
Method Article
ドットブロットによるRループ解析
要約
このプロトコルでは、RNA-DNAハイブリッドと置換DNA鎖で構成される3本鎖核酸構造であるRループを定量化する簡単な方法を詳しく説明しています。
要約
3本鎖核酸構造であるRループは、遺伝子調節におけるその役割についてますます認識されています。当初、Rループは転写の副産物であると考えられていました。しかし、罹患細胞におけるRループの減少に関する最近の発見により、Rループがさまざまなヒト細胞で機能的な役割を果たしていることが明らかになりました。次に、Rループの役割と、細胞がその存在量をどのようにバランスさせるかを理解することが重要です。この分野での課題は、Rループの定量化であり、その研究の多くがS9.6モノクローナル抗体に依存しており、そのRNA-DNAハイブリッドに対する特異性が疑問視されています。ここでは、S9.6抗体を用いたドットブロットを用いてRループを定量し、RNA-DNAハイブリッド、一本鎖RNA、二本鎖RNAをそれぞれ切断するRNase H、RNase T1、およびRNase IIIを用いたこのアッセイの感度と特異性を示します。この方法は再現性が高く、一般的な実験装置と試薬を使用し、2日以内に結果が得られます。このアッセイは、研究や臨床の現場でRループを定量化し、セナタキシンなどの遺伝子の突然変異がRループの存在量に及ぼす影響を評価するために使用できます。
概要
このプロトコールは、3本鎖核酸構造であるRループの存在量を迅速に比較評価できるドットブロットアッセイのステップバイステップガイドを提供します。Rループは、RNAが二本鎖DNAに侵入してRNA-DNAハイブリッドを生成し、もう一方のDNA鎖を置き換えるときに形成されます。Rループは、RNAのライフサイクルのさまざまな段階で見られます。転写複合体では、新生RNAはテンプレートDNAに相補的に合成され、非テンプレート鎖は置換されます。短いRNA-DNAハイブリッド(<10 bp)は、新生RNAを遊離するように分離されるため、出口チャネル1,2を介してRNAポリメラーゼ複合体を離れることができます。転写複合体の外側では、新生RNAはそのDNAテンプレートに近く、コピーされることからまだわずかにほどかれているため、RNAはそのテンプレートDNAと再ハイブリダイズしてRループを形成することができます3。さらに、Rループは、複製複合体と転写複合体が衝突した場合4、アンチセンス転写の場合5に形成されます。その形成には多くの機会があるため、Rループは珍しくなく、細胞の転写状態にもよりますが、ヒトゲノム6の3〜5%に見られます。Rループは、mRNAの遺伝子プロモーター7および終末5部位、ならびにリボソームRNA8およびトランスファーRNA9に沿って見出される。Rループは、染色体のテロメア領域にもあります。
Rループは規制の役割を果たします。それらは、プロモーター10,11での転写に影響を与え、クラススイッチ組換え12を媒介し、CRISPRベースのゲノム編集13,14,15を促進することにより、遺伝子発現を調節する。多くの細胞イベントと同様に、Rループの存在量は厳密に滴定されています。Rループが多すぎたり少なすぎたりすると、正常な細胞機能に影響を与える16,17.Rループは、RNase H、セナタキシン、およびRNA-DNAハイブリッド18,19,20,21,22を巻き戻す他のヘリカーゼを含む様々なタンパク質によって調節されている。
Rループの存在量をモニターするために、ゲノムワイドな方法は、まず抗体S9.6 8,23,24またはRNase H 10,26,27を含む他のヌクレアーゼ25でRループを濃縮し、次にシーケンシングによって濃縮されたRループの数を評価する。これらのシーケンシングベースの方法の初期のバージョンでは、正確な定量を可能にするのに十分なシーケンスカバレッジを達成できませんでしたが、シーケンシング技術の急速な進歩により、遺伝子座ごとのRループ解析が可能になりました。免疫蛍光法は、Rループ10,17の定量および局在化にも使用されています。これらの方法は包括的ですが、高価な機器と専門的な分析が必要なため、多くの臨床現場では、または初期評価として実用的ではありません。
臨床現場の検査室をまたいで一律に実施できる手技が求められています。ドットブロットは、特定の機器や計算解析なしで実施できるため、このようなオプションを提供します。Rループに対する突然変異の影響を評価するための予備的なステップとして、または臨床現場で、これらのドットブロットは感度の高い特異的な結果を提供する必要があります。ここでは、Rループを具体的に同定するアッセイについて説明します。二本鎖(ds)DNA、二本鎖RNA、および一本鎖RNAからのシグナルは除外されます。私たちのプロトコルは、S9.6抗体27を使用してRループ内のRNA-DNAハイブリッドを特定し、RNaseH、切断してRNA-DNAハイブリッド20,28のRNAの分解につながるエンドリボヌクレアーゼを組み込んで、検出されたシグナルがハイブリッドのシグナルであることを確認します。また、グアニン29,30で一本鎖RNAを切断するRNase T1と、ステムループ31,32を含む二本鎖RNAを切断するRNase IIIを組み込み、非特異的なシグナルをチェックしました。S9.6抗体は、長さがわずか8ヌクレオチドのものであっても、さまざまな長さのRNA-DNAハイブリッドを認識する33。
ここでは、核酸の単離から始まり、ドットブロット調製、およびS9.6抗体によるRループ検出までのプロトコールを紹介します。当社のプロトコルには、同量のサンプルがロードされ、信号が特異的であることを確認するための手順が含まれています。オリゴヌクレオチドを提供し、ポジティブコントロールとネガティブコントロールとして機能します。これは、Rループの存在量を評価するための迅速、簡単、かつユーザーフレンドリーな方法です。
プロトコル
1. 核分画のための細胞溶解
- 細胞を1倍リン酸緩衝生理食塩水(PBS)で2回洗浄します。トリプシンなどの標準的な細胞解離技術を用いて、組織培養皿から細胞を取り出します。血球計算盤を使用して細胞をカウントします。
注:以下に説明するステップは、一次ヒト皮膚線維芽細胞の分析に使用されましたが、一連の細胞タイプをアッセイすることができます。線維芽細胞は、10%のウシ胎児血清を含む基礎培地で増殖させた。あるいは、細胞溶解バッファー(表1)を洗浄後に細胞培養物に直接添加することもできます。 - 細胞懸濁液を1.5mLチューブに移し、細胞をペレット化します。
- サンプルを300 x g で4°Cで5分間遠心分離します。 メディアを吸引します。
- ステップ1.3の遠心分離設定を使用して、氷冷した1x PBSで2回洗浄します。
- コールドセル溶解バッファー(表1)を細胞ペレット(2 x 106 細胞あたり300 μL)に加えます。ピペットを上下に動かして、ペレットを再懸濁します。
- 氷上で10分間インキュベートします。
- 500 x g で5分間回転させ、核をペレット化します。
- 上清を捨て、核ペレットを400 μLの冷核溶解バッファーに再懸濁します(表1)。
- 氷上で10分間インキュベートします。
注:細胞の核および細胞質コンパートメントの分画により、シグナルの特異性が確保されます。核分離と細胞質分離の品質は、進行する前に評価できます(表2)。 - 20 mg/mL プロテイナーゼ K 3 μL を添加し、55 °C で 3-5 時間インキュベートします。
注:表示されている容量は2 x 106 セル用で、必要に応じてスケールアップまたはスケールダウンします。
2. ゲノムDNA(RNA-DNAハイブリッドを含む)の精製
- DNAが粘性がある場合は、粘度を下げるために超音波処理を行います(例えば、高出力で超音波処理を行い、30秒ON/30秒OFFで、4°Cのウォーターバスを使用して2分間)。
- 溶出バッファー400 μL(表1)とフェノール:クロロホルム:イソアミルアルコール400 μL(25:24:1 pH 8.0)を加えます。
- 10秒間渦巻きます。
- 12,000 x g で4°Cで5分間スピンダウンします。
- 水相(約350μL)を新しいチューブに移します。
- 1容量のクロロホルムを使用して1回抽出し、ボルテックスで10秒間渦巻き、次いで12,000 x g で4°Cで5分間スピンダウンします。 水相を新しいチューブ(約300μL)に移します。
- 35 μLの3 M酢酸ナトリウム(pH 5.2)、1 μLのグリコーゲン、および700 μLの氷冷100%エタノールを加えます。
- 10秒間ボルテックスし、4°Cで12,000 x g で30分間スピンダウンします。
- ペレットを1mLの70%エタノールで洗浄します。
- 10秒間ボルテックスし、4°Cで12,000 x g で15分間スピンダウンします。
- 上清を捨て、ペレットを自然乾燥させます。
- 12 μLの溶出バッファーとボルテックスを10秒間加えて再懸濁します。サンプルを37°Cで30分間インキュベートし、撹拌するか、4°Cで一晩インキュベートしてペレットを再懸濁します。
- 標準的な分光光度法を使用してDNA濃度を測定します。
注:表示されている容量は2 x 106 セル用で、必要に応じてスケールアップまたはスケールダウンします。DNA(RNA-DNAハイブリッドを含む)は、必要に応じて-20°Cで保存できます。
3. DNAサンプル(RNA-DNAハイブリッドを含む)をナイロンメンブレンにブロッティングします
- 溶出バッファー(50 ng/μL、25 ng/μL、または12.5 ng/μL)で核酸を所望の濃度に希釈します。これらのサンプルは、濃度の範囲(200、100、50、25、12.5 ng)で、線形範囲内に信号が存在することを保証します。
注:技術的および生物学的な複製、およびさまざまなRNase処理については、ステップ5を参照してください。 - 2 μLのサンプルごとに0.5 cm2の領域を占めるスペースが空くように、正に帯電したナイロンメンブレンを準備します。
- 各サンプルの2 μLを2つのメンブレン(1つはS9.6抗体用、もう1つはdsDNA抗体用)にスポットします。あるいは、大量のサンプルをロードできるドットブロットまたはスロットブロット装置を使用することもできます。
- サンプルがメンブレンに飽和するのを待ちます。少なくとも2分待ってから、メンブレンをUV光で架橋してください。
- メンブレンをUVデバイスの中央に置き、UVクロスリンカーで「Auto Crosslink」設定(1,200 μJ x 100)でメンブレンを架橋します。
4. S9.6抗体によるRNA-DNAハイブリッド検出
- メンブレンをブロッキング溶液(0.05% Tween-20(TBST)を含むトリス緩衝生理食塩水溶液5%牛乳)で、シェーカーで室温で1時間インキュベートします。
注:メンブレンを覆うのに十分なブロッキング溶液が必要です。 - メンブレンを一次抗体(TBST中の5%ミルク中)で4°Cで一晩、振盪しながらインキュベートします。1つのメンブレンに抗dsDNA抗体(1:10,000希釈)を添加します。1μg/mL S9.6抗体を第2のメンブレン(1:1,000希釈)に添加します。
注:S9.6抗体は、市販されているか、国立衛生研究所のS.Leppla博士、NIAIDから入手できます。 - 一次抗体を取り出し、TBSTで3回洗浄します。室温で振とうしながら、各洗浄を5〜10分間行います。
- 西洋ワサビペルオキシダーゼ(HRP)標識二次抗体(抗マウス、1:5,000希釈)とTBST中の5%ミルクと室温で振とうしながらインキュベートします。
注:抗dsDNAおよび抗RNA-DNAハイブリッドはどちらもマウス抗体です。 - 二次抗体を取り出し、TBSTで3回、室温で振とうしながら5〜10分間洗浄します。
- ECL(Enhanced Chemiluminescence)試薬を用いて、イメージング用のシグナルを取得してください。
- ImageJなどの標準的な画像処理ツールを使用して、信号強度を定量化します。
メモ: トラブルシューティングの詳細については 、表 2 を参照してください。
5. シグナル特異性を評価するためのリボヌクレアーゼ治療
注:RNase処理は、S9.6結合の特異性を実証するために、核酸サンプルに対して実施する必要があります。RNase T1またはRNase IIIではなくRNase Hで治療すると、S9.6免疫染色が減少するはずです。
- RNA-DNAハイブリッドを含むサンプルを4つの別々のチューブで調製して分解します。4つのサンプルのそれぞれを、5 U RNase H、1000 U RNase T1、0.5 U RNase III、またはモックのいずれかで処理します。サンプルを37°Cで15分間、20 μLの容量でインキュベートします。
- セクション3で説明されているように、各サンプルの2 μLをメンブレンにロードします。
6. シグナル特異性評価のためのオリゴヌクレオチドコントロールの調製
注:オリゴヌクレオチドコントロールは、S9.6結合の特異性を実証するために使用できます。S9.6は、RNA-DNAハイブリッドを認識するが、以前に報告されているようにdsDNAまたはdsRNAコントロールは認識しない34。
- オリゴヌクレオチド(表3)をアニーリングバッファー(10 mM Tris、pH 8.0、50 mM NaCl、1 mM EDTA)に100 μMまで溶解します。
- 4つの反応チューブを準備します
- RNA-DNAハイブリッド#1:10 μLのssRNAトップストランドを10 μLのssDNAボトムストランドおよび80 μLのアニーリングバッファーと混合します。
- RNA-DNAハイブリッド#2:10 μLのssDNAトップストランドを10 μLのssRNAボトムストランドおよび80 μLのアニーリングバッファーと混合します。
- dsRNA:10 μLのssRNAトップストランドを10 μLのssRNAボトムストランドおよび80 μLのアニーリングバッファーと混合します。
- dsDNA:10 μLのssDNAトップストランドを10 μLのssDNAボトムストランドおよび80 μLのアニーリングバッファーと混合します。
- ステップ6.2の4つの混合物を95°Cで10分間加熱します。
- チューブをゆっくりと室温まで冷まして、ストランドの再焼鈍を可能にします。アニーリングされた標準試料は、後で使用するために-20°Cで保存できます。
注:アニーリング効率は、非変性ゲル電気泳動で確認する必要があります。二本鎖は、アニーリングされていないオリゴヌクレオチドよりもゆっくりと移動します(表2)。 - セクション3で説明したように、各サンプルの2 μLを2つのメンブレン(1つはS9.6抗体用、もう1つはdsDNA抗体用)にロードします。
- セクション 4 で説明されている手順を実行します。
7. ImageJを用いたS9.6 Rループ信号強度の定量化と正規化
- S9.6、dsDNA染色の画像をTIFF形式で保存し、ImageJソフトウェア(https://imagej.nih.gov/ij/)を使用して解析します。
- 画像の反転オプションを選択します(編集|Invert)をクリックします。反転後、各ドットは暗い背景に対して白く表示されます。
- 楕円画像選択ツールを使用して、画像上の最大のドットを囲むのに十分な大きさの楕円を選択します。
- ROIマネージャーを使用して、定量化のために選択した領域を追加します。[Show All] と [Labels] のオプションが選択されていることを確認して、関心領域を視覚化できるようにします。
- ステップ 7.3 で使用したのと同じ楕円形の選択領域を使用して、定量化する各ドットの周囲に関心領域を追加します。 Command + Shift + E ショートカットを使用して、ステップ7.3で選択した領域を後続の各ドットにコピーします。
- 各関心領域の積分密度を測定します。
- 各サンプルのS9.6信号強度をdsDNAの測定値で除算して、S9.6/dsDNA信号比を求めます。実験を繰り返して結果を確認します(S9.6とdsDNAシグナル取得の両方で少なくとも3回)。平均の標準誤差は、S9.6/dsDNAの信号比から計算できます。
結果
S9.6(RNA-DNA)抗体の特異性を評価するための酵素治療。
一次ヒト皮膚線維芽細胞が成長した17。RNA-DNAハイブリッドを含むDNAを単離し、定量しました。2 μg のサンプルを RNase T1、RNase H、または RNase III で 37 °C で 15 分間分解しました。 また、RNase処理サンプルとの比較のために模擬サンプルも分析しました。サンプル(200、100、50、25、12.5、または6.25 ng)を、セクション3で説明したように、2つの異なるメンブレンにブロットしました。メンブレンは架橋され、ブロックされ、そのうちの1つがS9.6抗体でプローブされました(図1A)。
その結果、S9.6信号はロードされたサンプルの存在量と相関していることが示されました。RNase Hによる治療は行うが、RNase T1またはRNase IIIによる治療は行わないと、S9.6染色が減少します。
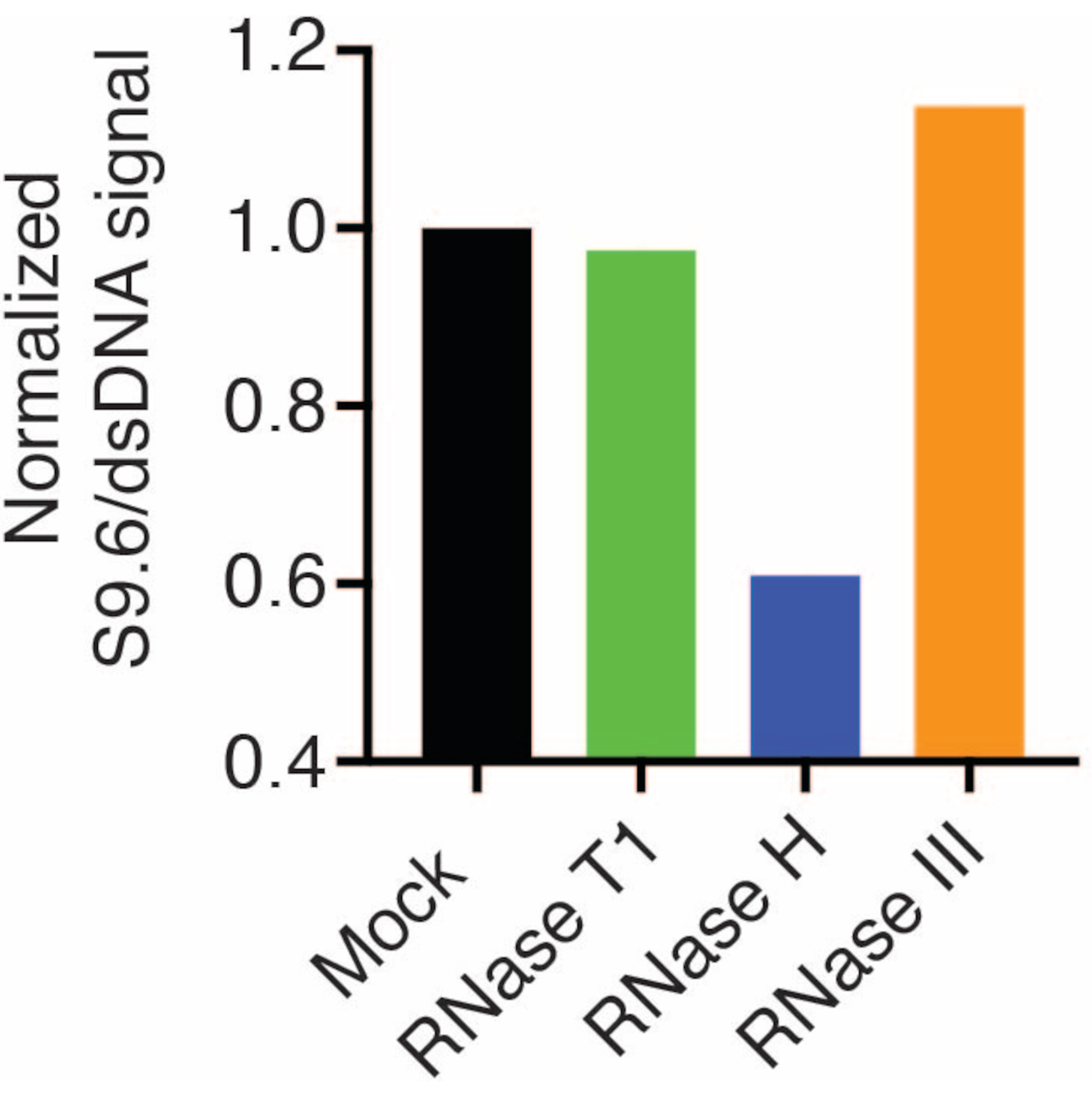
標準化のために、第2のメンブレンをdsDNA抗体(図1B)でプローブしました。画像Jを使用して、信号強度を解析しました。S9.6およびdsDNA抗体からのシグナル強度がダイナミックレンジ内にあったため、50 ngのサンプルを定量用に選択しました。信号強度は、模擬サンプルの強度に正規化されました。 データを図 2 に示します。
合成ヌクレオチドコントロールを用いたS9.6抗体ドットブロット。
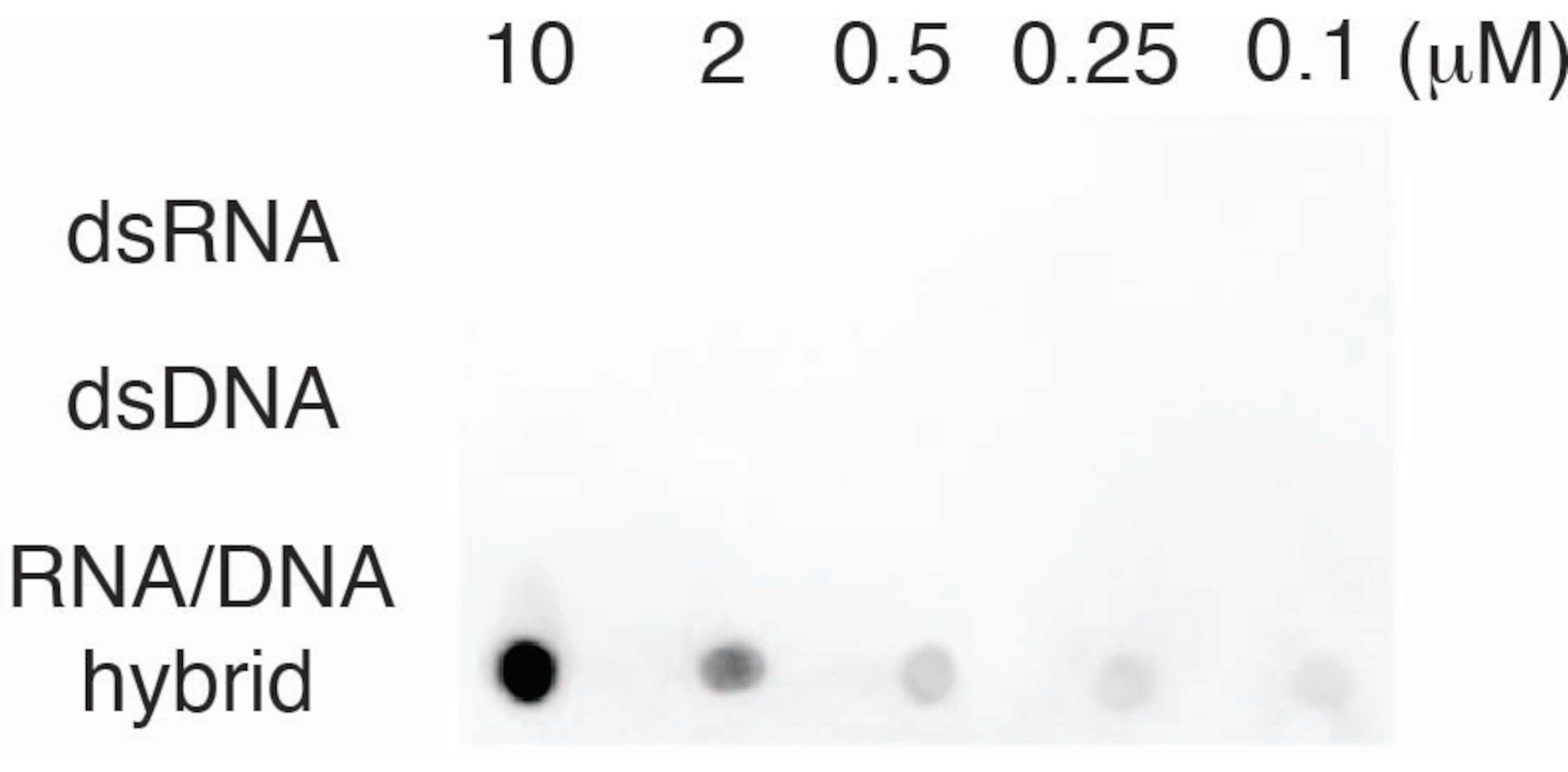
S9.6抗体の特異性を評価するために、セクション6で説明したように、dsRNA、dsDNA、およびRNA-DNAに対応するオリゴヌクレオチドを使用しました。RNA-DNA、dsRNA、およびdsDNAヌクレオチドの希釈系列を調製し、セクション3に記載したようにナイロンメンブレン上にブロットした。メンブレンをS9.6抗体でプローブしました(図3)。その結果、S9.6抗体は用量依存的にRNA-DNAハイブリッドに特異的に結合し、dsRNAおよびdsDNAに対する交差反応性は最小限であることが示されました。

図1:ヒト線維芽細胞由来の核酸を充填したドットブロットで示されるS9.6の特異性。 ヒト線維芽細胞由来の核酸サンプルを模擬処理するか、RNase T1、RNase H、またはRNase IIIで処理した後、2 μLドットあたり200、100、50、25、12.5、および6.25 ngの希釈シリーズでナイロンメンブレンにロードしました。次に、メンブレンをS9.6抗体(A)またはdsDNA抗体(B)でプローブしました。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図2:S9.6 Rループ染色の定量 図1 の50 ngサンプルをImageJによる定量用に選択しました。S9.6 シグナルを dsDNA シグナル強度で除算し、セクション 7 で概説した手順に従って模擬サンプルに正規化しました。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図3:オリゴヌクレオチドコントロールを用いたS9.6ドットブロット。 s9.6抗体は、dsRNA、dsDNA、またはRNA-DNAハイブリッドとして希釈された一連の合成オリゴヌクレオチドに対してドットブロットします。S9.6は、用量依存的にRNA-DNAハイブリッドに特異的に結合します。この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
| 細胞溶解バッファー | 10mL用 | 200mL用 | ファイナルコンク |
| 水、ヌクレアーゼフリー | 9mL | 180mL | - |
| NP-40 10% | 0.5mL | 10mL | 0.5% |
| 2M KCl | 0.4mL | 8mL | 80mM |
| 0.5Mパイプ(pH 8.0) | 100uL | 2mLの | 5mM |
| 核溶解バッファー | 10mL用 | 200mL用 | ファイナルコンク |
| 水、ヌクレアーゼフリー | 8.65ミリリットル | 173mL | - |
| 10%のSDS | 1mLの | 20mL | 1% |
| 1M Tris-HCl (pH 8.0) | 0.25mL | 5mL | 25メートル |
| 0.5M EDTAの | 100uL | 2mLの | 5メートルメートル |
| 溶出バッファー | 10mL用 | 200mL用 | ファイナルコンク |
| 1M Tris-Cl、pH 8.5 | 0.1ミリリットル | 2ミリリットル | 10 mM |
| 水、ヌクレアーゼフリー | 9.9ミリリットル | 198ミリリットル | - |
テーブル 1.バッファーの調製
| 歩 | 問題 | 考えられる理由 | 解決 |
| 1.9 | 該当なし | 細胞分画の検証 | 核分離および細胞質分離の品質は、標準的なプロテアーゼ阻害剤カクテルを細胞および核溶解バッファーに添加することで評価できます(表1)。 細胞質画分および核画分は、ウェスタンブロッティング分析によって評価し、細胞質画分における細胞質マーカー(GAPDHまたはHSP90など)による標識および核画分における核マーカー(HDAC1またはHistone H3など)による標識の適切な制限を確認できます。核分画におけるミトコンドリア汚染の寄与は、ミトコンドリアDNAに特異的なプローブを用いたqPCR分析によって評価できます。 |
| 2.1 | サンプルが粘性が高すぎる | セル番号が多すぎます。 | DNAを半分に減らし、超音波処理ステップ2.1を続けます |
| 2.12 | 目に見えるペレットはありません | 出発物質が不十分であるか、抽出中に失われます。 | より多くのセルを使用して最初から開始します。 |
| 2.13 | 低DNA濃度 | ||
| 3.1 | 希釈に十分なDNAがない | ||
| 3.4 | サンプルがメンブレンに飽和しない | メンブレンを1x TBSTに浸します。余分なバッファーを乾燥させ、ステップ3.4から開始します | |
| 4.6 | 斑点のあるパターンまたは斑点のあるパターンが検出される | 0.1% ドデシル硫酸ナトリウム(SDS)をサンプルに添加し、ステップ 3.1 に進みます | |
| 4.6 | ドットは「コーヒーリング」の外観をしています | 0.01% サルコシルをサンプルに添加し、ステップ 3.1 から続行します | |
| 5.2 | RNaseHコントロールはシグナルの減少を示していません | リボヌクレアーゼの消化が不完全である。 | インキュベーションを増やすか、酵素濃度を上げます。 |
| 6.5 | オリゴコントロールの信号がありません | デュプレックスは形成されませんでした。オリゴヌクレオチドが適切にアニールされていなかった。 | オリゴヌクレオチドとアニーリングバッファーの比率を確認します。 |
| 6.5 | S9.6 シグナルはハイブリッドに特有のものではありません | S9.6抗体バッチは非特異的結合を有する | S9.6抗体の新しいバッチの感度と特異性を、RNase酵素処理または合成オリゴヌクレオチド分析のいずれかを使用して検証します。 |
表 2:トラブルシューティング
| ssRNA、トップストランド | 5'-UGGGGGCUCGUCCGGGAUGGAUGGGAACCACUGAUCCCCC-3' |
| ssDNA、トップストランド | 5'-TGGGGGCTCGTCCGGGATATGGGAACCACTGATCCC-3' |
| ssRNA、ボトムストランド | 5'-GGGAUCAGUGGUUCCCAUAUCCCGGACGAGCCCCCA-3' |
| ssDNA、ボトムストランド | 5'-GGGATCAGTGGTTCCCATCATTCCCGGACGAGCCCCCA-3' |
テーブル3。オリゴヌクレオチド配列の制御
ディスカッション
3本鎖核酸であるRループは、RNAのライフサイクルのさまざまな段階で形成され、細胞プロセスを調節することがますます明らかになっています。Rループを完全に理解するには、Rループ検出のための信頼性の高い手法が必要です。ここでは、S9.6抗体8,23,24を用いてRループの存在量を調べるアプローチについて述べます。この方法により、細胞や組織培養サンプルから得られるRループの存在量を迅速に評価することができます。特別な機器や大量の出発材料は必要ありません。RNase処理の組み合わせにより、特異的で再現性のある結果が得られます。
S9.6抗体の特異性について懸念を抱いているという報告もあります。他の試薬と同様に、S9.6抗体にはバッチ間のばらつきがあります。私たちのプロトコルには、シグナルの特異性を確認するためのRNase H、RNase T1、およびRNase IIIが含まれています。さらに、S9.6抗体の各バッチの特異性を確保するために、合成オリゴヌクレオチドを使用しています。
R-loop生物学は成長分野です。今回紹介したような信頼性の高い検出・定量法の開発は、Rループがいつ形成され、どのように制御され、何を制御しているのかを解明するための機構研究を促進するでしょう。適切なコントロールにより、このドットブロットアッセイは、臨床および研究環境でRループの存在量をスクリーニングする簡単な方法です。
開示事項
著者は何も開示していません。
謝辞
この研究は、ハワードヒューズ医学研究所と国立神経障害および脳卒中研究所の学内研究によって支援されました。
S9.6抗体のバッチを解析用に提供してくださったStephen Leppla博士に感謝します。また、リボヌクレアーゼ治療にご協力いただいたDongjun Li博士にも感謝いたします。
資料
| Name | Company | Catalog Number | Comments |
| Anti-dsDNA antibody | Abcam | ab27156 | |
| Anti-RNA-DNA hybrid antibody (S9.6) | Kerafast | ENH001 | |
| Biorupter sonicator | Diagenode | UCD-200 | |
| EB Buffer | Qiagen | 19086 | |
| EDTA (0.5M) | Invitrogen | AM9261 | |
| Hybond N+ nylon membrane | GE healthcare Life Sciences | RPN203B | |
| KCl (2M) | Invitrogen | AM9640G | |
| NP-40 (Igepal CA-630) | Sigma | I8896 | |
| PBS | Invitrogen | 10010-023 | |
| Phenol:chloroform:isoamyl alcohol | Invitrogen | 15593031 | |
| PIPES (0.5M, pH 8.0) | VWR | AAJ61406-AE | |
| Proteinase K | Qiagen | 19131 | |
| RNase III | Invitrogen | AM2290 | |
| RNase H | New England Biolabs | M0297 | |
| RNase T1 | ThermoFisher Sci. | EN0541 | |
| SDS (10%) | Invitrogen | 15553027 | |
| sodium acetate (3M, pH 5.2) | Invitrogen | AM9740 | |
| Tris-buffered saline (10X) | Corning | 46-012-CM | |
| Tris-HCl (1M, pH 8.0) | KD Medical | RGF-3360 | |
| TrypLE | Invitrogen | 12605010 | |
| Tween-20 | Sigma | P9416 | |
| UV Stratalinker 2400 | Stratagene | Stratalinker 2400 | |
| Whatman marking pen | Sigma | WHA10499001 |
参考文献
- Daube, S. S., von Hippel, P. H. RNA displacement pathways during transcription from synthetic RNA-DNA bubble duplexes. Biochemistry. 33 (1), 340-347 (1994).
- Westover, K. D., Buschnell, D. A., Kornberg, R. D. Structural basis of transcription: separation of RNA from DNA by RNA polymerase II. Science. 303 (5660), 1014-1016 (2004).
- Itoh, T., Tomizawa, J. Formation of an RNA primer for initiation of replication of ColE1 DNA by ribonuclease H. Proceedings of the National Academy of Science USA. 77 (5), 2450-2454 (1980).
- Hamperl, S., Bocek, M. J., Saldivar, J. C., Swigut, T., Cimprich, K. A. Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell. 170 (4), 774-786 (2017).
- Skourti-Stathaki, K., Kamieniarz-Gdula, K., Proudfoot, N. J. R-loops induce repressive chromatin marks over mammalian gene terminators. Nature. 516 (7531), 436-439 (2014).
- Sanz, L. A., et al. conserved R-loop structures associate with specific epigenomic signatures in mammals. Molecular Cell. 63 (1), 167-178 (2016).
- Chen, L., et al. R-ChIP using inactive RNase H reveals dynamic coupling of R-loops with transcriptional pausing at gene promoters. Molecular Cell. 68 (4), 745-757 (2017).
- El Hage, A., French, S. L., Beyer, A. L., Tollervey, D. Loss of topoisomerase I leads to R-loop mediated transcriptional blocks during ribosomal RNA synthesis. Genes and Development. 24 (14), 1546-1558 (2010).
- Tran, P., et al. PIF1 family DNA helicases suppress R-loop mediated genome instability at tRNA genes. Nature Communication. 8, 15025(2017).
- Ginno, P. A., Lott, P. L., Christensen, H. C., Korf, I., Chedin, F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Molecular Cell. 45 (6), 814-825 (2012).
- Colak, D., et al. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science. 343 (6174), 1002-1005 (2014).
- Yu, K., Chedin, F., Hsieh, C., Wilson, T. E., Lieber, M. R. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nature Immunology. 4 (5), 442-451 (2003).
- Gasiunas, G., Barrangou, R., Horvath, P., Siksnys, V. Cas9-crRNA ribonucleotide complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proceedings of the National Academy of Science USA. 109 (39), 2579-2586 (2012).
- Westra, E. R., et al. CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by Cascade and Cas3. Molecular Cell. 46 (5), 595-605 (2012).
- Jiang, F., et al. Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science. 351 (6275), 867-871 (2016).
- Huertas, P., Aguilera, A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Molecular Cell. 12 (3), 711-721 (2003).
- Grunseich, C., et al. Senataxin mutation reveals how R-loops promote transcription by blocking DNA methylation at gene promoters. Molecular Cell. 69 (3), 426-437 (2018).
- Kim, H. D., Choe, J., Seo, Y. S. The sen1(+) gene of Schizosaccharomyces pombe, a homologue of budding yeast SEN1, encodes an RNA and DNA helicase. Biochemistry. 38 (44), 14697-14710 (1999).
- Stein, H., Hausen, P. Enzyme from calf thymus degrading the RNA moiety of DNA-RNA hybrids: effect on DNA-dependent RNA polymerase. Science. 166 (3903), 393-395 (1969).
- Cerritelli, S. M., Crouch, R. J. Ribonuclease H: the enzymes in eukaryotes. FEBS Journal. 276 (6), 1494-1505 (2009).
- Hyjek, M., Figiel, M., Nowotny, M. RNases H: Structure and mechanism. DNA Repair. 84, 102672(2019).
- Pohl, T. J., Zakian, V. A. Pif1 family DNA helicases: A helpmate to RNase H. DNA Repair. 84, 102633(2019).
- Pohjoismaki, J. L., et al. Mammalian mitochondrial DNA replication intermediates are essentially duplex but contain extensive tracts of RNA/DNA hybrid. Journal of Molecular Biology. 397 (5), 1144-1155 (2010).
- Sanz, L. A., Chedin, F. High-resolution, strand-specific R-loop mapping via S9.6-based DNA-RNA immunoprecipitation and high-throughput sequencing. Nature Protocols. 14 (6), 1734-1755 (2019).
- Wahba, L., Costantino, L., Tan, F. J., Zimmer, A., Koshland, D. S1-DRIP-seq identifies high expression and polyA tracts as major contributors to R-loop formation. Genes and Development. 30 (11), 1327-1338 (2016).
- Yan, Q., Shields, E. J., Bonasio, R., Sarma, K. Mapping native r-loops genome-wide using a targeted nuclease approach. Cell Reports. 29 (5), 1369-1380 (2019).
- Hu, Z., Zhang, A., Storz, G., Gottesman, S., Leppla, S. H. An antibody-based microarray assay for small RNA detection. Nucleic Acids Research. 34 (7), 52(2006).
- Nowotny, M., Gaidamakov, S. A., Crouch, R. J., Yang, W. Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell. 121 (7), 1005-1016 (2005).
- Sato, K., Egami, F. Studies on ribonucleases in takadiastase. Journal of Biochemistry. 44 (11), Tokyo. 753-767 (1957).
- Takahashi, K., Moore, S. Ribonuclease T1. Enzymes. 15, 435-468 (1982).
- Dunn, J. J. RNase III cleavage of single-stranded RNA: Effect of ionic strength in the fidelity of cleavage. Journal of Biological Chemistry. 251 (12), 3807-3814 (1976).
- Pertzev, A., Nicholson, A. W. Characterization of RNA sequence determinants and antideterminants of processing reactivity for a minimal substrate of Escherichia coli ribonuclease III. Nucleic Acids Research. 34 (13), 3708-3721 (2006).
- Phillips, D. D., et al. The sub-nanomolar binding of DNA-RNA hybrids by the single chain Fv fragment of antibody S9.6. Journal of Molecular Recognition. 26 (8), 376-381 (2013).
- Haruki, M., Noguchi, E., Kanaya, S., Crouch, R. J. Kinetic and stoichiometric analysis for the binding of Escherichia coli ribonuclease H1 to RNA-DNA hybrids using surface plasmon resonance. Journal of Biological Chemistry. 272 (35), 22015-22022 (1997).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved