
Method Article
Dot-Blot에 의한 R-Loop 분석
요약
이 프로토콜은 RNA-DNA 하이브리드와 변위된 DNA 가닥으로 구성된 3가닥 핵산 구조인 R-루프를 정량화하는 간단한 방법을 자세히 설명합니다.
초록
3가닥 핵산 구조인 R-loop는 유전자 조절에 대한 역할로 점점 더 인식되고 있습니다. 처음에 R-loops는 전사의 부산물로 생각되었습니다. 그러나 병든 세포에서 R-loops가 더 적다는 최근의 발견은 R-loops가 다양한 인간 세포에서 기능적 역할을 한다는 것을 분명히했습니다. 다음으로, R-루프의 역할과 세포가 풍부함의 균형을 맞추는 방법을 이해하는 것이 중요합니다. 이 분야의 과제는 R-loops의 정량화인데, 이는 대부분의 연구가 RNA-DNA 하이브리드에 대한 특이성에 의문이 제기된 S9.6 단클론 항체에 의존하기 때문입니다. 여기에서는 S9.6 항체와 함께 도트 블롯을 사용하여 R-loop를 정량화하고 RNA-DNA 하이브리드, 단일 가닥 RNA 및 이중 가닥 RNA를 각각 절단하는 RNase H, RNase T1 및 RNase III를 사용하여 이 분석의 민감도와 특이성을 보여줍니다. 이 방법은 재현성이 높고 일반 실험실 장비 및 시약을 사용하며 2일 이내에 결과를 제공합니다. 이 분석은 연구 및 임상 환경에서 R-loop를 정량화하고 senataxin과 같은 유전자의 돌연변이가 R-loop 존재비에 미치는 영향을 평가하는 데 사용할 수 있습니다.
서문
이 프로토콜은 3가닥 핵산 구조인 R-loop의 존재량을 빠르게 비교할 수 있는 dot-blot assay에 대한 단계별 가이드를 제공합니다. R-루프는 RNA가 이중 가닥 DNA에 침입하여 RNA-DNA 하이브리드를 생성하고 다른 DNA 가닥을 대체할 때 형성됩니다. R-loop는 RNA 수명 주기의 여러 단계에서 발견됩니다. 전사 복합체(transcriptional complex)에서 초기 RNA는 주형 DNA에 상보적으로 합성되고 비주형 가닥은 치환됩니다. 짧은 RNA-DNA 하이브리드(<10 bp)는 초기 RNA를 방출하여 출구 채널 1,2를 통해 RNA 중합효소 복합체를 떠날 수 있도록 분해됩니다. 전사 복합체 외부에서, 초기 RNA는 DNA 주형에 가까우며, 아직 복사되지 않은 상태로 약간 감겨 있기 때문에 RNA는 R-루프3을 형성하는 주형 DNA와 재혼성화할 수 있습니다. 또한 R-루프는 복제와 전사 복합체가 충돌할 때 형성될 수 있으며4 반센스전사5에서 형성될 수 있습니다. R-loops는 형성 기회가 많다는 점을 감안할 때, R-loop는 드물지 않으며, 세포의 전사 상태에 따라 human genome6의 3-5%에서 발견될 수 있습니다. R-loops는 mRNA의 유전자 프로모터7 및 종결5 부위, 리보솜 RNA8 및 전달 RNA9를 따라 발견됩니다. R-루프는 염색체의 텔로머 영역에도 있습니다.
R-루프는 규제 역할을 합니다. 이들은 프로모터 10,11에서 전사에 영향을 미치고, 클래스-스위치 재조합12을 매개하며, CRISPR 기반 게놈 편집 13,14,15를 촉진함으로써 유전자 발현을 조절합니다. 많은 세포 이벤트와 마찬가지로 R-루프 풍부도는 단단히 적정됩니다. R-루프가 너무 많거나 너무 적으면 정상적인 세포 기능에 영향을 미칩니다16,17. R-루프는 RNase H, senataxin 및 RNA-DNA 하이브리드를 푸는 기타 헬리카제를 포함한 다양한 단백질에 의해 조절됩니다 18,19,20,21,22.
R-loops의 풍부함을 모니터링하기 위해 게놈 전체 방법은 먼저 항체 S9.6 8,23,24 또는 RNase H 10,26,27을 포함한 다른 nuclease25로 R-loop를 농축한 다음 염기서열분석을 통해 농축된 R-loops의 수를 평가합니다. 이러한 염기서열분석 기반 방법의 초기 버전에서는 정확한 정량 분석을 가능하게 하는 적절한 염기서열 범위를 달성하지 못했지만, 염기서열분석 기술의 급속한 발전으로 이제 유전자좌별 R-루프 분석이 가능합니다. 면역형광 기술은 R-루프를 정량화하고 국소화하는 데에도 사용되었습니다10,17. 이러한 방법은 포괄적이지만 값비싼 장비와 전문 분석이 필요하기 때문에 많은 임상 환경이나 초기 평가로 실용적이지 않습니다.
임상 환경에서 여러 실험실에서 균일하게 수행할 수 있는 절차가 필요합니다. Dot-blot은 특정 장비나 컴퓨터 분석 없이 수행할 수 있기 때문에 이러한 옵션을 제공합니다. R-loop에 대한 돌연변이의 효과를 평가하기 위한 예비 단계 또는 임상 환경에서 이러한 dot-blot은 민감하고 특이적인 결과를 제공해야 합니다. 여기에서는 R-루프를 구체적으로 식별하는 분석에 대해 설명합니다. 이중 가닥(ds) DNA, 이중 가닥 RNA 및 단일 가닥 RNA의 신호를 제외합니다. 우리의 프로토콜은 S9.6 항체27을 사용하여 R-루프에서 RNA-DNA 하이브리드를 식별하고 RNA-DNA 하이브리드에서 RNA의 분해를 유도하는 엔도리보뉴클레아제인 RNase H를 통합합니다20,28, 검출된 신호가 하이브리드의 신호인지 확인합니다. 또한 guanine 29,30에서 single-stranded RNA를 절단하는 RNase T1과 stem-loops 31,32를 포함한 double-stranded RNA를 절단하는 RNase III를 통합하여 비특이적 신호를 확인했습니다. S9.6 항체는 다양한 길이의 RNA-DNA 하이브리드를 인식하며, 심지어 길이가 8 뉴클레오티드에 불과한 하이브리드도인식합니다 33.
여기에서는 핵산 분리로 시작하여 dot-blot 준비, S9.6 항체를 사용한 R-loop 검출로 시작하는 프로토콜을 제시합니다. 당사의 프로토콜에는 동일한 양의 샘플이 로드되고 신호가 구체적인지 확인하는 단계가 포함되어 있습니다. 그것은 양성 및 음성 대조군 역할을 하는 올리고뉴클레오티드를 제공합니다. 이것은 R-루프 존재도를 평가하는 빠르고 쉽고 사용자 친화적인 방법입니다.
프로토콜
1. 핵 분획을 위한 세포 용해
- 1x 인산염 완충 식염수(PBS)로 세포를 두 번 세척합니다. 트립신(trypsin)과 같은 표준 세포 해리 기술을 사용하여 조직 배양 접시에서 세포를 제거합니다. 혈구계를 사용하여 세포를 계산합니다.
참고: 아래에 설명된 단계는 1차 인간 피부 섬유아세포의 분석에 사용되었지만, 다양한 세포 유형을 분석할 수 있습니다. 섬유아세포는 10%의 소 태아 혈청을 함유하는 기저 배지에서 성장하였다. 대안적으로, 세포 용해 완충액(표 1)을 세척 후 세포 배양액에 직접 첨가할 수 있습니다. - 세포 현탁액을 1.5mL 튜브로 옮겨 세포를 펠렛화합니다.
- 300 x g 에서 4°C에서 5분 동안 시료를 원심분리합니다. 미디어를 흡인합니다.
- 1.3단계에서 원심분리 설정을 사용하여 얼음처럼 차가운 1x PBS로 두 번 세척합니다.
- 세포 펠릿에 저온 세포 용해 완충액(표 1)을 추가합니다(2 x 106 세포당 300μL). 펠릿을 다시 현탁시키기 위해 위아래로 피펫을 움직입니다.
- 얼음 위에서 10분 동안 배양합니다.
- 500 x g 에서 5분 동안 회전하여 핵을 펠릿화합니다.
- 상층액을 버리고 핵 펠릿을 400μL의 차가운 핵 용해 완충액에 다시 현탁시킵니다(표 1).
- 얼음 위에서 10분 동안 배양합니다.
참고: 세포의 핵 및 세포질 구획의 분별은 신호 특이성을 보장합니다. 진행하기 전에 핵 및 세포질 분리의 품질을 평가할 수 있습니다(표 2). - 3 μL의 20 mg/mL proteinase K를 첨가하고 55 °C에서 3-5시간 동안 배양합니다.
참고: 표시된 볼륨은 2 x 106 셀에 대한 것이며 필요에 따라 확대 또는 축소합니다.
2. 게놈 DNA(RNA-DNA 하이브리드 포함)의 정제
- DNA가 점성이있는 경우 초음파 처리를 수행하여 점도를 낮 춥니 다(예 : 4 ° C 수조를 사용하여 2 분 동안 고출력 출력, 30 초 켜기 / 30 초 끄기에서 초음파 처리).
- 400μL의 용출 완충액(표 1)과 400μL의 페놀:클로로포름:이소아밀 알코올(25:24:1, pH 8.0)을 추가합니다.
- 10초 동안 소용돌이.
- 12,000 x g 에서 4°C에서 5분간 스핀다운합니다.
- 수성상(약 350μL)을 새 튜브로 옮깁니다.
- 클로로포름 1 부피를 사용하여 한 번 추출하고 10 초 동안 와류 한 다음 4 ° C에서 5 분 동안 12,000 x g 에서 스핀 다운합니다. 수성상을 새 튜브(약 300μL)로 옮깁니다.
- 3M 아세트산나트륨(pH 5.2) 35μL, 글리코겐 1μL 및 얼음처럼 차가운 100% 에탄올 700μL를 추가합니다.
- 10초 동안 소용돌이치고 12,000 x g 에서 4°C에서 30분 동안 스핀 다운합니다.
- 펠릿을 1mL의 70% 에탄올로 세척합니다.
- 10초 동안 소용돌이치고 4°C에서 15분 동안 12,000 x g 에서 스핀 다운합니다.
- 상등액을 버리고 펠릿을 자연 건조시킵니다.
- 12μL의 용리 완충액을 추가하고 10초 동안 와류하여 재현탁합니다. 37°C에서 교반으로 샘플을 30분 동안 배양하거나 4°C에서 하룻밤 동안 배양하여 펠릿을 다시 현탁시킵니다.
- 표준 분광 광도계를 사용하여 DNA 농도를 측정합니다.
참고: 표시된 볼륨은 2 x 106 셀에 대한 것이며 필요에 따라 확대 또는 축소합니다. DNA(RNA-DNA 하이브리드 포함)는 필요한 경우 -20°C에서 보관할 수 있습니다.
3. DNA 샘플(RNA-DNA 하이브리드 포함)을 나일론 멤브레인에 블로팅
- 용리 완충액에서 원하는 농도(즉, 50ng/μL, 25ng/μL 또는 12.5ng/μL)로 핵산을 희석합니다. 농도 범위(200, 100, 50, 25, 12.5ng)의 이러한 샘플은 선형 범위 내에 신호가 있음을 보장합니다.
참고: 기술 및 생물학적 복제를 위해 충분한 샘플을 준비해야 하며 다양한 RNase 처리에 대해서는 5단계를 참조하십시오. - 각 2μL 샘플이 0.5cm2 면적을 차지할 수 있는 공간이 있도록 양전하를 띤 나일론 멤브레인을 준비합니다.
- 각 샘플의 2μL를 2개의 멤브레인(하나는 S9.6 항체용, 다른 하나는 dsDNA 항체용)에 배치합니다. 또는 더 큰 부피의 샘플을 로드할 수 있는 dot-blot 또는 slot-blot 장치를 사용할 수 있습니다.
- 샘플이 멤브레인으로 포화되도록 합니다. 멤브레인을 자외선으로 가교결합하기 전에 최소 2분을 기다리십시오.
- 멤브레인을 UV 장치의 중앙에 놓고 "Auto Crosslink" 설정(1,200 μJ x 100)을 사용하여 UV 가교제를 사용하여 멤브레인을 가교합니다.
4. S9.6 항체를 가진 RNA DNA 잡종 탐지
- 차단 용액(0.05% Tween-20(TBST)과 함께 Tris-buffered saline에 5% 우유와 함께 셰이커에서 실온에서 1시간 동안 멤브레인을 배양합니다.
알림: 멤브레인을 덮을 수 있는 충분한 차단 용액이 있어야 합니다. - 1차 항체(TBST의 5% 우유 중)에서 4°C에서 진탕과 함께 밤새 멤브레인을 배양합니다. 한쪽 멤브레인에 anti-dsDNA 항체(1:10,000 희석)를 추가합니다. 두 번째 멤브레인에 1μg/mL S9.6 항체를 추가합니다(1:1,000 희석).
참고: S9.6 항체는 시판하거나 미국 국립보건원(National Institutes of Health)의 NIAID의 S. Leppla 박사에서 구입할 수 있습니다. - 1차 항체를 제거하고 TBST로 3회 세척합니다. 실온에서 흔들면서 5-10분 동안 세탁할 때마다 수행하십시오.
- 양고추냉이 과산화효소(HRP) 접합 2차 항체(항 마우스, 1:5,000 희석)를 TBST의 5% 우유에 넣고 실온에서 진탕하여 배양합니다.
참고: Anti-dsDNA 및 anti-RNA-DNA hybrid는 모두 마우스 항체입니다. - 2차 항체를 제거하고 실온에서 흔들면서 TBST로 5-10분 동안 3회 세척합니다.
- ECL(Enhanced-Chemiluminescence) 시약으로 개발하여 이미징을 위한 신호를 획득합니다.
- ImageJ와 같은 표준 이미지 프로세싱 툴을 사용하여 신호 강도를 정량화할 수 있습니다.
참고: 문제 해결은 표 2에 자세히 설명되어 있습니다.
5. 신호 특이성을 평가하기 위한 리보뉴클레아제 처리
참고: S9.6 결합의 특이성을 입증하기 위해 핵산 샘플에 RNase 처리를 수행해야 합니다. RNase T1 또는 RNase III가 아닌 RNase H로 처리하면 S9.6 면역염색이 감소해야 합니다.
- RNA-DNA 하이브리드가 포함된 샘플을 4개의 별도 튜브에 준비하여 분해합니다. 4개의 시료 각각을 5 U RNase H, 1000 U RNase T1, 0.5 U RNase III 또는 모의로 처리합니다. 37°C에서 20μL 부피로 15분 동안 샘플을 배양합니다.
- 섹션 3에 설명된 대로 멤브레인에 각 샘플의 2μL를 로드합니다.
6. 신호 특이성을 평가하기 위한 올리고뉴클레오티드 대조군 준비
참고: 올리고뉴클레오티드 대조군은 S9.6 결합의 특이성을 입증하는 데 사용할 수 있습니다. S9.6은 RNA-DNA 하이브리드를 인식하지만, 이전에 보고된 바와 같이 dsDNA 또는 dsRNA 대조군은 인식하지 못한다34.
- 올리고뉴클레오티드(표 3)를 어닐링 완충액(10mM Tris, pH 8.0, 50mM NaCl, 1mM EDTA)에 100μM로 용해합니다.
- 다음을 위한 4개의 반응 튜브를 준비합니다.
- RNA-DNA 하이브리드 #1: 10 μL의 ssRNA 상단 가닥과 10 μL의 ssDNA 하단 가닥 및 80 μL의 어닐링 완충액을 혼합합니다.
- RNA-DNA 하이브리드 #2: 10 μL의 ssDNA 상단 가닥과 10 μL의 ssRNA 하단 가닥 및 80 μL의 어닐링 완충액을 혼합합니다.
- dsRNA: 10 μL의 ssRNA 상단 가닥과 10 μL의 ssRNA 하단 가닥 및 80 μL의 어닐링 완충액을 혼합합니다.
- dsDNA: 10 μL의 ssDNA 상단 가닥과 10 μL의 ssDNA 하단 가닥 및 80 μL의 어닐링 완충액을 혼합합니다.
- 6.2 단계의 4 가지 혼합물을 95 ° C에서 10 분 동안 가열합니다.
- 가닥을 재어닐링할 수 있도록 튜브를 실온으로 천천히 냉각시킵니다. 어닐링된 표준은 나중에 사용할 수 있도록 -20°C에서 보관할 수 있습니다.
주: 어닐링 효율성은 non-denaturing 젤 전기 이동법으로 검사되어야 합니다. 듀플렉스는 어닐링되지 않은 올리고뉴클레오티드보다 더 느리게 이동합니다(표 2). - 섹션 3에 설명된 대로 S9.6 항체와 dsDNA 항체에 각각 하나씩 총 2개의 멤브레인에 각 시료 2μL를 로드합니다.
- 섹션 4에 설명된 단계를 수행합니다.
7. ImageJ를 사용한 S9.6 R-loop 신호 강도의 정량화 및 정규화.
- S9.6, dsDNA 염색 이미지를 TIFF 형식으로 저장하고 ImageJ 소프트웨어(https://imagej.nih.gov/ij/)를 사용하여 분석합니다.
- 이미지 반전 옵션(Edit(편집) | Invert)를 클릭합니다. 반전 후 각 점은 어두운 배경에 흰색으로 표시됩니다.
- 타원형 이미지 선택 도구를 사용하여 이미지에서 가장 큰 점을 둘러쌀 수 있을 만큼 큰 타원을 선택합니다.
- ROI 관리자를 사용하여 정량화를 위해 선택한 영역을 추가합니다. 관심 영역을 시각화할 수 있도록 "Show All(모두 표시)" 및 "Labels(레이블)" 옵션이 선택되어 있는지 확인합니다.
- 7.3단계에서 사용된 것과 동일한 타원형 선택 영역을 사용하여 정량화할 각 점 주위에 관심 영역을 추가합니다. Command + Shift + E 단축키를 사용하여 7.3단계에서 선택한 영역을 이후의 각 점에 복사합니다.
- 각 관심 영역의 적분 밀도를 측정합니다.
- 각 샘플에 대한 S9.6 신호 강도를 dsDNA 측정으로 나누어 S9.6/dsDNA 신호 비율을 구합니다. 실험을 반복하여 결과를 확인합니다(S9.6 및 dsDNA 신호 획득 모두에 대해 최소 3회). 평균의 표준 오차는 S9.6/dsDNA 신호 비율에서 계산할 수 있습니다.
결과
S9.6(RNA-DNA) 항체의 특이성을 평가하기 위한 효소 처리.
1차 인간 피부 섬유아세포는 성장하였다17. RNA-DNA 하이브리드를 가진 DNA를 분리하고 정량화했습니다. 샘플 중 2μg을 37°C에서 15분 동안 RNase T1, RNase H 또는 RNase III로 절단했습니다. RNase 처리된 샘플과 비교하기 위해 모의 샘플도 분석했습니다. 샘플(200, 100, 50, 25, 12.5 또는 6.25ng)을 섹션 3에 설명된 대로 두 개의 서로 다른 멤브레인에 블롯팅했습니다. 멤브레인은 가교 결합되고 차단되었으며 그 중 하나는 S9.6 항체로 프로브되었습니다(그림 1A).
그 결과 S9.6 신호가 로드된 샘플의 풍부함과 상관관계가 있음을 보여주었습니다. RNase T1 또는 RNase III가 아닌 RNase H로 처리하면 S9.6 염색이 감소합니다.
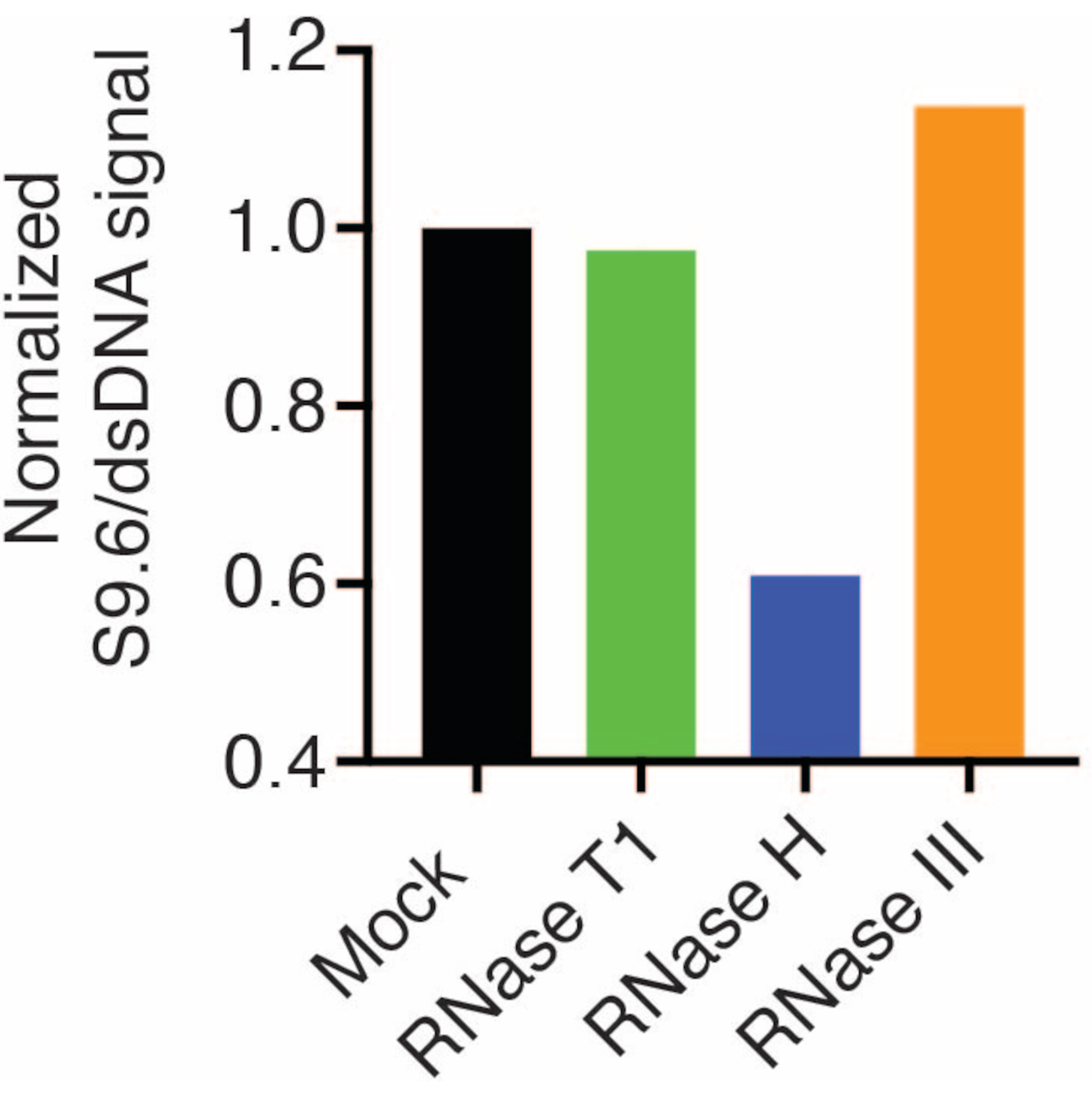
두 번째 멤브레인은 정상화를 위해 dsDNA 항체(그림 1B)로 조사되었습니다. 신호 강도를 분석하기 위해 영상 J를 사용했습니다. 50ng 샘플은 S9.6 및 dsDNA 항체의 신호 강도가 동적 범위 내에 있었기 때문에 정량화를 위해 선택되었습니다. 신호 강도는 모의 샘플의 강도로 정규화되었습니다. 데이터는 그림 2에 나와 있습니다.
합성 뉴클레오티드 대조군을 사용한 S9.6 항체 도트 블롯.
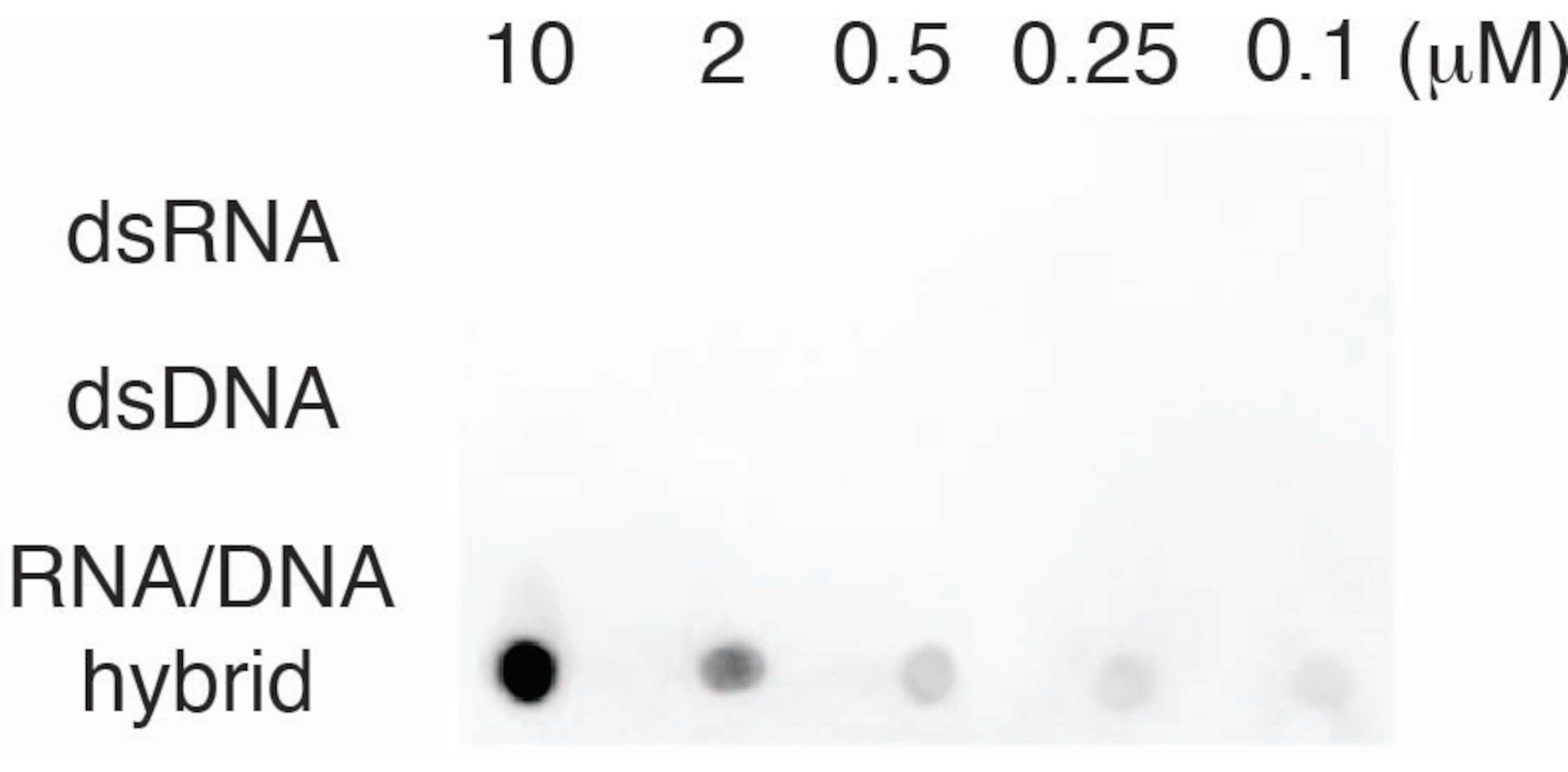
S9.6 항체의 특이성을 평가하기 위해 섹션 6에 설명된 대로 dsRNA, dsDNA 및 RNA-DNA에 해당하는 올리고뉴클레오티드를 사용했습니다. RNA-DNA, dsRNA 및 dsDNA 뉴클레오티드의 희석 시리즈를 준비하고 섹션 3에 설명된 대로 나일론 멤브레인 상에 블롯팅했습니다. 멤브레인은 S9.6 항체로 프로브되었습니다(그림 3). 결과는 S9.6 항체가 용량 의존적 방식으로 RNA-DNA 하이브리드에 특이적으로 결합하고 dsRNA 및 dsDNA에 대한 최소한의 교차 반응을 보였다는 것을 보여주었습니다.

그림 1: 인간 섬유아세포의 핵산이 로드된 점블롯으로 나타난 S9.6의 특이성. 인간 섬유아세포의 핵산 샘플을 모의 처리하거나 RNase T1, RNase H 또는 RNase III로 처리한 다음 2μL 도트당 200, 100, 50, 25, 12.5 및 6.25ng의 희석 시리즈로 나일론 멤브레인에 로드했습니다. 그런 다음 S9.6 항체(A) 또는 dsDNA 항체(B)로 멤브레인을 조사했습니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}

그림 2: S9.6 R-루프 염색의 정량화.그림 1의 50ng 샘플은 ImageJ를 사용한 정량화를 위해 선택되었습니다. S9.6 신호를 dsDNA 신호 강도로 나눈 다음 섹션 7에 설명된 단계에 따라 모의 샘플로 정규화했습니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}

그림 3: 올리고뉴클레오티드 대조군을 사용한 S9.6 점-블롯. dsRNA, dsDNA 또는 RNA-DNA 하이브리드로 희석된 일련의 합성 올리고뉴클레오티드에 대한 S9.6 항체 도트 블롯. S9.6은 용량 의존적 방식으로 RNA-DNA 하이브리드에 특이적으로 결합합니다. 이 그림의 더 큰 버전을 보려면 여기를 클릭하십시오.
{kind=link}
| 세포 용해 완충액 | 10mL용 | 200mL용 | 파이널 콘크. |
| 물, 뉴클레아제 무함유 | 9mL | 180밀리리터 | - |
| 10% NP-40 | 0.5mL | 10mL | 0.5% |
| 2M 케이클 | 0.4mL | 8mL | 80미디엄 |
| 0.5M 파이프(pH 8.0) | 100 미룰라 | 2mL | 5중 미터 |
| 핵 용해 완충액 | 10mL용 | 200mL용 | 파이널 콘크. |
| 물, 뉴클레아제 무함유 | 8.65mL | 173mL | - |
| 10% SDS | 1mL | 20mL | 1% |
| 1M 트리스-HCl(pH 8.0) | 0.25mL | 5 밀리리터 | 25 밀리미터미터 |
| 0.5M 에다 | 100 미룰라 | 2mL | 5 밀리미터미터 |
| 용리 버퍼 | 10mL용 | 200mL용 | 파이널 콘크. |
| 1M 트리스-Cl, pH 8.5 | 0.1 밀리리터 | 2 mL | 10 밀리미터미터 |
| 물, 뉴클레아제 무함유 | 9.9 밀리리터 | 198 밀리리터 | - |
표 1. 완충액 준비
| 걸음 | 문제 | 가능한 이유 | 용액 |
| 1.9 | 해당 사항 없음 | 세포 분획 확인 | 핵 및 세포질 분리의 품질은 세포와 핵 용해 완충액에 표준 단백질 분해효소 억제제 칵테일을 첨가하여 평가할 수 있습니다(표 1). 세포질 및 핵 분획은 웨스턴 블로팅 분석으로 평가하여 세포질 분획에서 세포질 마커(예: GAPDH 또는 HSP90)를 사용한 라벨링과 핵 분획에서 핵 마커(예: HDAC1 또는 Histone H3)를 사용한 라벨링의 적절한 제한을 확인할 수 있습니다. 핵 분획에서 미토콘드리아 오염의 기여도는 미토콘드리아 DNA에 특이적인 프로브를 사용한 qPCR 분석으로 평가할 수 있습니다. |
| 2.1 | 샘플이 너무 점성이 있습니다. | 셀 번호가 너무 높습니다. | DNA를 절반으로 줄이고 초음파 처리 단계 2.1을 계속합니다. |
| 2.12 | 펠릿이 보이지 않음 | 출발 물질이 충분하지 않거나 추출 중 손실이 발생합니다. | 더 많은 셀을 사용하여 처음부터 시작하십시오. |
| 2.13 | 낮은 DNA 농도 | ||
| 3.1 | 희석을 위한 DNA가 충분하지 않음 | ||
| 3.4 | 샘플이 멤브레인으로 포화되지 않습니다. | 멤브레인을 1x TBST에 담그십시오. 초과 버퍼를 건조시키고 3.4단계에서 시작합니다. | |
| 4.6 | 고르지 못하거나 얼룩덜룩한 패턴이 감지됩니다. | 샘플에 0.1% SDS(Sodium dodecyl sulfate)를 추가하고 3.1단계에서 계속합니다. | |
| 4.6 | 점들은 "커피 링" 모양을 가지고 있습니다 | 샘플에 0.01% Sarkosyl을 추가하고 3.1단계에서 계속합니다. | |
| 5.2 | RNaseH 컨트롤은 신호 감소를 보이지 않습니다. | 리보뉴클레아제 소화가 불완전합니다. | 배양을 늘리거나 효소 농도를 증가시킵니다. |
| 6.5 | 올리고 컨트롤에 대한 신호 없음 | 듀플렉스가 형성되지 않았습니다. 올리고뉴클레오티드가 제대로 어닐링되지 않았습니다. | 올리고뉴클레오티드와 어닐링 완충액의 비율을 확인합니다. |
| 6.5 | S9.6 신호는 하이브리드에만 국한되지 않습니다. | S9.6 항체 배치는 비특이적 결합을 가짐 | RNase 효소 처리 또는 합성 올리고뉴클레오티드 분석을 사용하여 새로운 S9.6 항체 배치의 민감도와 특이성을 검증합니다. |
표 2: 문제 해결
| ssRNA, 상단 가닥 | 5' - UGGGGGCUCGUCCGGGAUAUGGGAACCACUGAUCCC-3' |
| ssDNA, 톱 스트랜드 | 5'-TGGGGGCTCGTCCGGGATATGGGAACCACTGATCCC-3' |
| ssRNA, 하단 가닥 | 5' - GGGAUCAGUGGUUCCCCAAUAUCCCGGACGAGCCCCCA-3' |
| ssDNA, 하단 가닥 | 5'-GGGATCAGTGGTTCCCATATCCCGGACGAGCCCCCA-3' |
표 3. 올리고뉴클레오타이드 염기서열 제어
토론
3가닥 핵산인 R-루프는 RNA의 수명 주기 동안 여러 단계에서 형성되며 세포 과정을 조절하는 것으로 점점 더 많이 발견되고 있습니다. R-루프를 완전히 이해하려면 R-루프 감지를 위한 신뢰할 수 있는 기술이 필요합니다. 여기에서는 S9.6 항체 8,23,24를 사용하여 R-loops의 풍부함을 조사하는 접근 방식을 설명합니다. 이 방법을 사용하면 세포 및 조직 배양 샘플의 R-loop 존재량을 빠르게 평가할 수 있습니다. 특별한 장비나 많은 양의 출발 물질이 필요하지 않습니다. RNase 처리의 조합을 사용하여 구체적이고 재현 가능한 결과를 보장합니다.
일부에서는 S9.6 항체의 특이성에 대한 우려를 보고했습니다. 다른 시약과 마찬가지로 S9.6 항체에 대한 배치 간 변동성이 있을 수 있습니다. 당사의 프로토콜에는 신호 특이성을 확인하기 위한 RNase H, RNase T1 및 RNase III가 포함됩니다. 또한 합성 올리고뉴클레오티드를 사용하여 S9.6 항체의 각 배치의 특이성을 보장합니다.
R-loop 생물학은 성장하는 분야입니다. 여기에 제시된 것과 같은 신뢰할 수 있는 검출 및 정량화 방법의 개발은 R-루프가 형성되는 시기, R-루프가 어떻게 조절되는지, 그리고 무엇을 조절하는지 밝히기 위한 기계론적 연구를 촉진할 것입니다. 적절한 대조군을 통해 이 dot-blot assay는 임상 및 연구 환경에서 R-loop 존재량을 스크리닝하는 간단한 방법입니다.
공개
저자는 공개할 내용이 없습니다.
감사의 말
이 연구는 하워드 휴즈 의학 연구소(Howard Hughes Medical Institute)와 국립 신경 장애 및 뇌졸중 연구소(National Institute of Neurological Disorders and Stroke)의 교내 연구(Intramural Research)의 지원을 받았습니다.
분석을 위해 S9.6 항체 배치를 제공해 주신 Stephen Leppla 박사님께 감사드립니다. 또한 리보뉴클레아제 치료에 도움을 주신 Dr. Dongjun Li에게도 감사드립니다.
자료
| Name | Company | Catalog Number | Comments |
| Anti-dsDNA antibody | Abcam | ab27156 | |
| Anti-RNA-DNA hybrid antibody (S9.6) | Kerafast | ENH001 | |
| Biorupter sonicator | Diagenode | UCD-200 | |
| EB Buffer | Qiagen | 19086 | |
| EDTA (0.5M) | Invitrogen | AM9261 | |
| Hybond N+ nylon membrane | GE healthcare Life Sciences | RPN203B | |
| KCl (2M) | Invitrogen | AM9640G | |
| NP-40 (Igepal CA-630) | Sigma | I8896 | |
| PBS | Invitrogen | 10010-023 | |
| Phenol:chloroform:isoamyl alcohol | Invitrogen | 15593031 | |
| PIPES (0.5M, pH 8.0) | VWR | AAJ61406-AE | |
| Proteinase K | Qiagen | 19131 | |
| RNase III | Invitrogen | AM2290 | |
| RNase H | New England Biolabs | M0297 | |
| RNase T1 | ThermoFisher Sci. | EN0541 | |
| SDS (10%) | Invitrogen | 15553027 | |
| sodium acetate (3M, pH 5.2) | Invitrogen | AM9740 | |
| Tris-buffered saline (10X) | Corning | 46-012-CM | |
| Tris-HCl (1M, pH 8.0) | KD Medical | RGF-3360 | |
| TrypLE | Invitrogen | 12605010 | |
| Tween-20 | Sigma | P9416 | |
| UV Stratalinker 2400 | Stratagene | Stratalinker 2400 | |
| Whatman marking pen | Sigma | WHA10499001 |
참고문헌
- Daube, S. S., von Hippel, P. H. RNA displacement pathways during transcription from synthetic RNA-DNA bubble duplexes. Biochemistry. 33 (1), 340-347 (1994).
- Westover, K. D., Buschnell, D. A., Kornberg, R. D. Structural basis of transcription: separation of RNA from DNA by RNA polymerase II. Science. 303 (5660), 1014-1016 (2004).
- Itoh, T., Tomizawa, J. Formation of an RNA primer for initiation of replication of ColE1 DNA by ribonuclease H. Proceedings of the National Academy of Science USA. 77 (5), 2450-2454 (1980).
- Hamperl, S., Bocek, M. J., Saldivar, J. C., Swigut, T., Cimprich, K. A. Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell. 170 (4), 774-786 (2017).
- Skourti-Stathaki, K., Kamieniarz-Gdula, K., Proudfoot, N. J. R-loops induce repressive chromatin marks over mammalian gene terminators. Nature. 516 (7531), 436-439 (2014).
- Sanz, L. A., et al. conserved R-loop structures associate with specific epigenomic signatures in mammals. Molecular Cell. 63 (1), 167-178 (2016).
- Chen, L., et al. R-ChIP using inactive RNase H reveals dynamic coupling of R-loops with transcriptional pausing at gene promoters. Molecular Cell. 68 (4), 745-757 (2017).
- El Hage, A., French, S. L., Beyer, A. L., Tollervey, D. Loss of topoisomerase I leads to R-loop mediated transcriptional blocks during ribosomal RNA synthesis. Genes and Development. 24 (14), 1546-1558 (2010).
- Tran, P., et al. PIF1 family DNA helicases suppress R-loop mediated genome instability at tRNA genes. Nature Communication. 8, 15025(2017).
- Ginno, P. A., Lott, P. L., Christensen, H. C., Korf, I., Chedin, F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Molecular Cell. 45 (6), 814-825 (2012).
- Colak, D., et al. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science. 343 (6174), 1002-1005 (2014).
- Yu, K., Chedin, F., Hsieh, C., Wilson, T. E., Lieber, M. R. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nature Immunology. 4 (5), 442-451 (2003).
- Gasiunas, G., Barrangou, R., Horvath, P., Siksnys, V. Cas9-crRNA ribonucleotide complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proceedings of the National Academy of Science USA. 109 (39), 2579-2586 (2012).
- Westra, E. R., et al. CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by Cascade and Cas3. Molecular Cell. 46 (5), 595-605 (2012).
- Jiang, F., et al. Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science. 351 (6275), 867-871 (2016).
- Huertas, P., Aguilera, A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Molecular Cell. 12 (3), 711-721 (2003).
- Grunseich, C., et al. Senataxin mutation reveals how R-loops promote transcription by blocking DNA methylation at gene promoters. Molecular Cell. 69 (3), 426-437 (2018).
- Kim, H. D., Choe, J., Seo, Y. S. The sen1(+) gene of Schizosaccharomyces pombe, a homologue of budding yeast SEN1, encodes an RNA and DNA helicase. Biochemistry. 38 (44), 14697-14710 (1999).
- Stein, H., Hausen, P. Enzyme from calf thymus degrading the RNA moiety of DNA-RNA hybrids: effect on DNA-dependent RNA polymerase. Science. 166 (3903), 393-395 (1969).
- Cerritelli, S. M., Crouch, R. J. Ribonuclease H: the enzymes in eukaryotes. FEBS Journal. 276 (6), 1494-1505 (2009).
- Hyjek, M., Figiel, M., Nowotny, M. RNases H: Structure and mechanism. DNA Repair. 84, 102672(2019).
- Pohl, T. J., Zakian, V. A. Pif1 family DNA helicases: A helpmate to RNase H. DNA Repair. 84, 102633(2019).
- Pohjoismaki, J. L., et al. Mammalian mitochondrial DNA replication intermediates are essentially duplex but contain extensive tracts of RNA/DNA hybrid. Journal of Molecular Biology. 397 (5), 1144-1155 (2010).
- Sanz, L. A., Chedin, F. High-resolution, strand-specific R-loop mapping via S9.6-based DNA-RNA immunoprecipitation and high-throughput sequencing. Nature Protocols. 14 (6), 1734-1755 (2019).
- Wahba, L., Costantino, L., Tan, F. J., Zimmer, A., Koshland, D. S1-DRIP-seq identifies high expression and polyA tracts as major contributors to R-loop formation. Genes and Development. 30 (11), 1327-1338 (2016).
- Yan, Q., Shields, E. J., Bonasio, R., Sarma, K. Mapping native r-loops genome-wide using a targeted nuclease approach. Cell Reports. 29 (5), 1369-1380 (2019).
- Hu, Z., Zhang, A., Storz, G., Gottesman, S., Leppla, S. H. An antibody-based microarray assay for small RNA detection. Nucleic Acids Research. 34 (7), 52(2006).
- Nowotny, M., Gaidamakov, S. A., Crouch, R. J., Yang, W. Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell. 121 (7), 1005-1016 (2005).
- Sato, K., Egami, F. Studies on ribonucleases in takadiastase. Journal of Biochemistry. 44 (11), Tokyo. 753-767 (1957).
- Takahashi, K., Moore, S. Ribonuclease T1. Enzymes. 15, 435-468 (1982).
- Dunn, J. J. RNase III cleavage of single-stranded RNA: Effect of ionic strength in the fidelity of cleavage. Journal of Biological Chemistry. 251 (12), 3807-3814 (1976).
- Pertzev, A., Nicholson, A. W. Characterization of RNA sequence determinants and antideterminants of processing reactivity for a minimal substrate of Escherichia coli ribonuclease III. Nucleic Acids Research. 34 (13), 3708-3721 (2006).
- Phillips, D. D., et al. The sub-nanomolar binding of DNA-RNA hybrids by the single chain Fv fragment of antibody S9.6. Journal of Molecular Recognition. 26 (8), 376-381 (2013).
- Haruki, M., Noguchi, E., Kanaya, S., Crouch, R. J. Kinetic and stoichiometric analysis for the binding of Escherichia coli ribonuclease H1 to RNA-DNA hybrids using surface plasmon resonance. Journal of Biological Chemistry. 272 (35), 22015-22022 (1997).
재인쇄 및 허가
JoVE'article의 텍스트 или 그림을 다시 사용하시려면 허가 살펴보기
허가 살펴보기This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. 판권 소유