
Method Article
R-Loop Analysis by Dot-Blot
In This Article
Summary
This protocol details a simple method that quantifies R-loop, a three-stranded nucleic acid structure that comprises of an RNA-DNA hybrid and a displaced DNA strand.
Abstract
The three-stranded nucleic acid structure, R-loop, is increasingly recognized for its role in gene regulation. Initially, R-loops were thought to be the by-products of transcription; but recent findings of fewer R-loops in diseased cells made it clear that R-loops have functional roles in a variety of human cells. Next, it is critical to understand the roles of R-loops and how cells balance their abundance. A challenge in the field is the quantitation of R-loops since much of the work relies on the S9.6 monoclonal antibody whose specificity for RNA-DNA hybrids has been questioned. Here, we use dot-blots with the S9.6 antibody to quantify R-loops and show the sensitivity and specificity of this assay with RNase H, RNase T1, and RNase III that cleave RNA-DNA hybrids, single-stranded RNA, and double-stranded RNA, respectively. This method is highly reproducible, uses general laboratory equipment and reagents, and provides results within two days. This assay can be used in research and clinical settings to quantify R-loops and assess the effect of mutations in genes such as senataxin on R-loop abundance.
Introduction
This protocol provides a step-by-step guide to a dot-blot assay that allows a quick comparative assessment of the abundance of R-loop, a three-stranded nucleic acid structure. R-loop forms when RNA invades a double-stranded DNA to generate an RNA-DNA hybrid and displaces the other DNA strand. R-loops are found in different stages of the lifecycle of RNA. In the transcriptional complex, the nascent RNA is synthesized complementary to the template DNA, and the non-template strand is displaced. The short RNA-DNA hybrid (<10 bp) is resolved to free the nascent RNA so it can leave the RNA polymerase complex through the exit channel1,2. Outside of the transcriptional complex, the nascent RNA is close to its DNA template, which is still slightly unwound from being copied, thus the RNA can rehybridize with its template DNA forming R-loops3. Additionally, R-loops can form when replication and transcription complexes collide4, and in antisense transcription5. Given the many opportunities for their formation, R-loops are not rare, and can be found in 3-5% of the human genome6, depending on the cell's transcription status. R-loops are found in gene promoters7 and termination5 sites in mRNA, and along ribosomal RNA8 as well as transfer RNA9. R-loops are also in telomeric regions of chromosomes.
R-loops play a regulatory role. They regulate gene expression by affecting transcription at promoters10,11, mediating class-switch recombination12, and facilitating CRISPR-based genome editing13,14,15. Like many cellular events, R-loop abundance is tightly titrated; too many or too few R-loops impact normal cell function16,17. R-loops are regulated by a variety of proteins including RNase H, senataxin, and other helicases that unwind the RNA-DNA hybrids18,19,20,21,22.
To monitor the abundance of R-loops, genome-wide methods first enrich for R-loops with the antibody S9.68,23,24 or with other nucleases25 including RNase H10,26,27, and then assess the number of enriched R-loops by sequencing. Early versions of these sequencing-based methods did not achieve adequate sequence coverage to allow precise quantitation, but rapid improvement in sequencing technologies now allows locus-by-locus R-loop analysis. Immunofluorescence techniques have also been used to quantify and localize R-loops10,17. These methods are comprehensive, but they are not practical in many clinical settings or as initial assessments since they require expensive equipment and specialized analysis.
A procedure that can be done uniformly across laboratories in clinical settings is needed. Dot-blots provide such an option since they can be carried out without any specific equipment or computational analysis. As a preliminary step or in clinical settings to evaluate the effects of mutations on R-loops, these dot-blots must provide sensitive and specific results. Here, we describe our assay that identifies R-loops specifically; it excludes signals from double-stranded (ds) DNA, double-stranded RNA, and single-stranded RNA. Our protocol uses the S9.6 antibody27 to identify RNA-DNA hybrids in R-loops and incorporates RNase H, an endoribonuclease that cleaves and therefore leads to the degradation of the RNA in an RNA-DNA hybrid20,28, to ensure that the detected signals are those of hybrids. We also incorporated RNase T1 that cleaves single-stranded RNA at guanine29,30, and RNase III that cleaves double-stranded RNA including stem-loops31,32 to check for nonspecific signals. The S9.6 antibody recognizes RNA-DNA hybrids of varying lengths, even those that are only 8 nucleotides long33.
Here, we present the protocol that begins with nucleic acid isolation followed by dot-blot preparation, and R-loop detection with S9.6 antibody. Our protocol includes steps to ensure that equal amounts of samples are loaded, and the signals are specific. It provides oligonucleotides to serve as positive and negative controls. This is a quick, easy, and user-friendly method to assess R-loop abundance.
Protocol
1. Cell lysis for nuclear fractionation
- Wash cells with 1x phosphate-buffered saline (PBS) twice. Remove cells from tissue culture dishes using standard cell dissociation techniques such as trypsin. Count cells using a hemocytometer.
NOTE: The steps described below were used for the analysis of primary human skin fibroblasts, although an array of cell types can be assayed. Fibroblasts were grown in basal media containing 10% fetal bovine serum. Alternatively, cell lysis buffer (Table 1) can be added directly to the cell-culture after washing. - Transfer the cell suspension to a 1.5 mL tube to pellet the cells.
- Centrifuge the sample at 300 x g for 5 min at 4 °C. Aspirate the media.
- Wash twice with ice-cold 1x PBS using centrifugation settings in step 1.3.
- Add cold cell lysis buffer (Table 1) to the cell pellet (300 µL per 2 x 106 cells). Pipette up and down to resuspend the pellet.
- Incubate on ice for 10 min.
- Spin at 500 x g for 5 min to pellet the nuclei.
- Discard supernatant and re-suspend the nuclear pellet in 400 µL of cold nuclear lysis buffer (Table 1).
- Incubate on ice for 10 min.
NOTE: Fractionation of the nuclear and cytoplasmic compartments of the cells ensures signal specificity. The quality of nuclear and cytoplasmic separation can be evaluated before proceeding (Table 2). - Add 3 µL of 20 mg/mL proteinase K and incubate for 3-5 h at 55 °C.
NOTE: Volumes indicated are for 2 x 106 cells, scale up or down as necessary.
2. Purification of genomic DNA (which includes RNA-DNA hybrids)
- If DNA is viscous, perform sonication to reduce viscosity (e.g., sonication at high power output, 30 s ON/ 30 s OFF, for 2 min using a 4 °C water bath).
- Add 400 µL of elution buffer (Table 1) and 400 µL of phenol:chloroform:isoamyl alcohol (25:24:1 pH 8.0).
- Vortex for 10 s.
- Spin down at 12,000 x g for 5 min at 4 °C.
- Transfer the aqueous phase (approximately 350 µL) to a new tube.
- Extract once using 1 volume of chloroform, vortex for 10 s, then spin down at 12,000 x g for 5 min at 4 °C. Transfer the aqueous phase to a new tube (approximately 300 µL).
- Add 35 µL of 3 M sodium acetate (pH 5.2), 1 µL glycogen and 700 µL of ice-cold 100% ethanol.
- Vortex for 10 s and spin down at 12,000 x g for 30 min at 4 °C.
- Wash the pellet with 1 mL of 70% ethanol.
- Vortex for 10 s and spin down at 12,000 x g for 15 min at 4 °C.
- Discard the supernatant and let the pellet air dry.
- Add 12 µL of elution buffer and vortex for 10 s to resuspend. Incubate the sample for 30 min at 37 °C with agitation or at 4 °C overnight to re-suspend the pellet.
- Measure the DNA concentration using standard spectrophotometry.
NOTE: Volumes indicated are for 2 x 106 cells, scale up or down as necessary. DNA (with RNA-DNA hybrids) may be stored at -20 °C, if needed.
3. Blotting DNA samples (which include RNA-DNA hybrids) onto nylon membranes
- Prepare dilutions of nucleic acids to desired concentrations in elution buffer (i.e., 50 ng/µL, 25 ng/µL, or 12.5 ng/µL). These samples with a range of concentrations (200, 100, 50, 25, 12.5 ng) ensure that there will be signals within the linear range.
NOTE: Be sure to prepare enough sample for technical and biological replicates, and for the various RNase treatments, see Step 5. - Prepare a positively charged nylon membrane so that there is room for each 2 µL sample to occupy a 0.5 cm2 area.
- Spot 2 µL of each sample onto 2 membranes: one for the S9.6 antibody and the other for dsDNA antibody. Alternatively, a dot-blot or slot-blot apparatus which allows the loading of samples with larger volumes can be used.
- Allow the samples to saturate into the membrane. Wait at least 2 min before crosslinking the membrane with UV light.
- Place the membrane into the center of the UV device and crosslink the membrane using a UV crosslinker using the "Auto Crosslink" setting (1,200 µJ x 100).
4. RNA-DNA hybrid detection with S9.6 antibody
- Incubate the membrane in blocking solution (5% milk in Tris-buffered saline with 0.05% Tween-20 (TBST) for 1 h at room temperature on a shaker.
NOTE: There should enough blocking solution to cover the membrane. - Incubate the membranes overnight in primary antibody (in 5% milk in TBST) at 4 °C with shaking. Add anti-dsDNA antibody (1:10,000 dilution) to one membrane. Add 1µg/mL S9.6 antibody to the second membrane (1:1,000 dilution).
NOTE: S9.6 antibody is available commercially or from Dr. S. Leppla, NIAID, National Institutes of Health. - Remove primary antibody and wash 3x with TBST. Perform each wash for 5-10 min with shaking at room temperature.
- Incubate with horseradish peroxidase (HRP) conjugated secondary antibody (anti-mouse, 1:5,000 dilution) in 5% milk in TBST with shaking at room temperature.
NOTE: Anti-dsDNA and anti-RNA-DNA hybrid are both mouse antibodies. - Remove the secondary antibody and wash 3x with TBST for 5-10 min with shaking at room temperature.
- Develop with enhanced-chemiluminescence (ECL) reagents to acquire signals for imaging.
- Quantify signal intensity using standard image processing tools such as ImageJ.
NOTE: Troubleshooting is detailed in Table 2.
5. Ribonuclease treatments to evaluate signal specificity
NOTE: RNase treatment should be performed on the nucleic acid samples to demonstrate the specificity of S9.6 binding. Treatment with RNase H, but not RNase T1 or RNase III should result in a reduction in S9.6 immunostaining.
- Digest the samples containing RNA-DNA hybrids by preparing them in four separate tubes. Treat each of the 4 samples with either 5 U RNase H, 1000 U RNase T1, 0.5 U RNase III, or mock. Incubate samples at 37 °C for 15 min in 20 µL volumes.
- Load 2 µL of each sample on a membrane as described in section 3.
6. Preparation of oligonucleotide controls to evaluate signal specificity
NOTE: Oligonucleotide controls can be used to demonstrate the specificity of S9.6 binding. S9.6 recognizes RNA-DNA hybrids, but not dsDNA or dsRNA controls, as has been previously reported34.
- Dissolve oligonucleotides (Table 3) in annealing buffer (10 mM Tris, pH 8.0; 50 mM NaCl, 1 mM EDTA) to 100 µM.
- Prepare 4 reaction tubes for
- RNA-DNA hybrid #1: Mix 10 µL of ssRNA top strand with 10 µL of ssDNA bottom strand and 80 µL of annealing buffer.
- RNA-DNA hybrid #2: Mix 10 µL of ssDNA top strand with 10 µL of ssRNA bottom strand and 80 µL of annealing buffer.
- dsRNA: Mix 10 µL of ssRNA top strand with 10 µL of ssRNA bottom strand and 80 µL of annealing buffer.
- dsDNA: Mix 10 µL of ssDNA top strand with 10 µL of ssDNA bottom strand and 80 µL of annealing buffer.
- Heat the 4 mixtures from step 6.2 at 95 °C for 10 min.
- Allow tubes to cool slowly to room temperature to allow reannealing of the strands. Annealed standards can be stored at -20 °C for later use.
NOTE: Annealing efficiency should be checked by non-denaturing gel electrophoresis. Duplexes migrate more slowly than the unannealed oligonucleotides (Table 2). - Load 2 µL of each sample on 2 membranes, one for S9.6 antibody and one for dsDNA antibody, as described in section 3.
- Perform steps described in section 4.
7. Quantification and normalization of S9.6 R-loop signal intensity using ImageJ.
- Save images of S9.6, dsDNA staining in TIFF format, and analyze them using the ImageJ software (https://imagej.nih.gov/ij/).
- Select the image invert option (Edit | Invert). After inversion, each dot will be visible as white against a dark background.
- Use the oval image selection tool to select an oval that is large enough to surround the largest dot on the image.
- Use the ROI manager to add the selected area for quantification. Ensure that the "Show All" and "Labels" options are selected so that the regions of interest can be visualized.
- Use the same oval selection area used during step 7.3 to add additional regions of interest around each dot to be quantified. Use Command + Shift + E shortcut to copy the selected area from step 7.3 to each of the subsequent dots.
- Measure the integrated density of each of the regions of interest.
- Divide the S9.6 signal intensity for each sample by the measurement of dsDNA to obtain the S9.6/dsDNA signal ratio. Verify the results by repeating the experiments (at least triplicates for both S9.6 and dsDNA signal acquisition). Standard error of the mean can be calculated from the S9.6/dsDNA signal ratios.
Results
Enzymatic treatment to evaluate the specificity of S9.6 (RNA-DNA) antibody.
Primary human skin fibroblasts were grown17. DNA with RNA-DNA hybrids was isolated and quantified. Two µg of the samples were digested with RNase T1, RNase H, or RNase III for 15 min at 37 °C. A mock sample was also analyzed for comparison to the RNase-treated samples. Samples (200, 100, 50, 25, 12.5, or 6.25 ng) were blotted onto two different membranes as described in section 3. The membranes were crosslinked, blocked and one of them was probed with S9.6 antibody (Figure 1A).
The results showed that the S9.6 signal correlates with the abundance of the loaded sample. Treatment with RNase H, but not RNase T1 or RNase III results in a reduction in S9.6 staining.
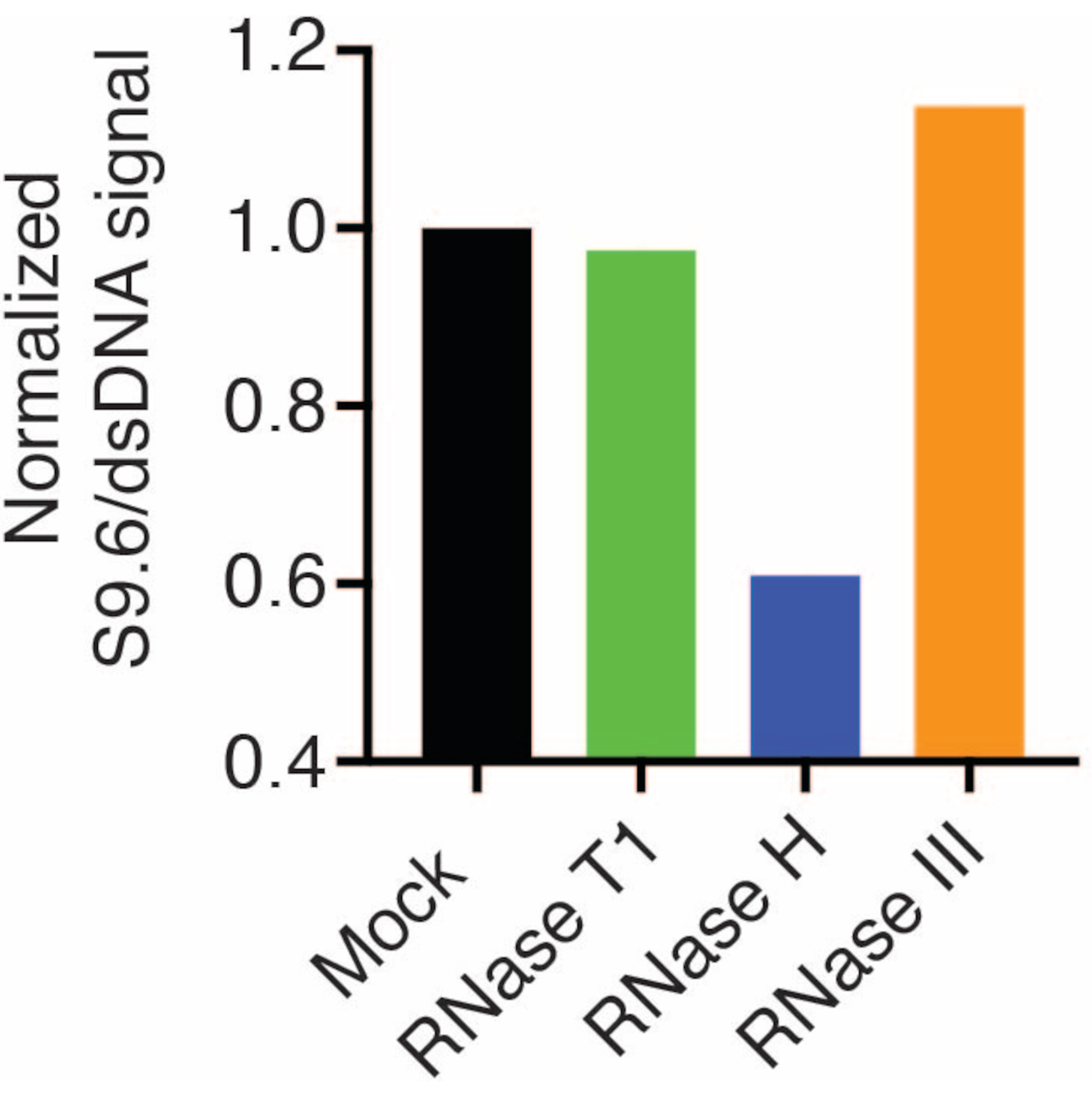
A second membrane was probed with a dsDNA antibody (Figure 1B) for the normalization. Image J was used to analyze the signal intensities. The 50 ng samples were selected for quantification as the signal intensities from the S9.6 and dsDNA antibodies were within the dynamic range. Signal intensities were normalized to those in mock samples. Data are shown in Figure 2.
S9.6 antibody dot-blot using synthetic nucleotide controls.
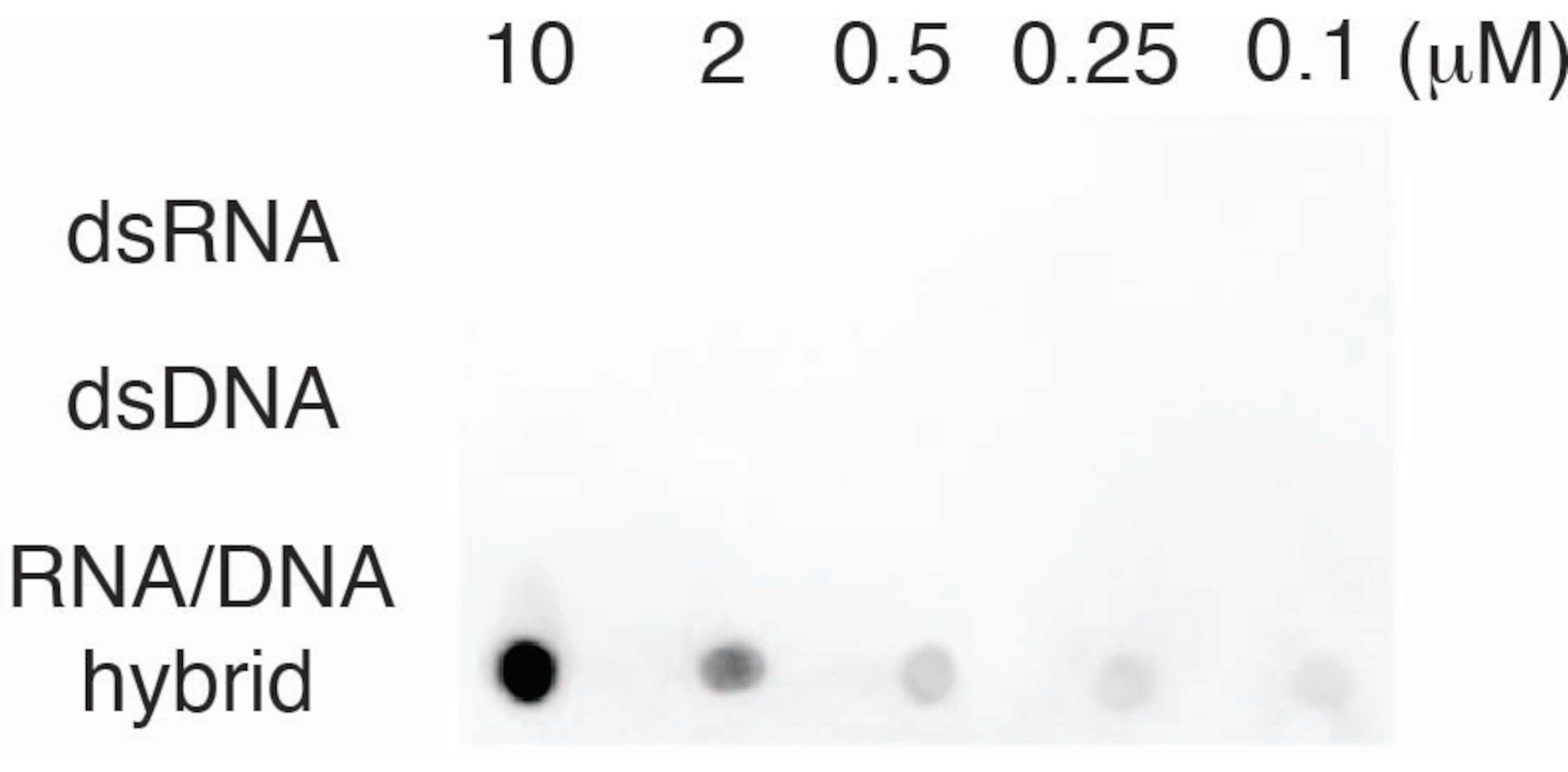
To evaluate the specificity of the S9.6 antibody, we used oligonucleotides corresponding to dsRNA, dsDNA, and RNA-DNA as described in section 6. A dilution series of RNA-DNA, dsRNA, and dsDNA nucleotides were prepared and blotted onto the nylon membrane as described in section 3. The membrane was probed with the S9.6 antibody (Figure 3). Results showed that the S9.6 antibody binds specifically to RNA-DNA hybrids in a dose-dependent manner and showed minimal cross-reactivity to dsRNAs and dsDNAs.

Figure 1: Specificity of S9.6 as shown by dot-blot loaded with nucleic acids from human fibroblasts. Nucleic acid samples from human fibroblasts were either mock treated or treated with RNase T1, RNase H, or RNase III and then loaded onto nylon membranes in a dilution series of 200, 100, 50, 25, 12.5, and 6.25 ng per 2 µL dot. Membranes were then probed with S9.6 antibody (A), or dsDNA antibody (B). Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Quantification of S9.6 R-loop staining. 50 ng samples from Figure 1 were selected for quantification with ImageJ. S9.6 signal was divided by dsDNA signal intensity, then normalized to the mock sample following the steps outlined in section 7. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: S9.6 dot-blot using oligonucleotide controls. S9.6 antibody dot-blot against a dilution series of synthetic oligonucleotides as dsRNA, dsDNA, or RNA-DNA hybrid. S9.6 binds specifically to RNA-DNA hybrids in a dose-dependent manner. Please click here to view a larger version of this figure.
{kind=link}
| Cell lysis buffer | For 10mL | For 200mL | Final Conc. |
| Water, nuclease-free | 9mL | 180mL | - |
| 10% NP-40 | 0.5mL | 10mL | 0.5% |
| 2M KCl | 0.4mL | 8mL | 80mM |
| 0.5M PIPES (pH 8.0) | 100uL | 2mL | 5mM |
| Nuclear lysis buffer | For 10mL | For 200mL | Final Conc. |
| Water, nuclease-free | 8.65mL | 173mL | - |
| 10% SDS | 1mL | 20mL | 1% |
| 1M Tris-HCl (pH 8.0) | 0.25mL | 5mL | 25 mM |
| 0.5M EDTA | 100uL | 2mL | 5 mM |
| Elution Buffer | For 10mL | For 200mL | Final Conc. |
| 1M Tris-Cl, pH 8.5 | 0.1 mL | 2 mL | 10 mM |
| Water, nuclease-free | 9.9 mL | 198 mL | - |
Table 1. Preparation of buffers
| Step | Problem | Possible reason | Solution |
| 1.9 | N/A | Verify cell fractionation | The quality of nuclear and cytoplasmic separation can be evaluated by adding standard protease inhibitor cocktails to the cell and nuclear lysis buffers (Table 1). The cytoplasmic and nuclear fractions can be evaluated by western blotting analysis to confirm adequate restriction of labeling with cytoplasmic markers (for example, GAPDH or HSP90) in cytoplasmic fractions and labeling with nuclear markers (for example, HDAC1 or Histone H3) in nuclear fractions. The contribution of mitochondrial contamination in the nuclear fraction can be evaluated by qPCR analysis with probes specific for mitochondrial DNA. |
| 2.1 | Sample is too viscous | Cell number is too high. | Reduce the DNA by half and continue with the sonication step 2.1 |
| 2.12 | No visible pellet | Insufficient starting material or loss during extraction. | Start from the beginning using more cells. |
| 2.13 | Low DNA concentration | ||
| 3.1 | Not enough DNA for dilutions | ||
| 3.4 | Sample won't saturate into the membrane | Soak the membrane in 1x TBST. Allow excess buffer to dry and start at step 3.4 | |
| 4.6 | Patchy or speckled pattern is detected | Add 0.1% Sodium dodecyl sulfate (SDS) to the sample and continue at step 3.1 | |
| 4.6 | The dots have a "coffee ring" appearance | Add 0.01% Sarkosyl to the sample and continue at step 3.1 | |
| 5.2 | The RNaseH control shows no decrease in signal | The ribonuclease digestion is incomplete. | Increase incubation or increase enzyme concentration. |
| 6.5 | No signal for the oligo controls | Duplex wasn't formed. Oligonucleotides weren't properly annealed. | Verify the ratios of oligonucleotides and annealing buffer. |
| 6.5 | S9.6 signal isn't specific to hybrids | S9.6 antibody batch has non-specific binding | Validate the sensitivity and specificity of new batches of S9.6 antibody with the use of either RNase enzyme treatments or synthetic oligonucleotide analysis. |
Table 2: Troubleshooting
| ssRNA, top strand | 5’-UGGGGGCUCGUCCGGGAUAUGGGAACCACUGAUCCC-3’ |
| ssDNA, top strand | 5’-TGGGGGCTCGTCCGGGATATGGGAACCACTGATCCC-3’ |
| ssRNA, bottom strand | 5’-GGGAUCAGUGGUUCCCAUAUCCCGGACGAGCCCCCA-3’ |
| ssDNA, bottom strand | 5’-GGGATCAGTGGTTCCCATATCCCGGACGAGCCCCCA-3’ |
Table 3. Control oligonucleotide sequences
Discussion
The 3-stranded nucleic acids, R-loops, form in different stages during the lifecycle of RNA and are increasingly found to regulate cellular processes. To fully understand R-loops, reliable techniques for R-loop detection are necessary. Here, we describe an approach to interrogate the abundance of R-loops using S9.6 antibody8,23,24. This method allows for a quick assessment of R-loop abundance from cells and tissue-culture samples. It does not require special equipment, or a large quantity of starting material. It ensures specific and reproducible results using a combination of RNase treatments.
Some have reported concerns about the specificity of the S9.6 antibody. As with any reagent, there may be batch to batch variability with the S9.6 antibody. Our protocol includes RNase H, RNase T1 and RNase III to check signal specificity. In addition, we use synthetic oligonucleotides to ensure the specificity of each batch of S9.6 antibody.
R-loop biology is a growing field; the development of reliable detection and quantification methods, such as the one presented here, will facilitate mechanistic studies to elucidate when R-loops form, how they are regulated, and what they regulate. With appropriate controls, this dot-blot assay is a simple method to screen for R-loop abundance in clinical and research settings.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was supported by the Howard Hughes Medical Institute and Intramural Research at the National Institute of Neurological Disorders and Stroke.
We thank Dr. Stephen Leppla for providing batches of S9.6 antibody for analysis. We also thank Dr. Dongjun Li for his assistance with ribonuclease treatments.
Materials
| Name | Company | Catalog Number | Comments |
| Anti-dsDNA antibody | Abcam | ab27156 | |
| Anti-RNA-DNA hybrid antibody (S9.6) | Kerafast | ENH001 | |
| Biorupter sonicator | Diagenode | UCD-200 | |
| EB Buffer | Qiagen | 19086 | |
| EDTA (0.5M) | Invitrogen | AM9261 | |
| Hybond N+ nylon membrane | GE healthcare Life Sciences | RPN203B | |
| KCl (2M) | Invitrogen | AM9640G | |
| NP-40 (Igepal CA-630) | Sigma | I8896 | |
| PBS | Invitrogen | 10010-023 | |
| Phenol:chloroform:isoamyl alcohol | Invitrogen | 15593031 | |
| PIPES (0.5M, pH 8.0) | VWR | AAJ61406-AE | |
| Proteinase K | Qiagen | 19131 | |
| RNase III | Invitrogen | AM2290 | |
| RNase H | New England Biolabs | M0297 | |
| RNase T1 | ThermoFisher Sci. | EN0541 | |
| SDS (10%) | Invitrogen | 15553027 | |
| sodium acetate (3M, pH 5.2) | Invitrogen | AM9740 | |
| Tris-buffered saline (10X) | Corning | 46-012-CM | |
| Tris-HCl (1M, pH 8.0) | KD Medical | RGF-3360 | |
| TrypLE | Invitrogen | 12605010 | |
| Tween-20 | Sigma | P9416 | |
| UV Stratalinker 2400 | Stratagene | Stratalinker 2400 | |
| Whatman marking pen | Sigma | WHA10499001 |
References
- Daube, S. S., von Hippel, P. H. RNA displacement pathways during transcription from synthetic RNA-DNA bubble duplexes. Biochemistry. 33 (1), 340-347 (1994).
- Westover, K. D., Buschnell, D. A., Kornberg, R. D. Structural basis of transcription: separation of RNA from DNA by RNA polymerase II. Science. 303 (5660), 1014-1016 (2004).
- Itoh, T., Tomizawa, J. Formation of an RNA primer for initiation of replication of ColE1 DNA by ribonuclease H. Proceedings of the National Academy of Science USA. 77 (5), 2450-2454 (1980).
- Hamperl, S., Bocek, M. J., Saldivar, J. C., Swigut, T., Cimprich, K. A. Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell. 170 (4), 774-786 (2017).
- Skourti-Stathaki, K., Kamieniarz-Gdula, K., Proudfoot, N. J. R-loops induce repressive chromatin marks over mammalian gene terminators. Nature. 516 (7531), 436-439 (2014).
- Sanz, L. A., et al. conserved R-loop structures associate with specific epigenomic signatures in mammals. Molecular Cell. 63 (1), 167-178 (2016).
- Chen, L., et al. R-ChIP using inactive RNase H reveals dynamic coupling of R-loops with transcriptional pausing at gene promoters. Molecular Cell. 68 (4), 745-757 (2017).
- El Hage, A., French, S. L., Beyer, A. L., Tollervey, D. Loss of topoisomerase I leads to R-loop mediated transcriptional blocks during ribosomal RNA synthesis. Genes and Development. 24 (14), 1546-1558 (2010).
- Tran, P., et al. PIF1 family DNA helicases suppress R-loop mediated genome instability at tRNA genes. Nature Communication. 8, 15025(2017).
- Ginno, P. A., Lott, P. L., Christensen, H. C., Korf, I., Chedin, F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Molecular Cell. 45 (6), 814-825 (2012).
- Colak, D., et al. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science. 343 (6174), 1002-1005 (2014).
- Yu, K., Chedin, F., Hsieh, C., Wilson, T. E., Lieber, M. R. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nature Immunology. 4 (5), 442-451 (2003).
- Gasiunas, G., Barrangou, R., Horvath, P., Siksnys, V. Cas9-crRNA ribonucleotide complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proceedings of the National Academy of Science USA. 109 (39), 2579-2586 (2012).
- Westra, E. R., et al. CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by Cascade and Cas3. Molecular Cell. 46 (5), 595-605 (2012).
- Jiang, F., et al. Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science. 351 (6275), 867-871 (2016).
- Huertas, P., Aguilera, A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Molecular Cell. 12 (3), 711-721 (2003).
- Grunseich, C., et al. Senataxin mutation reveals how R-loops promote transcription by blocking DNA methylation at gene promoters. Molecular Cell. 69 (3), 426-437 (2018).
- Kim, H. D., Choe, J., Seo, Y. S. The sen1(+) gene of Schizosaccharomyces pombe, a homologue of budding yeast SEN1, encodes an RNA and DNA helicase. Biochemistry. 38 (44), 14697-14710 (1999).
- Stein, H., Hausen, P. Enzyme from calf thymus degrading the RNA moiety of DNA-RNA hybrids: effect on DNA-dependent RNA polymerase. Science. 166 (3903), 393-395 (1969).
- Cerritelli, S. M., Crouch, R. J. Ribonuclease H: the enzymes in eukaryotes. FEBS Journal. 276 (6), 1494-1505 (2009).
- Hyjek, M., Figiel, M., Nowotny, M. RNases H: Structure and mechanism. DNA Repair. 84, 102672(2019).
- Pohl, T. J., Zakian, V. A. Pif1 family DNA helicases: A helpmate to RNase H. DNA Repair. 84, 102633(2019).
- Pohjoismaki, J. L., et al. Mammalian mitochondrial DNA replication intermediates are essentially duplex but contain extensive tracts of RNA/DNA hybrid. Journal of Molecular Biology. 397 (5), 1144-1155 (2010).
- Sanz, L. A., Chedin, F. High-resolution, strand-specific R-loop mapping via S9.6-based DNA-RNA immunoprecipitation and high-throughput sequencing. Nature Protocols. 14 (6), 1734-1755 (2019).
- Wahba, L., Costantino, L., Tan, F. J., Zimmer, A., Koshland, D. S1-DRIP-seq identifies high expression and polyA tracts as major contributors to R-loop formation. Genes and Development. 30 (11), 1327-1338 (2016).
- Yan, Q., Shields, E. J., Bonasio, R., Sarma, K. Mapping native r-loops genome-wide using a targeted nuclease approach. Cell Reports. 29 (5), 1369-1380 (2019).
- Hu, Z., Zhang, A., Storz, G., Gottesman, S., Leppla, S. H. An antibody-based microarray assay for small RNA detection. Nucleic Acids Research. 34 (7), 52(2006).
- Nowotny, M., Gaidamakov, S. A., Crouch, R. J., Yang, W. Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell. 121 (7), 1005-1016 (2005).
- Sato, K., Egami, F. Studies on ribonucleases in takadiastase. Journal of Biochemistry. 44 (11), Tokyo. 753-767 (1957).
- Takahashi, K., Moore, S. Ribonuclease T1. Enzymes. 15, 435-468 (1982).
- Dunn, J. J. RNase III cleavage of single-stranded RNA: Effect of ionic strength in the fidelity of cleavage. Journal of Biological Chemistry. 251 (12), 3807-3814 (1976).
- Pertzev, A., Nicholson, A. W. Characterization of RNA sequence determinants and antideterminants of processing reactivity for a minimal substrate of Escherichia coli ribonuclease III. Nucleic Acids Research. 34 (13), 3708-3721 (2006).
- Phillips, D. D., et al. The sub-nanomolar binding of DNA-RNA hybrids by the single chain Fv fragment of antibody S9.6. Journal of Molecular Recognition. 26 (8), 376-381 (2013).
- Haruki, M., Noguchi, E., Kanaya, S., Crouch, R. J. Kinetic and stoichiometric analysis for the binding of Escherichia coli ribonuclease H1 to RNA-DNA hybrids using surface plasmon resonance. Journal of Biological Chemistry. 272 (35), 22015-22022 (1997).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved