
Method Article
Análise R-Loop por Dot-Blot
Neste Artigo
Resumo
Este protocolo detalha um método simples que quantifica o R-loop, uma estrutura de ácido nucleico de três fitas que compreende um híbrido de RNA-DNA e uma fita de DNA deslocada.
Resumo
A estrutura de ácido nucleico de três fitas, R-loop, é cada vez mais reconhecida por seu papel na regulação gênica. Inicialmente, pensava-se que os R-loops eram os subprodutos da transcrição; mas descobertas recentes de menos R-loops em células doentes deixaram claro que os R-loops têm papéis funcionais em uma variedade de células humanas. Em seguida, é fundamental entender os papéis dos R-loops e como as células equilibram sua abundância. Um desafio no campo é a quantificação de R-loops, uma vez que grande parte do trabalho depende do anticorpo monoclonal S9.6, cuja especificidade para híbridos de RNA-DNA foi questionada. Aqui, usamos dot-blots com o anticorpo S9.6 para quantificar R-loops e mostrar a sensibilidade e especificidade deste ensaio com RNase H, RNase T1 e RNase III que clivam híbridos de RNA-DNA, RNA de fita simples e RNA de fita dupla, respectivamente. Este método é altamente reprodutível, usa equipamentos e reagentes gerais de laboratório e fornece resultados em dois dias. Este ensaio pode ser usado em ambientes clínicos e de pesquisa para quantificar R-loops e avaliar o efeito de mutações em genes como a senataxina na abundância de R-loop.
Introdução
Este protocolo fornece um guia passo a passo para um ensaio de dot-blot que permite uma rápida avaliação comparativa da abundância de R-loop, uma estrutura de ácido nucleico de três fitas. O R-loop se forma quando o RNA invade um DNA de fita dupla para gerar um híbrido de RNA-DNA e desloca a outra fita de DNA. Os R-loops são encontrados em diferentes estágios do ciclo de vida do RNA. No complexo transcricional, o RNA nascente é sintetizado complementar ao DNA molde e a fita não molde é deslocada. O híbrido RNA-DNA curto (<10 pb) é resolvido para liberar o RNA nascente para que ele possa deixar o complexo RNA polimerase através do canal de saída 1,2. Fora do complexo transcricional, o RNA nascente está próximo de seu molde de DNA, que ainda está ligeiramente desenrolado de ser copiado, portanto, o RNA pode rehibridizar com seu DNA molde formando R-loops3. Além disso, os R-loops podem se formar quando os complexos de replicação e transcrição colidem4 e na transcrição antisense5. Dadas as muitas oportunidades para sua formação, os R-loops não são raros e podem ser encontrados em 3-5% do genoma humano6, dependendo do status de transcrição da célula. As alças R são encontradas nos locais dos promotores do gene7 e da terminação5 no mRNA e ao longo do RNAribossômico 8, bem como do RNAde transferência 9. As alças R também estão nas regiões teloméricas dos cromossomos.
Os loops R desempenham um papel regulador. Eles regulam a expressão gênica afetando a transcrição nos promotores10,11, mediando a recombinação de troca de classe12 e facilitando a edição do genoma baseada em CRISPR 13,14,15. Como muitos eventos celulares, a abundância do R-loop é bem titulada; muitos ou poucos R-loops afetam a função celular normal16,17. Os R-loops são regulados por uma variedade de proteínas, incluindo RNase H, senataxina e outras helicases que desenrolam os híbridos RNA-DNA 18,19,20,21,22.
Para monitorar a abundância de R-loops, os métodos de todo o genoma primeiro enriquecem os R-loops com o anticorpo S9.6 8,23,24 ou com outras nucleases25, incluindo RNase H 10,26,27, e então avaliam o número de R-loops enriquecidos por sequenciamento. As primeiras versões desses métodos baseados em sequenciamento não alcançaram uma cobertura de sequência adequada para permitir uma quantificação precisa, mas a rápida melhoria nas tecnologias de sequenciamento agora permite a análise de loop R locus por locus. Técnicas de imunofluorescência também têm sido utilizadas para quantificar e localizar as alças R10,17. Esses métodos são abrangentes, mas não são práticos em muitos ambientes clínicos ou como avaliações iniciais, pois exigem equipamentos caros e análises especializadas.
É necessário um procedimento que possa ser feito uniformemente em todos os laboratórios em ambientes clínicos. Os pontos-borrões fornecem essa opção, pois podem ser realizados sem nenhum equipamento específico ou análise computacional. Como uma etapa preliminar ou em ambientes clínicos para avaliar os efeitos das mutações nas alças R, essas manchas de pontos devem fornecer resultados sensíveis e específicos. Aqui, descrevemos nosso ensaio que identifica especificamente os R-loops; exclui sinais de DNA de fita dupla (DS), RNA de fita dupla e RNA de fita simples. Nosso protocolo usa o anticorpo S9.627 para identificar híbridos de RNA-DNA em R-loops e incorpora RNase H, uma endorribonuclease que cliva e, portanto, leva à degradação do RNA em um híbrido RNA-DNA20,28, para garantir que os sinais detectados sejam os de híbridos. Também incorporamos RNase T1 que cliva RNA de fita simples em guanina29,30 e RNase III que cliva RNA de fita dupla, incluindo alças de haste 31,32 para verificar sinais inespecíficos. O anticorpo S9.6 reconhece híbridos de RNA-DNA de comprimentos variados, mesmo aqueles com apenas 8 nucleotídeosde comprimento 33.
Aqui, apresentamos o protocolo que começa com o isolamento de ácido nucleico seguido de preparação de dot-blot e detecção de R-loop com anticorpo S9.6. Nosso protocolo inclui etapas para garantir que quantidades iguais de amostras sejam carregadas e que os sinais sejam específicos. Ele fornece oligonucleotídeos para servir como controles positivos e negativos. Este é um método rápido, fácil e fácil de usar para avaliar a abundância do R-loop.
Protocolo
1. Lise celular para fracionamento nuclear
- Lave as células com 1x solução salina tamponada com fosfato (PBS) duas vezes. Remova as células das placas de cultura de tecidos usando técnicas padrão de dissociação celular, como tripsina. Conte as células usando um hemocitômetro.
NOTA: As etapas descritas abaixo foram usadas para a análise de fibroblastos primários da pele humana, embora uma variedade de tipos de células possa ser analisada. Os fibroblastos foram cultivados em meio basal contendo 10% de soro fetal bovino. Em alternativa, o tampão de lise celular (Quadro 1) pode ser adicionado directamente à cultura celular após a lavagem. - Transfira a suspensão celular para um tubo de 1,5 mL para pellet as células.
- Centrifugar a amostra a 300 x g durante 5 min a 4 °C. Aspire a mídia.
- Lave duas vezes com 1x PBS gelado usando as configurações de centrifugação na etapa 1.3.
- Adicione tampão de lise de células frias (Tabela 1) ao pellet celular (300 μL por 2 x 106 células). Pipete para cima e para baixo para ressuspender o pellet.
- Incube no gelo por 10 min.
- Gire a 500 x g por 5 min para pellet os núcleos.
- Rejeitar o sobrenadante e ressuspender o sedimento nuclear em 400 μL de tampão de lise nuclear frio (Quadro 1).
- Incube no gelo por 10 min.
NOTA: O fracionamento dos compartimentos nuclear e citoplasmático das células garante a especificidade do sinal. A qualidade da separação nuclear e citoplasmática pode ser avaliada antes de prosseguir (Tabela 2). - Adicione 3 μL de proteinase K a 20 mg / mL e incube por 3-5 h a 55 ° C.
NOTA: Os volumes indicados são para 2 x 106 células, aumente ou diminua conforme necessário.
2. Purificação do DNA genômico (que inclui híbridos de RNA-DNA)
- Se o DNA for viscoso, execute a sonicação para reduzir a viscosidade (por exemplo, sonicação em alta potência, 30 s ON/30 s OFF, por 2 min usando um banho-maria a 4 °C).
- Adicionar 400 μL de tampão de eluição (Tabela 1) e 400 μL de fenol:clorofórmio:álcool isoamílico (25:24:1 pH 8,0).
- Vórtice por 10 s.
- Gire para baixo a 12.000 x g por 5 min a 4 ° C.
- Transferir a fase aquosa (cerca de 350 μL) para um novo tubo.
- Extrair uma vez com 1 volume de clorofórmio, vórtice durante 10 s, depois centrifugar a 12 000 x g durante 5 min a 4 °C. Transferir a fase aquosa para um novo tubo (cerca de 300 μL).
- Adicione 35 μL de acetato de sódio 3 M (pH 5,2), 1 μL de glicogênio e 700 μL de etanol 100% gelado.
- Vórtice por 10 s e gire para baixo a 12.000 x g por 30 min a 4 ° C.
- Lave o pellet com 1 mL de etanol a 70%.
- Vórtice por 10 s e gire para baixo a 12.000 x g por 15 min a 4 ° C.
- Descarte o sobrenadante e deixe o pellet secar ao ar.
- Adicione 12 μL de tampão de eluição e vórtice por 10 s para ressuspender. Incubar a amostra durante 30 min a 37 °C com agitação ou a 4 °C durante a noite para ressuspender o sedimento.
- Meça a concentração de DNA usando espectrofotometria padrão.
NOTA: Os volumes indicados são para 2 x 106 células, aumente ou diminua conforme necessário. O ADN (com híbridos RNA-ADN) pode ser armazenado a -20 °C, se necessário.
3. Amostras de DNA de blotting (que incluem híbridos de RNA-DNA) em membranas de náilon
- Prepare diluições de ácidos nucléicos para as concentrações desejadas no tampão de eluição (ou seja, 50 ng/μL, 25 ng/μL ou 12,5 ng/μL). Essas amostras com uma faixa de concentrações (200, 100, 50, 25, 12,5 ng) garantem que haverá sinais dentro da faixa linear.
NOTA: Certifique-se de preparar amostra suficiente para réplicas técnicas e biológicas e para os vários tratamentos de RNase, consulte a Etapa 5. - Prepare uma membrana de nylon carregada positivamente para que haja espaço para cada amostra de 2 μL ocupar uma área de 0,5 cm2.
- Localize 2 μL de cada amostra em 2 membranas: uma para o anticorpo S9.6 e outra para o anticorpo dsDNA. Alternativamente, um aparelho de dot-blot ou slot-blot que permite o carregamento de amostras com volumes maiores pode ser usado.
- Deixe as amostras saturarem na membrana. Aguarde pelo menos 2 minutos antes de reticular a membrana com luz UV.
- Coloque a membrana no centro do dispositivo UV e reticule a membrana usando um reticulador UV usando a configuração "Auto Crosslink" (1,200 μJ x 100).
4. Detecção híbrida de RNA-DNA com anticorpo S9.6
- Incube a membrana em solução de bloqueio (5% de leite em solução salina tamponada com Tris com 0,05% de Tween-20 (TBST) por 1 h em temperatura ambiente em um shaker.
NOTA: Deve haver solução de bloqueio suficiente para cobrir a membrana. - Incubar as membranas durante a noite em anticorpo primário (em leite a 5% em TBST) a 4 °C com agitação. Adicione anticorpo anti-dsDNA (diluição 1:10.000) a uma membrana. Adicione 1μg/mL de anticorpo S9.6 à segunda membrana (diluição 1:1.000).
NOTA: O anticorpo S9.6 está disponível comercialmente ou no Dr. S. Leppla, NIAID, National Institutes of Health. - Remova o anticorpo primário e lave 3x com TBST. Realize cada lavagem por 5-10 min com agitação em temperatura ambiente.
- Incubar com anticorpo secundário conjugado com peroxidase de rábano (HRP) (anti-camundongo, diluição 1:5.000) em leite a 5% em TBST com agitação à temperatura ambiente.
NOTA: Anti-dsDNA e anti-RNA-DNA híbrido são anticorpos de camundongo. - Remova o anticorpo secundário e lave 3x com TBST por 5-10 min com agitação em temperatura ambiente.
- Desenvolva com reagentes de quimioluminescência aprimorada (ECL) para adquirir sinais para imagens.
- Quantifique a intensidade do sinal usando ferramentas de processamento de imagem padrão, como o ImageJ.
NOTA: A solução de problemas é detalhada na Tabela 2.
5. Tratamentos com ribonuclease para avaliar a especificidade do sinal
NOTA: O tratamento com RNase deve ser realizado nas amostras de ácido nucleico para demonstrar a especificidade da ligação de S9.6. O tratamento com RNase H, mas não com RNase T1 ou RNase III, deve resultar em uma redução na imunomarcação de S9.6.
- Digerir as amostras contendo híbridos de RNA-ADN, preparando-as em quatro tubos separados. Trate cada uma das 4 amostras com 5 U RNase H, 1000 U RNase T1, 0,5 U RNase III ou simulado. Incubar amostras a 37 °C durante 15 min em volumes de 20 μL.
- Carregar 2 μL de cada amostra numa membrana, conforme descrito no ponto 3.
6. Preparação de controles de oligonucleotídeos para avaliar a especificidade do sinal
NOTA: Os controles de oligonucleotídeos podem ser usados para demonstrar a especificidade da ligação S9.6. S9.6 reconhece híbridos de RNA-DNA, mas não controles de dsDNA ou dsRNA, como foi relatado anteriormente34.
- Dissolver os oligonucleótidos (quadro 3) em tampão de recozimento (Tris 10 mM, pH 8,0; NaCl 50 mM, EDTA 1 mM) a 100 μM.
- Prepare 4 tubos de reação para
- Híbrido RNA-DNA # 1: Misture 10 μL de fita superior de ssRNA com 10 μL de fita inferior de ssDNA e 80 μL de tampão de recozimento.
- Híbrido de RNA-DNA # 2: Misture 10 μL de fita superior de ssDNA com 10 μL de fita inferior de ssRNA e 80 μL de tampão de recozimento.
- dsRNA: Misture 10 μL de fita superior de ssRNA com 10 μL de fita inferior de ssRNA e 80 μL de tampão de recozimento.
- dsDNA: Misture 10 μL de fita superior de ssDNA com 10 μL de fita inferior de ssDNA e 80 μL de tampão de recozimento.
- Aqueça as 4 misturas da etapa 6.2 a 95 °C por 10 min.
- Deixe os tubos esfriarem lentamente até a temperatura ambiente para permitir o recozimento dos fios. Os padrões recozidos podem ser armazenados a -20 °C para uso posterior.
NOTA: A eficiência do recozimento deve ser verificada por eletroforese em gel não desnaturante. Os duplex migram mais lentamente do que os oligonucleotídeos não recozidos (Tabela 2). - Carregar 2 μL de cada amostra em 2 membranas, uma para o anticorpo S9.6 e outra para o anticorpo dsDNA, conforme descrito na secção 3.
- Execute as etapas descritas na seção 4.
7. Quantificação e normalização da intensidade do sinal S9.6 R-loop usando ImageJ.
- Salve imagens de S9.6, coloração dsDNA em formato TIFF e analise-as usando o software ImageJ (https://imagej.nih.gov/ij/).
- Selecione a opção de inversão de imagem (Editar | Inverter). Após a inversão, cada ponto ficará visível como branco contra um fundo escuro.
- Use a ferramenta de seleção de imagem oval para selecionar um oval que seja grande o suficiente para envolver o maior ponto na imagem.
- Use o gerenciador de ROI para adicionar a área selecionada para quantificação. Certifique-se de que as opções "Mostrar tudo" e "Rótulos" estejam selecionadas para que as regiões de interesse possam ser visualizadas.
- Use a mesma área de seleção oval usada durante a etapa 7.3 para adicionar regiões de interesse adicionais ao redor de cada ponto a ser quantificado. Use o atalho Command + Shift + E para copiar a área selecionada da etapa 7.3 para cada um dos pontos subsequentes.
- Meça a densidade integrada de cada uma das regiões de interesse.
- Divida a intensidade do sinal S9.6 para cada amostra pela medição do dsDNA para obter a relação do sinal S9.6/dsDNA. Verifique os resultados repetindo os experimentos (pelo menos triplicados para aquisição de sinal S9.6 e dsDNA). O erro padrão da média pode ser calculado a partir das razões de sinal S9.6 / dsDNA.
Resultados
Tratamento enzimático para avaliar a especificidade do anticorpo S9.6 (RNA-DNA).
Fibroblastos primários da pele humana foram cultivados17. O DNA com híbridos RNA-DNA foi isolado e quantificado. Dois μg das amostras foram digeridos com RNase T1, RNase H ou RNase III por 15 min a 37 °C. Uma amostra simulada também foi analisada para comparação com as amostras tratadas com RNase. As amostras (200, 100, 50, 25, 12,5 ou 6,25 ng) foram borradas em duas membranas diferentes, conforme descrito na seção 3. As membranas foram reticuladas, bloqueadas e uma delas foi sondada com anticorpo S9.6 (Figura 1A).
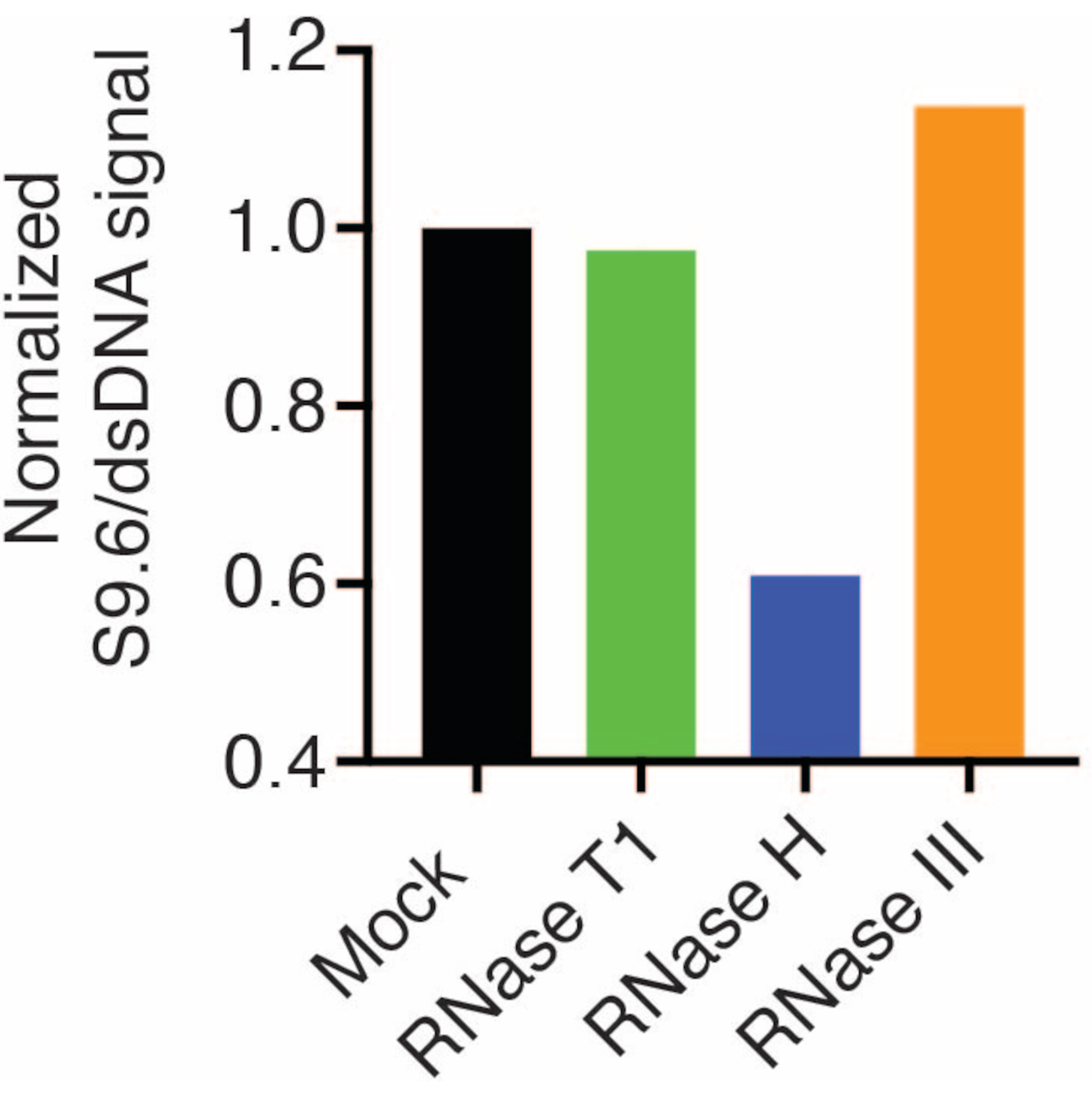
Os resultados mostraram que o sinal S9.6 se correlaciona com a abundância da amostra carregada. O tratamento com RNase H, mas não com RNase T1 ou RNase III, resulta em uma redução na coloração S9.6.
Uma segunda membrana foi sondada com um anticorpo dsDNA (Figura 1B) para a normalização. A imagem J foi usada para analisar as intensidades do sinal. As amostras de 50 ng foram selecionadas para quantificação, pois as intensidades de sinal dos anticorpos S9.6 e dsDNA estavam dentro da faixa dinâmica. As intensidades de sinal foram normalizadas para aquelas em amostras simuladas. Os dados são mostrados na Figura 2.
S9.6 anticorpo dot-blot usando controles de nucleotídeos sintéticos.
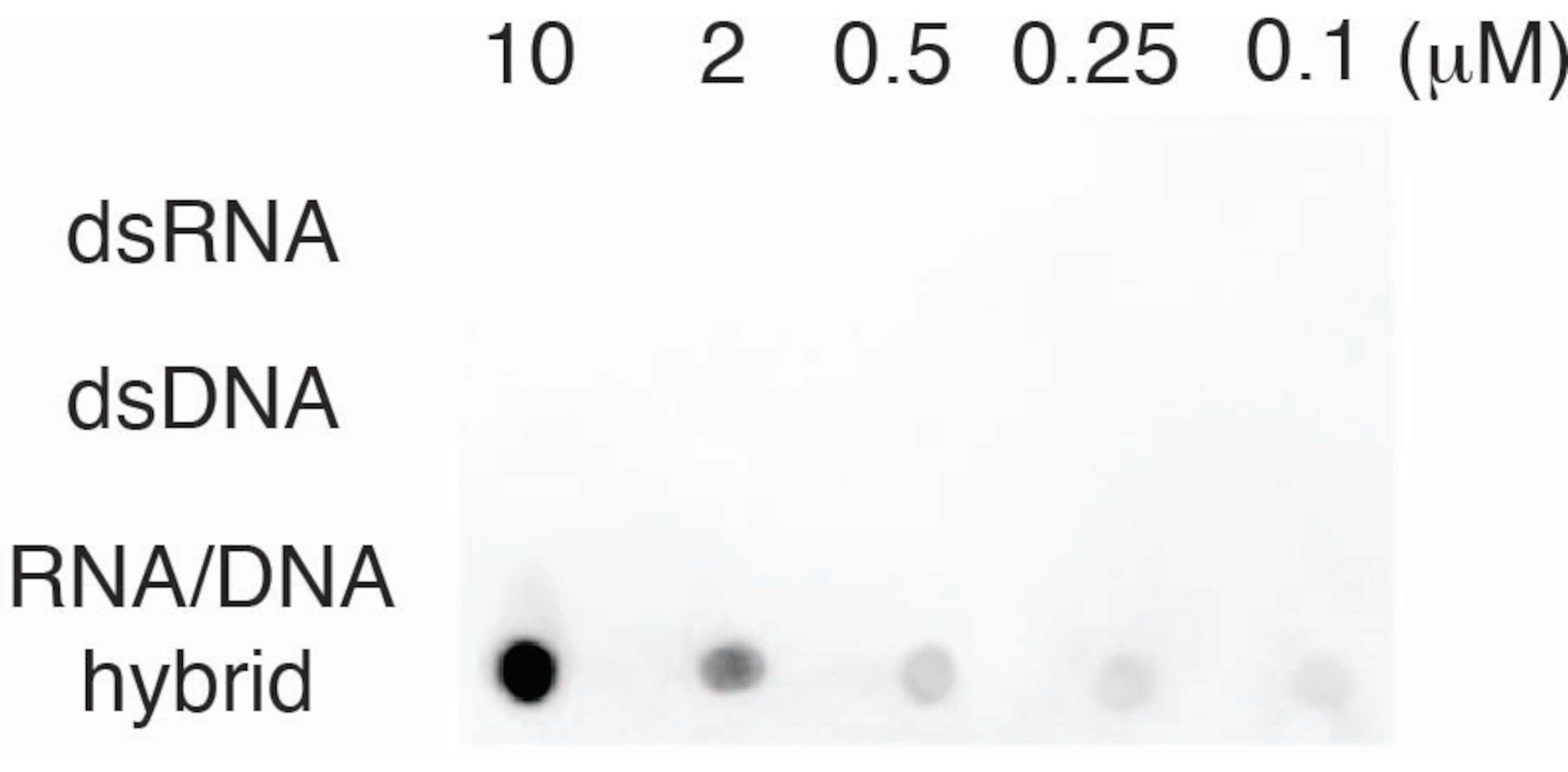
Para avaliar a especificidade do anticorpo S9.6, usamos oligonucleotídeos correspondentes a dsRNA, dsDNA e RNA-DNA, conforme descrito na seção 6. Uma série de diluições de nucleotídeos RNA-DNA, dsRNA e dsDNA foi preparada e borrada na membrana de náilon, conforme descrito na seção 3. A membrana foi sondada com o anticorpo S9.6 (Figura 3). Os resultados mostraram que o anticorpo S9.6 se liga especificamente a híbridos de RNA-DNA de maneira dose-dependente e mostrou reatividade cruzada mínima a dsRNAs e dsDNAs.

Figura 1: Especificidade de S9.6 conforme mostrado por dot-blot carregado com ácidos nucléicos de fibroblastos humanos. Amostras de ácido nucleico de fibroblastos humanos foram tratadas simuladamente ou tratadas com RNase T1, RNase H ou RNase III e, em seguida, carregadas em membranas de náilon em uma série de diluição de 200, 100, 50, 25, 12,5 e 6,25 ng por ponto de 2 μL. As membranas foram então sondadas com anticorpo S9.6 (A) ou anticorpo dsDNA (B). Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: Quantificação da coloração S9.6 R-loop. Amostras de 50 ng da Figura 1 foram selecionadas para quantificação com ImageJ. O sinal S9.6 foi dividido pela intensidade do sinal dsDNA e, em seguida, normalizado para a amostra simulada seguindo as etapas descritas na seção 7. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: S9.6 dot-blot usando controles de oligonucleotídeos. S9.6 contra uma série de diluição de oligonucleotídeos sintéticos como dsRNA, dsDNA ou híbrido de RNA-DNA. S9.6 liga-se especificamente a híbridos de RNA-DNA de maneira dose-dependente. Clique aqui para ver uma versão maior desta figura.
{kind=link}
| Tampão de lise celular | Para 10mL | Para 200mL | Final Conc. |
| Água, sem nuclease | 9mL | 180mL | - |
| 10% NP-40 | 0,5mL | 10mL | 0.5% |
| 2M KCl | 0,4mL | 8mL | 80mM |
| TUBOS DE 0,5 M (pH 8,0) | 100uL | 2mL | 5mM |
| Tampão de lise nuclear | Para 10mL | Para 200mL | Final Conc. |
| Água, sem nuclease | 8,65mL | 173mL | - |
| 10% SDS | 1mL | 20mL | 1% |
| 1M Tris-HCl (pH 8,0) | 0,25mL | 5mL | 25 mM |
| EDTA de 0,5 milhões | 100uL | 2mL | 5 mM |
| Tampão de eluição | Para 10mL | Para 200mL | Final Conc. |
| 1M Tris-Cl, pH 8,5 | 0,1 mL | 2 mL | 10 mM |
| Água, sem nuclease | 9,9 ml | 198 ml | - |
Tabela 1. Preparação de tampões
| Passo | Problema | Possível razão | Solução |
| 1.9 | N/A | Verificar o fracionamento da célula | A qualidade da separação nuclear e citoplasmática pode ser avaliada adicionando coquetéis inibidores de protease padrão aos tampões de lise celular e nuclear (Tabela 1). As frações citoplasmáticas e nucleares podem ser avaliadas por análise de western blotting para confirmar a restrição adequada da marcação com marcadores citoplasmáticos (por exemplo, GAPDH ou HSP90) em frações citoplasmáticas e marcação com marcadores nucleares (por exemplo, HDAC1 ou histona H3) em frações nucleares. A contribuição da contaminação mitocondrial na fração nuclear pode ser avaliada por análise de qPCR com sondas específicas para DNA mitocondrial. |
| 2.1 | A amostra é muito viscosa | O número de células é muito alto. | Reduza o DNA pela metade e continue com a etapa de sonicação 2.1 |
| 2.12 | Nenhum pellet visível | Material de partida insuficiente ou perda durante a extração. | Comece do início usando mais células. |
| 2.13 | Baixa concentração de DNA | ||
| 3.1 | DNA insuficiente para diluições | ||
| 3.4 | A amostra não satura na membrana | Mergulhe a membrana em 1x TBST. Deixe o excesso de tampão secar e comece na etapa 3.4 | |
| 4.6 | Padrão irregular ou salpicado é detectado | Adicione 0,1% de dodecil sulfato de sódio (SDS) à amostra e continue na etapa 3.1 | |
| 4.6 | Os pontos têm uma aparência de "anel de café" | Adicione 0,01% de Sarkosyl à amostra e continue na etapa 3.1 | |
| 5.2 | O controle RNaseH não mostra diminuição no sinal | A digestão da ribonuclease é incompleta. | Aumente a incubação ou aumente a concentração da enzima. |
| 6.5 | Sem sinal para os controles oligo | Duplex não foi formado. Os oligonucleotídeos não foram recozidos adequadamente. | Verificar as proporções de oligonucleotídeos e tampão de recozimento. |
| 6.5 | O sinal S9.6 não é específico para híbridos | O lote de anticorpos S9.6 tem ligação não específica | Valide a sensibilidade e especificidade de novos lotes de anticorpos S9.6 com o uso de tratamentos com enzimas RNase ou análise de oligonucleotídeos sintéticos. |
Tabela 2: Solução de problemas
| ssRNA, fita superior | 5'-UGGGGGCUCGUCCGGGAUAUAUGGGAACCACUGAUCCC-3' |
| ssDNA, fita superior | 5'-TGGGGGCTCGTCCGGGATATGGGAACCACTGATCCC-3' |
| ssRNA, fita inferior | 5'-GGGAUCAGUGGUUCCCAUAUCCCGGACGAGCCCCCA-3' |
| ssDNA, fita inferior | 5'-GGGATCAGTGGTTCCCATATCCCGGACGAGCCCCCA-3' |
Tabela 3. Sequências de oligonucleotídeos de controle
Discussão
Os ácidos nucléicos de fita 3, R-loops, formam-se em diferentes estágios durante o ciclo de vida do RNA e são cada vez mais encontrados para regular os processos celulares. Para entender completamente os R-loops, são necessárias técnicas confiáveis para detecção de R-loop. Aqui, descrevemos uma abordagem para interrogar a abundância de R-loops usando o anticorpo S9.6 8,23,24. Este método permite uma avaliação rápida da abundância de R-loop de células e amostras de cultura de tecidos. Não requer equipamento especial ou grande quantidade de material de partida. Garante resultados específicos e reprodutíveis usando uma combinação de tratamentos com RNase.
Alguns relataram preocupações sobre a especificidade do anticorpo S9.6. Como acontece com qualquer reagente, pode haver variabilidade de lote para lote com o anticorpo S9.6. Nosso protocolo inclui RNase H, RNase T1 e RNase III para verificar a especificidade do sinal. Além disso, usamos oligonucleotídeos sintéticos para garantir a especificidade de cada lote de anticorpo S9.6.
A biologia R-loop é um campo em crescimento; o desenvolvimento de métodos confiáveis de detecção e quantificação, como o apresentado aqui, facilitará estudos mecanísticos para elucidar quando os R-loops se formam, como são regulados e o que regulam. Com controles apropriados, este ensaio de mancha de pontos é um método simples para rastrear a abundância de R-loop em ambientes clínicos e de pesquisa.
Divulgações
Os autores não têm nada a divulgar.
Agradecimentos
Este trabalho foi apoiado pelo Howard Hughes Medical Institute e Intramural Research no National Institute of Neurological Disorders and Stroke.
Agradecemos ao Dr. Stephen Leppla por fornecer lotes de anticorpos S9.6 para análise. Também agradecemos ao Dr. Dongjun Li por sua assistência nos tratamentos com ribonuclease.
Materiais
| Name | Company | Catalog Number | Comments |
| Anti-dsDNA antibody | Abcam | ab27156 | |
| Anti-RNA-DNA hybrid antibody (S9.6) | Kerafast | ENH001 | |
| Biorupter sonicator | Diagenode | UCD-200 | |
| EB Buffer | Qiagen | 19086 | |
| EDTA (0.5M) | Invitrogen | AM9261 | |
| Hybond N+ nylon membrane | GE healthcare Life Sciences | RPN203B | |
| KCl (2M) | Invitrogen | AM9640G | |
| NP-40 (Igepal CA-630) | Sigma | I8896 | |
| PBS | Invitrogen | 10010-023 | |
| Phenol:chloroform:isoamyl alcohol | Invitrogen | 15593031 | |
| PIPES (0.5M, pH 8.0) | VWR | AAJ61406-AE | |
| Proteinase K | Qiagen | 19131 | |
| RNase III | Invitrogen | AM2290 | |
| RNase H | New England Biolabs | M0297 | |
| RNase T1 | ThermoFisher Sci. | EN0541 | |
| SDS (10%) | Invitrogen | 15553027 | |
| sodium acetate (3M, pH 5.2) | Invitrogen | AM9740 | |
| Tris-buffered saline (10X) | Corning | 46-012-CM | |
| Tris-HCl (1M, pH 8.0) | KD Medical | RGF-3360 | |
| TrypLE | Invitrogen | 12605010 | |
| Tween-20 | Sigma | P9416 | |
| UV Stratalinker 2400 | Stratagene | Stratalinker 2400 | |
| Whatman marking pen | Sigma | WHA10499001 |
Referências
- Daube, S. S., von Hippel, P. H. RNA displacement pathways during transcription from synthetic RNA-DNA bubble duplexes. Biochemistry. 33 (1), 340-347 (1994).
- Westover, K. D., Buschnell, D. A., Kornberg, R. D. Structural basis of transcription: separation of RNA from DNA by RNA polymerase II. Science. 303 (5660), 1014-1016 (2004).
- Itoh, T., Tomizawa, J. Formation of an RNA primer for initiation of replication of ColE1 DNA by ribonuclease H. Proceedings of the National Academy of Science USA. 77 (5), 2450-2454 (1980).
- Hamperl, S., Bocek, M. J., Saldivar, J. C., Swigut, T., Cimprich, K. A. Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell. 170 (4), 774-786 (2017).
- Skourti-Stathaki, K., Kamieniarz-Gdula, K., Proudfoot, N. J. R-loops induce repressive chromatin marks over mammalian gene terminators. Nature. 516 (7531), 436-439 (2014).
- Sanz, L. A., et al. conserved R-loop structures associate with specific epigenomic signatures in mammals. Molecular Cell. 63 (1), 167-178 (2016).
- Chen, L., et al. R-ChIP using inactive RNase H reveals dynamic coupling of R-loops with transcriptional pausing at gene promoters. Molecular Cell. 68 (4), 745-757 (2017).
- El Hage, A., French, S. L., Beyer, A. L., Tollervey, D. Loss of topoisomerase I leads to R-loop mediated transcriptional blocks during ribosomal RNA synthesis. Genes and Development. 24 (14), 1546-1558 (2010).
- Tran, P., et al. PIF1 family DNA helicases suppress R-loop mediated genome instability at tRNA genes. Nature Communication. 8, 15025(2017).
- Ginno, P. A., Lott, P. L., Christensen, H. C., Korf, I., Chedin, F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Molecular Cell. 45 (6), 814-825 (2012).
- Colak, D., et al. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science. 343 (6174), 1002-1005 (2014).
- Yu, K., Chedin, F., Hsieh, C., Wilson, T. E., Lieber, M. R. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nature Immunology. 4 (5), 442-451 (2003).
- Gasiunas, G., Barrangou, R., Horvath, P., Siksnys, V. Cas9-crRNA ribonucleotide complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proceedings of the National Academy of Science USA. 109 (39), 2579-2586 (2012).
- Westra, E. R., et al. CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by Cascade and Cas3. Molecular Cell. 46 (5), 595-605 (2012).
- Jiang, F., et al. Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science. 351 (6275), 867-871 (2016).
- Huertas, P., Aguilera, A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Molecular Cell. 12 (3), 711-721 (2003).
- Grunseich, C., et al. Senataxin mutation reveals how R-loops promote transcription by blocking DNA methylation at gene promoters. Molecular Cell. 69 (3), 426-437 (2018).
- Kim, H. D., Choe, J., Seo, Y. S. The sen1(+) gene of Schizosaccharomyces pombe, a homologue of budding yeast SEN1, encodes an RNA and DNA helicase. Biochemistry. 38 (44), 14697-14710 (1999).
- Stein, H., Hausen, P. Enzyme from calf thymus degrading the RNA moiety of DNA-RNA hybrids: effect on DNA-dependent RNA polymerase. Science. 166 (3903), 393-395 (1969).
- Cerritelli, S. M., Crouch, R. J. Ribonuclease H: the enzymes in eukaryotes. FEBS Journal. 276 (6), 1494-1505 (2009).
- Hyjek, M., Figiel, M., Nowotny, M. RNases H: Structure and mechanism. DNA Repair. 84, 102672(2019).
- Pohl, T. J., Zakian, V. A. Pif1 family DNA helicases: A helpmate to RNase H. DNA Repair. 84, 102633(2019).
- Pohjoismaki, J. L., et al. Mammalian mitochondrial DNA replication intermediates are essentially duplex but contain extensive tracts of RNA/DNA hybrid. Journal of Molecular Biology. 397 (5), 1144-1155 (2010).
- Sanz, L. A., Chedin, F. High-resolution, strand-specific R-loop mapping via S9.6-based DNA-RNA immunoprecipitation and high-throughput sequencing. Nature Protocols. 14 (6), 1734-1755 (2019).
- Wahba, L., Costantino, L., Tan, F. J., Zimmer, A., Koshland, D. S1-DRIP-seq identifies high expression and polyA tracts as major contributors to R-loop formation. Genes and Development. 30 (11), 1327-1338 (2016).
- Yan, Q., Shields, E. J., Bonasio, R., Sarma, K. Mapping native r-loops genome-wide using a targeted nuclease approach. Cell Reports. 29 (5), 1369-1380 (2019).
- Hu, Z., Zhang, A., Storz, G., Gottesman, S., Leppla, S. H. An antibody-based microarray assay for small RNA detection. Nucleic Acids Research. 34 (7), 52(2006).
- Nowotny, M., Gaidamakov, S. A., Crouch, R. J., Yang, W. Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell. 121 (7), 1005-1016 (2005).
- Sato, K., Egami, F. Studies on ribonucleases in takadiastase. Journal of Biochemistry. 44 (11), Tokyo. 753-767 (1957).
- Takahashi, K., Moore, S. Ribonuclease T1. Enzymes. 15, 435-468 (1982).
- Dunn, J. J. RNase III cleavage of single-stranded RNA: Effect of ionic strength in the fidelity of cleavage. Journal of Biological Chemistry. 251 (12), 3807-3814 (1976).
- Pertzev, A., Nicholson, A. W. Characterization of RNA sequence determinants and antideterminants of processing reactivity for a minimal substrate of Escherichia coli ribonuclease III. Nucleic Acids Research. 34 (13), 3708-3721 (2006).
- Phillips, D. D., et al. The sub-nanomolar binding of DNA-RNA hybrids by the single chain Fv fragment of antibody S9.6. Journal of Molecular Recognition. 26 (8), 376-381 (2013).
- Haruki, M., Noguchi, E., Kanaya, S., Crouch, R. J. Kinetic and stoichiometric analysis for the binding of Escherichia coli ribonuclease H1 to RNA-DNA hybrids using surface plasmon resonance. Journal of Biological Chemistry. 272 (35), 22015-22022 (1997).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados