befindet. Medien mit einem Bicarbonat-Puffersystem sind auf das Vorhandensein von CO2 angewiesen, um den pH-Wert aufrechtzuerhalten. Gele, die sich zu lange außerhalb des Inkubators befunden haben, driften im pH-Wert, was sich auf die rheologische Beurteilung auswirken kann. Es kann von Vorteil sein, die Gele mit 10 mM HEPES zu pflegen, um diesen Effekt zu verhindern. - Übertragen Sie das Gel vorsichtig auf die Rheometerplatte. Kürzen Sie dann mit einem Skalpell die Höhe des Gels auf ca. 1 mm, um die Verformung des Gels zu minimieren, wenn es unter die Rheometerplatte geladen wird.
HINWEIS: Achten Sie darauf, die Rheometerplatte nicht zu berühren oder zu beschädigen, wenn Sie das Skalpell verwenden. - Stellen Sie den parallelen Plattenabstand auf 1 mm ein. Schneiden Sie überschüssiges Gel ab, das nicht von den Rheometerplatten verdeckt wird.
- Führen Sie die gewünschten Testeinstellungen am Rheometer gemäß den Anweisungen des Herstellers durch.
HINWEIS: Für jede neue Probenbedingung wird empfohlen, einen Amplitudensweep von 0,1 bis 100 % Dehnung durchzuführen, um sicherzustellen, dass alle Tests auf einem Dehnungsniveau innerhalb des linearen viskoelastischen Bereichs der Probe durchgeführt werden.
8. Lebend-/Totfärbung von verkapselten Zellen
- Nehmen Sie das Medium aus den Vertiefungen und waschen Sie die Peptidgele zweimal mit 1x PBS, wobei Sie die gleiche Technik wie bei einem Medienwechsel anwenden (Schritt 5.7).
- Entfernen Sie Peptidgele, die gemäß Schritt 7.2 in 24-Well-Platteneinsätzen kultiviert wurden. Bewahren Sie die Gele in 1x PBS in der Original-Well-Platte auf, bis sie zum Färben bereit sind.
HINWEIS: Seien Sie vorsichtig, da die Gele zu diesem Zeitpunkt zerbrechlich sein können, insbesondere nach längerer Kultur. - Bereiten Sie eine Lebend-/Totfärbelösung vor, die eine Färbung von 500 μl pro 24-Well-Platte oder 50 μl pro Well einer 96-Well-Platte ermöglicht. Eine typische Färbung ist 4 μM Ethidium-Homodimer und 2 μM Calcein AM in 1x PBS. Schützen Sie die resultierende Lösung vor Licht.
HINWEIS: Die Konzentrationen von lebenden/toten Reagenzien können je nach verwendetem Zelltyp und Reagenzienlieferant weitere Optimierungen erfordern. - Entfernen Sie vorsichtig PBS von jedem Gel und ersetzen Sie es durch ein paar Tropfen der Färbelösung, wobei Sie darauf achten müssen, dass jedes Gel gut bedeckt ist.
- Inkubieren Sie die Gele in der Färbelösung im Dunkeln für 10–15 Minuten und visualisieren Sie sie dann mit einem Konfokal-/Fluoreszenzmikroskop.
HINWEIS: Für Bilder mit höherer Qualität kann es von Vorteil sein, die Gele in Glasbodenschalen mit Deckglasstärke zu übertragen.
9. Fixierung von Peptidgelen für die Endpunkt-Bildgebung
- Waschen Sie die Peptidgele nach Schritt 8.1.
- Fügen Sie 4 % Paraformaldehyd (PFA) in 1x PBS hinzu: 100 μl für jede Vertiefung einer 96-Well-Platte und 1 ml für jedes Gel in einer 24-Well-Platteneinlage (einige Tropfen sollten auf das Gel in der Einlage gegeben werden).
ACHTUNG: Paraformaldehyd (PFA) ist hochgiftig und wird leicht über die Haut aufgenommen. Es ist extrem zerstörerisch für Haut, Augen, Schleimhäute und die oberen Atemwege. PFA sollte in einem Abzug gehandhabt werden, und die Benutzer sollten Schutzkleidung und Handschuhe tragen. Alternative chemische Fixiermittel können je nach Endanwendung auf die gleiche Weise verwendet werden. - Inkubieren Sie Peptidgele 1 h lang bei Raumtemperatur in PFA-Fixiermittel.
- Entfernen Sie das PFA-Fixiermittel und waschen Sie die Peptidgele zweimal mit 1x PBS.
HINWEIS: Das Protokoll kann hier pausiert werden, fixierte Peptidgele bei 4 °C in 1x PBS bis zu 4 Wochen lagern, dabei darauf achten, dass die Platte gut mit Paraffinfolie verschlossen ist.
10. Einbetten von Peptidgelen zum Trennen
HINWEIS: Die Einbettung von Peptidgelen in 4% Agar ist ein entscheidender Schritt vor der Paraffineinbettung für die Immunhistochemie. Alternativ können Gele in 2%-Agar eingebettet und mit einem Vibratom geschnitten werden (typischerweise liefern 500-μm-Schnitte gute Ergebnisse). Dies ist ein optionaler Schritt, bei dem hydratisierte Gelabschnitte hergestellt werden, die für die Färbung der extrazellulären Matrixlokalisation im Gel unter Verwendung der Methoden in Abschnitt 11 vorteilhaft sein können.
- Bereiten Sie geschmolzene 2%ige oder 4%ige Agarlösung in 1x PBS (siehe Hinweis oben) zu, indem Sie sie in der Mikrowelle kochen. Vor Gebrauch einige Minuten abkühlen lassen.
HINWEIS: Geschmolzener Agar stellt eine Hitzegefahr dar – vorsichtig mit Hand- und Gesichtsschutz behandeln. Nach der Zubereitung kann die Agarlösung bis zur Verwendung bei 4 °C gelagert werden. - Entfernen Sie das Peptidgel aus dem Zellkultureinsatz nach Schritt 7.2.
- Bedecken Sie mit einer Pasteurpipette aus Kunststoff den Boden einer histologischen Einbettungsform mit einer dünnen Schicht Agarlösung. Einige Sekunden bei 20 °C abkühlen lassen.
- Geben Sie das Peptidgel mit einem Spatel in die Mitte des Agars. Bedecken Sie dann das Peptidgel vollständig mit Agar.
HINWEIS: Das Gel sollte nicht in den Agar einsinken. Wenn dies der Fall ist, entfernen Sie das Gel und warten Sie noch einige Sekunden, bis sich der Agar stärker verfestigt hat, und versuchen Sie es erneut. Versuchen Sie, nicht zu viel Verfestigung stattfinden zu lassen, da es sonst zu einer schwachen Verbindung zwischen den beiden Schichten kommt. - Lassen Sie das eingebettete Gel 1 h bei 4 °C abkühlen, bevor Sie es aus der histologischen Form entfernen.
HINWEIS: Das Protokoll kann hier pausiert werden, eingebettete Gele bei 4 °C in 1x PBS bis zu 4 Wochen lagern. - Wenn eine Immunhistochemie durchgeführt werden soll, geben Sie eingebettete Gele in einen Gewebeprozessor und verfahren Sie mit Standardlabormethoden.
HINWEIS: Alternativ können eingebettete Gele mit einem Vibratom in hydratisierte Abschnitte geschnitten werden. Hydratisierte Schnitte sollten in verschlossenen Platten bei 4 °C in 1x PBS bis zu 4 Wochen gelagert werden.
11. Färbung von Zellen in Gelen mittels Immunzytochemie
- Entfernen Sie das 1x PBS, das die Peptidgele/Gelabschnitte bedeckt. Entfernen Sie alle Gele, die sich noch in 24-Well-Platteneinsätzen befinden, nach Schritt 7.2.
- Die Gele/Gelabschnitte in einen Blockierungspuffer legen und 30 min bei 20 °C inkubieren.
HINWEIS: Ein typischer Blockierungspuffer besteht aus 0,5 % Rinderserumalbumin (BSA) in 1x PBS und 0,1 % Triton X-100. Triton X-100 ist giftig und verursacht schwere Augenschäden, Hautreizungen und ist sehr giftig für Wasserlebewesen. Die Benutzer sollten Schutzkleidung, Augenschutz und Handschuhe tragen. - Bereiten Sie Primärantikörper in Blockpuffer bei optimierten Arbeitskonzentrationen vor. Erlauben Sie 200 μl pro 24-Well-Plattengel, 100 μl pro Gelabschnitt und 50 μl pro 96-Well-Plattenvertiefung.
HINWEIS: In der Regel sollten die Antikörperkonzentrationen, die für die 3D-Färbung in Gelen verwendet werden, doppelt so hoch sein wie die Konzentration, die in 2D verwendet wird. - Entfernen Sie den Blockierungspuffer und fügen Sie den Gelen tropfenweise Antikörperlösung hinzu.
- Die Platte mit Paraffinfolie versiegeln und über Nacht bei 4 °C inkubieren.
- Entfernen Sie die Antikörperlösung und waschen Sie sie zweimal mit Blockierungspuffer.
- Fügen Sie einen Sekundärantikörper hinzu, indem Sie die gleichen Verfahren anwenden, die in den Schritten 11.3 und 11.4 beschrieben wurden.
- Inkubieren Sie im Dunkeln über Nacht bei 4 °C oder 3 Stunden bei 20 °C.
- Antikörperlösung entfernen und zweimal mit 1x PBS waschen.
- Die Proben werden in eine 1:1.000 DAPI-Lösung eingelegt und bei 4 °C im Dunkeln 1 h inkubiert.
- Übertragen Sie das Gel auf ein Deckglas und nehmen Sie es durch Fluoreszenz-/Konfokalmikroskopie auf.
12. RNA-Extraktion
HINWEIS: Die bei dieser Methode verwendeten Volumina sind anwendbar, wenn die Peptidgele in 24-Well-Platteneinsätze ausgesät werden. Es können auch andere Gelformate verwendet und die Volumina entsprechend angepasst werden.
- Nehmen Sie das Medium aus den Vertiefungen und waschen Sie die Peptidgele zweimal mit 1x PBS, wobei Sie die gleiche Technik wie bei einem Medienwechsel anwenden (Schritt 5.7).
- Entfernen Sie Peptidgele, die gemäß Schritt 7.2 in 24-Well-Platteneinsätzen kultiviert wurden. Geben Sie jedes Gel in ein separates 15-ml-Zentrifugenröhrchen.
HINWEIS: Seien Sie vorsichtig, da die Gele zu diesem Zeitpunkt zerbrechlich sein können, insbesondere nach längerer Kultur. - Mit einem P1000 500 μl Trypsin-EDTA (0,25 %) in jedes Röhrchen geben und zum Mischen auf und ab pipettieren und das Gel aufbrechen.
- Inkubieren Sie die Gele im Trypsin-EDTA bei 37 °C für 3-5 min.
HINWEIS: Die Inkubationszeiten können je nach verwendetem Zelltyp optimiert werden müssen. - Fügen Sie 5 ml 1x PBS hinzu, um das Trypsin-EDTA zu verdünnen.
- Zentrifugieren Sie bei 200 x g für 5 min die Pelletzellen.
- Entfernen Sie den Überstand.
HINWEIS: Seien Sie vorsichtig, da sich zwischen dem Zellpellet und dem Überstand eine Gelschicht gebildet haben kann. - Resuspendieren Sie das Zellpellet gemäß den Anweisungen des Herstellers in Lysepuffer und fahren Sie gemäß den Standardprotokollen für die RNA-Extraktion fort.
Ergebnisse
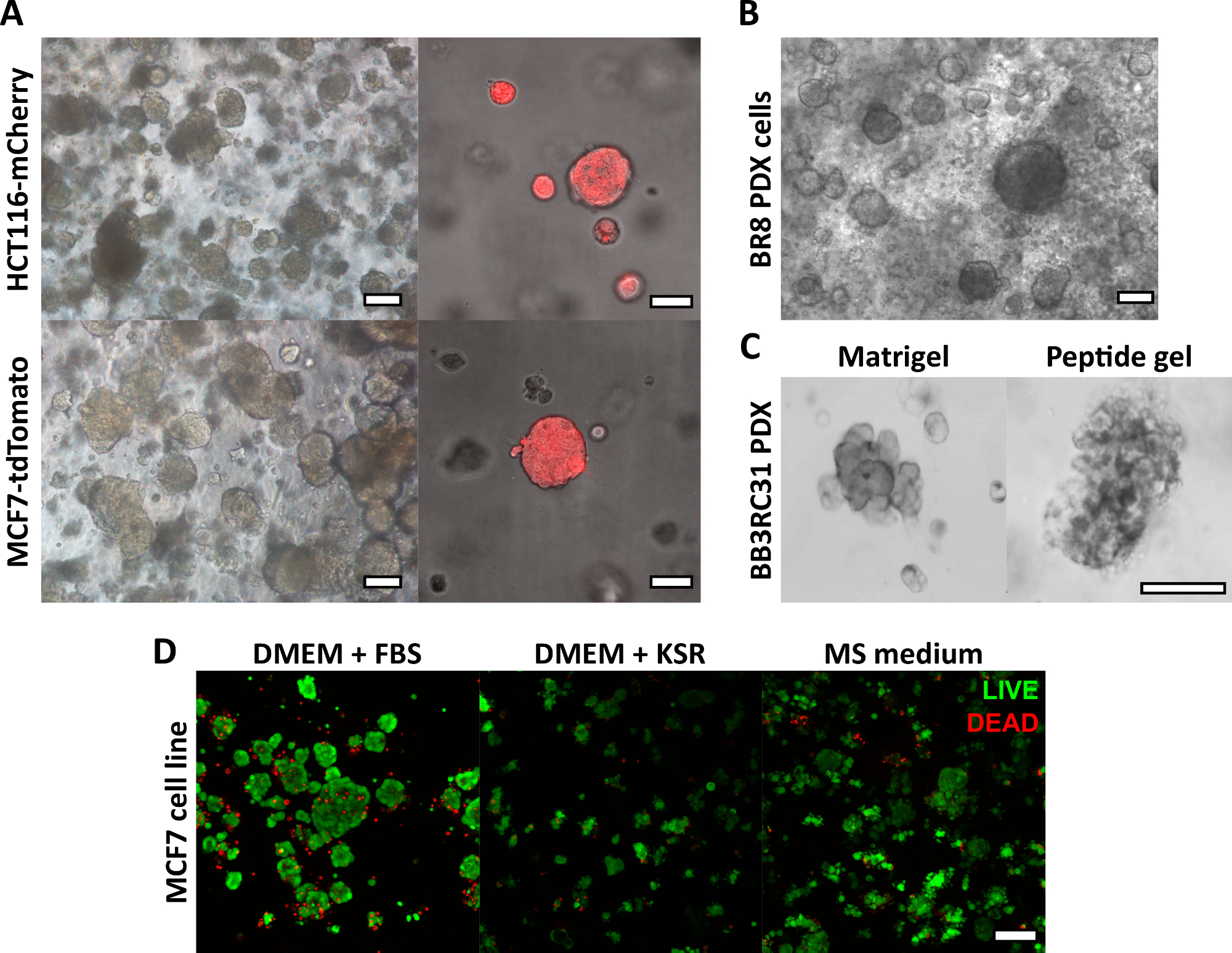
Die hier beschriebene Herstellungsmethode für Peptidgele ermöglicht es dem Benutzer, eine maßgeschneiderte 3D-Kulturumgebung zu definieren und zu erstellen. Während die mechanische Umgebung in erster Linie durch die Peptidkonzentration bestimmt wird, können interessante Matrixkomponenten auch bei kontrollierten Dichten hinzugefügt werden, wie die Beispielrechnung in Abbildung 1 zeigt. In seiner einfachsten Form bietet das Peptid-Gel-Protokoll jedoch eine Methode zur Verkapselung von Zellen in einer matrixfreien 3D-Umgebung. Abbildung 2 zeigt, wie dieser Ansatz mit einer Vielzahl von Krebsmodellen kombiniert werden kann, einschließlich fluoreszenzmarkierter Krebszelllinien (Abbildung 2A) und patientenabgeleitetem Xenotransplantatmaterial (PDX) (Abbildung 2B,C). Wichtig ist, dass sowohl Zelllinien als auch PDX-Material in den Gelen unter serumfreien Bedingungen kultiviert werden können (Abbildung 2C,D), wodurch ein 3D-Kultursystem mit vollständig definierter Zusammensetzung entsteht.
Da das Peptid selbst keine zellbindenden Motive enthält, weisen verkapselte Zellen typischerweise eine abgerundete Morphologie in den unmodifizierten Peptidgelen auf. Abbildung 3A zeigt dies für humane Mamma-Fibroblasten in einem 6 mg/ml-Peptidgel, verglichen mit ihrer klassischen länglichen Morphologie, die in reinem Matrigel und einem reinen Kollagengel zu sehen ist. Wichtig ist jedoch, dass das Peptid-Gel-Protokoll den Einbau von Matrixkomponenten von Interesse ermöglicht. Abbildung 3A zeigt, wie die Zugabe von 200 μg/ml Kollagen I die längliche Fibroblastenmorphologie in den Peptidgelen wiederherstellen kann.
Matrixzusätze können auch das Wachstum und die Organisation anderer Zelltypen unterstützen, z. B. MCF10A, wie in Abbildung 3B gezeigt. In diesem Fall ermöglicht die Zugabe von 100 μg/ml Kollagen I zu einem 6 mg/ml-Peptidgel die Bildung von Azinusstrukturen bis zum 7. Tag. Weitere Komplexität kann auch durch den Einbau einer unterstützenden Zellschicht in indirekter Co-Kultur eingeführt werden. Abbildung 3C zeigt, wie der kombinierte Ansatz von Matrix-Inkorporation und indirekter Co-Kultur mit humanen Mamma-Fibroblasten das Wachstum und die Organisation von MCF10A verbessern kann.
Ein weiterer wichtiger Parameter ist die Konzentration des Peptids, das bei der Herstellung von Peptidgelen verwendet wird. Abbildung 4A zeigt ein Beispiel dafür, wie die Kontrolle der Peptidkonzentration, in diesem Fall zwischen 4 und 10 mg/ml, zu einer Steifigkeit zwischen 100 und 1000 μm Pa führt. Diese Gele können matrixfrei hergestellt oder mit Matrixzusätzen hergestellt werden, um eine gleichzeitige Kontrolle von Steifigkeit und Zusammensetzung zu ermöglichen. Peptidgele mit Matrixzusätzen können geschnitten und gefärbt werden, um die Verteilung dieser Zusätze sichtbar zu machen. Abbildung 4B, C zeigen zwei Ansätze dafür: Einbettung in 4%-Agar, gefolgt von Standardgewebeverarbeitung und Paraffineinbettung für die Immunhistochemie (Abbildung 4B) oder Einbettung in 2%-Agar, gefolgt von Vibratom-Schnitten und Fluoreszenzfärbung (Abbildung 4C).
Bei der Modifikation der Zusammensetzung der Peptidgele ist es entscheidend sicherzustellen, dass diese Veränderungen die mechanische Umgebung, die den Zellen ursprünglich präsentiert wird, nicht beeinträchtigen. Abbildung 4D zeigt, wie Modifikationen der Peptidkonzentration verwendet werden können, um Änderungen der Steifigkeit des Peptidgels beim Matrixeinbau auszugleichen. Oszillatorische rheologische Messungen der Gelsteifigkeit (Speichermodul, G') können dann zwischen den Auswirkungen der Gelzusammensetzung und der Steifigkeit auf die Zellmorphologie unterscheiden. Wie in den Hellfeldbildern gezeigt, entwickeln MDA MB 231-Zellen eine längliche Morphologie bei der Zugabe von Kollagen zu Peptidgelen von 10 mg/ml oder 15 mg/ml. Abbildung 4E zeigt, dass diese länglichen Zellen positiv für pFAK gefärbt sind, was auf eine Wechselwirkung mit ihrer umgebenden Matrix hinweist. Die anfänglich matrixfreie Umgebung der Peptidgele macht sie auch zu einer idealen Plattform für die Untersuchung der zellulären Synthese und Abscheidung von Matrixkomponenten von Interesse. Abbildung 4F zeigt die lokalisierte Ablagerung von Kollagen I durch MCF7-Zellen, die in 10 mg/mL Peptidgelen verkapselt sind.
Einer der Hauptvorteile der Peptidgele ist die Leichtigkeit, mit der Standardlabormethoden auf ihre Analyse angewendet werden können. Material kann für die qRT-PCR extrahiert werden, um Genexpressionsprofile zu bestimmen (wie in unserer jüngsten Veröffentlichung5 gezeigt). Die Bildgebung mittels Hellfeldmikroskopie ermöglicht zusätzlich die Echtzeit-Visualisierung des Zellwachstums. Abbildung 5 zeigt einige der typischen Probleme bei der Fehlerbehebung, die bei erfolglosen Peptidgelen auftreten können: unvollständiges Mischen des Gelvorläufers (Abbildung 5A,B); falsche Optimierung der Peptidkonzentration (Abbildung 5C,D) oder der Seeding-Dichte (Abbildung 5E,F); und falsche Neutralisation von saurem Kollagen vor dem Einbau in die Peptidgele (Abbildung 5G,H). Insbesondere die Peptidkonzentration und die Aussaatdichte müssen für jede Zelllinie und Peptidquelle optimiert werden, um sicherzustellen, dass die Kulturumgebung angemessen definiert und repräsentativ für die betreffende Anwendung ist.

Abbildung 1: Eine Beispielrechnung für die Zusammensetzung der Matrix und die Seeding-Dichte. Dieser Beispiel-Arbeitsablauf beschreibt das Verfahren, das befolgt würde, um zwei Peptidgel-Vorläufer mit Zusätzen von 100 μg/ml Kollagen bei einer endgültigen Zelldichte von 1 x 105 Zellen/ml zu säen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2: Matrixfreie Peptidgele bieten eine geeignete 3D-Kulturplattform für Zelllinien- und patientenabgeleitete Krebsmodelle. (A) HCT116-Darmkrebs- und MCF7-Brustkrebszelllinien, die konstitutiv die Fluoreszenzmarker mCherry bzw. tdTomato exprimieren, bilden bis zum 9. Tag Zellcluster in 6 mg/ml-Gelen (links) und können mit Hilfe der Fluoreszenzmikroskopie live abgebildet werden (rechts, Maßstabsbalken 50 μm); (B) Patientinnen-stammende Xenotransplantatzellen (PDX) von einer dreifach negativen Brustkrebspatientin (BR8) bilden bis zum 7. Tag Zellcluster in 10 mg/ml-Peptidgelen; (C) PDX-Zellen von Östrogenrezeptor-positiven Brusttumoren (BB3RC31) können unter serumfreien Bedingungen18 gezüchtet werden, gezeigt mit einer Basalmembranmatrix-Kontrolle (z. B. Matrigel) an passender Passage zum Vergleich; (D) MCF7-Brustkrebszellen sind in 6 mg/ml-Peptidgelen unter matrix- und serumfreien Bedingungen lebensfähig, wie mit einem LEBEND/TOT-Zell-Assay an Tag 7 beurteilt wurde. KSR = Knockout-Serumersatz, MS medium = Mammosphärenmedium19. Maßstabsleiste 100 μm, sofern nicht anders angegeben. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 3: Die Komplexität des Peptidgels kann durch die Einführung von Matrixzusätzen und Co-Kultur erhöht werden. (A) Die humane Mamma-Fibroblasten-Zelllinie HMFU19 erfordert Kollagenzusätze, um eine längliche Morphologie in einem 6 mg/ml-Peptidgel wiederherzustellen, dargestellt mit reiner Basalmembranmatrix (z. B. Matrigel) und 1,5 mg/ml Rattenschwanz-Kollagen-I-Gel zum Vergleich, Maßstabsbalken 50 μm; (B) MCF10A-normale Brustzellen bilden bis zum 7. Tag Azinusstrukturen in 6 mg/ml-Peptidgelen unter Zugabe von 100 μg/ml humanem Kollagen I, Skalenbalken 100 μm; (C) Die kombinierte Zugabe der Matrixkomponenten Fibronektin/HA (Hyaluronsäure, Molekulargewicht 804 kDa) und HMFU19 in indirekter Co-Kultur erhöht die Größe und Organisation von MCF10A acini in 10 mg/ml-Peptidgelen, beurteilt durch gespaltene Caspase-3-Färbung, Maßstabsbalken 50 μm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 4: Peptidgele ermöglichen die unabhängige Kontrolle von Steifigkeit und Zusammensetzung sowie die Beurteilung der zellabgelagerten Matrix. (A) Bulk-Rheologie-Messungen, die einen typischen Steifigkeitsbereich (Speichermodul, G') zeigen, der durch Kontrolle der Peptidkonzentration erreicht werden kann, * zeigt p < 0,05 an; (B) Immunhistochemie mit Färbung von 150 μg/ml Kollagen I in einem 10 mg/ml-Peptidgel mit verkapseltem MCF7 (Tag 7, Maßstabsbalken 100 μm); (C) Immunfluoreszenz der Kollagen-I-Verteilung in einem 6 mg/ml-Peptidgel mit 200 μg/ml humanem Kollagen I, durch Agar-Einbettung und Vibratom-Schnitt, Maßstabsbalken 25 μm; (D) Die Zugabe von 200 μg/ml Kollagen I führt zu einer geringfügigen Verringerung des Speichermoduls G' von 10 mg/ml-Peptidgelen (oszillatorische Rheologie in großen Mengen), die durch eine Erhöhung der Peptidkonzentration auf 15 mg/ml ausgeglichen wird. MDA MB 231 dreifach negative Brustkrebszellen werden in jeder Erkrankung gezeigt (Tag 7, Skalenbalken 50 μm); (E) MDA MB 231 in 15 mg/ml-Peptidgelen mit 200 μg/ml humanem Kollagen I zeigen eine Verlängerung und Wechselwirkung mit der Matrix über pFAK-Färbung (Tag 14, Maßstabsbalken 50 μm); (F) In situ Färbung der MCF7-Kollagen-I-Abscheidung in einem initial matrixfreien 10 mg/mL Peptidgel (Tag 10, Skalenbalken 100 μm). Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 5: Häufige Probleme bei der Fehlerbehebung bei Peptidgelen können mithilfe der Hellfeldmikroskopie behoben werden. Bei den gezeigten Zellen handelt es sich um normale MCF10A-Brustepithelzellen an Tag 7, sofern nicht anders angegeben. (A) Ein korrekt gemischter Gelvorläufer sollte optisch klar und ohne Inkonsistenzen sein, während (B) unzureichendes Mischen/Neutralisieren sichtbare Inhomogenitäten/Schlieren im Peptidgel verursachen kann (weiße Pfeile); (C) MCF10A bildet azinäre Strukturen in 6 mg/ml-Peptidgelen unter Zugabe von indirekter HMFU19-Cokultur, jedoch (D) bei 15 mg/ml ist die Peptidkonzentration zu hoch, um eine Azinusbildung zu ermöglichen; (E) MCF10A, das in einer Menge von 5 x 105 Zellen/ml ausgesät wird, bildet bei Zugabe von 100 μg/ml Kollagen I azinäre Strukturen in 6 mg/ml-Gelen, jedoch ist (F) bei einer Zelldichte von 2 x 105 Zellen/ml zu gering, um eine Azinusbildung zu ermöglichen; (G) Kollagenzusätze können bis zum 14. Tag große Zellcluster erzeugen, jedoch kann (H) eine falsche Zugabe (Kollagenneutralisation zu früh im Prozess) das Clusterwachstum verhindern. Maßstabsleiste 100 μm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Diskussion
Wir haben festgestellt, dass die hier beschriebenen Peptidgele eine einfache, kostengünstige und flexible Lösung zur Unterstützung der 3D-Kultur mehrerer Zelltypen sind. Durch die vollständige Kontrolle über die Konzentration des verwendeten Peptids und die vorgenommenen Protein- oder Glykanzusätze ermöglicht diese Methode, die Peptidgele sorgfältig auf ihre Anwendung abzustimmen.
Der entscheidende Vorteil der Peptidgele gegenüber bestehenden Verfahren besteht darin, dass die Matrixzusammensetzung und die mechanischen Eigenschaften unabhängig voneinander gesteuert werden können, wobei ein einfaches Verfahren verwendet wird, das keine komplexen chemischen Verfahren erfordert. Die mechanischen Eigenschaften des Peptidgels werden in erster Linie durch die Peptidkonzentration im Ausgangsvorläufer des Gels bestimmt. Die anschließende Zugabe von Zellen und/oder Matrixkomponenten ermöglicht dann die Erstellung einer vollständig benutzerdefinierten in vitro-Umgebung. Obwohl Matrixzusätze die anfänglichen mechanischen Eigenschaften des Gels verändern können, kann dies leicht durch die unabhängige Variation der Peptidkonzentration5 ausgeglichen werden. Dies bietet einen spürbaren Vorteil gegenüber bestehenden Systemen, beispielsweise Kollagengelen, bei denen Parameter, die die Steifigkeit steuern, häufig auch zu einer Veränderung der Integrin-Bindungsmotive führen20,21.
Wir haben die Anwendung des Peptidgels für die In-vitro-Kultur von Krebszelllinien und patientengewonnenem Material demonstriert5. Der mit dem Peptidgel zugängliche Steifigkeitsbereich (im Bereich von 100 bis 1000 Pa) ist ideal geeignet, um normale und Tumormatrixumgebungen in Weichteilen wie der Brust zu replizieren. Wir sind uns jedoch bewusst, dass andere Anwendungen wesentlich steifere Umgebungen erfordern, z. B. im Bereich von 10 bis 20 kPa für die Knochenregeneration. Eine weitere Modifikation des hier vorgestellten Protokolls wäre erforderlich, um die erreichbare Steifigkeit in diesen Bereich zu erweitern, der eher für alternative Ansätze wie Alginatgele22 typisch ist. In ähnlicher Weise haben wir hier ein einfaches Verfahren zur Funktionalisierung durch physikalischen Einschluss von Matrixproteinen/Glykanen innerhalb des Peptidgels beschrieben. Für die hier beschriebenen Anwendungen funktioniert dieser Ansatz gut und lässt sich leicht für die Verwendung durch nicht-spezialisierte Gruppen anpassen, die 3D-In-vitro-Modelle von Krankheiten verwenden möchten. Wie viele andere Hydrogele11 kann das hier verwendete Peptid erweitert werden, um zellbindende oder andere biologische Motive einzuschließen, und für einige Anwendungen kann dieser Ansatz bevorzugt sein.
Wir haben einige wichtige Punkte identifiziert, die besondere Aufmerksamkeit erfordern, um den Erfolg zu gewährleisten. Die Bildung des Gelvorläufers ist ein kritischer Zwischenschritt, der es dem Benutzer ermöglicht, die Richtigkeit der verwendeten Bedingungen zu überprüfen, bevor Zellen eingebaut werden. Dieser Vorläufer kann mehrere Wochen (bei 4 °C) gelagert werden, muss jedoch vor der Verwendung bei 80 °C und anschließend bei 37 °C inkubiert werden. Ein geeigneter Vorläufer ist bei 80 °C vollständig flüssig und bei 37 °C selbsttragend. Diese Kontrollen sind unerlässlich, um sicherzustellen, dass die Gelierung korrekt erfolgt. Zellen und/oder Matrix können dann unter physiologischen Bedingungen eingebaut werden.
Labore, die bereits 3D-Matrizen verwenden, werden mit der sorgfältigen Handhabung vertraut sein, die erforderlich ist, um Zellen in die Peptidgele einzukapseln. Es muss darauf geachtet werden, dass die Unruhe der Zellen vor und während der Verkapselungsschritte begrenzt wird. Wir haben festgestellt, dass bestimmte Zelltypen während dieses Prozesses unterschiedlich anfällig für Schäden sind, die vom Benutzer sorgfältig bewertet werden müssen. Die hier beschriebenen Konzentrationen des Peptidgels ermöglichen es, die Gelierung in einem Zeitrahmen abzulaufen, der es für die genannten Zellen ermöglicht, Zellen einzukapseln, bevor sie auf den Boden der Gussvertiefung sinken, aber langsam genug, dass sie durch diesen Prozess nicht beschädigt werden. Es ist jedoch zu beachten, dass einige empfindliche Zelltypen eine schnellere Neutralisation erfordern können, um eine längere Exposition gegenüber einem erhöhten pH-Wert zu vermeiden. In diesem Fall kann die Zugabe von 10 mM HEPES zu dem Medium, das das Peptidgel umgibt, von Vorteil sein.
Bei der Anwendung der in diesem Protokoll beschriebenen Methode ist es sehr wichtig, die Qualität der Peptidquelle sorgfältig zu berücksichtigen. Anstatt als funktionelles Motiv oder Beschichtung verwendet zu werden, ist das Peptid hier die Gesamtheit des unlöslichen Teils des Hydrogels. Daher haben Verunreinigungen oder Variationen in der Peptidstruktur wahrscheinlich einen signifikanten Einfluss auf die Integrität oder Fähigkeit, die Zellviabilität im endgültigen Hydrogel zu unterstützen. Bei der Umstellung auf eine neue Peptidcharge muss darauf geachtet werden, dass der Lieferant eine gute Konsistenz von Charge zu Charge aufweist und das Verhalten des Peptids bei der Bildung des Gelvorläufers überprüft wird.
Zusammenfassend lässt sich sagen, dass dieses Protokoll ein 3D-Kultursystem beschreibt, bei dem der Schwerpunkt auf der unabhängigen Kontrolle mechanischer und biologischer Eigenschaften liegt. Die Einfachheit und Anpassungsfähigkeit der Methode macht sie für die Anwendung durch jedes Zellkulturlabor und für eine Vielzahl von Anwendungen geeignet5. In Zukunft könnte dieses Protokoll erweitert werden, um die kovalente Modifikation der Peptidsequenz zu ermöglichen. Dies könnte mit fortschrittlichen Mikroskopiemethoden kombiniert werden, um die Zugkräfte zu untersuchen, die Zellen auf ihre umgebende Matrix ausüben. Von entscheidender Bedeutung ist jedoch die Fähigkeit, zwischen künstlich eingebauter Matrix und Matrix, die von den verkapselten Zellen selbst synthetisiert wird, zu unterscheiden. Diese Fähigkeit, Matrixveränderungen im Laufe der Zeit zu kontrollieren und zu überwachen, wird beispiellose Einblicke in die Rolle von Zell-Matrix-Interaktionen bei der Entwicklung von Krebs und anderen Krankheiten ermöglichen.
Offenlegungen
Die Autoren haben nichts offenzulegen.
Danksagungen
Wir danken dem National Centre for the Replacement, Refinement and Reduction of Animals in Research NC/N0015831/1 für JCA, GF und CLRM, NC/T001267/1 für RBC, CLRM, JCA, KL-S und KS, NC/T001259/1 für JCA, KL-S und CLRM und NC/P002285/1 für AMG, SJ und CLRM. Außerdem Finanzierung durch den Engineering and Physical Sciences Research Council EP/R035563/1 an KL-S und CLRM und EP/N006615/1 an JLT und CLRM. Abbildung 1 wurde mit adaptierten Grafiken von Servier Medical Art erstellt. Servier Medical Art von Servier ist lizenziert unter einer Creative Commons Namensnennung 3.0 Unported Lizenz.
Materialien
| Name | Company | Catalog Number | Comments |
| Gel fabrication - Reagents | |||
| FEFEFKFK | Pepceuticals | n/a | Polypeptide; available from various suppliers. Pepceuticals is our recommended supplier due to the quality of the product. |
| PBS 10X | Gibco | 70011-036 | |
| Sodium hydroxide (1 M) | Sigma-Aldrich | S2770 | NaOH; dilute to 0.5 M prior to use |
| Water | Sigma-Aldrich | W3500 | |
| Gel fabrication - Equipment and Consumables | |||
| 15 mL falcon tubes | Greiner | 188261 | If using different brand ensure the material withstands temperatures of up to 90°C |
| 24 well plate | Corning Costar | 3524 | Alternative brands/suppliers can be used as long as there is a gap between the insert base and the plate surface |
| Centrifuge | Any | 200 x g for 3 minutes | |
| Class II Microbiological Safety Cabinet | Any | ||
| Fine balance | Any | Readability 0.1 mg | |
| Hanging insert for 24 well plate | Millipore | MCRP24H48 | Alternative brands/suppliers can be used as long as there is a gap between the insert base and the plate surface |
| Incubator | Any | 37°C, 5% CO2, humidified environment | |
| Oven | Any | set to 80°C | |
| P1000/200/20/10 pipette | Any | It is essential the pipettes used for the procedure are calibrated | |
| P1000/200/20/10 tips | Any | ||
| pH meter with microprobe | Any | ||
| Spatula | Any | ||
| Vortex | Any | ||
| Matrix addition | |||
| Collagen I (human) | Stem Cell Technologies | 07005 | |
| Collagen I (rat tail) | Gibco | A10483 | |
| Fibronectin | Stem Cell Technologies | 07159 | |
| Hyaluronic Acid | Iduron | HA804 | |
| Matrigel | Corning | 354234 | |
| Cell encapsulation/culture | |||
| B27 Supplement (no retinoic acid) | Gibco | 12587010 | Media additions for serum free cultures (Figure 2D) |
| Cholera toxin | Sigma-Aldrich | C-8052 | Media additions for MCF10A cells (Figure 3, 5) |
| DMEM | Gibco | 21969-035 | |
| DMEM/F12 | Sigma-Aldrich | D8062 | Media additions for MCF10A cells (Figure 3, 5) |
| DMEM/F12 Phenol Red Free | Gibco | 21041-025 | Media additions for serum free cultures (Figure 2D) |
| DPBS | Gibco | 14190-094 | |
| EGF | SourceBiosciences | ABC016 | Media additions for MCF10A cells (Figure 3, 5) |
| Fetal Bovine Serum | Gibco | 10500-064 | |
| Horse serum | Gibco | 26050-070 | Media additions for MCF10A cells (Figure 3, 5) |
| Human cancer/epithelial cell lines | e.g. MCF7/tdTomato MCF7/MCF10a/HCT116-mCherry | ||
| Human mammary fibroblasts | e.g. HMFU19 | ||
| Hydrocortisone | Sigma-Aldrich | H-0888 | Media additions for MCF10A cells (Figure 3, 5) |
| Insulin | Sigma-Aldrich | I9278 | Media additions for MCF10A cells (Figure 3, 5) |
| Knockout serum replacement | Gibco | 10828-028 | Media additions for serum free cultures (Figure 2D) |
| L-glutamine | Gibco | 25030-024 | |
| RPMI | Gibco | 21875-034 | |
| RPMI Phenol Red Free | Sigma-Aldrich | R7509 | |
| Imaging and other assays | |||
| 4% paraformaldehyde | Polysciences | 18814 | |
| Agar | SLS | CHE1070 | |
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | 5482 | |
| Confocal and/or fluorescent microscope | Any | e.g. Leica TCS SPE confocal laser scanning microscope (Figures 2-4) | |
| DAPI solution | Invitrogen | D3571 | 300 uM working solution |
| DPX mounting medium | ThermoFisher Scientific | ||
| Glass cover slips | Any | No1 coverslips 0.13 - 0.17 mm thickness | |
| Glass-bottom dishes | MatTek | ||
| Goat Anti-Rabbit IgG H&L (HRP polymer) | Abcam | ab214880 | |
| Haematoxylin and Eosin | Any | ||
| Histology molds (disposable, plastic) | Any | ||
| Image analysis software | ImageJ | ||
| Live/Dead assay kit | Invitrogen | L3224 | |
| Microtome | Any | ||
| Phalloidin | Life Technologies | F432/R415 | |
| Pierce Peroxidase IHC Detection Kit | ThermoFisher Scientific | 36000 | |
| Primary Ab Caspase 3 | Abcam | ab34710 | Shown in Figure 3C |
| Primary Ab Collagen I | Cell Signalling Technology | 9661 | Shown in Figure 4B, C, F |
| Primary Ab pFAK Tyr 397 | ThermoFisher Scientific | 44-624G | Shown in Figure 4E |
| Prolong gold/diamond anti-fade mountant with DAPI | Molecular Probes | S36939 | |
| Rheometer Physica MCR 301 | Anton Paar | ||
| Scalpel | Any | ||
| Secondary antibody Goat anti Rabbit AF488 | nvitrogen | a11034 | |
| Secondary antibody Goat anti Rabbit AF546 | Invitrogen | a11010 | |
| SuperFrost slides | ThermoFisher Scientific | Coating e.g. APES can help to retain microtome sections on slides. | |
| Triton X 100 | Sigma-Aldrich | X100 | |
| Trypsin-EDTA (0.25%) | Gibco | 25300054 | |
| Vibratome | Leica |
Referenzen
- Hynes, R. The extracellular matrix: not just pretty fibrils. Science. 326 (5957), 1216-1219 (2009).
- Tian, C., et al. Cancer-cell-derived matrisome proteins promote metastasis in pancreatic ductal adenocarcinoma. Cancer Research. 80 (7), 1461-1474 (2020).
- Hebert, J. D., et al. Proteomic profiling of the ECM of xenograft breast cancer metastases in different organs reveals distinct metastatic niches. Cancer Research. 80 (7), 1475-1485 (2020).
- Vennin, C., et al. CAF hierarchy driven by pancreatic cancer cell p53-status creates a pro-metastatic and chemoresistant environment via perlecan. Nature Communication. 10 (1), 3637(2019).
- Ashworth, J. C., et al. Peptide gels of fully-defined composition and mechanics for probing cell-cell and cell-matrix interactions in vitro. Matrix Biology. 85, 15-33 (2020).
- Toniatti, C., Jones, P., Graham, H., Pagliara, B., Draetta, G. Oncology drug discovery: Planning a turnaround. Cancer Discovery. 4 (4), 397-404 (2014).
- Mak, I. W., Evaniew, N., Ghert, M. Lost in translation: animal models and clinical trials in cancer treatment. American Journal of Translational Research. 6 (2), 114-118 (2014).
- Aisenbrey, E. A., Murphy, W. L. Synthetic alternatives to Matrigel. Nature Reviews Materials. 5, 539-551 (2020).
- Onion, D., et al. 3-Dimensional patient-derived lung cancer assays reveal resistance to standards-of-care promoted by stromal cells but sensitivity to histone deacetylase inhibitors. Molecular Cancer Therapy. 15 (4), 753-763 (2016).
- Saunders, J. H., et al. Individual patient oesophageal cancer 3D models for tailored treatment. Oncotarget. 8 (15), 24224-24236 (2017).
- Caliari, S. R., Burdick, J. A. A practical guide to hydrogels for cell culture. Nature Methods. 13 (5), 405-414 (2016).
- Kühn, S., et al. Cell-instructive multiphasic gel-in-gel materials. Advanced Functional Materials. 30, 1908857(2020).
- Gjorevski, N., et al. Designer matrices for intestinal stem cell and organoid culture. Nature. 539 (7630), 560-564 (2016).
- Gjorevski, N., Lutolf, M. P. Synthesis and characterization of well- defined hydrogel matrices and their application to intestinal stem cell and organoid culture. Nature Protocols. 12 (11), 2263-2274 (2017).
- Saiani, A., et al. Self assembly and gelation properties of α-helix versus β-sheet forming peptides. Soft Matter. 5 (1), 193-202 (2008).
- Wan, S., et al. Self-assembling peptide hydrogel for intervertebral disc tissue engineering. Acta Biomaterialia. 46, 29-40 (2016).
- Blache, U., Stevens, M. M., Gentleman, E. Harnessing the secreted extracellular matrix to engineer tissues. Nature Biomedical Engineering. 4 (4), 357-363 (2020).
- Sachs, N., et al. A living biobank of breast cancer organoids captures disease heterogeneity. Cell. 172 (1-2), 373-386 (2018).
- Shaw, F. L., et al. A detailed mammosphere assay protocol for the quantification of breast stem cell activity. Journal of Mammary Gland Biology and Neoplasia. 17 (2), 111-117 (2012).
- Barcus, C. E., Keely, P. J., Eliceiri, K. W., Schule, L. A. Stiff collagen matrices increase tumorigenic prolactin signaling in breast cancer cells. Journal of Biological Chemistry. 288, 12722-12732 (2013).
- Bax, D. V., et al. Impact of UV- and carbodiimide-based crosslinking on the Integrin-binding properties of collagen-based materials. Acta Biomaterialia. 100, 280(2019).
- Huang, B. P., et al. Multi-peptide presentation and hydrogel mechanics jointly enhance therapeutic duo-potential of entrapped stromal cells. Biomaterials. 245, 119973(2020).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten