
Method Article
R-Loop-Analyse mittels Dot-Blot
In diesem Artikel
Zusammenfassung
Dieses Protokoll beschreibt eine einfache Methode zur Quantifizierung der R-Schleife, einer dreisträngigen Nukleinsäurestruktur, die aus einem RNA-DNA-Hybrid und einem verschobenen DNA-Strang besteht.
Zusammenfassung
Die dreisträngige Nukleinsäurestruktur, die R-Schleife, wird zunehmend für ihre Rolle bei der Genregulation anerkannt. Ursprünglich dachte man, dass R-Schleifen die Nebenprodukte der Transkription sind; Jüngste Befunde von weniger R-Schleifen in erkrankten Zellen machten jedoch deutlich, dass R-Schleifen in einer Vielzahl von menschlichen Zellen eine funktionelle Rolle spielen. Als nächstes ist es wichtig, die Rolle der R-Schleifen zu verstehen und zu verstehen, wie Zellen ihre Häufigkeit ausgleichen. Eine Herausforderung in diesem Bereich ist die Quantifizierung von R-Schleifen, da ein Großteil der Arbeit auf dem monoklonalen Antikörper S9.6 beruht, dessen Spezifität für RNA-DNA-Hybride in Frage gestellt wurde. Hier verwenden wir Dot-Blots mit dem S9.6-Antikörper, um R-Schleifen zu quantifizieren und die Sensitivität und Spezifität dieses Assays mit RNase H, RNase T1 und RNase III zu zeigen, die RNA-DNA-Hybride, einzelsträngige RNA bzw. doppelsträngige RNA spalten. Diese Methode ist hochgradig reproduzierbar, verwendet allgemeine Laborgeräte und Reagenzien und liefert Ergebnisse innerhalb von zwei Tagen. Dieser Assay kann in der Forschung und im klinischen Umfeld eingesetzt werden, um R-Schleifen zu quantifizieren und die Wirkung von Mutationen in Genen wie Senataxin auf die R-Schleifen-Häufigkeit zu bewerten.
Einleitung
Dieses Protokoll bietet eine Schritt-für-Schritt-Anleitung für einen Dot-Blot-Assay, der eine schnelle vergleichende Bewertung der Häufigkeit von R-Loop, einer dreisträngigen Nukleinsäurestruktur, ermöglicht. Eine R-Schleife entsteht, wenn RNA in eine doppelsträngige DNA eindringt, um einen RNA-DNA-Hybrid zu erzeugen, und den anderen DNA-Strang verdrängt. R-Schleifen kommen in verschiedenen Stadien des Lebenszyklus von RNA vor. Im Transkriptionskomplex wird die naszierende RNA komplementär zur Matrizen-DNA synthetisiert, und der Nicht-Matrizenstrang wird verdrängt. Der kurze RNA-DNA-Hybrid (<10 bp) wird aufgelöst, um die naszierende RNA zu befreien, so dass sie den RNA-Polymerase-Komplex durch den Austrittskanal 1,2 verlassen kann. Außerhalb des Transkriptionskomplexes befindet sich die entstehende RNA in der Nähe ihres DNA-Matrizes, der noch leicht vom Kopieren abgewickelt ist, so dass die RNA mit ihrer Matrizen-DNA rehybridisieren und R-Schleifenbilden kann 3. Darüber hinaus können sich R-Schleifen bilden, wenn Replikations- und Transkriptionskomplexe kollidieren4 und bei der Antisense-Transkription5. Angesichts der vielen Möglichkeiten ihrer Bildung sind R-Schleifen nicht selten und können je nach Transkriptionsstatus der Zelle in 3-5% des menschlichen Genoms gefunden werden6. R-Schleifen finden sich in den Genpromotoren7 und Terminierungsstellen5 in der mRNA sowie entlang der ribosomalen RNA8 sowie der Transfer-RNA9. R-Schleifen befinden sich auch in telomeren Regionen von Chromosomen.
R-Loops spielen eine regulatorische Rolle. Sie regulieren die Genexpression, indem sie die Transkription an den Promotoren10,11 beeinflussen, die Klassenschalter-Rekombinationvermitteln 12 und die CRISPR-basierte Genomeditierung erleichtern 13,14,15. Wie bei vielen zellulären Ereignissen ist die R-Schleifen-Abundanz eng titriert; Zu viele oder zu wenige R-Schleifen beeinträchtigen die normale Zellfunktion16,17. R-Schleifen werden von einer Vielzahl von Proteinen reguliert, darunter RNase H, Senataxin und andere Helikasen, die die RNA-DNA-Hybride 18,19,20,21,22 abwickeln.
Um die Häufigkeit von R-Schleifen zu überwachen, reichern genomweite Methoden zunächst für R-Schleifen mit dem Antikörper S9.6 8,23,24 oder mit anderen Nukleasen25 einschließlich RNase H 10,26,27 an und bestimmen dann die Anzahl der angereicherten R-Schleifen durch Sequenzierung. Frühe Versionen dieser sequenzierungsbasierten Methoden erreichten keine ausreichende Sequenzabdeckung, um eine präzise Quantifizierung zu ermöglichen, aber die schnelle Verbesserung der Sequenzierungstechnologien ermöglicht nun eine R-Loop-Analyse von Locus zu Locus. Immunfluoreszenztechniken wurden auch verwendet, um R-Schleifen zu quantifizieren und zu lokalisieren 10,17. Diese Methoden sind umfassend, aber in vielen klinischen Umgebungen oder als Ersteinschätzung nicht praktikabel, da sie teure Geräte und spezielle Analysen erfordern.
Es wird ein Verfahren benötigt, das in allen Laboren im klinischen Umfeld einheitlich durchgeführt werden kann. Dot-Blots bieten eine solche Option, da sie ohne spezielle Ausrüstung oder computergestützte Analyse durchgeführt werden können. Als vorbereitender Schritt oder im klinischen Umfeld, um die Auswirkungen von Mutationen auf R-Schleifen zu bewerten, müssen diese Dot-Blots empfindliche und spezifische Ergebnisse liefern. Hier beschreiben wir unseren Assay, der R-Schleifen spezifisch identifiziert; Es schließt Signale von doppelsträngiger (ds) DNA, doppelsträngiger RNA und einzelsträngiger RNA aus. Unser Protokoll verwendet den S9.6-Antikörper27 zur Identifizierung von RNA-DNA-Hybriden in R-Schleifen und enthält RNase H, eine Endoribonuklease, die spaltet und daher zum Abbau der RNA in einem RNA-DNA-Hybriden 20,28 führt, um sicherzustellen, dass die detektierten Signale die von Hybriden sind. Wir haben auch RNase T1 eingebaut, die einzelsträngige RNA an Guanin29,30 spaltet, und RNase III, die doppelsträngige RNA einschließlich der Stammschleifen31,32 spaltet, um auf unspezifische Signale zu prüfen. Der S9.6-Antikörper erkennt RNA-DNA-Hybride unterschiedlicher Länge, auch solche, die nur 8 Nukleotide lang sind33.
Hier stellen wir das Protokoll vor, das mit der Nukleinsäureisolierung beginnt, gefolgt von der Dot-Blot-Vorbereitung und dem R-Loop-Nachweis mit S9.6-Antikörpern. Unser Protokoll enthält Schritte, um sicherzustellen, dass gleiche Mengen an Proben geladen werden und die Signale spezifisch sind. Es liefert Oligonukleotide, die als Positiv- und Negativkontrollen dienen. Dies ist eine schnelle, einfache und benutzerfreundliche Methode, um die R-Loop-Häufigkeit zu bewerten.
Protokoll
1. Zelllyse für die Kernfraktionierung
- Waschen Sie die Zellen zweimal mit 1x phosphatgepufferter Kochsalzlösung (PBS). Entnehmen Sie Zellen aus Gewebekulturschalen mit Standard-Zelldissoziationstechniken wie Trypsin. Zählen Sie Zellen mit einem Hämozytometer.
HINWEIS: Die unten beschriebenen Schritte wurden für die Analyse primärer menschlicher Hautfibroblasten verwendet, obwohl eine Reihe von Zelltypen getestet werden kann. Die Fibroblasten wurden in Basalmedien gezüchtet, die 10% fötales Rinderserum enthielten. Alternativ kann Zelllysepuffer (Tabelle 1) nach dem Waschen direkt in die Zellkultur gegeben werden. - Übertragen Sie die Zellsuspension in ein 1,5-ml-Röhrchen, um die Zellen zu pelletieren.
- Die Probe wird bei 300 x g für 5 min bei 4 °C zentrifugiert. Saugen Sie das Medium an.
- Zweimal mit eiskaltem 1x PBS unter Verwendung der Zentrifugationseinstellungen in Schritt 1.3 waschen.
- Geben Sie dem Zellpellet kalten Zelllysepuffer (Tabelle 1) hinzu (300 μl pro 2 x 106 Zellen). Pipettieren Sie auf und ab, um das Pellet wieder aufzuhängen.
- 10 min auf Eis inkubieren.
- Bei 500 x g für 5 min drehen, um die Kerne zu pelletieren.
- Der Überstand wird verworfen und das Kernpellet in 400 μl kaltem Kernlysepuffer wieder suspendiert (Tabelle 1).
- 10 min auf Eis inkubieren.
HINWEIS: Die Fraktionierung der nuklearen und zytoplasmatischen Kompartimente der Zellen gewährleistet die Signalspezifität. Die Qualität der nuklearen und zytoplasmatischen Trennung kann vor dem Fortfahren bewertet werden (Tabelle 2). - 3 μl 20 mg/ml Proteinase K zugeben und 3-5 h bei 55 °C inkubieren.
HINWEIS: Die angegebenen Volumina gelten für 2 x 106 Zellen, skalieren Sie nach Bedarf nach oben oder unten.
2. Reinigung von genomischer DNA (einschließlich RNA-DNA-Hybriden)
- Wenn die DNA viskos ist, führen Sie eine Beschallung durch, um die Viskosität zu verringern (z. B. Beschallung bei hoher Ausgangsleistung, 30 s EIN/ 30 s AUS, für 2 Minuten mit einem 4 °C Wasserbad).
- 400 μl Elutionspuffer (Tabelle 1) und 400 μl Phenol:Chloroform:Isoamylalkohol (25:24:1 pH 8,0) zugeben.
- Vortex für 10 s.
- Bei 12.000 x g für 5 min bei 4 °C runterschleudern.
- Die wässrige Phase (ca. 350 μl) wird in ein neues Röhrchen überführt.
- Einmal mit 1 Volumen Chloroform extrahieren, 10 s vortexen, dann 5 min bei 4 °C bei 12.000 x g herunterschleudern. Die wässrige Phase in ein neues Röhrchen (ca. 300 μl) überführen.
- Fügen Sie 35 μl 3 M Natriumacetat (pH 5,2), 1 μl Glykogen und 700 μl eiskaltes 100%iges Ethanol hinzu.
- 10 s vortexen und 30 min bei 4 °C bei 12.000 x g herunterschleudern.
- Waschen Sie das Pellet mit 1 ml 70%igem Ethanol.
- 10 s vortexen und 15 min bei 4 °C bei 12.000 x g herunterschleudern.
- Entsorgen Sie den Überstand und lassen Sie das Pellet an der Luft trocknen.
- 12 μl Elutionspuffer zugeben und 10 s lang vortexen, um zu resuspendieren. Die Probe wird 30 Minuten lang bei 37 °C unter Rühren oder über Nacht bei 4 °C inkubiert, um das Pellet wieder zu suspendieren.
- Messen Sie die DNA-Konzentration mit Hilfe der Standard-Spektrophotometrie.
HINWEIS: Die angegebenen Volumina gelten für 2 x 106 Zellen, skalieren Sie nach Bedarf nach oben oder unten. DNA (mit RNA-DNA-Hybriden) kann bei Bedarf bei -20 °C gelagert werden.
3. Blotten von DNA-Proben (die RNA-DNA-Hybride enthalten) auf Nylonmembranen
- Bereiten Sie Verdünnungen von Nukleinsäuren auf die gewünschten Konzentrationen im Elutionspuffer vor (d. h. 50 ng/μL, 25 ng/μL oder 12,5 ng/μL). Diese Proben mit einem Konzentrationsbereich (200, 100, 50, 25, 12,5 ng) stellen sicher, dass Signale innerhalb des linearen Bereichs vorhanden sind.
HINWEIS: Stellen Sie sicher, dass Sie genügend Proben für technische und biologische Replikate vorbereiten, und für die verschiedenen RNase-Behandlungen siehe Schritt 5. - Bereiten Sie eine positiv geladene Nylonmembran so vor, dass jede 2-μl-Probe Platz für eine Fläche von 0,5 cm2 hat.
- 2 μl jeder Probe auf 2 Membranen aufleuchten: eine für den S9.6-Antikörper und die andere für den dsDNA-Antikörper. Alternativ kann eine Dot-Blot- oder Slot-Blot-Apparatur verwendet werden, die das Laden von Proben mit größeren Volumina ermöglicht.
- Lassen Sie die Proben in die Membran einziehen. Warten Sie mindestens 2 Minuten, bevor Sie die Membran mit UV-Licht vernetzen.
- Platzieren Sie die Membran in der Mitte des UV-Gerätes und vernetzen Sie die Membran mit einem UV-Vernetzer mit der Einstellung "Auto Crosslink" (1.200 μJ x 100).
4. RNA-DNA-Hybrid-Nachweis mit S9.6-Antikörper
- Die Membran wird in einer Blockierungslösung (5 % Milch in Tris-gepufferter Kochsalzlösung mit 0,05 % Tween-20 (TBST)) 1 h bei Raumtemperatur auf einem Shaker inkubiert.
HINWEIS: Es sollte genügend Verstopfungslösung vorhanden sein, um die Membran zu bedecken. - Die Membranen werden über Nacht in Primärantikörpern (in 5 % Milch in TBST) bei 4 °C unter Schütteln inkubiert. Geben Sie einen Anti-dsDNA-Antikörper (1:10.000 Verdünnung) zu einer Membran. Geben Sie 1 μg/ml S9.6-Antikörper in die zweite Membran (1:1.000-Verdünnung).
HINWEIS: Der S9.6-Antikörper ist im Handel oder bei Dr. S. Leppla, NIAID, National Institutes of Health, erhältlich. - Primären Antikörper entfernen und 3x mit TBST waschen. Führen Sie jede Wäsche 5-10 Minuten lang unter Schütteln bei Raumtemperatur durch.
- Inkubieren Sie mit Meerrettichperoxidase (HRP)-konjugierten Sekundärantikörpern (Anti-Maus, 1:5.000 Verdünnung) in 5% Milch in TBST unter Schütteln bei Raumtemperatur.
HINWEIS: Anti-dsDNA und anti-RNA-DNA-Hybrid sind beide Maus-Antikörper. - Entfernen Sie den Sekundärantikörper und waschen Sie 3x mit TBST für 5-10 min unter Schütteln bei Raumtemperatur.
- Entwickeln Sie mit ECL-Reagenzien (Enhanced-Chemilumineszenz), um Signale für die Bildgebung zu erfassen.
- Quantifizieren Sie die Signalintensität mit Standard-Bildverarbeitungstools wie ImageJ.
HINWEIS: Die Fehlerbehebung ist in Tabelle 2 beschrieben.
5. Ribonuklease-Behandlungen zur Bewertung der Signalspezifität
HINWEIS: Die RNase-Behandlung sollte an den Nukleinsäureproben durchgeführt werden, um die Spezifität der S9.6-Bindung nachzuweisen. Die Behandlung mit RNase H, aber nicht mit RNase T1 oder RNase III sollte zu einer Verringerung der S9.6-Immunfärbung führen.
- Verdauen Sie die Proben, die RNA-DNA-Hybride enthalten, indem Sie sie in vier separaten Röhrchen vorbereiten. Behandeln Sie jede der 4 Proben entweder mit 5 U RNase H, 1000 U RNase T1, 0,5 U RNase III oder Mock. Inkubieren Sie die Proben 15 Minuten lang bei 37 °C in einem Volumen von 20 μl.
- Laden Sie 2 μl jeder Probe auf eine Membran, wie in Abschnitt 3 beschrieben.
6. Vorbereitung von Oligonukleotidkontrollen zur Bewertung der Signalspezifität
HINWEIS: Oligonukleotidkontrollen können verwendet werden, um die Spezifität der S9.6-Bindung zu demonstrieren. S9.6 erkennt RNA-DNA-Hybride, aber keine dsDNA- oder dsRNA-Kontrollen, wie bereits berichtet wurde34.
- Oligonukleotide (Tabelle 3) in Annealing-Puffer (10 mM Tris, pH 8,0; 50 mM NaCl, 1 mM EDTA) auf 100 μM auflösen.
- Bereiten Sie 4 Reaktionsgefäße für
- RNA-DNA-Hybrid #1: Mischen Sie 10 μl ssRNA-Oberstrang mit 10 μl ssDNA-Unterstrang und 80 μl Annealing-Puffer.
- RNA-DNA-Hybrid #2: Mischen Sie 10 μl ssDNA-Oberstrang mit 10 μl ssRNA-Unterstrang und 80 μl Annealing-Puffer.
- dsRNA: Mischen Sie 10 μl ssRNA-Oberstrang mit 10 μl ssRNA-Unterstrang und 80 μl Annealing-Puffer.
- dsDNA: Mischen Sie 10 μl ssDNA-Oberstrang mit 10 μl ssDNA-Unterstrang und 80 μl Annealing-Puffer.
- Die 4 Mischungen aus Schritt 6.2 bei 95 °C 10 Min. erhitzen.
- Lassen Sie die Rohre langsam auf Raumtemperatur abkühlen, damit die Litzen wieder geglüht werden können. Geglühte Standards können für eine spätere Verwendung bei -20 °C gelagert werden.
HINWEIS: Die Glüheffizienz sollte durch eine nicht denaturierende Gelelektrophorese überprüft werden. Duplexe wandern langsamer als die ungetemperten Oligonukleotide (Tabelle 2). - 2 μl jeder Probe werden auf 2 Membranen geladen, eine für den S9.6-Antikörper und eine für den dsDNA-Antikörper, wie in Abschnitt 3 beschrieben.
- Führen Sie die in Abschnitt 4 beschriebenen Schritte aus.
7. Quantifizierung und Normalisierung der Signalintensität von S9.6 R-Schleifen mit ImageJ.
- Speichern Sie Bilder von S9.6- und dsDNA-Färbungen im TIFF-Format und analysieren Sie sie mit der ImageJ-Software (https://imagej.nih.gov/ij/).
- Wählen Sie die Option zum Umkehren des Bildes (Bearbeiten | Invertieren). Nach der Umkehrung ist jeder Punkt weiß auf dunklem Hintergrund sichtbar.
- Verwenden Sie das Auswahlwerkzeug für ovale Bilder, um ein Oval auszuwählen, das groß genug ist, um den größten Punkt auf dem Bild zu umgeben.
- Verwenden Sie den ROI-Manager, um den ausgewählten Bereich für die Quantifizierung hinzuzufügen. Stellen Sie sicher, dass die Optionen "Alle anzeigen" und "Beschriftungen" ausgewählt sind, damit die interessierenden Bereiche visualisiert werden können.
- Verwenden Sie den gleichen ovalen Auswahlbereich, der in Schritt 7.3 verwendet wurde, um zusätzliche interessante Bereiche um jeden zu quantifizierenden Punkt hinzuzufügen. Verwenden Sie die Tastenkombination Befehl + Umschalt + E , um den ausgewählten Bereich aus Schritt 7.3 in jeden der folgenden Punkte zu kopieren.
- Messen Sie die integrierte Dichte der einzelnen interessierenden Regionen.
- Teilen Sie die S9.6-Signalintensität für jede Probe durch die Messung der dsDNA, um das S9.6/dsDNA-Signalverhältnis zu erhalten. Überprüfen Sie die Ergebnisse, indem Sie die Experimente wiederholen (mindestens dreifache Wiederholung sowohl für die S9.6- als auch für die dsDNA-Signalerfassung). Der Standardfehler des Mittelwerts kann aus den Signalverhältnissen S9.6/dsDNA berechnet werden.
Ergebnisse
Enzymatische Behandlung zur Bewertung der Spezifität von S9.6 (RNA-DNA) Antikörpern.
Primäre menschliche Hautfibroblasten wurden gezüchtet17. DNA mit RNA-DNA-Hybriden wurde isoliert und quantifiziert. Zwei μg der Proben wurden mit RNase T1, RNase H oder RNase III für 15 min bei 37 °C aufgeschlossen. Eine Scheinprobe wurde ebenfalls zum Vergleich mit den RNase-behandelten Proben analysiert. Die Proben (200, 100, 50, 25, 12,5 oder 6,25 ng) wurden auf zwei verschiedene Membranen geblottet, wie in Abschnitt 3 beschrieben. Die Membranen wurden vernetzt, blockiert und eine von ihnen mit S9.6-Antikörpern untersucht (Abbildung 1A).
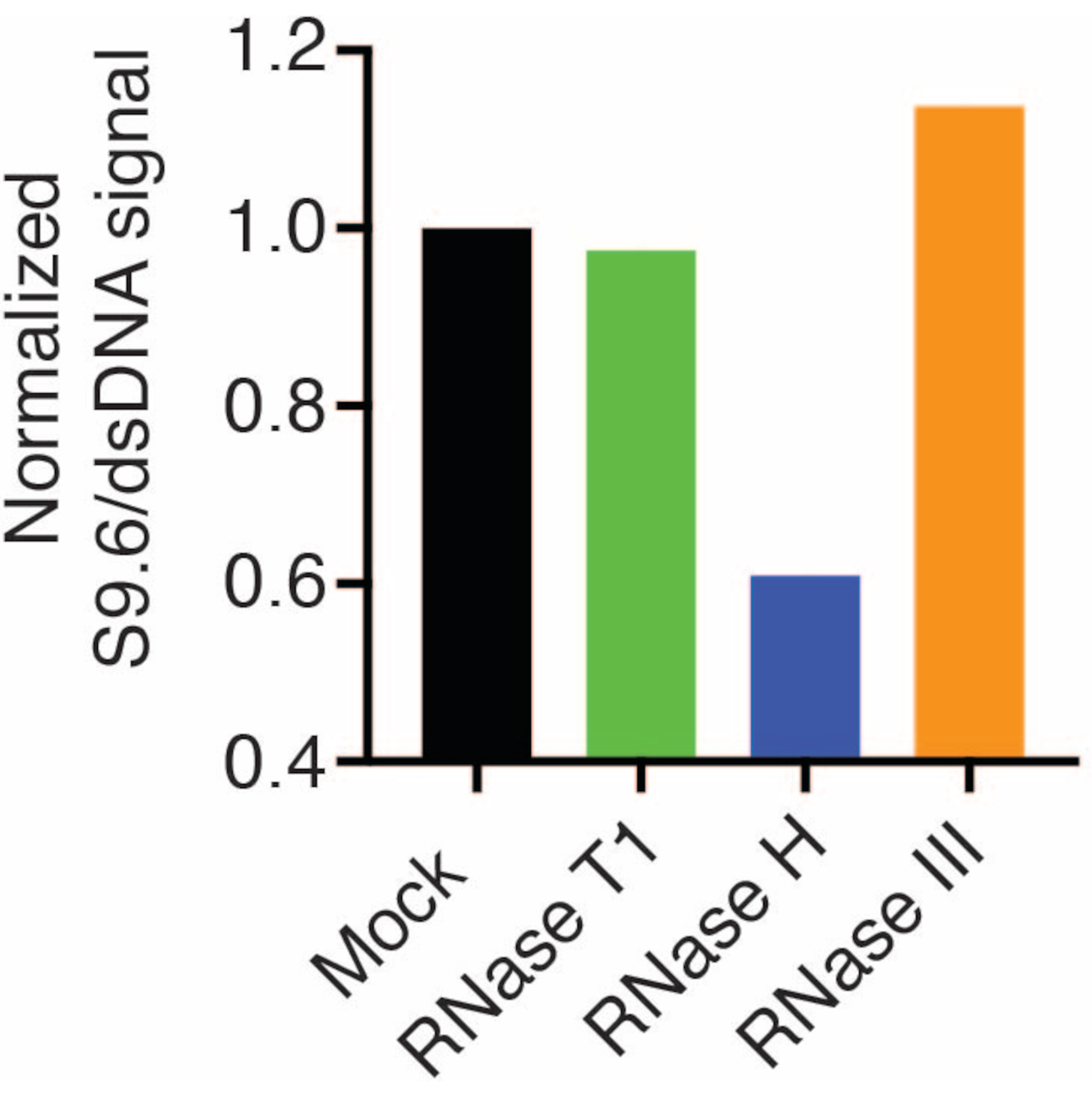
Die Ergebnisse zeigten, dass das S9.6-Signal mit der Häufigkeit der geladenen Probe korreliert. Die Behandlung mit RNase H, jedoch nicht mit RNase T1 oder RNase III, führt zu einer Verringerung der S9.6-Färbung.
Eine zweite Membran wurde zur Normalisierung mit einem dsDNA-Antikörper (Abbildung 1B) untersucht. Bild J wurde verwendet, um die Signalintensitäten zu analysieren. Die 50 ng-Proben wurden für die Quantifizierung ausgewählt, da die Signalintensitäten der S9.6- und dsDNA-Antikörper innerhalb des dynamischen Bereichs lagen. Die Signalintensitäten wurden auf die in Scheinproben normiert. Die Daten sind in Abbildung 2 dargestellt.
S9.6-Antikörper-Dot-Blot unter Verwendung synthetischer Nukleotidkontrollen.
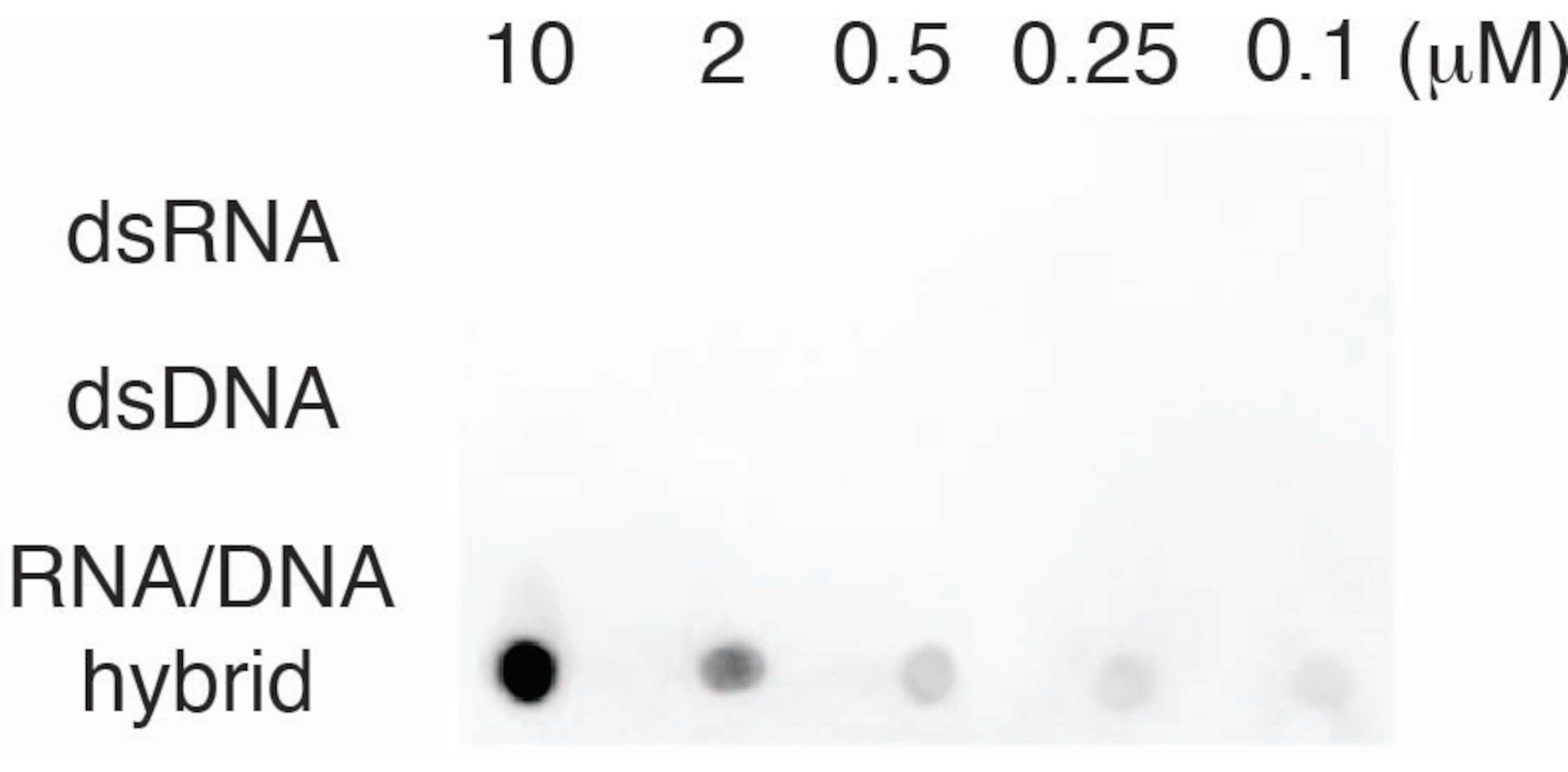
Um die Spezifität des S9.6-Antikörpers zu bewerten, verwendeten wir Oligonukleotide, die dsRNA, dsDNA und RNA-DNA entsprechen, wie in Abschnitt 6 beschrieben. Eine Verdünnungsreihe von RNA-DNA-, dsRNA- und dsDNA-Nukleotiden wurde hergestellt und auf die Nylonmembran geblottet, wie in Abschnitt 3 beschrieben. Die Membran wurde mit dem S9.6-Antikörper untersucht (Abbildung 3). Die Ergebnisse zeigten, dass der S9.6-Antikörper dosisabhängig spezifisch an RNA-DNA-Hybride bindet und eine minimale Kreuzreaktivität zu dsRNAs und dsDNAs zeigte.

Abbildung 1: Spezifität von S9.6, gezeigt durch einen Dot-Blot, der mit Nukleinsäuren aus humanen Fibroblasten beladen ist. Nukleinsäureproben von humanen Fibroblasten wurden entweder simuliert oder mit RNase T1, RNase H oder RNase III behandelt und dann in einer Verdünnungsreihe von 200, 100, 50, 25, 12,5 und 6,25 ng pro 2 μL Punkt auf Nylonmembranen geladen. Die Membranen wurden dann mit dem S9.6-Antikörper (A) oder dem dsDNA-Antikörper (B) untersucht. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2: Quantifizierung der S9.6 R-Schleifen-Färbung. 50 ng Proben aus Abbildung 1 wurden für die Quantifizierung mit ImageJ ausgewählt. Das S9.6-Signal wurde durch die dsDNA-Signalintensität dividiert und dann gemäß den in Abschnitt 7 beschriebenen Schritten auf die Scheinprobe normiert. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 3: S9.6 Dot-Blot unter Verwendung von Oligonukleotidkontrollen. S9.6 Antikörper-Dot-Blot gegen eine Verdünnungsreihe synthetischer Oligonukleotide als dsRNA, dsDNA oder RNA-DNA-Hybrid. S9.6 bindet spezifisch dosisabhängig an RNA-DNA-Hybride. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
| Puffer für die Zelllyse | Für 10 ml | Für 200 ml | Finale Konz. |
| Wasser, nukleasefrei | 9 ml | 180 ml | - |
| 10 % NP-40 | 0,5 ml | 10 ml | 0.5% |
| 2M KCl | 0,4 ml | 8 ml | 80mM |
| 0,5M ROHRE (pH 8,0) | 100 μl | 2 ml | 5 Mio. |
| Puffer für die nukleare Lyse | Für 10 ml | Für 200 ml | Finale Konz. |
| Wasser, nukleasefrei | 8,65 ml | 173 mL | - |
| 10% Sicherheitsdatenblatt | 1 ml | 20 ml | 1% |
| 1M Tris-HCl (pH 8,0) | 0,25 ml | 5 ml | ca. 25 mM |
| 0,5 M EDTA | 100 μl | 2 ml | 5 mM |
| Elutions-Puffer | Für 10 ml | Für 200 ml | Finale Konz. |
| 1M Tris-Cl, pH 8,5 | 0,1 mL | 2 mL | ca. 10 mM |
| Wasser, nukleasefrei | 9,9 mL | 198 mL | - |
Tabelle 1. Vorbereitung von Puffern
| Schritt | Problem | Mögliche Ursache | Lösung |
| 1.9 | N/A | Überprüfen der Zellfraktionierung | Die Qualität der nuklearen und zytoplasmatischen Trennung kann durch Zugabe von Standard-Proteaseinhibitor-Cocktails zu den Zell- und Kernlysepuffern beurteilt werden (Tabelle 1). Die zytoplasmatischen und nuklearen Fraktionen können durch Western-Blotting-Analyse ausgewertet werden, um eine adäquate Einschränkung der Markierung mit zytoplasmatischen Markern (z. B. GAPDH oder HSP90) in zytoplasmatischen Fraktionen und die Markierung mit nukleären Markern (z. B. HDAC1 oder Histon H3) in nuklearen Fraktionen zu bestätigen. Der Beitrag der mitochondrialen Kontamination in der Kernfraktion kann durch qPCR-Analyse mit Sonden bewertet werden, die spezifisch für mitochondriale DNA sind. |
| 2.1 | Probe ist zu viskos | Die Handynummer ist zu hoch. | Reduzieren Sie die DNA um die Hälfte und fahren Sie mit dem Beschallungsschritt 2.1 fort |
| 2.12 | Kein sichtbares Pellet | Unzureichendes Ausgangsmaterial oder Verlust bei der Extraktion. | Beginnen Sie von Anfang an mit mehr Zellen. |
| 2.13 | Niedrige DNA-Konzentration | ||
| 3.1 | Nicht genug DNA für Verdünnungen | ||
| 3.4 | Die Probe sättigt nicht in der Membran | Die Membran in 1x TBST einweichen. Lassen Sie den überschüssigen Puffer trocknen und beginnen Sie mit Schritt 3.4 | |
| 4.6 | Fleckiges oder gesprenkeltes Muster wird erkannt | Geben Sie 0,1 % Natriumdodecylsulfat (SDS) in die Probe und fahren Sie mit Schritt 3.1 fort | |
| 4.6 | Die Punkte haben das Aussehen eines "Kaffeerings" | 0,01 % Sarkosyl in die Probe geben und mit Schritt 3.1 fortfahren | |
| 5.2 | Die RNaseH-Steuerung zeigt keinen Signalabfall an | Die Ribonuklease-Verdauung ist unvollständig. | Erhöhen Sie die Inkubation oder erhöhen Sie die Enzymkonzentration. |
| 6.5 | Kein Signal für die Oligo-Steuerung | Duplex wurde nicht gebildet. Oligonukleotide wurden nicht richtig geglüht. | Überprüfen Sie das Verhältnis von Oligonukleotiden und Annealing-Puffer. |
| 6.5 | Das S9.6-Signal ist nicht spezifisch für Hybride | S9.6-Antikörpercharge hat eine unspezifische Bindung | Validieren Sie die Sensitivität und Spezifität neuer Chargen von S9.6-Antikörpern entweder mit RNase-Enzymbehandlungen oder synthetischer Oligonukleotidanalyse. |
Tabelle 2: Fehlerbehebung
| ssRNA, oberer Strang | 5'-UGGGGGCUCGUCCGGGAUAUGGGAACCACUGAUCCC-3' |
| ssDNA, oberer Strang | 5'-TGGGGGCTCGTCCGGGATATGGGAACCACTGATCCC-3' |
| ssRNA, unterer Strang | 5'-GGGAUCAGUGGUUCCCAUAUCCCGGACGAGCCCCCA-3' |
| ssDNA, unterer Strang | 5'-GGGATCAGTGGTTCCCATATCCCGGACGAGCCCCCA-3' |
Tabelle 3. Kontrolle von Oligonukleotidsequenzen
Diskussion
Die 3-strängigen Nukleinsäuren, R-Schleifen, bilden sich in verschiedenen Stadien während des Lebenszyklus der RNA und werden zunehmend zur Regulierung zellulärer Prozesse eingesetzt. Um R-Schleifen vollständig zu verstehen, sind zuverlässige Techniken zur R-Schleifen-Detektion erforderlich. Hier beschreiben wir einen Ansatz zur Untersuchung der Häufigkeit von R-Schleifen mit Hilfe des S9.6-Antikörpers 8,23,24. Diese Methode ermöglicht eine schnelle Beurteilung der R-Loop-Abundanz aus Zellen und Gewebekulturproben. Es erfordert keine spezielle Ausrüstung oder eine große Menge an Ausgangsmaterial. Es gewährleistet spezifische und reproduzierbare Ergebnisse durch eine Kombination von RNase-Behandlungen.
Einige haben Bedenken hinsichtlich der Spezifität des S9.6-Antikörpers geäußert. Wie bei jedem Reagenz kann es auch beim S9.6-Antikörper zu Chargenschwankungen kommen. Unser Protokoll umfasst RNase H, RNase T1 und RNase III, um die Signalspezifität zu überprüfen. Darüber hinaus verwenden wir synthetische Oligonukleotide, um die Spezifität jeder Charge von S9.6-Antikörpern sicherzustellen.
Die R-Loop-Biologie ist ein wachsendes Gebiet; Die Entwicklung zuverlässiger Nachweis- und Quantifizierungsmethoden, wie die hier vorgestellte, wird mechanistische Studien erleichtern, um aufzuklären, wann sich R-Schleifen bilden, wie sie reguliert werden und was sie regulieren. Mit geeigneten Kontrollen ist dieser Dot-Blot-Assay eine einfache Methode zum Screening auf R-Loop-Häufigkeit in klinischen und Forschungsumgebungen.
Offenlegungen
Die Autoren haben nichts offenzulegen.
Danksagungen
Diese Arbeit wurde vom Howard Hughes Medical Institute und der Intramural Research am National Institute of Neurological Disorders and Stroke unterstützt.
Wir danken Dr. Stephen Leppla für die Bereitstellung von Chargen von S9.6-Antikörpern für die Analyse. Wir danken auch Dr. Dongjun Li für seine Unterstützung bei der Ribonuklease-Behandlung.
Materialien
| Name | Company | Catalog Number | Comments |
| Anti-dsDNA antibody | Abcam | ab27156 | |
| Anti-RNA-DNA hybrid antibody (S9.6) | Kerafast | ENH001 | |
| Biorupter sonicator | Diagenode | UCD-200 | |
| EB Buffer | Qiagen | 19086 | |
| EDTA (0.5M) | Invitrogen | AM9261 | |
| Hybond N+ nylon membrane | GE healthcare Life Sciences | RPN203B | |
| KCl (2M) | Invitrogen | AM9640G | |
| NP-40 (Igepal CA-630) | Sigma | I8896 | |
| PBS | Invitrogen | 10010-023 | |
| Phenol:chloroform:isoamyl alcohol | Invitrogen | 15593031 | |
| PIPES (0.5M, pH 8.0) | VWR | AAJ61406-AE | |
| Proteinase K | Qiagen | 19131 | |
| RNase III | Invitrogen | AM2290 | |
| RNase H | New England Biolabs | M0297 | |
| RNase T1 | ThermoFisher Sci. | EN0541 | |
| SDS (10%) | Invitrogen | 15553027 | |
| sodium acetate (3M, pH 5.2) | Invitrogen | AM9740 | |
| Tris-buffered saline (10X) | Corning | 46-012-CM | |
| Tris-HCl (1M, pH 8.0) | KD Medical | RGF-3360 | |
| TrypLE | Invitrogen | 12605010 | |
| Tween-20 | Sigma | P9416 | |
| UV Stratalinker 2400 | Stratagene | Stratalinker 2400 | |
| Whatman marking pen | Sigma | WHA10499001 |
Referenzen
- Daube, S. S., von Hippel, P. H. RNA displacement pathways during transcription from synthetic RNA-DNA bubble duplexes. Biochemistry. 33 (1), 340-347 (1994).
- Westover, K. D., Buschnell, D. A., Kornberg, R. D. Structural basis of transcription: separation of RNA from DNA by RNA polymerase II. Science. 303 (5660), 1014-1016 (2004).
- Itoh, T., Tomizawa, J. Formation of an RNA primer for initiation of replication of ColE1 DNA by ribonuclease H. Proceedings of the National Academy of Science USA. 77 (5), 2450-2454 (1980).
- Hamperl, S., Bocek, M. J., Saldivar, J. C., Swigut, T., Cimprich, K. A. Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell. 170 (4), 774-786 (2017).
- Skourti-Stathaki, K., Kamieniarz-Gdula, K., Proudfoot, N. J. R-loops induce repressive chromatin marks over mammalian gene terminators. Nature. 516 (7531), 436-439 (2014).
- Sanz, L. A., et al. conserved R-loop structures associate with specific epigenomic signatures in mammals. Molecular Cell. 63 (1), 167-178 (2016).
- Chen, L., et al. R-ChIP using inactive RNase H reveals dynamic coupling of R-loops with transcriptional pausing at gene promoters. Molecular Cell. 68 (4), 745-757 (2017).
- El Hage, A., French, S. L., Beyer, A. L., Tollervey, D. Loss of topoisomerase I leads to R-loop mediated transcriptional blocks during ribosomal RNA synthesis. Genes and Development. 24 (14), 1546-1558 (2010).
- Tran, P., et al. PIF1 family DNA helicases suppress R-loop mediated genome instability at tRNA genes. Nature Communication. 8, 15025(2017).
- Ginno, P. A., Lott, P. L., Christensen, H. C., Korf, I., Chedin, F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Molecular Cell. 45 (6), 814-825 (2012).
- Colak, D., et al. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science. 343 (6174), 1002-1005 (2014).
- Yu, K., Chedin, F., Hsieh, C., Wilson, T. E., Lieber, M. R. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nature Immunology. 4 (5), 442-451 (2003).
- Gasiunas, G., Barrangou, R., Horvath, P., Siksnys, V. Cas9-crRNA ribonucleotide complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proceedings of the National Academy of Science USA. 109 (39), 2579-2586 (2012).
- Westra, E. R., et al. CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by Cascade and Cas3. Molecular Cell. 46 (5), 595-605 (2012).
- Jiang, F., et al. Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science. 351 (6275), 867-871 (2016).
- Huertas, P., Aguilera, A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Molecular Cell. 12 (3), 711-721 (2003).
- Grunseich, C., et al. Senataxin mutation reveals how R-loops promote transcription by blocking DNA methylation at gene promoters. Molecular Cell. 69 (3), 426-437 (2018).
- Kim, H. D., Choe, J., Seo, Y. S. The sen1(+) gene of Schizosaccharomyces pombe, a homologue of budding yeast SEN1, encodes an RNA and DNA helicase. Biochemistry. 38 (44), 14697-14710 (1999).
- Stein, H., Hausen, P. Enzyme from calf thymus degrading the RNA moiety of DNA-RNA hybrids: effect on DNA-dependent RNA polymerase. Science. 166 (3903), 393-395 (1969).
- Cerritelli, S. M., Crouch, R. J. Ribonuclease H: the enzymes in eukaryotes. FEBS Journal. 276 (6), 1494-1505 (2009).
- Hyjek, M., Figiel, M., Nowotny, M. RNases H: Structure and mechanism. DNA Repair. 84, 102672(2019).
- Pohl, T. J., Zakian, V. A. Pif1 family DNA helicases: A helpmate to RNase H. DNA Repair. 84, 102633(2019).
- Pohjoismaki, J. L., et al. Mammalian mitochondrial DNA replication intermediates are essentially duplex but contain extensive tracts of RNA/DNA hybrid. Journal of Molecular Biology. 397 (5), 1144-1155 (2010).
- Sanz, L. A., Chedin, F. High-resolution, strand-specific R-loop mapping via S9.6-based DNA-RNA immunoprecipitation and high-throughput sequencing. Nature Protocols. 14 (6), 1734-1755 (2019).
- Wahba, L., Costantino, L., Tan, F. J., Zimmer, A., Koshland, D. S1-DRIP-seq identifies high expression and polyA tracts as major contributors to R-loop formation. Genes and Development. 30 (11), 1327-1338 (2016).
- Yan, Q., Shields, E. J., Bonasio, R., Sarma, K. Mapping native r-loops genome-wide using a targeted nuclease approach. Cell Reports. 29 (5), 1369-1380 (2019).
- Hu, Z., Zhang, A., Storz, G., Gottesman, S., Leppla, S. H. An antibody-based microarray assay for small RNA detection. Nucleic Acids Research. 34 (7), 52(2006).
- Nowotny, M., Gaidamakov, S. A., Crouch, R. J., Yang, W. Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell. 121 (7), 1005-1016 (2005).
- Sato, K., Egami, F. Studies on ribonucleases in takadiastase. Journal of Biochemistry. 44 (11), Tokyo. 753-767 (1957).
- Takahashi, K., Moore, S. Ribonuclease T1. Enzymes. 15, 435-468 (1982).
- Dunn, J. J. RNase III cleavage of single-stranded RNA: Effect of ionic strength in the fidelity of cleavage. Journal of Biological Chemistry. 251 (12), 3807-3814 (1976).
- Pertzev, A., Nicholson, A. W. Characterization of RNA sequence determinants and antideterminants of processing reactivity for a minimal substrate of Escherichia coli ribonuclease III. Nucleic Acids Research. 34 (13), 3708-3721 (2006).
- Phillips, D. D., et al. The sub-nanomolar binding of DNA-RNA hybrids by the single chain Fv fragment of antibody S9.6. Journal of Molecular Recognition. 26 (8), 376-381 (2013).
- Haruki, M., Noguchi, E., Kanaya, S., Crouch, R. J. Kinetic and stoichiometric analysis for the binding of Escherichia coli ribonuclease H1 to RNA-DNA hybrids using surface plasmon resonance. Journal of Biological Chemistry. 272 (35), 22015-22022 (1997).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten