
Method Article
Análisis de R-Loop por Dot-Blot
En este artículo
Resumen
Este protocolo detalla un método simple que cuantifica el R-loop, una estructura de ácido nucleico de tres cadenas que se compone de un híbrido de ARN-ADN y una cadena de ADN desplazada.
Resumen
La estructura de ácido nucleico tricatenario, R-loop, es cada vez más reconocida por su papel en la regulación génica. Inicialmente, se pensó que los bucles R eran los subproductos de la transcripción; pero los hallazgos recientes de menos bucles R en las células enfermas dejaron claro que los bucles R tienen roles funcionales en una variedad de células humanas. A continuación, es fundamental comprender las funciones de los bucles R y cómo las células equilibran su abundancia. Un desafío en el campo es la cuantificación de los bucles R, ya que gran parte del trabajo se basa en el anticuerpo monoclonal S9.6, cuya especificidad para los híbridos de ARN-ADN ha sido cuestionada. Aquí, utilizamos dot-blots con el anticuerpo S9.6 para cuantificar los bucles R y mostrar la sensibilidad y especificidad de este ensayo con RNasa H, RNasa T1 y RNasa III que escinden híbridos ARN-ADN, ARN monocatenario y ARN bicatenario, respectivamente. Este método es altamente reproducible, utiliza equipos y reactivos generales de laboratorio y proporciona resultados en dos días. Este ensayo se puede utilizar en entornos clínicos y de investigación para cuantificar los bucles R y evaluar el efecto de las mutaciones en genes como la senataxina en la abundancia de bucles R.
Introducción
Este protocolo proporciona una guía paso a paso para un ensayo de transferencia de puntos que permite una evaluación comparativa rápida de la abundancia de R-loop, una estructura de ácido nucleico de tres cadenas. El bucle R se forma cuando el ARN invade un ADN bicatenario para generar un híbrido ARN-ADN y desplaza a la otra cadena de ADN. Los bucles R se encuentran en diferentes etapas del ciclo de vida del ARN. En el complejo transcripcional, el ARN naciente se sintetiza de forma complementaria al ADN molde y la hebra no molde se desplaza. El híbrido corto ARN-ADN (<10 pb) está resuelto a liberar el ARN naciente para que pueda salir del complejo ARN polimerasa a través del canal de salida 1,2. Fuera del complejo transcripcional, el ARN naciente está cerca de su molde de ADN, que todavía está ligeramente desenrollado para ser copiado, por lo que el ARN puede rehibridarse con su molde de ADN formando bucles R3. Además, se pueden formar bucles R cuando los complejos de replicación y transcripción colisionan4, y en la transcripción antisentido5. Dadas las muchas oportunidades para su formación, los R-loops no son raros, y se pueden encontrar en el 3-5% del genoma humano6, dependiendo del estado de transcripción de la célula. Los bucles R se encuentran en los promotores de genes7 y en los sitios de terminación5 en el ARNm, y a lo largo del ARN ribosómico8, así como en el ARNde transferencia 9. Los bucles R también se encuentran en las regiones teloméricas de los cromosomas.
Los bucles R desempeñan un papel regulador. Regulan la expresión génica afectando la transcripción en los promotores10,11, mediando la recombinación de cambio de clase12 y facilitando la edición del genoma basada en CRISPR 13,14,15. Al igual que muchos eventos celulares, la abundancia del bucle R está estrictamente titulada; demasiados o muy pocos bucles R afectan la función normal de la célula16,17. Los bucles R están regulados por una variedad de proteínas, como la ARNasa H, la senataxina y otras helicasas que desenrollan los híbridos ARN-ADN 18,19,20,21,22.
Para monitorizar la abundancia de R-loops, los métodos de todo el genoma primero enriquecen los R-loops con el anticuerpo S9.6 8,23,24 o con otras nucleasas25, incluida la RNasa H 10,26,27, y luego evalúan el número de R-loops enriquecidos mediante secuenciación. Las primeras versiones de estos métodos basados en la secuenciación no lograron una cobertura de secuencia adecuada para permitir una cuantificación precisa, pero la rápida mejora en las tecnologías de secuenciación ahora permite el análisis de bucle R locus por locus. También se han utilizado técnicas de inmunofluorescencia para cuantificar y localizar los bucles R10,17. Estos métodos son integrales, pero no son prácticos en muchos entornos clínicos ni como evaluaciones iniciales, ya que requieren equipos costosos y análisis especializados.
Se necesita un procedimiento que se pueda realizar de manera uniforme en todos los laboratorios en entornos clínicos. Las manchas de puntos ofrecen esta opción, ya que pueden llevarse a cabo sin ningún equipo específico ni análisis computacional. Como paso previo o en entornos clínicos para evaluar los efectos de las mutaciones en los R-loops, estos dot-blots deben proporcionar resultados sensibles y específicos. Aquí, describimos nuestro ensayo que identifica específicamente los bucles R; excluye las señales del ADN bicatenario (ds), el ARN bicatenario y el ARN monocatenario. Nuestro protocolo utiliza el anticuerpo S9.627 para identificar híbridos ARN-ADN en R-loops e incorpora RNasa H, una endorribonucleasa que escinde y, por lo tanto, conduce a la degradación del ARN en un híbrido ARN-ADN 20,28, para asegurar que las señales detectadas son las de los híbridos. También incorporamos la RNasa T1 que escinde el ARN monocatenario a la guanina29,30, y la RNasa III que escinde el ARN bicatenario, incluidos los bucles de tallo31,32 para comprobar si hay señales inespecíficas. El anticuerpo S9.6 reconoce híbridos de ARN-ADN de diferentes longitudes, incluso aquellos que tienen solo 8 nucleótidos de largo33.
Aquí, presentamos el protocolo que comienza con el aislamiento de ácido nucleico seguido de la preparación de dot-blot y la detección de R-loop con el anticuerpo S9.6. Nuestro protocolo incluye pasos para garantizar que se carguen cantidades iguales de muestras y que las señales sean específicas. Proporciona oligonucleótidos para que sirvan como controles positivos y negativos. Este es un método rápido, fácil y fácil de usar para evaluar la abundancia de R-loop.
Protocolo
1. Lisis celular para fraccionamiento nuclear
- Lave las células con 1x solución salina tamponada con fosfato (PBS) dos veces. Extraiga las células de las placas de cultivo de tejidos utilizando técnicas estándar de disociación celular como la tripsina. Cuente las células con un hemocitómetro.
NOTA: Los pasos que se describen a continuación se utilizaron para el análisis de fibroblastos primarios de piel humana, aunque se puede analizar una variedad de tipos de células. Los fibroblastos se cultivaron en medios basales que contenían un 10% de suero fetal bovino. Alternativamente, el tampón de lisis celular (Tabla 1) se puede agregar directamente al cultivo celular después del lavado. - Transfiera la suspensión celular a un tubo de 1,5 mL para granular las células.
- Centrifugar la muestra a 300 x g durante 5 min a 4 °C. Aspirar los medios de comunicación.
- Lave dos veces con 1x PBS helado usando la configuración de centrifugación en el paso 1.3.
- Agregue tampón de lisis celular fría (Tabla 1) al pellet de celda (300 μL por 2 x 106 celdas). Pipetear hacia arriba y hacia abajo para volver a suspender el pellet.
- Incubar en hielo durante 10 min.
- Girar a 500 x g durante 5 min para pellet los núcleos.
- Deseche el sobrenadante y vuelva a suspender el pellet nuclear en 400 μL de tampón de lisis nuclear fría (Tabla 1).
- Incubar en hielo durante 10 min.
NOTA: El fraccionamiento de los compartimentos nuclear y citoplasmático de las células garantiza la especificidad de la señal. La calidad de la separación nuclear y citoplasmática puede evaluarse antes de proceder (Tabla 2). - Añadir 3 μL de 20 mg/mL de proteinasa K e incubar durante 3-5 h a 55 °C.
NOTA: Los volúmenes indicados son para 2 x 106 celdas, amplíe o reduzca según sea necesario.
2. Purificación de ADN genómico (que incluye híbridos ARN-ADN)
- Si el ADN es viscoso, realizar la sonicación para reducir la viscosidad (por ejemplo, sonicación a alta potencia de salida, 30 s ON/30 s OFF, durante 2 min utilizando un baño de agua a 4 °C).
- Añadir 400 μL de tampón de elución (Tabla 1) y 400 μL de fenol:cloroformo:alcohol isoamílico (25:24:1 pH 8,0).
- Vórtice durante 10 s.
- Centrifugar a 12.000 x g durante 5 min a 4 °C.
- Transfiera la fase acuosa (aproximadamente 350 μL) a un tubo nuevo.
- Extraiga una vez usando 1 volumen de cloroformo, vórtice durante 10 s, luego gire hacia abajo a 12,000 x g durante 5 min a 4 °C. Transfiera la fase acuosa a un nuevo tubo (aproximadamente 300 μL).
- Añadir 35 μL de acetato de sodio 3 M (pH 5,2), 1 μL de glucógeno y 700 μL de etanol 100% helado.
- Vórtice durante 10 s y gire hacia abajo a 12.000 x g durante 30 min a 4 °C.
- Lavar el pellet con 1 mL de etanol al 70%.
- Vórtice durante 10 s y centrifugado a 12.000 x g durante 15 min a 4 °C.
- Deseche el sobrenadante y deje que el pellet se seque al aire.
- Añadir 12 μL de tampón de elución y vórtice durante 10 s para resuspender. Incubar la muestra durante 30 min a 37 °C con agitación o a 4 °C durante la noche para volver a suspender el pellet.
- Mida la concentración de ADN utilizando espectrofotometría estándar.
NOTA: Los volúmenes indicados son para 2 x 106 celdas, amplíe o reduzca según sea necesario. El ADN (con híbridos ARN-ADN) puede almacenarse a -20 °C, si es necesario.
3. Transferencia de muestras de ADN (que incluyen híbridos de ARN-ADN) en membranas de nailon
- Prepare diluciones de ácidos nucleicos a las concentraciones deseadas en el tampón de elución (es decir, 50 ng/μL, 25 ng/μL o 12,5 ng/μL). Estas muestras con un rango de concentraciones (200, 100, 50, 25, 12,5 ng) aseguran que habrá señales dentro del rango lineal.
NOTA: Asegúrese de preparar suficiente muestra para las réplicas técnicas y biológicas, y para los diversos tratamientos con ARNasa, consulte el Paso 5. - Prepare una membrana de nailon cargada positivamente de modo que haya espacio para que cada muestra de 2 μL ocupe un área de 0,5 cm2.
- Coloque 2 μL de cada muestra en 2 membranas: una para el anticuerpo S9.6 y la otra para el anticuerpo dsDNA. Alternativamente, se puede utilizar un aparato de transferencia de puntos o de ranura que permite la carga de muestras con volúmenes más grandes.
- Permita que las muestras se saturen en la membrana. Espere al menos 2 minutos antes de reticular la membrana con luz ultravioleta.
- Coloque la membrana en el centro del dispositivo UV y reticule la membrana usando un reticulante UV usando la configuración "Auto Crosslink" (1,200 μJ x 100).
4. Detección de híbridos ARN-ADN con anticuerpo S9.6
- Incubar la membrana en solución bloqueante (5% de leche en solución salina tamponada con Tris con 0,05% de Tween-20 (TBST) durante 1 h a temperatura ambiente en un agitador.
NOTA: Debe haber suficiente solución de bloqueo para cubrir la membrana. - Incubar las membranas durante la noche en anticuerpo primario (en leche al 5% en TBST) a 4 °C con agitación. Agregue un anticuerpo anti-dsDNA (dilución 1:10,000) a una membrana. Añadir 1 μg/ml de anticuerpo S9.6 a la segunda membrana (dilución 1:1.000).
NOTA: El anticuerpo S9.6 está disponible comercialmente o a través del Dr. S. Leppla, NIAID, Institutos Nacionales de Salud. - Retire el anticuerpo primario y lave 3 veces con TBST. Realice cada lavado durante 5-10 minutos agitando a temperatura ambiente.
- Incubar con anticuerpo secundario conjugado con peroxidasa de rábano picante (HRP) (anti-ratón, dilución 1:5.000) en leche al 5% en TBST con agitación a temperatura ambiente.
NOTA: Tanto el anti-dsDNA como el híbrido anti-ARN-ADN son anticuerpos de ratón. - Retire el anticuerpo secundario y lave 3 veces con TBST durante 5-10 minutos agitando a temperatura ambiente.
- Desarrolle con reactivos de quimioluminiscencia mejorada (ECL) para adquirir señales para la obtención de imágenes.
- Cuantifique la intensidad de la señal utilizando herramientas estándar de procesamiento de imágenes como ImageJ.
NOTA: La solución de problemas se detalla en la Tabla 2.
5. Tratamientos con ribonucleasas para evaluar la especificidad de la señal
NOTA: El tratamiento con RNasa debe realizarse en las muestras de ácido nucleico para demostrar la especificidad de la unión a S9.6. El tratamiento con RNasa H, pero no con RNasa T1 o RNasa III, debería dar lugar a una reducción de la inmunotinción S9.6.
- Digiera las muestras que contienen híbridos de ARN-ADN preparándolas en cuatro tubos separados. Trate cada una de las 4 muestras con 5 U de RNasa H, 1000 U de RNasa T1, 0,5 U de RNasa III o simulado. Incubar muestras a 37 °C durante 15 min en volúmenes de 20 μL.
- Cargue 2 μL de cada muestra en una membrana como se describe en la sección 3.
6. Preparación de controles de oligonucleótidos para evaluar la especificidad de la señal
NOTA: Los controles de oligonucleótidos se pueden utilizar para demostrar la especificidad de la unión a S9.6. S9.6 reconoce híbridos ARN-ADN, pero no controles de dsDNA o dsRNA, como se ha informado previamente34.
- Disuelva los oligonucleótidos (Tabla 3) en tampón de recocido (10 mM Tris, pH 8.0; 50 mM NaCl, 1 mM EDTA) a 100 μM.
- Prepare 4 tubos de reacción para
- Híbrido ARN-ADN #1: Mezcle 10 μL de hebra superior de ssRNA con 10 μL de hebra inferior de ssDNA y 80 μL de tampón de recocido.
- Híbrido ARN-ADN #2: Mezcle 10 μL de hebra superior de ssDNA con 10 μL de hebra inferior de ssRNA y 80 μL de tampón de recocido.
- dsRNA: Mezcle 10 μL de hebra superior de ssRNA con 10 μL de hebra inferior de ssRNA y 80 μL de tampón de recocido.
- dsDNA: Mezcle 10 μL de hebra superior de ssDNA con 10 μL de hebra inferior de ssDNA y 80 μL de tampón de recocido.
- Calentar las 4 mezclas del paso 6.2 a 95 °C durante 10 min.
- Deje que los tubos se enfríen lentamente a temperatura ambiente para permitir el recocido de las hebras. Los patrones recocidos se pueden almacenar a -20 °C para su uso posterior.
NOTA: La eficiencia del recocido debe verificarse mediante electroforesis en gel no desnaturalizante. Los dúplex migran más lentamente que los oligonucleótidos no recocidos (Tabla 2). - Cargue 2 μL de cada muestra en 2 membranas, una para el anticuerpo S9.6 y otra para el anticuerpo dsDNA, como se describe en la sección 3.
- Realice los pasos descritos en la sección 4.
7. Cuantificación y normalización de la intensidad de la señal de bucle R S9.6 utilizando ImageJ.
- Guarde imágenes de S9.6, tinción de dsDNA en formato TIFF y analícelas con el software ImageJ (https://imagej.nih.gov/ij/).
- Seleccione la opción invertir imagen (Editar | Invertir). Después de la inversión, cada punto será visible como blanco sobre un fondo oscuro.
- Utilice la herramienta de selección de imagen ovalada para seleccionar un óvalo que sea lo suficientemente grande como para rodear el punto más grande de la imagen.
- Utilice el administrador de ROI para agregar el área seleccionada para la cuantificación. Asegúrese de que las opciones "Mostrar todo" y "Etiquetas" estén seleccionadas para que se puedan visualizar las regiones de interés.
- Utilice la misma área de selección ovalada utilizada durante el paso 7.3 para añadir regiones de interés adicionales alrededor de cada punto que se va a cuantificar. Utilice el atajo Comando + Mayús + E para copiar el área seleccionada del paso 7.3 en cada uno de los puntos siguientes.
- Mida la densidad integrada de cada una de las regiones de interés.
- Divida la intensidad de la señal S9.6 para cada muestra por la medición de dsDNA para obtener la relación de señal S9.6/dsDNA. Verifique los resultados repitiendo los experimentos (al menos por triplicado para la adquisición de señales de S9.6 y dsDNA). El error estándar de la media se puede calcular a partir de las proporciones de señal S9.6/dsDNA.
Resultados
Tratamiento enzimático para evaluar la especificidad del anticuerpo S9.6 (ARN-ADN).
Se cultivaron fibroblastos primarios de piel humana17. Se aisló y cuantificó el ADN con híbridos ARN-ADN. Dos μg de las muestras se digierieron con RNasa T1, RNasa H o RNasa III durante 15 min a 37 °C. También se analizó una muestra simulada para compararla con las muestras tratadas con RNasa. Las muestras (200, 100, 50, 25, 12,5 o 6,25 ng) se secaron en dos membranas diferentes como se describe en la sección 3. Las membranas fueron reticuladas, bloqueadas y una de ellas fue sondeada con el anticuerpo S9.6 (Figura 1A).
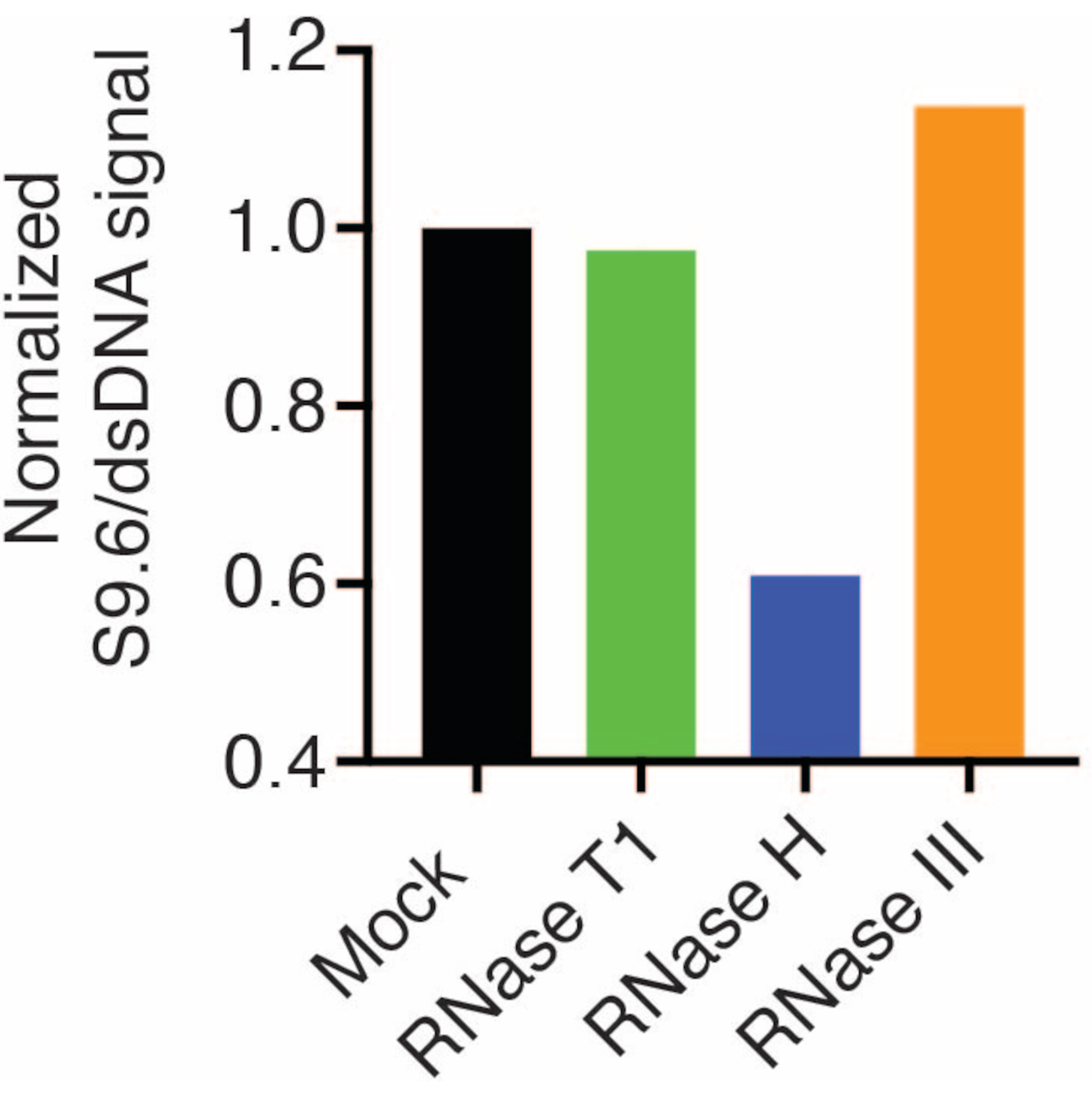
Los resultados mostraron que la señal S9.6 se correlaciona con la abundancia de la muestra cargada. El tratamiento con RNasa H, pero no con RNasa T1 o RNasa III, produce una reducción de la tinción S9.6.
Se sondeó una segunda membrana con un anticuerpo dsDNA (Figura 1B) para la normalización. Se utilizó la imagen J para analizar las intensidades de la señal. Las muestras de 50 ng se seleccionaron para la cuantificación ya que las intensidades de señal de los anticuerpos S9.6 y dsDNA estaban dentro del rango dinámico. Las intensidades de la señal se normalizaron a las de las muestras simuladas. Los datos se muestran en la Figura 2.
Dot-blot de anticuerpos S9.6 mediante controles de nucleótidos sintéticos.
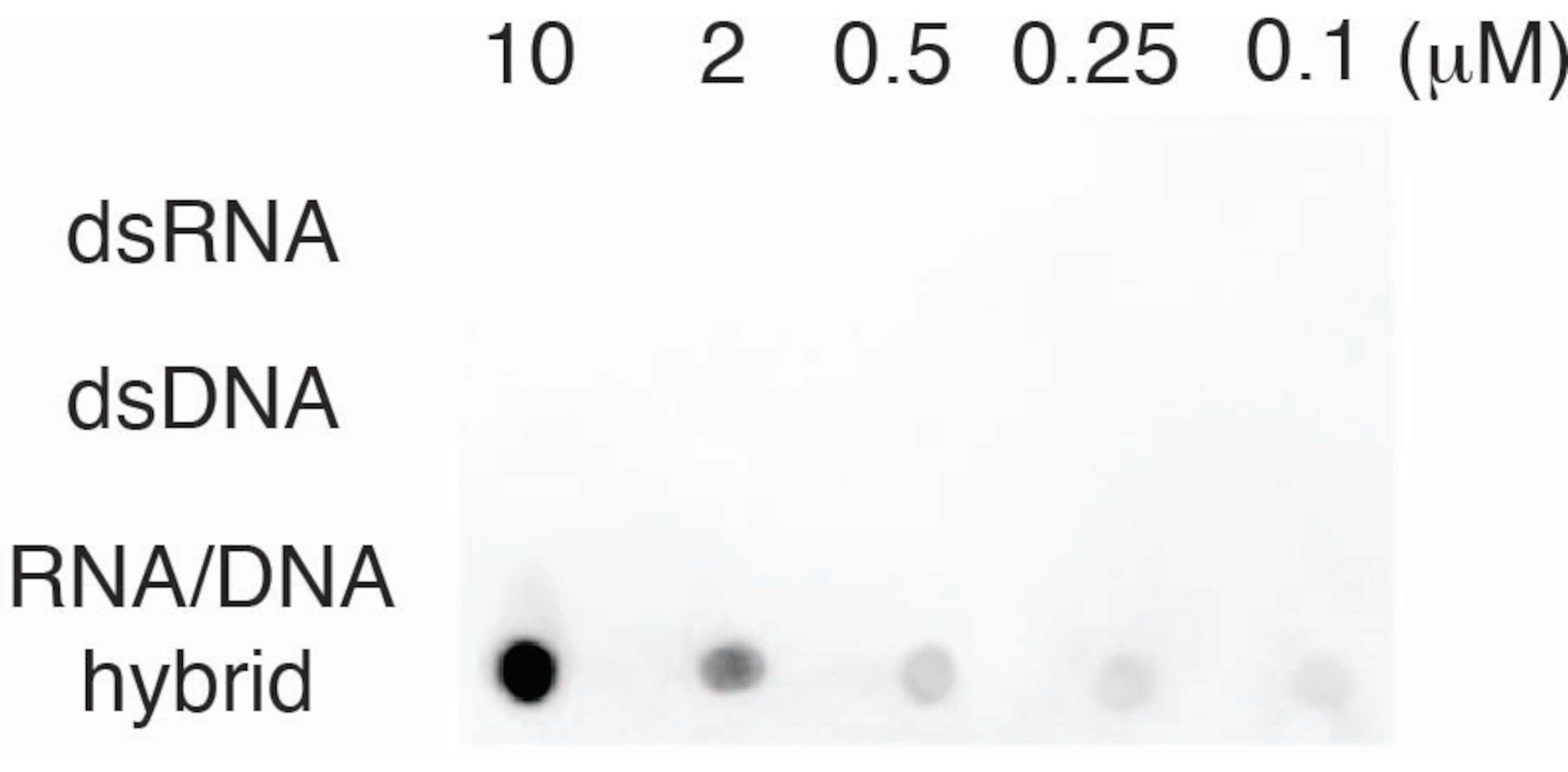
Para evaluar la especificidad del anticuerpo S9.6, utilizamos oligonucleótidos correspondientes a dsRNA, dsDNA y RNA-DNA como se describe en la sección 6. Se preparó una serie de dilución de nucleótidos de ARN-ADN, dsARN y dsDNA y se secó en la membrana de nailon como se describe en la sección 3. La membrana se sondeó con el anticuerpo S9.6 (Figura 3). Los resultados mostraron que el anticuerpo S9.6 se une específicamente a los híbridos de ARN-ADN de una manera dependiente de la dosis y mostró una reactividad cruzada mínima a los dsRNAs y dsDNAs.

Figura 1: Especificidad de S9.6 mostrada por dot-blot cargado con ácidos nucleicos de fibroblastos humanos. Las muestras de ácido nucleico de fibroblastos humanos se trataron simuladamente o se trataron con RNasa T1, RNasa H o RNasa III y luego se cargaron en membranas de nailon en una serie de diluciones de 200, 100, 50, 25, 12,5 y 6,25 ng por punto de 2 μL. A continuación, las membranas se sondearon con el anticuerpo S9.6 (A) o el anticuerpo dsDNA (B). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Cuantificación de la tinción con S9.6 R-loop. Se seleccionaron muestras de 50 ng de la Figura 1 para su cuantificación con ImageJ. La señal S9.6 se dividió por la intensidad de la señal de dsDNA, luego se normalizó a la muestra simulada siguiendo los pasos descritos en la sección 7. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: S9.6 dot-blot usando controles de oligonucleótidos. Anticuerpo S9.6 dot-blot contra una serie de dilución de oligonucleótidos sintéticos como dsRNA, dsDNA o híbrido ARN-ADN. S9.6 se une específicamente a los híbridos de ARN-ADN de una manera dependiente de la dosis. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
| Tampón de lisis celular | Para 10mL | Para 200mL | Final Conc. |
| Agua, sin nucleasas | 9 ml | 180mL | - |
| 10% NP-40 | 0.5mL | 10 ml | 0.5% |
| 2M KCl | 0.4mL | 8 ml | 80mM |
| TUBOS DE 0,5 M (pH 8,0) | 100uL | 2 ml | 5 mM |
| Tampón de lisis nuclear | Para 10mL | Para 200mL | Final Conc. |
| Agua, sin nucleasas | 8.65mL | 173 ml | - |
| 10% SDS | 1 ml | 20mL | 1% |
| 1M Tris-HCl (pH 8.0) | 0.25mL | 5 ml | 25 mM |
| 0,5 M de EDTA | 100uL | 2 ml | 5 mM |
| Tampón de elución | Para 10mL | Para 200mL | Final Conc. |
| 1M Tris-Cl, pH 8.5 | 0,1 mL | 2 mL | 10 mM |
| Agua, sin nucleasas | 9.9 mL | 198 mL | - |
Tabla 1. Preparación de tampones
| Paso | Problema | Posible razón | Solución |
| 1.9 | N/A | Verificación del fraccionamiento de celdas | La calidad de la separación nuclear y citoplasmática puede evaluarse mediante la adición de cócteles de inhibidores de proteasa estándar a los tampones de lisis celular y nuclear (Tabla 1). Las fracciones citoplasmáticas y nucleares pueden evaluarse mediante análisis de Western blot para confirmar la restricción adecuada del marcaje con marcadores citoplasmáticos (por ejemplo, GAPDH o HSP90) en las fracciones citoplasmáticas y el marcaje con marcadores nucleares (por ejemplo, HDAC1 o histona H3) en las fracciones nucleares. La contribución de la contaminación mitocondrial en la fracción nuclear puede evaluarse mediante análisis de qPCR con sondas específicas para el ADN mitocondrial. |
| 2.1 | La muestra es demasiado viscosa | El número de celda es demasiado alto. | Reducir el ADN a la mitad y continuar con el paso de sonicación 2.1 |
| 2.12 | Sin pellet visible | Material de partida insuficiente o pérdida durante la extracción. | Comience desde el principio usando más celdas. |
| 2.13 | Baja concentración de ADN | ||
| 3.1 | No hay suficiente ADN para las diluciones | ||
| 3.4 | La muestra no se saturará en la membrana | Remoje la membrana en 1x TBST. Deje que el exceso de tampón se seque y comience en el paso 3.4 | |
| 4.6 | Se detecta un patrón irregular o moteado | Añada dodecil sulfato de sodio (SDS) al 0,1% a la muestra y continúe en el paso 3.1 | |
| 4.6 | Los puntos tienen una apariencia de "anillo de café" | Agregue 0.01% de Sarkosyl a la muestra y continúe en el paso 3.1 | |
| 5.2 | El control RNaseH no muestra ninguna disminución de la señal | La digestión de la ribonucleasa es incompleta. | Aumentar la incubación o aumentar la concentración de enzimas. |
| 6.5 | No hay señal para los controles de oligonucleótidos | El dúplex no se formó. Los oligonucleótidos no se recocieron correctamente. | Verifique las proporciones de oligonucleótidos y tampón de recocido. |
| 6.5 | La señal S9.6 no es específica de los híbridos | El lote de anticuerpos S9.6 tiene una unión inespecífica | Validar la sensibilidad y especificidad de nuevos lotes de anticuerpos S9.6 con el uso de tratamientos con enzimas RNasa o análisis de oligonucleótidos sintéticos. |
Tabla 2: Solución de problemas
| ssRNA, hebra superior | 5'-UGGGGGCUCGUCCGGGAUAUGGGAACCACUGAUCCC-3' |
| ssDNA, hebra superior | 5'-TGGGGGCTCGTCCGGGATATGGGAACCACTGATCCC-3' |
| ssRNA, hebra inferior | 5'-GGGAUCAGUGGUUCCCAUAUCCCGGACGAGCCCCCA-3' |
| ssDNA, hebra inferior | 5'-GGGATCAGTGGTTCCCATATCCCGGACGAGCCCCCA-3' |
Tabla 3. Secuencias de oligonucleótidos de control
Discusión
Los ácidos nucleicos de 3 hebras, R-loops, se forman en diferentes etapas durante el ciclo de vida del ARN y cada vez se descubre más que regulan los procesos celulares. Para comprender completamente los bucles R, se necesitan técnicas confiables para la detección de bucles R. Aquí, describimos un enfoque para interrogar la abundancia de R-loops utilizando el anticuerpo S9.6 8,23,24. Este método permite una evaluación rápida de la abundancia de R-loop a partir de células y muestras de cultivo de tejidos. No requiere un equipo especial, ni una gran cantidad de material de partida. Garantiza resultados específicos y reproducibles mediante una combinación de tratamientos con ARNasa.
Algunos han informado de preocupaciones sobre la especificidad del anticuerpo S9.6. Al igual que con cualquier reactivo, puede haber variabilidad de lote a lote con el anticuerpo S9.6. Nuestro protocolo incluye RNasa H, RNasa T1 y RNasa III para comprobar la especificidad de la señal. Además, utilizamos oligonucleótidos sintéticos para garantizar la especificidad de cada lote de anticuerpo S9.6.
La biología del bucle R es un campo en crecimiento; el desarrollo de métodos fiables de detección y cuantificación, como el que se presenta aquí, facilitará los estudios mecanicistas para dilucidar cuándo se forman los bucles R, cómo se regulan y qué regulan. Con los controles adecuados, este ensayo de transferencia de puntos es un método sencillo para detectar la abundancia de bucle R en entornos clínicos y de investigación.
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Este trabajo fue apoyado por el Instituto Médico Howard Hughes y la Investigación Intramuros del Instituto Nacional de Trastornos Neurológicos y Accidentes Cerebrovasculares.
Agradecemos al Dr. Stephen Leppla por proporcionar lotes de anticuerpos S9.6 para su análisis. También agradecemos al Dr. Dongjun Li por su ayuda con los tratamientos con ribonucleasas.
Materiales
| Name | Company | Catalog Number | Comments |
| Anti-dsDNA antibody | Abcam | ab27156 | |
| Anti-RNA-DNA hybrid antibody (S9.6) | Kerafast | ENH001 | |
| Biorupter sonicator | Diagenode | UCD-200 | |
| EB Buffer | Qiagen | 19086 | |
| EDTA (0.5M) | Invitrogen | AM9261 | |
| Hybond N+ nylon membrane | GE healthcare Life Sciences | RPN203B | |
| KCl (2M) | Invitrogen | AM9640G | |
| NP-40 (Igepal CA-630) | Sigma | I8896 | |
| PBS | Invitrogen | 10010-023 | |
| Phenol:chloroform:isoamyl alcohol | Invitrogen | 15593031 | |
| PIPES (0.5M, pH 8.0) | VWR | AAJ61406-AE | |
| Proteinase K | Qiagen | 19131 | |
| RNase III | Invitrogen | AM2290 | |
| RNase H | New England Biolabs | M0297 | |
| RNase T1 | ThermoFisher Sci. | EN0541 | |
| SDS (10%) | Invitrogen | 15553027 | |
| sodium acetate (3M, pH 5.2) | Invitrogen | AM9740 | |
| Tris-buffered saline (10X) | Corning | 46-012-CM | |
| Tris-HCl (1M, pH 8.0) | KD Medical | RGF-3360 | |
| TrypLE | Invitrogen | 12605010 | |
| Tween-20 | Sigma | P9416 | |
| UV Stratalinker 2400 | Stratagene | Stratalinker 2400 | |
| Whatman marking pen | Sigma | WHA10499001 |
Referencias
- Daube, S. S., von Hippel, P. H. RNA displacement pathways during transcription from synthetic RNA-DNA bubble duplexes. Biochemistry. 33 (1), 340-347 (1994).
- Westover, K. D., Buschnell, D. A., Kornberg, R. D. Structural basis of transcription: separation of RNA from DNA by RNA polymerase II. Science. 303 (5660), 1014-1016 (2004).
- Itoh, T., Tomizawa, J. Formation of an RNA primer for initiation of replication of ColE1 DNA by ribonuclease H. Proceedings of the National Academy of Science USA. 77 (5), 2450-2454 (1980).
- Hamperl, S., Bocek, M. J., Saldivar, J. C., Swigut, T., Cimprich, K. A. Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell. 170 (4), 774-786 (2017).
- Skourti-Stathaki, K., Kamieniarz-Gdula, K., Proudfoot, N. J. R-loops induce repressive chromatin marks over mammalian gene terminators. Nature. 516 (7531), 436-439 (2014).
- Sanz, L. A., et al. conserved R-loop structures associate with specific epigenomic signatures in mammals. Molecular Cell. 63 (1), 167-178 (2016).
- Chen, L., et al. R-ChIP using inactive RNase H reveals dynamic coupling of R-loops with transcriptional pausing at gene promoters. Molecular Cell. 68 (4), 745-757 (2017).
- El Hage, A., French, S. L., Beyer, A. L., Tollervey, D. Loss of topoisomerase I leads to R-loop mediated transcriptional blocks during ribosomal RNA synthesis. Genes and Development. 24 (14), 1546-1558 (2010).
- Tran, P., et al. PIF1 family DNA helicases suppress R-loop mediated genome instability at tRNA genes. Nature Communication. 8, 15025(2017).
- Ginno, P. A., Lott, P. L., Christensen, H. C., Korf, I., Chedin, F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Molecular Cell. 45 (6), 814-825 (2012).
- Colak, D., et al. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science. 343 (6174), 1002-1005 (2014).
- Yu, K., Chedin, F., Hsieh, C., Wilson, T. E., Lieber, M. R. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nature Immunology. 4 (5), 442-451 (2003).
- Gasiunas, G., Barrangou, R., Horvath, P., Siksnys, V. Cas9-crRNA ribonucleotide complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proceedings of the National Academy of Science USA. 109 (39), 2579-2586 (2012).
- Westra, E. R., et al. CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by Cascade and Cas3. Molecular Cell. 46 (5), 595-605 (2012).
- Jiang, F., et al. Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science. 351 (6275), 867-871 (2016).
- Huertas, P., Aguilera, A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Molecular Cell. 12 (3), 711-721 (2003).
- Grunseich, C., et al. Senataxin mutation reveals how R-loops promote transcription by blocking DNA methylation at gene promoters. Molecular Cell. 69 (3), 426-437 (2018).
- Kim, H. D., Choe, J., Seo, Y. S. The sen1(+) gene of Schizosaccharomyces pombe, a homologue of budding yeast SEN1, encodes an RNA and DNA helicase. Biochemistry. 38 (44), 14697-14710 (1999).
- Stein, H., Hausen, P. Enzyme from calf thymus degrading the RNA moiety of DNA-RNA hybrids: effect on DNA-dependent RNA polymerase. Science. 166 (3903), 393-395 (1969).
- Cerritelli, S. M., Crouch, R. J. Ribonuclease H: the enzymes in eukaryotes. FEBS Journal. 276 (6), 1494-1505 (2009).
- Hyjek, M., Figiel, M., Nowotny, M. RNases H: Structure and mechanism. DNA Repair. 84, 102672(2019).
- Pohl, T. J., Zakian, V. A. Pif1 family DNA helicases: A helpmate to RNase H. DNA Repair. 84, 102633(2019).
- Pohjoismaki, J. L., et al. Mammalian mitochondrial DNA replication intermediates are essentially duplex but contain extensive tracts of RNA/DNA hybrid. Journal of Molecular Biology. 397 (5), 1144-1155 (2010).
- Sanz, L. A., Chedin, F. High-resolution, strand-specific R-loop mapping via S9.6-based DNA-RNA immunoprecipitation and high-throughput sequencing. Nature Protocols. 14 (6), 1734-1755 (2019).
- Wahba, L., Costantino, L., Tan, F. J., Zimmer, A., Koshland, D. S1-DRIP-seq identifies high expression and polyA tracts as major contributors to R-loop formation. Genes and Development. 30 (11), 1327-1338 (2016).
- Yan, Q., Shields, E. J., Bonasio, R., Sarma, K. Mapping native r-loops genome-wide using a targeted nuclease approach. Cell Reports. 29 (5), 1369-1380 (2019).
- Hu, Z., Zhang, A., Storz, G., Gottesman, S., Leppla, S. H. An antibody-based microarray assay for small RNA detection. Nucleic Acids Research. 34 (7), 52(2006).
- Nowotny, M., Gaidamakov, S. A., Crouch, R. J., Yang, W. Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell. 121 (7), 1005-1016 (2005).
- Sato, K., Egami, F. Studies on ribonucleases in takadiastase. Journal of Biochemistry. 44 (11), Tokyo. 753-767 (1957).
- Takahashi, K., Moore, S. Ribonuclease T1. Enzymes. 15, 435-468 (1982).
- Dunn, J. J. RNase III cleavage of single-stranded RNA: Effect of ionic strength in the fidelity of cleavage. Journal of Biological Chemistry. 251 (12), 3807-3814 (1976).
- Pertzev, A., Nicholson, A. W. Characterization of RNA sequence determinants and antideterminants of processing reactivity for a minimal substrate of Escherichia coli ribonuclease III. Nucleic Acids Research. 34 (13), 3708-3721 (2006).
- Phillips, D. D., et al. The sub-nanomolar binding of DNA-RNA hybrids by the single chain Fv fragment of antibody S9.6. Journal of Molecular Recognition. 26 (8), 376-381 (2013).
- Haruki, M., Noguchi, E., Kanaya, S., Crouch, R. J. Kinetic and stoichiometric analysis for the binding of Escherichia coli ribonuclease H1 to RNA-DNA hybrids using surface plasmon resonance. Journal of Biological Chemistry. 272 (35), 22015-22022 (1997).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados