
Method Article
Analisi R-Loop mediante Dot-Blot
In questo articolo
Riepilogo
Questo protocollo descrive in dettaglio un metodo semplice che quantifica l'R-loop, una struttura di acido nucleico a tre filamenti che comprende un ibrido RNA-DNA e un filamento di DNA spostato.
Abstract
La struttura dell'acido nucleico a tre filamenti, R-loop, è sempre più riconosciuta per il suo ruolo nella regolazione genica. Inizialmente, si pensava che gli R-loop fossero i sottoprodotti della trascrizione; ma recenti scoperte di un minor numero di R-loop nelle cellule malate hanno chiarito che gli R-loop hanno ruoli funzionali in una varietà di cellule umane. Successivamente, è fondamentale comprendere i ruoli dei cicli R e il modo in cui le cellule bilanciano la loro abbondanza. Una sfida nel campo è la quantificazione degli R-loops, poiché gran parte del lavoro si basa sull'anticorpo monoclonale S9.6, la cui specificità per gli ibridi RNA-DNA è stata messa in discussione. Qui, utilizziamo dot-blot con l'anticorpo S9.6 per quantificare gli R-loop e mostrare la sensibilità e la specificità di questo test con RNasi H, RNasi T1 e RNasi III che scindono rispettivamente gli ibridi RNA-DNA, l'RNA a singolo filamento e l'RNA a doppio filamento. Questo metodo è altamente riproducibile, utilizza attrezzature di laboratorio e reagenti generici e fornisce risultati entro due giorni. Questo test può essere utilizzato in ambito clinico e di ricerca per quantificare gli R-loop e valutare l'effetto delle mutazioni in geni come la senatassina sull'abbondanza dell'R-loop.
Introduzione
Questo protocollo fornisce una guida passo passo a un test dot-blot che consente una rapida valutazione comparativa dell'abbondanza di R-loop, una struttura di acido nucleico a tre filamenti. L'R-loop si forma quando l'RNA invade un DNA a doppio filamento per generare un ibrido RNA-DNA e sposta l'altro filamento di DNA. Gli R-loop si trovano in diverse fasi del ciclo di vita dell'RNA. Nel complesso trascrizionale, l'RNA nascente viene sintetizzato in modo complementare al DNA stampo e il filamento non stampo viene spostato. L'ibrido RNA-DNA corto (<10 bp) è deciso a liberare l'RNA nascente in modo che possa lasciare il complesso RNA polimerasi attraverso il canale di uscita 1,2. Al di fuori del complesso trascrizionale, l'RNA nascente è vicino al suo stampo di DNA, che è ancora leggermente srotolato dall'essere copiato, quindi l'RNA può riibridarsi con il suo DNA stampo formando R-loop3. Inoltre, i R-loop possono formarsi quando i complessi di replicazione e trascrizione si scontrano4 e nella trascrizione antisenso5. Date le numerose opportunità per la loro formazione, gli R-loop non sono rari e possono essere trovati nel 3-5% del genoma umano6, a seconda dello stato di trascrizione della cellula. Gli R-loop si trovano nei promotori genici7 e nei siti di terminazione5 nell'mRNA e lungo l'RNA ribosomiale8 e l'RNAdi trasferimento 9. Gli R-loop si trovano anche nelle regioni telomeriche dei cromosomi.
Gli R-loop svolgono un ruolo regolatore. Regolano l'espressione genica influenzando la trascrizione ai promotori10,11, mediando la ricombinazione class-switch12 e facilitando l'editing genomico basato su CRISPR 13,14,15. Come molti eventi cellulari, l'abbondanza dell'R-loop è strettamente titolata; troppi o troppo pochi R-loop influiscono sulla normale funzione cellulare16,17. Gli R-loop sono regolati da una varietà di proteine tra cui la RNasi H, la senataxina e altre elicasi che svolgono gli ibridi RNA-DNA 18,19,20,21,22.
Per monitorare l'abbondanza di R-loops, i metodi genome-wide arricchiscono prima gli R-loop con l'anticorpo S9.6 8,23,24 o con altre nucleasi25 tra cui la RNasi H 10,26,27, quindi valutano il numero di R-loop arricchiti mediante sequenziamento. Le prime versioni di questi metodi basati sul sequenziamento non raggiungevano un'adeguata copertura della sequenza per consentire una quantificazione precisa, ma il rapido miglioramento delle tecnologie di sequenziamento consente ora l'analisi R-loop locus-by-locus. Le tecniche di immunofluorescenza sono state utilizzate anche per quantificare e localizzare i loop R 10,17. Questi metodi sono completi, ma non sono pratici in molti contesti clinici o come valutazioni iniziali poiché richiedono attrezzature costose e analisi specializzate.
È necessaria una procedura che possa essere eseguita in modo uniforme in tutti i laboratori in ambito clinico. I dot-blot forniscono tale opzione poiché possono essere eseguiti senza alcuna attrezzatura specifica o analisi computazionale. Come fase preliminare o in ambito clinico per valutare gli effetti delle mutazioni sui R-loops, questi dot-blot devono fornire risultati sensibili e specifici. Qui, descriviamo il nostro saggio che identifica specificamente gli R-loop; esclude i segnali dal DNA a doppio filamento (ds), dall'RNA a doppio filamento e dall'RNA a singolo filamento. Il nostro protocollo utilizza l'anticorpo S9.627 per identificare gli ibridi RNA-DNA nei R-loop e incorpora la RNasi H, un'endoribonucleasi che si scinde e quindi porta alla degradazione dell'RNA in un ibrido RNA-DNA 20,28, per garantire che i segnali rilevati siano quelli degli ibridi. Abbiamo anche incorporato la RNasi T1 che slega l'RNA a singolo filamento alla guanina29,30 e la RNasi III che scinde l'RNA a doppio filamento, inclusi gli stem-loop31,32 per verificare la presenza di segnali aspecifici. L'anticorpo S9.6 riconosce ibridi RNA-DNA di varie lunghezze, anche quelli che sono lunghi solo 8 nucleotidi33.
Qui, presentiamo il protocollo che inizia con l'isolamento dell'acido nucleico seguito dalla preparazione del dot-blot e dal rilevamento dell'R-loop con l'anticorpo S9.6. Il nostro protocollo include passaggi per garantire che vengano caricate quantità uguali di campioni e che i segnali siano specifici. Fornisce oligonucleotidi che fungono da controlli positivi e negativi. Questo è un metodo rapido, facile e intuitivo per valutare l'abbondanza dell'R-loop.
Protocollo
1. Lisi cellulare per il frazionamento nucleare
- Lavare due volte le celle con 1 soluzione salina tamponata con fosfato (PBS). Rimuovere le cellule dai piatti di coltura tissutale utilizzando tecniche standard di dissociazione cellulare come la tripsina. Contare le cellule utilizzando un emocitometro.
NOTA: Le fasi descritte di seguito sono state utilizzate per l'analisi dei fibroblasti primari della pelle umana, sebbene sia possibile analizzare una serie di tipi di cellule. I fibroblasti sono stati coltivati in terreni basali contenenti il 10% di siero fetale bovino. In alternativa, il tampone di lisi cellulare (Tabella 1) può essere aggiunto direttamente alla coltura cellulare dopo il lavaggio. - Trasferire la sospensione cellulare in una provetta da 1,5 mL per pellettare le cellule.
- Centrifugare il campione a 300 x g per 5 minuti a 4 °C. Aspirare i supporti.
- Lavare due volte con 1x PBS ghiacciato utilizzando le impostazioni di centrifugazione al punto 1.3.
- Aggiungere il tampone di lisi delle celle fredde (Tabella 1) al pellet cellulare (300 μl per 2 x 106 cellule). Pipettare su e giù per risospendere il pellet.
- Incubare con ghiaccio per 10 min.
- Centrifugare a 500 x g per 5 minuti per pellettare i nuclei.
- Scartare il surnatante e risospendere il pellet nucleare in 400 μL di tampone di lisi nucleare fredda (Tabella 1).
- Incubare con ghiaccio per 10 min.
NOTA: Il frazionamento dei compartimenti nucleari e citoplasmatici delle cellule garantisce la specificità del segnale. La qualità della separazione nucleare e citoplasmatica può essere valutata prima di procedere (Tabella 2). - Aggiungere 3 μL di 20 mg/mL di proteinasi K e incubare per 3-5 ore a 55 °C.
NOTA: I volumi indicati sono per 2 x 106 celle, aumentare o diminuire in base alle esigenze.
2. Purificazione del DNA genomico (che include ibridi RNA-DNA)
- Se il DNA è viscoso, eseguire la sonicazione per ridurre la viscosità (ad esempio, sonicazione ad alta potenza, 30 s ON/ 30 s OFF, per 2 minuti utilizzando un bagno d'acqua a 4 °C).
- Aggiungere 400 μl di tampone di eluizione (Tabella 1) e 400 μl di fenolo:cloroformio:alcol isoamilico (25:24:1 pH 8,0).
- Vortice per 10 s.
- Centrifugare a 12.000 x g per 5 minuti a 4 °C.
- Trasferire la fase acquosa (circa 350 μl) in una nuova provetta.
- Estrarre una volta con 1 volume di cloroformio, agitare per 10 s, quindi centrifugare a 12.000 x g per 5 minuti a 4 °C. Trasferire la fase acquosa in una nuova provetta (circa 300 μl).
- Aggiungere 35 μl di acetato di sodio 3 M (pH 5,2), 1 μl di glicogeno e 700 μl di etanolo al 100% ghiacciato.
- Vortice per 10 s e centrifuga a 12.000 x g per 30 min a 4 °C.
- Lavare il pellet con 1 mL di etanolo al 70%.
- Vortice per 10 s e centrifuga a 12.000 x g per 15 min a 4 °C.
- Scartare il surnatante e lasciare asciugare il pellet all'aria.
- Aggiungere 12 μL di tampone di eluizione e vorticare per 10 s per risuspendare. Incubare il campione per 30 minuti a 37 °C con agitazione o a 4 °C durante la notte per risospendere il pellet.
- Misurare la concentrazione di DNA utilizzando la spettrofotometria standard.
NOTA: I volumi indicati sono per 2 x 106 celle, aumentare o diminuire in base alle esigenze. Il DNA (con ibridi RNA-DNA) può essere conservato a -20 °C, se necessario.
3. Tamponare campioni di DNA (che includono ibridi RNA-DNA) su membrane di nylon
- Preparare le diluizioni degli acidi nucleici alle concentrazioni desiderate nel tampone di eluizione (ad esempio, 50 ng/μL, 25 ng/μL o 12,5 ng/μL). Questi campioni con un intervallo di concentrazioni (200, 100, 50, 25, 12,5 ng) assicurano che ci saranno segnali all'interno dell'intervallo lineare.
NOTA: Assicurarsi di preparare un campione sufficiente per le repliche tecniche e biologiche e per i vari trattamenti con RNasi, vedere il passaggio 5. - Preparare una membrana di nylon caricata positivamente in modo che ci sia spazio per ogni campione da 2 μl per occupare un'area di 0,5 cm2.
- Posizionare 2 μL di ciascun campione su 2 membrane: una per l'anticorpo S9.6 e l'altra per l'anticorpo dsDNA. In alternativa, è possibile utilizzare un apparecchio dot-blot o slot-blot che consente il caricamento di campioni con volumi maggiori.
- Lasciare che i campioni si saturino nella membrana. Attendere almeno 2 minuti prima di reticolare la membrana con la luce UV.
- Posizionare la membrana al centro del dispositivo UV e reticolare la membrana utilizzando un reticolante UV utilizzando l'impostazione "Reticolazione automatica" (1.200 μJ x 100).
4. Rilevamento ibrido RNA-DNA con anticorpo S9.6
- Incubare la membrana in una soluzione bloccante (5% di latte in soluzione salina tamponata con Tris con 0,05% di Tween-20 (TBST) per 1 ora a temperatura ambiente su uno shaker.
NOTA: Dovrebbe esserci abbastanza soluzione bloccante per coprire la membrana. - Incubare le membrane per una notte in anticorpi primari (in latte al 5% in TBST) a 4 °C con agitazione. Aggiungere l'anticorpo anti-dsDNA (diluizione 1:10.000) a una membrana. Aggiungere 1μg/mL di anticorpo S9.6 alla seconda membrana (diluizione 1:1.000).
NOTA: L'anticorpo S9.6 è disponibile in commercio o presso il Dr. S. Leppla, NIAID, National Institutes of Health. - Rimuovere l'anticorpo primario e lavare 3 volte con TBST. Eseguire ogni lavaggio per 5-10 minuti agitando a temperatura ambiente.
- Incubare con anticorpo secondario coniugato con perossidasi di rafano (HRP) (anti-topo, diluizione 1:5.000) in latte al 5% in TBST con agitazione a temperatura ambiente.
NOTA: L'anti-dsDNA e l'ibrido anti-RNA-DNA sono entrambi anticorpi di topo. - Rimuovere l'anticorpo secondario e lavare 3 volte con TBST per 5-10 minuti agitando a temperatura ambiente.
- Sviluppare con reagenti a chemiluminescenza potenziata (ECL) per acquisire segnali per l'imaging.
- Quantifica l'intensità del segnale utilizzando strumenti di elaborazione delle immagini standard come ImageJ.
NOTA: La risoluzione dei problemi è descritta in dettaglio nella Tabella 2.
5. Trattamenti con ribonucleasi per valutare la specificità del segnale
NOTA: Il trattamento con RNasi deve essere eseguito sui campioni di acido nucleico per dimostrare la specificità del legame con S9.6. Il trattamento con RNasi H, ma non con RNasi T1 o RNasi III, dovrebbe portare a una riduzione dell'immunocolorazione S9.6.
- Digerire i campioni contenenti ibridi RNA-DNA preparandoli in quattro provette separate. Trattare ciascuno dei 4 campioni con 5 U RNasi H, 1000 U RNasi T1, 0,5 U RNasi III o simulare. Incubare i campioni a 37 °C per 15 minuti in volumi di 20 μl.
- Caricare 2 μl di ciascun campione su una membrana come descritto nella sezione 3.
6. Preparazione di controlli oligonucleotidici per valutare la specificità del segnale
NOTA: I controlli oligonucleotidici possono essere utilizzati per dimostrare la specificità del legame S9.6. S9.6 riconosce gli ibridi RNA-DNA, ma non i controlli dsDNA o dsRNA, come è stato precedentemente riportato34.
- Sciogliere gli oligonucleotidi (Tabella 3) in tampone di ricottura (10 mM Tris, pH 8,0; 50 mM NaCl, 1 mM EDTA) a 100 μM.
- Preparare 4 provette di reazione per
- Ibrido RNA-DNA #1: Mescolare 10 μL di filamento superiore di ssRNA con 10 μL di filamento inferiore di ssDNA e 80 μL di tampone di ricottura.
- Ibrido RNA-DNA #2: Mescolare 10 μL di filamento superiore di ssDNA con 10 μL di filamento inferiore di ssRNA e 80 μL di tampone di ricottura.
- dsRNA: Mescolare 10 μL di filamento superiore di ssRNA con 10 μL di filamento inferiore di ssRNA e 80 μL di tampone di ricottura.
- dsDNA: Miscelare 10 μL di filamento superiore di ssDNA con 10 μL di filamento inferiore di ssDNA e 80 μL di tampone di ricottura.
- Scaldare le 4 miscele del passaggio 6.2 a 95 °C per 10 min.
- Lasciare raffreddare lentamente i tubi a temperatura ambiente per consentire la ricottura dei fili. Gli standard ricotti possono essere conservati a -20 °C per un uso successivo.
NOTA: L'efficienza di ricottura deve essere verificata mediante elettroforesi su gel non denaturante. I duplex migrano più lentamente rispetto agli oligonucleotidi non ricotti (Tabella 2). - Caricare 2 μl di ciascun campione su 2 membrane, una per l'anticorpo S9.6 e una per l'anticorpo dsDNA, come descritto nella sezione 3.
- Eseguire i passaggi descritti nella sezione 4.
7. Quantificazione e normalizzazione dell'intensità del segnale S9.6 R-loop utilizzando ImageJ.
- Salvare le immagini di S9.6, colorazione dsDNA in formato TIFF e analizzarle utilizzando il software ImageJ (https://imagej.nih.gov/ij/).
- Seleziona l'opzione di inversione dell'immagine (Modifica | Inverti). Dopo l'inversione, ogni punto sarà visibile in bianco su uno sfondo scuro.
- Usa lo strumento di selezione dell'immagine ovale per selezionare un ovale abbastanza grande da circondare il punto più grande dell'immagine.
- Utilizza il ROI manager per aggiungere l'area selezionata per la quantificazione. Assicurarsi che le opzioni "Mostra tutto" ed "Etichette" siano selezionate in modo che le regioni di interesse possano essere visualizzate.
- Utilizzare la stessa area di selezione ovale utilizzata durante il passaggio 7.3 per aggiungere ulteriori regioni di interesse attorno a ciascun punto da quantificare. Usa la scorciatoia Comando + Maiusc + E per copiare l'area selezionata dal passaggio 7.3 a ciascuno dei punti successivi.
- Misurare la densità integrata di ciascuna delle regioni di interesse.
- Dividere l'intensità del segnale S9.6 per ciascun campione per la misurazione del dsDNA per ottenere il rapporto del segnale S9.6/dsDNA. Verificare i risultati ripetendo gli esperimenti (almeno triplicati sia per l'acquisizione del segnale S9.6 che per quello del dsDNA). L'errore standard della media può essere calcolato dai rapporti del segnale S9.6/dsDNA.
Risultati
Trattamento enzimatico per valutare la specificità dell'anticorpo S9.6 (RNA-DNA).
Sono stati coltivati fibroblasti primari della pelle umana17. Il DNA con ibridi RNA-DNA è stato isolato e quantificato. Due μg dei campioni sono stati digeriti con RNasi T1, RNasi H o RNasi III per 15 minuti a 37 °C. È stato analizzato anche un campione simulato per il confronto con i campioni trattati con RNasi. I campioni (200, 100, 50, 25, 12,5 o 6,25 ng) sono stati tamponati su due membrane diverse come descritto nella sezione 3. Le membrane sono state reticolate, bloccate e una di esse è stata sondata con l'anticorpo S9.6 (Figura 1A).
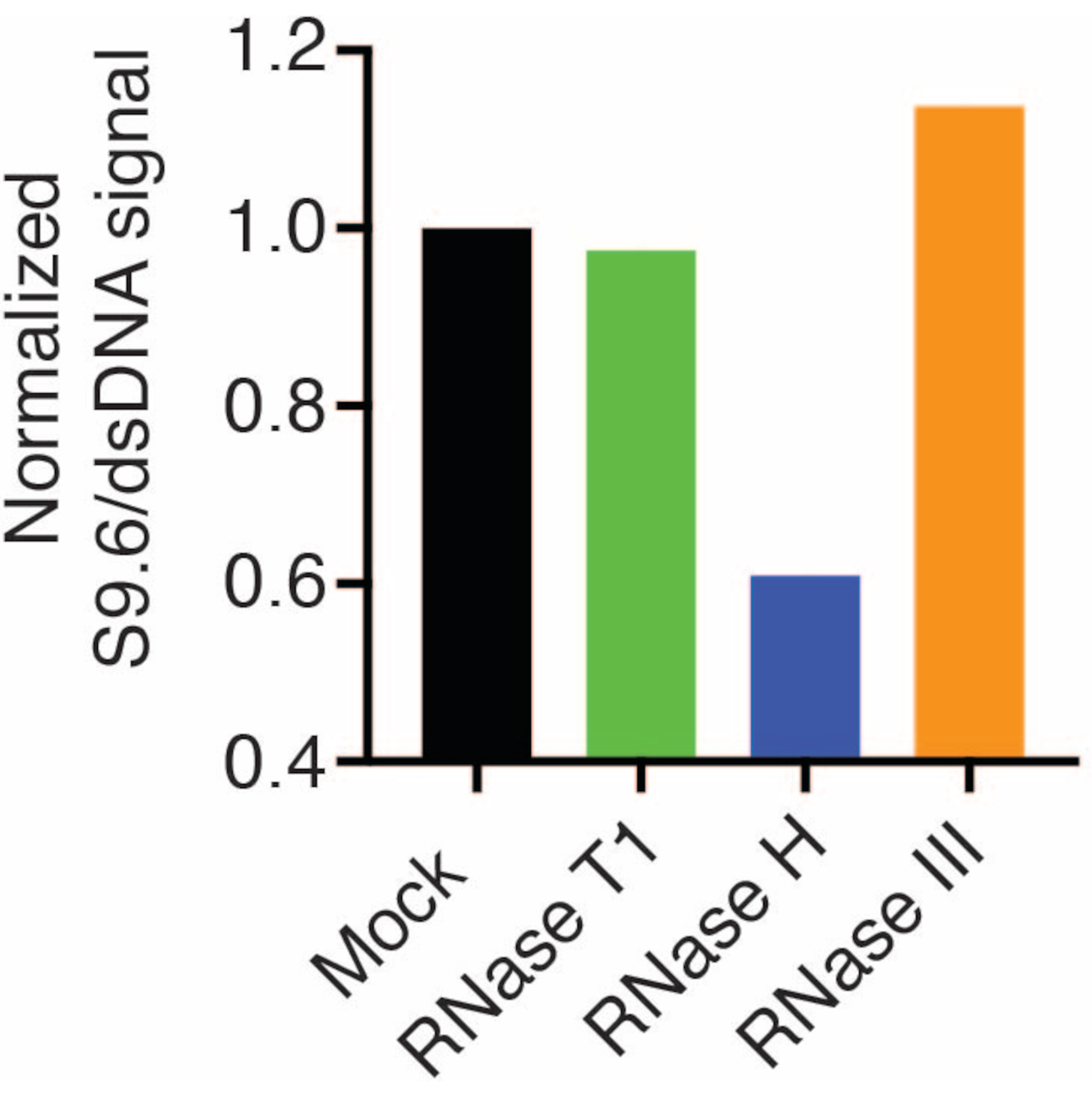
I risultati hanno mostrato che il segnale S9.6 è correlato all'abbondanza del campione caricato. Il trattamento con RNasi H, ma non con RNasi T1 o RNasi III, determina una riduzione della colorazione S9.6.
Una seconda membrana è stata sondata con un anticorpo dsDNA (Figura 1B) per la normalizzazione. L'immagine J è stata utilizzata per analizzare l'intensità del segnale. I campioni da 50 ng sono stati selezionati per la quantificazione poiché le intensità del segnale degli anticorpi S9.6 e dsDNA erano all'interno dell'intervallo dinamico. Le intensità del segnale sono state normalizzate a quelle dei campioni simulati. I dati sono mostrati nella Figura 2.
Dot-blot dell'anticorpo S9.6 utilizzando controlli nucleotidici sintetici.
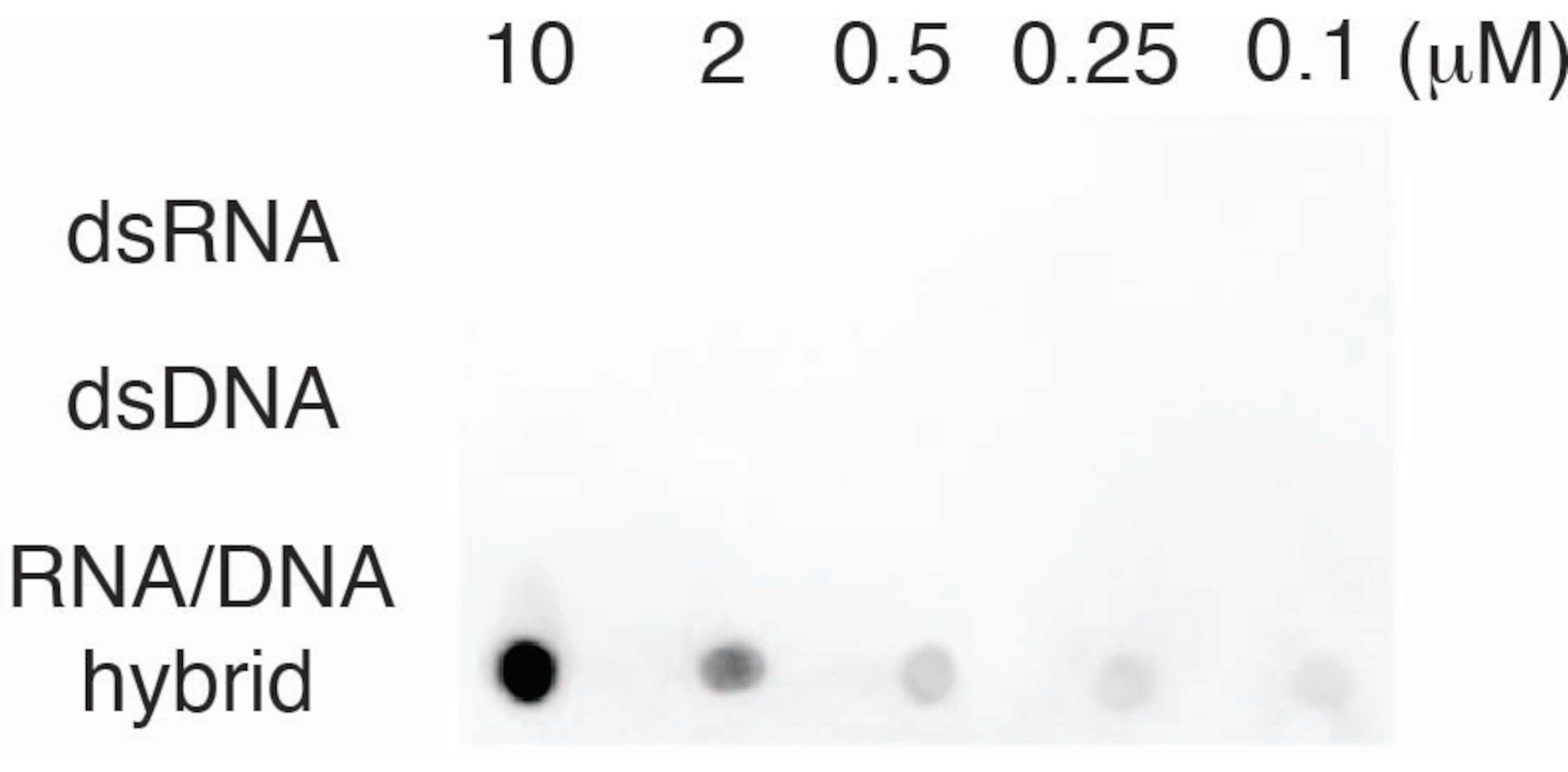
Per valutare la specificità dell'anticorpo S9.6, abbiamo utilizzato oligonucleotidi corrispondenti a dsRNA, dsDNA e RNA-DNA come descritto nella sezione 6. Una serie di diluizioni di nucleotidi RNA-DNA, dsRNA e dsDNA è stata preparata e tamponata sulla membrana di nylon come descritto nella sezione 3. La membrana è stata sondata con l'anticorpo S9.6 (Figura 3). I risultati hanno mostrato che l'anticorpo S9.6 si lega specificamente agli ibridi RNA-DNA in modo dose-dipendente e ha mostrato una minima reattività crociata ai dsRNA e ai dsDNA.

Figura 1: Specificità di S9.6 come mostrato dal dot-blot caricato con acidi nucleici da fibroblasti umani. I campioni di acido nucleico provenienti da fibroblasti umani sono stati trattati con simulazione o trattati con RNasi T1, RNasi H o RNasi III e quindi caricati su membrane di nylon in una serie di diluizioni di 200, 100, 50, 25, 12,5 e 6,25 ng per 2 μL di punto. Le membrane sono state quindi sondate con l'anticorpo S9.6 (A) o con l'anticorpo dsDNA (B). Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2: Quantificazione della colorazione R-loop S9.6. I campioni da 50 ng della Figura 1 sono stati selezionati per la quantificazione con ImageJ. Il segnale S9.6 è stato diviso per l'intensità del segnale dsDNA, quindi normalizzato al campione simulato seguendo i passaggi descritti nella sezione 7. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3: S9.6 dot-blot utilizzando controlli oligonucleotidici. S9.6 anticorpo dot-blot contro una serie di diluizioni di oligonucleotidi sintetici come dsRNA, dsDNA o ibrido RNA-DNA. S9.6 si lega specificamente agli ibridi RNA-DNA in modo dose-dipendente. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
| Tampone di lisi cellulare | Per 10 ml | Per 200 ml | Conc. finale |
| Acqua, senza nucleasi | 9 ml | 180 ml | - |
| 10% NP-40 | 0,5 ml | 10 ml | 0.5% |
| 2 milioni di KCl | 0,4 ml | 8 ml | 80mM |
| TUBI DA 0,5 M (pH 8,0) | 100uL | 2 ml | 5mM |
| Tampone di lisi nucleare | Per 10 ml | Per 200 ml | Conc. finale |
| Acqua, senza nucleasi | 8,65 ml | 173 ml | - |
| 10% SDS | 1 ml | 20 ml | 1% |
| 1M Tris-HCl (pH 8,0) | 0,25 ml | 5 ml | 25 mM |
| 0,5 milioni di EDTA | 100uL | 2 ml | 5 mM |
| Tampone di eluizione | Per 10 ml | Per 200 ml | Conc. finale |
| 1M Tris-Cl, pH 8,5 | 0,1 ml | 2 ml | 10 mM |
| Acqua, senza nucleasi | 9,9 ml | 198 ml | - |
Tabella 1. Preparazione dei tamponi
| Passo | Problema | Possibile motivo | Soluzione |
| 1.9 | N/A | Verificare il frazionamento cellulare | La qualità della separazione nucleare e citoplasmatica può essere valutata aggiungendo cocktail standard di inibitori della proteasi ai tamponi cellulari e di lisi nucleare (Tabella 1). Le frazioni citoplasmatiche e nucleari possono essere valutate mediante western blotting per confermare un'adeguata restrizione della marcatura con marcatori citoplasmatici (ad esempio, GAPDH o HSP90) nelle frazioni citoplasmatiche e la marcatura con marcatori nucleari (ad esempio, HDAC1 o Istone H3) nelle frazioni nucleari. Il contributo della contaminazione mitocondriale nella frazione nucleare può essere valutato mediante analisi qPCR con sonde specifiche per il DNA mitocondriale. |
| 2.1 | Il campione è troppo viscoso | Il numero di cellulare è troppo alto. | Ridurre il DNA della metà e continuare con la fase di sonicazione 2.1 |
| 2.12 | Nessun pallino visibile | Materiale di partenza insufficiente o perdita durante l'estrazione. | Inizia dall'inizio usando più celle. |
| 2.13 | Bassa concentrazione di DNA | ||
| 3.1 | Non abbastanza DNA per le diluizioni | ||
| 3.4 | Il campione non si satura nella membrana | Immergere la membrana in 1x TBST. Lasciare asciugare il tampone in eccesso e iniziare dal passaggio 3.4 | |
| 4.6 | Viene rilevato un motivo a chiazze o macchie. | Aggiungere lo 0,1% di sodio dodecil solfato (SDS) al campione e continuare dal passaggio 3.1 | |
| 4.6 | I punti hanno un aspetto ad "anello da caffè" | Aggiungere lo 0,01% di Sarkosyl al campione e continuare al passaggio 3.1 | |
| 5.2 | Il controllo RNaseH non mostra alcuna diminuzione del segnale | La digestione della ribonucleasi è incompleta. | Aumentare l'incubazione o aumentare la concentrazione enzimatica. |
| 6.5 | Nessun segnale per i controlli oligo | Il duplex non è stato formato. Gli oligonucleotidi non sono stati ricotti correttamente. | Verificare i rapporti tra oligonucleotidi e tampone di ricottura. |
| 6.5 | Il segnale S9.6 non è specifico per gli ibridi | Il lotto di anticorpi S9.6 ha un legame non specifico | Convalidare la sensibilità e la specificità di nuovi lotti di anticorpi S9.6 con l'uso di trattamenti enzimatici con RNasi o analisi con oligonucleotidi sintetici. |
Tabella 2: Risoluzione dei problemi
| ssRNA, filamento superiore | 5'-UGGGGGCUCGUCCGGGAUAUGGGAACCACUGAUCCC-3' |
| ssDNA, filamento superiore | 5'-TGGGGGCTCGTCCGGGATATGGGAACCACTGATCCC-3' |
| ssRNA, filamento inferiore | 5'-GGGAUCAGUGGUUCCCAUAUCCCGGACGAGCCCCCA-3' |
| ssDNA, filamento inferiore | 5'-GGGATCAGTGGTTCCCATATCCCGGACGAGCCCCCA-3' |
Tabella 3. Controllo delle sequenze oligonucleotidiche
Discussione
Gli acidi nucleici a 3 filamenti, R-loops, si formano in diverse fasi durante il ciclo di vita dell'RNA e si trovano sempre più spesso a regolare i processi cellulari. Per comprendere appieno i loop R, sono necessarie tecniche affidabili per il rilevamento dei loop R. Qui, descriviamo un approccio per interrogare l'abbondanza di R-loop utilizzando l'anticorpo S9.6 8,23,24. Questo metodo consente una rapida valutazione dell'abbondanza dell'R-loop da cellule e campioni di coltura tissutale. Non richiede attrezzature speciali o una grande quantità di materiale di partenza. Garantisce risultati specifici e riproducibili utilizzando una combinazione di trattamenti con RNasi.
Alcuni hanno segnalato preoccupazioni sulla specificità dell'anticorpo S9.6. Come con qualsiasi reagente, ci può essere variabilità da lotto a lotto con l'anticorpo S9.6. Il nostro protocollo include RNasi H, RNasi T1 e RNasi III per verificare la specificità del segnale. Inoltre, utilizziamo oligonucleotidi sintetici per garantire la specificità di ogni lotto di anticorpi S9.6.
La biologia R-loop è un campo in crescita; lo sviluppo di metodi di rilevamento e quantificazione affidabili, come quello qui presentato, faciliterà gli studi meccanicistici per chiarire quando si formano i loop R, come sono regolati e cosa regolano. Con controlli appropriati, questo test dot-blot è un metodo semplice per lo screening dell'abbondanza di R-loop in contesti clinici e di ricerca.
Divulgazioni
Gli autori non hanno nulla da rivelare.
Riconoscimenti
Questo lavoro è stato supportato dall'Howard Hughes Medical Institute e dalla ricerca intramurale presso il National Institute of Neurological Disorders and Stroke.
Ringraziamo il Dr. Stephen Leppla per aver fornito lotti di anticorpi S9.6 per l'analisi. Ringraziamo anche il Dr. Dongjun Li per la sua assistenza nei trattamenti con ribonucleasi.
Materiali
| Name | Company | Catalog Number | Comments |
| Anti-dsDNA antibody | Abcam | ab27156 | |
| Anti-RNA-DNA hybrid antibody (S9.6) | Kerafast | ENH001 | |
| Biorupter sonicator | Diagenode | UCD-200 | |
| EB Buffer | Qiagen | 19086 | |
| EDTA (0.5M) | Invitrogen | AM9261 | |
| Hybond N+ nylon membrane | GE healthcare Life Sciences | RPN203B | |
| KCl (2M) | Invitrogen | AM9640G | |
| NP-40 (Igepal CA-630) | Sigma | I8896 | |
| PBS | Invitrogen | 10010-023 | |
| Phenol:chloroform:isoamyl alcohol | Invitrogen | 15593031 | |
| PIPES (0.5M, pH 8.0) | VWR | AAJ61406-AE | |
| Proteinase K | Qiagen | 19131 | |
| RNase III | Invitrogen | AM2290 | |
| RNase H | New England Biolabs | M0297 | |
| RNase T1 | ThermoFisher Sci. | EN0541 | |
| SDS (10%) | Invitrogen | 15553027 | |
| sodium acetate (3M, pH 5.2) | Invitrogen | AM9740 | |
| Tris-buffered saline (10X) | Corning | 46-012-CM | |
| Tris-HCl (1M, pH 8.0) | KD Medical | RGF-3360 | |
| TrypLE | Invitrogen | 12605010 | |
| Tween-20 | Sigma | P9416 | |
| UV Stratalinker 2400 | Stratagene | Stratalinker 2400 | |
| Whatman marking pen | Sigma | WHA10499001 |
Riferimenti
- Daube, S. S., von Hippel, P. H. RNA displacement pathways during transcription from synthetic RNA-DNA bubble duplexes. Biochemistry. 33 (1), 340-347 (1994).
- Westover, K. D., Buschnell, D. A., Kornberg, R. D. Structural basis of transcription: separation of RNA from DNA by RNA polymerase II. Science. 303 (5660), 1014-1016 (2004).
- Itoh, T., Tomizawa, J. Formation of an RNA primer for initiation of replication of ColE1 DNA by ribonuclease H. Proceedings of the National Academy of Science USA. 77 (5), 2450-2454 (1980).
- Hamperl, S., Bocek, M. J., Saldivar, J. C., Swigut, T., Cimprich, K. A. Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell. 170 (4), 774-786 (2017).
- Skourti-Stathaki, K., Kamieniarz-Gdula, K., Proudfoot, N. J. R-loops induce repressive chromatin marks over mammalian gene terminators. Nature. 516 (7531), 436-439 (2014).
- Sanz, L. A., et al. conserved R-loop structures associate with specific epigenomic signatures in mammals. Molecular Cell. 63 (1), 167-178 (2016).
- Chen, L., et al. R-ChIP using inactive RNase H reveals dynamic coupling of R-loops with transcriptional pausing at gene promoters. Molecular Cell. 68 (4), 745-757 (2017).
- El Hage, A., French, S. L., Beyer, A. L., Tollervey, D. Loss of topoisomerase I leads to R-loop mediated transcriptional blocks during ribosomal RNA synthesis. Genes and Development. 24 (14), 1546-1558 (2010).
- Tran, P., et al. PIF1 family DNA helicases suppress R-loop mediated genome instability at tRNA genes. Nature Communication. 8, 15025(2017).
- Ginno, P. A., Lott, P. L., Christensen, H. C., Korf, I., Chedin, F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Molecular Cell. 45 (6), 814-825 (2012).
- Colak, D., et al. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science. 343 (6174), 1002-1005 (2014).
- Yu, K., Chedin, F., Hsieh, C., Wilson, T. E., Lieber, M. R. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nature Immunology. 4 (5), 442-451 (2003).
- Gasiunas, G., Barrangou, R., Horvath, P., Siksnys, V. Cas9-crRNA ribonucleotide complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proceedings of the National Academy of Science USA. 109 (39), 2579-2586 (2012).
- Westra, E. R., et al. CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by Cascade and Cas3. Molecular Cell. 46 (5), 595-605 (2012).
- Jiang, F., et al. Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science. 351 (6275), 867-871 (2016).
- Huertas, P., Aguilera, A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Molecular Cell. 12 (3), 711-721 (2003).
- Grunseich, C., et al. Senataxin mutation reveals how R-loops promote transcription by blocking DNA methylation at gene promoters. Molecular Cell. 69 (3), 426-437 (2018).
- Kim, H. D., Choe, J., Seo, Y. S. The sen1(+) gene of Schizosaccharomyces pombe, a homologue of budding yeast SEN1, encodes an RNA and DNA helicase. Biochemistry. 38 (44), 14697-14710 (1999).
- Stein, H., Hausen, P. Enzyme from calf thymus degrading the RNA moiety of DNA-RNA hybrids: effect on DNA-dependent RNA polymerase. Science. 166 (3903), 393-395 (1969).
- Cerritelli, S. M., Crouch, R. J. Ribonuclease H: the enzymes in eukaryotes. FEBS Journal. 276 (6), 1494-1505 (2009).
- Hyjek, M., Figiel, M., Nowotny, M. RNases H: Structure and mechanism. DNA Repair. 84, 102672(2019).
- Pohl, T. J., Zakian, V. A. Pif1 family DNA helicases: A helpmate to RNase H. DNA Repair. 84, 102633(2019).
- Pohjoismaki, J. L., et al. Mammalian mitochondrial DNA replication intermediates are essentially duplex but contain extensive tracts of RNA/DNA hybrid. Journal of Molecular Biology. 397 (5), 1144-1155 (2010).
- Sanz, L. A., Chedin, F. High-resolution, strand-specific R-loop mapping via S9.6-based DNA-RNA immunoprecipitation and high-throughput sequencing. Nature Protocols. 14 (6), 1734-1755 (2019).
- Wahba, L., Costantino, L., Tan, F. J., Zimmer, A., Koshland, D. S1-DRIP-seq identifies high expression and polyA tracts as major contributors to R-loop formation. Genes and Development. 30 (11), 1327-1338 (2016).
- Yan, Q., Shields, E. J., Bonasio, R., Sarma, K. Mapping native r-loops genome-wide using a targeted nuclease approach. Cell Reports. 29 (5), 1369-1380 (2019).
- Hu, Z., Zhang, A., Storz, G., Gottesman, S., Leppla, S. H. An antibody-based microarray assay for small RNA detection. Nucleic Acids Research. 34 (7), 52(2006).
- Nowotny, M., Gaidamakov, S. A., Crouch, R. J., Yang, W. Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell. 121 (7), 1005-1016 (2005).
- Sato, K., Egami, F. Studies on ribonucleases in takadiastase. Journal of Biochemistry. 44 (11), Tokyo. 753-767 (1957).
- Takahashi, K., Moore, S. Ribonuclease T1. Enzymes. 15, 435-468 (1982).
- Dunn, J. J. RNase III cleavage of single-stranded RNA: Effect of ionic strength in the fidelity of cleavage. Journal of Biological Chemistry. 251 (12), 3807-3814 (1976).
- Pertzev, A., Nicholson, A. W. Characterization of RNA sequence determinants and antideterminants of processing reactivity for a minimal substrate of Escherichia coli ribonuclease III. Nucleic Acids Research. 34 (13), 3708-3721 (2006).
- Phillips, D. D., et al. The sub-nanomolar binding of DNA-RNA hybrids by the single chain Fv fragment of antibody S9.6. Journal of Molecular Recognition. 26 (8), 376-381 (2013).
- Haruki, M., Noguchi, E., Kanaya, S., Crouch, R. J. Kinetic and stoichiometric analysis for the binding of Escherichia coli ribonuclease H1 to RNA-DNA hybrids using surface plasmon resonance. Journal of Biological Chemistry. 272 (35), 22015-22022 (1997).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati