
Method Article
ניתוח לולאת R על ידי Dot-Blot
In This Article
Summary
פרוטוקול זה מפרט שיטה פשוטה המכמתת את לולאת R, מבנה חומצת גרעין תלת-גדילי המורכב מהכלאה של RNA-DNA וגדיל DNA שנעקר.
Abstract
מבנה חומצת הגרעין התלת-גדילי, לולאת R, מוכר יותר ויותר בשל תפקידו בוויסות גנים. בתחילה, לולאות R נחשבו כתוצרי לוואי של שעתוק; אך ממצאים אחרונים של פחות לולאות R בתאים חולים הבהירו כי לולאות R יש תפקידים פונקציונליים במגוון תאים אנושיים. לאחר מכן, זה קריטי להבין את התפקידים של לולאות R וכיצד תאים מאזנים את השפע שלהם. אתגר בתחום הוא כימות לולאות R מכיוון שחלק גדול מהעבודה מסתמך על הנוגדן החד-שבטי S9.6 שהספציפיות שלו להכלאות RNA-DNA מוטלת בספק. כאן, אנו משתמשים בכתמי נקודות עם הנוגדן S9.6 כדי לכמת לולאות R ולהראות את הרגישות והספציפיות של בדיקה זו עם RNase H, RNase T1 ו-RNase III המבקעים היברידיות RNA-DNA, RNA חד-גדילי ו-RNA דו-גדילי, בהתאמה. שיטה זו ניתנת לשחזור רב, משתמשת בציוד מעבדה כללי וריאגנטים, ומספקת תוצאות תוך יומיים. ניתן להשתמש בבדיקה זו במחקר ובמסגרות קליניות כדי לכמת לולאות R ולהעריך את ההשפעה של מוטציות בגנים כגון סנאטקסין על שפע לולאת R.
Introduction
פרוטוקול זה מספק מדריך שלב אחר שלב לבדיקת כתם נקודות המאפשרת הערכה השוואתית מהירה של השפע של לולאת R, מבנה חומצת גרעין תלת-גדילית. לולאת R נוצרת כאשר RNA פולש ל-DNA דו-גדילי כדי ליצור הכלאה של RNA-DNA ומעביר את גדיל ה-DNA השני. לולאות R נמצאות בשלבים שונים של מחזור החיים של RNA. בקומפלקס השעתוק, ה-RNA המתהווה מסונתז משלים ל-DNA התבנית, והגדיל שאינו תבנית נעקר. הכלאה קצרה של RNA-DNA (<10 bp) נפתרת כדי לשחרר את ה-RNA המתהווה כך שיוכל לעזוב את קומפלקס ה-RNA פולימראז דרך תעלת היציאה 1,2. מחוץ לקומפלקס השעתוק, ה-RNA המתהווה קרוב לתבנית ה-DNA שלו, שעדיין מעט לא מפותלת מההעתקה, ולכן ה-RNA יכול להכלאה מחדש עם תבנית ה-DNA שלו ויוצר לולאות R3. בנוסף, לולאות R יכולות להיווצר כאשר מתחמי שכפול ושעתוק מתנגשים4, ובשעתוק אנטי-סנס5. בהתחשב בהזדמנויות הרבות להיווצרותן, לולאות R אינן נדירות, וניתן למצוא אותן ב-3-5% מהגנום האנושי6, תלוי במצב השעתוק של התא. לולאות R נמצאות במקדמי גנים7 ובאתרי סיום5 ב-mRNA, ולאורך RNA ריבוזומלי8 כמו גם RNAהעברה 9. לולאות R נמצאות גם באזורים טלומריים של כרומוזומים.
לולאות R ממלאות תפקיד רגולטורי. הם מווסתים את ביטוי הגנים על ידי השפעה על השעתוק במקדמים10,11, מתווכים רקומבינציה של מתג מחלקה12, ומקלים על עריכת גנום מבוססת CRISPR 13,14,15. כמו אירועים סלולריים רבים, שפע לולאת R הוא טיטרציה הדוקה; יותר מדי או מעט מדי לולאות R משפיעות על תפקוד תקין של התא16,17. לולאות R מווסתות על ידי מגוון חלבונים כולל RNase H, סנאטקסין והליקזים אחרים המשחררים את הכלאות RNA-DNA 18,19,20,21,22.
כדי לנטר את השפע של לולאות R, שיטות כלל-גנומיות מעשירות תחילה עבור לולאות R עם הנוגדן S9.6 8,23,24 או עם נוקלאזות אחרות25 כולל RNase H 10,26,27, ולאחר מכן מעריכות את מספר לולאות ה-R המועשרות על ידי ריצוף. גרסאות מוקדמות של שיטות מבוססות ריצוף אלה לא השיגו כיסוי רצף הולם כדי לאפשר כימות מדויק, אך שיפור מהיר בטכנולוגיות הריצוף מאפשר כעת ניתוח לולאת R לוקוס אחר לוקוס. טכניקות אימונופלואורסצנטיות שימשו גם לכימות ולמיקום לולאות R10,17. שיטות אלו הן מקיפות, אך הן אינן מעשיות במסגרות קליניות רבות או כהערכות ראשוניות מכיוון שהן דורשות ציוד יקר וניתוח מיוחד.
יש צורך בהליך שניתן לבצע באופן אחיד בין מעבדות במסגרות קליניות. כתמי נקודות מספקים אפשרות כזו מכיוון שניתן לבצע אותם ללא כל ציוד ספציפי או ניתוח חישובי. כצעד מקדים או במסגרות קליניות להערכת ההשפעות של מוטציות על לולאות R, כתמי נקודות אלה חייבים לספק תוצאות רגישות וספציפיות. כאן, אנו מתארים את הבדיקה שלנו המזהה לולאות R באופן ספציפי; הוא אינו כולל אותות מ-DNA דו-גדילי (ds), RNA דו-גדילי ו-RNA חד-גדילי. הפרוטוקול שלנו משתמש בנוגדניםS9.6 27 כדי לזהות הכלאות RNA-DNA בלולאות R ומשלב RNase H, אנדוריבונוקלאז שנבקע ולכן מוביל לפירוק ה-RNA בהכלאה של RNA-DNA20,28, כדי להבטיח שהאותות שזוהו הם של היברידיים. שילבנו גם RNase T1 המבקע RNA חד-גדילי בגואנין29,30, ו-RNase III המבקע RNA דו-גדילי כולל לולאות גזע31,32 כדי לבדוק אם יש אותות לא ספציפיים. הנוגדן S9.6 מזהה הכלאות RNA-DNA באורכים שונים, אפילו כאלה שאורכם רק 8 נוקלאוטידים33.
כאן, אנו מציגים את הפרוטוקול שמתחיל בבידוד חומצות גרעין ואחריו הכנת כתמי נקודות, וזיהוי לולאת R עם נוגדן S9.6. הפרוטוקול שלנו כולל שלבים כדי להבטיח שכמויות שוות של דגימות נטענות, והאותות ספציפיים. הוא מספק אוליגונוקלאוטידים כדי לשמש כבקרות חיוביות ושליליות. זוהי שיטה מהירה, קלה וידידותית למשתמש להערכת שפע לולאת R.
Protocol
1. ליזיס תאים לפיצול גרעיני
- שטפו תאים עם 1x מי מלח עם חוצץ פוספט (PBS) פעמיים. הסר תאים מכלי תרבית רקמה באמצעות טכניקות דיסוציאציה סטנדרטיות של תאים כגון טריפסין. ספור תאים באמצעות המוציטומטר.
הערה: השלבים המתוארים להלן שימשו לניתוח פיברובלסטים ראשוניים של עור אנושי, אם כי ניתן לבחון מערך של סוגי תאים. פיברובלסטים גודלו במצע בסיסי המכיל 10% סרום בקר עוברי. לחלופין, ניתן להוסיף מאגר ליזה של תאים (טבלה 1) ישירות לתרבית התאים לאחר הכביסה. - העבירו את תרחיף התאים לצינור של 1.5 מ"ל כדי לגלול את התאים.
- צנטריפוגה את הדגימה ב-300 x גרם למשך 5 דקות ב-4 מעלות צלזיוס. שאפו את התקשורת.
- שטפו פעמיים עם PBS 1x קר כקרח באמצעות הגדרות צנטריפוגה בשלב 1.3.
- הוסף מאגר ליזה של תאים קרים (טבלה 1) לגלולת התא (300 מיקרוליטר לכל 2 x 106 תאים). פיפטה למעלה ולמטה כדי להשעות את הכדור.
- דגירה על קרח למשך 10 דקות.
- סובב ב-500 x גרם למשך 5 דקות כדי לגלול את הגרעינים.
- השליכו את הסופרנטנט והשעו מחדש את הגלולה הגרעינית ב-400 מיקרוליטר של מאגר ליזה גרעיני קר (טבלה 1).
- דגירה על קרח למשך 10 דקות.
הערה: פיצול התאים הגרעיניים והציטופלזמיים של התאים מבטיח את ספציפיות האות. ניתן להעריך את איכות ההפרדה הגרעינית והציטופלזמית לפני שתמשיך (טבלה 2). - מוסיפים 3 מיקרוליטר של 20 מ"ג/מ"ל פרוטאינאז K ודוגרים במשך 3-5 שעות בטמפרטורה של 55 מעלות צלזיוס.
הערה: אמצעי האחסון המצוינים הם עבור 2 x 106 תאים, הגדלה או הקטנה לפי הצורך.
2. טיהור DNA גנומי (הכולל הכלאות RNA-DNA)
- אם ה-DNA צמיג, בצע סוניקציה כדי להפחית את הצמיגות (למשל, סוניקציה בתפוקת הספק גבוהה, 30 שניות ON/30 S OFF, למשך 2 דקות באמצעות אמבט מים של 4 מעלות צלזיוס).
- הוסף 400 מיקרוליטר של מאגר פליטה (טבלה 1) ו-400 מיקרוליטר של פנול: כלורופורם: אלכוהול איזואמיל (25:24:1 pH 8.0).
- מערבולת למשך 10 שניות.
- יש להסתובב בטמפרטורה של 12,000 x גרם למשך 5 דקות בטמפרטורה של 4 מעלות צלזיוס.
- העבירו את השלב המימי (כ -350 מיקרוליטר) לצינור חדש.
- יש לחלץ פעם אחת באמצעות נפח אחד של כלורופורם, מערבולת למשך 10 שניות, ואז לסובב כלפי מטה ב-12,000 x גרם למשך 5 דקות ב-4 מעלות צלזיוס. העבירו את השלב המימי לצינור חדש (כ -300 מיקרוליטר).
- הוסף 35 מיקרוליטר של 3 M נתרן אצטט (pH 5.2), 1 מיקרוליטר גליקוגן ו-700 מיקרוליטר 100% אתנול קר כקרח.
- מערבולת למשך 10 שניות ומסתובבת מטה ב-12,000 x g למשך 30 דקות ב-4 מעלות צלזיוס.
- שוטפים את הגלולה עם 1 מ"ל של 70% אתנול.
- מערבולת למשך 10 שניות ומסתובבת מטה ב-12,000 x g למשך 15 דקות ב-4 מעלות צלזיוס.
- השליכו את הסופרנטנט ותנו לגלולה להתייבש באוויר.
- הוסף 12 מיקרוליטר של מאגר פליטה ומערבולת למשך 10 שניות כדי להשעות מחדש. דגרו את הדגימה למשך 30 דקות ב-37 מעלות צלזיוס עם תסיסה או ב-4 מעלות צלזיוס למשך הלילה כדי להשעות מחדש את הגלולה.
- מדוד את ריכוז ה- DNA באמצעות ספקטרופוטומטריה סטנדרטית.
הערה: אמצעי האחסון המצוינים הם עבור 2 x 106 תאים, הגדלה או הקטנה לפי הצורך. DNA (עם הכלאות RNA-DNA) עשוי להיות מאוחסן ב-20 מעלות צלזיוס, במידת הצורך.
3. ספיגת דגימות DNA (הכוללות היברידיות RNA-DNA) על ממברנות ניילון
- הכן דילולים של חומצות גרעין לריכוזים הרצויים במאגר הפליטה (כלומר, 50 ננוגרם/מיקרוליטר, 25 ננוגרם/מיקרוליטר או 12.5 ננוגרם/מיקרוליטר). דגימות אלה עם טווח ריכוזים (200, 100, 50, 25, 12.5 ננוגרם) מבטיחות שיהיו אותות בטווח הליניארי.
הערה: הקפד להכין מספיק דגימה לשכפולים טכניים וביולוגיים, ולטיפולי RNase השונים, ראה שלב 5. - הכן קרום ניילון טעון חיובי כך שיהיה מקום לכל דגימה של 2 מיקרוליטר לתפוס שטח של 0.5 ס"מ.
- הבחין ב-2 מיקרוליטר מכל דגימה על 2 ממברנות: האחת לנוגדנים S9.6 והשנייה לנוגדני dsDNA. לחלופין, ניתן להשתמש במנגנון כתם נקודות או חריץ המאפשר טעינת דגימות בנפחים גדולים יותר.
- אפשר לדגימות להרוות לתוך הממברנה. המתן לפחות 2 דקות לפני הצלבת הממברנה עם אור UV.
- הנח את הממברנה במרכז מכשיר ה-UV וקשר את הממברנה באמצעות קישור צולב UV באמצעות ההגדרה "קישור צולב אוטומטי" (1,200 μJ x 100).
4. זיהוי היברידי RNA-DNA עם נוגדן S9.6
- דגרו את הממברנה בתמיסה חוסמת (5% חלב בתמיסת מלח Tris-buffered עם 0.05% Tween-20 (TBST) למשך שעה בטמפרטורת החדר על שייקר.
הערה: צריך להיות מספיק תמיסת חסימה כדי לכסות את הממברנה. - דגרו את הממברנות למשך הלילה בנוגדנים ראשוניים (בחלב 5% ב-TBST) בטמפרטורה של 4 מעלות צלזיוס עם ניעור. הוסף נוגדן נגד dsDNA (דילול 1:10,000) לממברנה אחת. הוסף נוגדן S9.6 של 1 מיקרוגרם/מ"ל לקרום השני (דילול של 1:1,000).
הערה: נוגדן S9.6 זמין מסחרית או אצל ד"ר ס. לפלה, NIAID, המכונים הלאומיים לבריאות. - יש להסיר את הנוגדן הראשוני ולשטוף 3 פעמים עם TBST. בצע כל שטיפה במשך 5-10 דקות עם ניעור בטמפרטורת החדר.
- דגירה עם נוגדן משני מצומד חזרת פרוקסידאז (HRP) (נגד עכברים, דילול 1:5,000) בחלב 5% ב-TBST עם ניעור בטמפרטורת החדר.
הערה: אנטי-dsDNA ואנטי-RNA-DNA היברידיים הם שניהם נוגדנים לעכברים. - הסר את הנוגדן המשני ושטוף 3 פעמים עם TBST למשך 5-10 דקות עם ניעור בטמפרטורת החדר.
- לפתח עם ריאגנטים כימיים משופרים (ECL) כדי לרכוש אותות להדמיה.
- כמת את עוצמת האות באמצעות כלי עיבוד תמונה סטנדרטיים כגון ImageJ.
הערה: פתרון הבעיות מפורט בטבלה 2.
5. טיפולי ריבונוקלאז להערכת ספציפיות האות
הערה: יש לבצע טיפול ב-RNase על דגימות חומצות הגרעין כדי להדגים את הספציפיות של קשירת S9.6. טיפול ב-RNase H, אך לא ב-RNase T1 או RNase III אמור לגרום להפחתה בצביעת חיסון S9.6.
- עכל את הדגימות המכילות הכלאות RNA-DNA על ידי הכנתן בארבעה צינורות נפרדים. טפל בכל אחת מ-4 הדגימות עם 5 U RNase H, 1000 U RNase T1, 0.5 U RNase III או מדומה. דגירה של דגימות בטמפרטורה של 37 מעלות צלזיוס למשך 15 דקות בנפחים של 20 מיקרוליטר.
- טען 2 מיקרוליטר מכל דגימה על ממברנה כמתואר בסעיף 3.
6. הכנת בקרות אוליגונוקלאוטידים להערכת ספציפיות האות
הערה: ניתן להשתמש בבקרות אוליגונוקלאוטידים כדי להדגים את הספציפיות של קשירת S9.6. S9.6 מזהה היברידיות של RNA-DNA, אך לא בקרות dsDNA או dsRNA, כפי שדווח בעבר34.
- ממיסים אוליגונוקלאוטידים (טבלה 3) במאגר חישול (10 מ"מ Tris, pH 8.0; 50 מ"מ NaCl, 1 מ"מ EDTA) עד 100 מיקרומטר.
- הכן 4 צינורות תגובה עבור
- RNA-DNA היברידי #1: מערבבים 10 מיקרוליטר של גדיל עליון ssRNA עם 10 מיקרוליטר של גדיל תחתון ssDNA ו-80 מיקרוליטר של מאגר חישול.
- RNA-DNA היברידי #2: מערבבים 10 מיקרוליטר של גדיל עליון ssDNA עם 10 מיקרוליטר של גדיל תחתון ssRNA ו-80 מיקרוליטר של מאגר חישול.
- dsRNA: מערבבים 10 מיקרוליטר של גדיל עליון ssRNA עם 10 מיקרוליטר של גדיל תחתון ssRNA ו-80 מיקרוליטר של מאגר חישול.
- dsDNA: מערבבים 10 מיקרוליטר של גדיל עליון ssDNA עם 10 מיקרוליטר של גדיל תחתון ssDNA ו-80 מיקרוליטר של מאגר חישול.
- מחממים את 4 התערובות משלב 6.2 בחום של 95 מעלות למשך 10 דקות.
- הניחו לצינורות להתקרר לאט לטמפרטורת החדר כדי לאפשר חישול מחדש של הגדילים. ניתן לאחסן תקנים מחוסמים בטמפרטורה של -20 מעלות צלזיוס לשימוש מאוחר יותר.
הערה: יש לבדוק את יעילות החישול על ידי אלקטרופורזה של ג'ל שאינו דנטור. דופלקסים נודדים לאט יותר מהאוליגונוקלאוטידים הלא מחוסמים (טבלה 2). - טען 2 מיקרוליטר מכל דגימה על 2 ממברנות, אחת לנוגדנים S9.6 ואחת לנוגדנים dsDNA, כמתואר בסעיף 3.
- בצע את השלבים המתוארים בסעיף 4.
7. כימות ונורמליזציה של עוצמת אות לולאת R S9.6 באמצעות ImageJ.
- שמור תמונות של S9.6, צביעת dsDNA בפורמט TIFF, ונתח אותן באמצעות תוכנת ImageJ (https://imagej.nih.gov/ij/).
- בחר באפשרות היפוך התמונה (Edit | הפוך). לאחר ההיפוך, כל נקודה תיראה בלבן על רקע כהה.
- השתמשו בכלי לבחירת תמונה אליפסה לבחירת אליפסה גדולה מספיק כדי להקיף את הנקודה הגדולה ביותר בתמונה.
- השתמש במנהל ההחזר על ההשקעה כדי להוסיף את האזור שנבחר לכימות. ודא שהאפשרויות "הצג הכל" ו"תוויות" נבחרו כך שניתן יהיה להציג באופן חזותי את אזורי העניין.
- השתמש באותו אזור בחירה אליפסה ששימש במהלך שלב 7.3 כדי להוסיף אזורי עניין נוספים סביב כל נקודה שיש לכמת. השתמש בקיצור המקשים Command + Shift + E כדי להעתיק את האזור שנבחר משלב 7.3 לכל אחת מהנקודות הבאות.
- מדוד את הצפיפות המשולבת של כל אחד מאזורי העניין.
- חלקו את עוצמת האות S9.6 עבור כל דגימה על ידי מדידת dsDNA כדי לקבל את יחס האות S9.6/dsDNA. אמת את התוצאות על ידי חזרה על הניסויים (לפחות משולש עבור רכישת אותות S9.6 ו-dsDNA). ניתן לחשב שגיאת תקן של הממוצע מיחסי האותות S9.6/dsDNA.
תוצאות
טיפול אנזימטי להערכת הספציפיות של נוגדן S9.6 (RNA-DNA).
פיברובלסטים ראשוניים של עור אנושי גודלו17. DNA עם הכלאות RNA-DNA בודד וכומת. שני מיקרוגרם מהדגימות עוכלו עם RNase T1, RNase H או RNase III למשך 15 דקות ב-37 מעלות צלזיוס. דגימה מדומה נותחה גם להשוואה לדגימות שטופלו ב-RNase. דגימות (200, 100, 50, 25, 12.5 או 6.25 ננוגרם) נמחקו על שני ממברנות שונות כמתואר בסעיף 3. הממברנות היו מקושרות, נחסמו, ואחת מהן נבדקה עם נוגדן S9.6 (איור 1A).
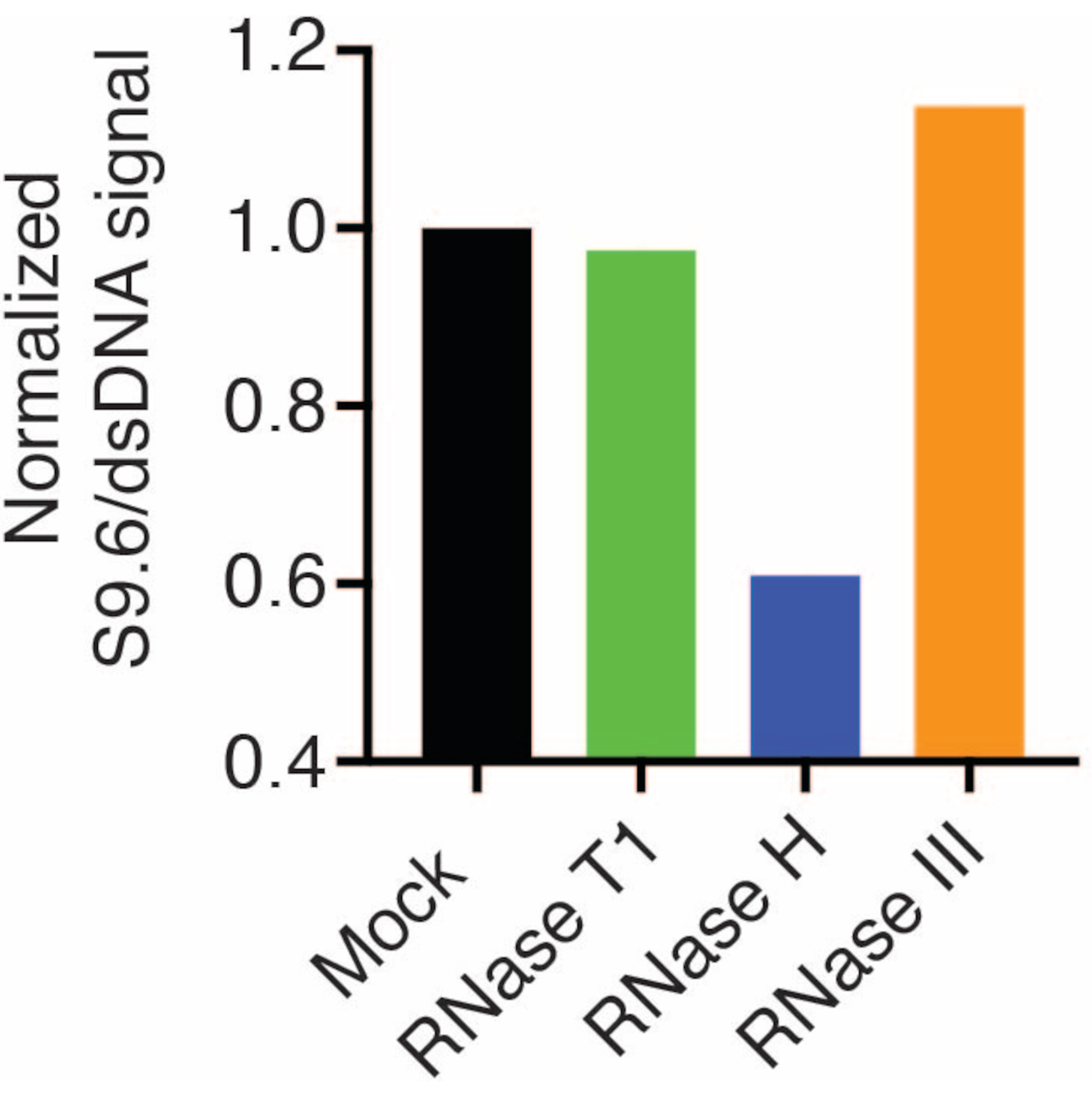
התוצאות הראו כי האות S9.6 מתאם עם שפע הדגימה הטעונה. טיפול ב-RNase H, אך לא ב-RNase T1 או RNase III מביא להפחתה בצביעת S9.6.
ממברנה שנייה נבדקה עם נוגדן dsDNA (איור 1B) לנורמליזציה. תמונה J שימשה לניתוח עוצמות האות. דגימות ה-50 ננוגרם נבחרו לכימות מכיוון שעוצמות האות מהנוגדנים S9.6 ו-dsDNA היו בטווח הדינמי. עוצמות האות נורמלו לאלו בדגימות מדומה. הנתונים מוצגים באיור 2.
כתם נקודות נוגדן S9.6 באמצעות בקרות נוקלאוטידים סינתטיות.
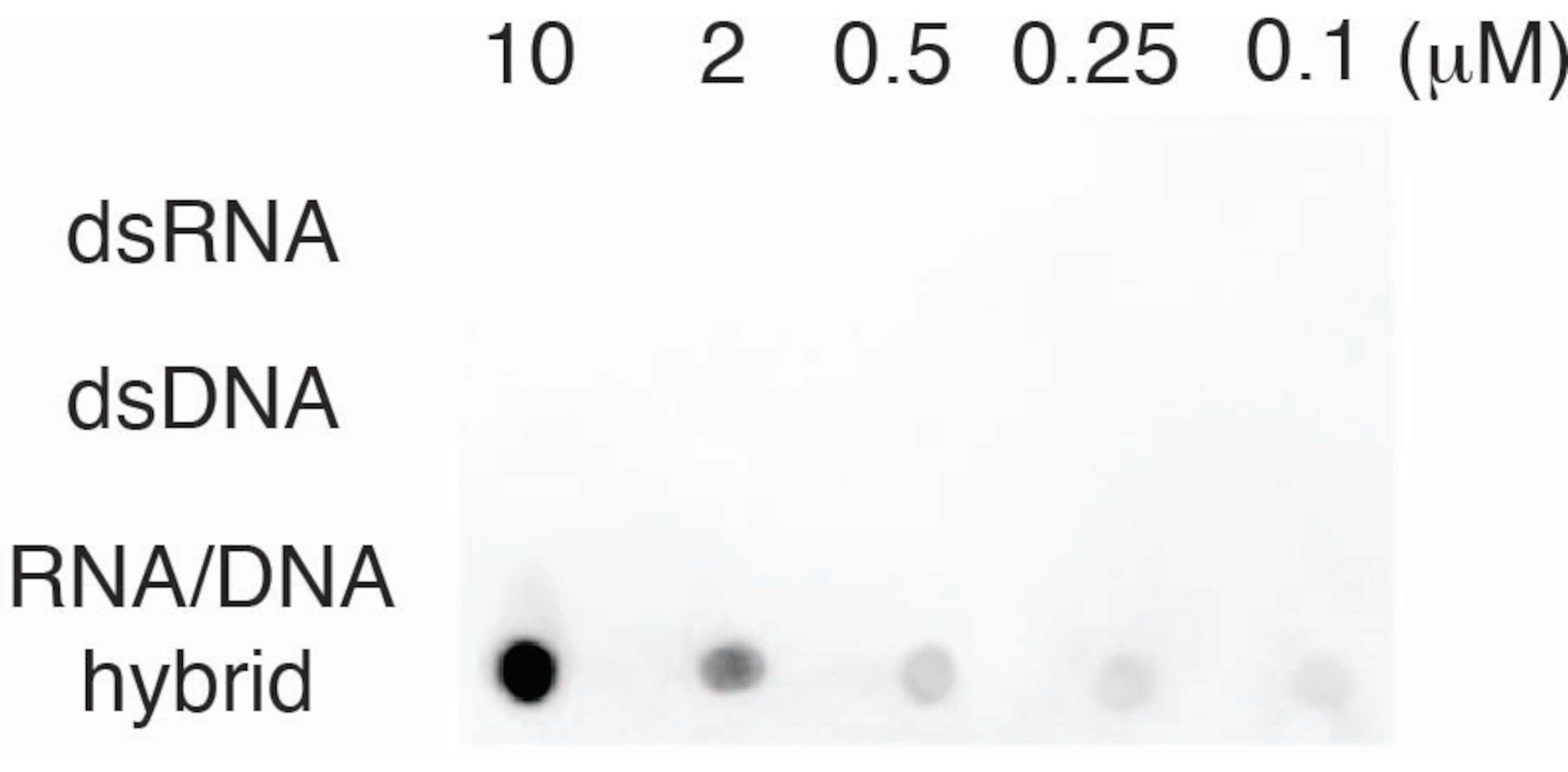
כדי להעריך את הספציפיות של נוגדן S9.6, השתמשנו באוליגונוקלאוטידים המתאימים ל-dsRNA, dsDNA ו-RNA-DNA כמתואר בסעיף 6. סדרת דילול של נוקלאוטידים של RNA-DNA, dsRNA ו-dsDNA הוכנו ונמחקו על קרום הניילון כמתואר בסעיף 3. הממברנה נבדקה עם נוגדן S9.6 (איור 3). התוצאות הראו כי הנוגדן S9.6 נקשר באופן ספציפי להכלאות RNA-DNA באופן תלוי מינון והראה תגובתיות צולבת מינימלית ל-dsRNAs ו-dsDNAs.

איור 1: הספציפיות של S9.6 כפי שמוצג על ידי כתם נקודות עמוס בחומצות גרעין מפיברובלסטים אנושיים. דגימות חומצות גרעין מפיברובלסטים אנושיים טופלו בדומה או טופלו ב-RNase T1, RNase H או RNase III ולאחר מכן הועמסו על ממברנות ניילון בסדרת דילול של 200, 100, 50, 25, 12.5 ו-6.25 ננוגרם לכל 2 מיקרוליטר נקודה. לאחר מכן נבדקו הממברנות עם נוגדן S9.6 (A), או נוגדן dsDNA (B). אנא לחץ כאן לצפייה בגרסה גדולה יותר של איור זה.
{kind=link}

איור 2: כימות של צביעת לולאת R S9.6. 50 דגימות נגרם מאיור 1 נבחרו לכימות עם ImageJ. אות S9.6 חולק בעוצמת האות dsDNA, ולאחר מכן נורמל לדגימה המדומה בעקבות השלבים המתוארים בסעיף 7. אנא לחץ כאן לצפייה בגרסה גדולה יותר של איור זה.
{kind=link}

איור 3: כתם נקודות S9.6 באמצעות בקרות אוליגונוקלאוטיד. כתם נקודות נוגדן S9.6 כנגד סדרת דילול של אוליגונוקלאוטידים סינתטיים כ-dsRNA, dsDNA או היברידית RNA-DNA. S9.6 נקשר במיוחד להכלאות RNA-DNA באופן תלוי מינון. אנא לחץ כאן לצפייה בגרסה גדולה יותר של איור זה.
{kind=link}
| מאגר ליזה תאים | עבור 10 מ"ל | עבור 200 מ"ל | קונק סופי. |
| מים, ללא נוקלאז | 9 מ"ל | 180 מ"ל | - |
| 10% NP-40 | 0.5 מ"ל | 10 מ"ל | 0.5% |
| 2 מיליון KCl | 0.4 מ"ל | 8 מ"ל | 80 מ"מ |
| צינורות 0.5 מ' (pH 8.0) | 100uL | 2 מ"ל | 5 מ"מ |
| מאגר ליזה גרעיני | עבור 10 מ"ל | עבור 200 מ"ל | קונק סופי. |
| מים, ללא נוקלאז | 8.65 מ"ל | 173 מ"ל | - |
| 10% SDS | 1 מ"ל | 20 מ"ל | 1% |
| 1M Tris-HCl (pH 8.0) | 0.25 מ"ל | 5 מ"ל | 25 מ"מ |
| 0.5 מיליון EDTA | 100uL | 2 מ"ל | 5 מ"מ |
| מאגר סילוק | עבור 10 מ"ל | עבור 200 מ"ל | קונק סופי. |
| 1M Tris-Cl, pH 8.5 | 0.1 מ"ל | 2 מ"ל | 10 מ"מ |
| מים, ללא נוקלאז | 9.9 מ"ל | 198 מ"ל | - |
טבלה 1. הכנת מאגרים
| צעד | בעיה | סיבה אפשרית | תמיסה |
| 1.9 | לא ישים | אימות פיצול תאים | ניתן להעריך את איכות ההפרדה הגרעינית והציטופלזמית על ידי הוספת קוקטיילים סטנדרטיים של מעכבי פרוטאז לתא ולמאגרי ליזה גרעיניים (טבלה 1). ניתן להעריך את השברים הציטופלזמיים והגרעיניים על ידי ניתוח כתמים מערביים כדי לאשר הגבלה נאותה של תיוג עם סמנים ציטופלזמיים (לדוגמה, GAPDH או HSP90) בשברים ציטופלזמיים ותיוג עם סמנים גרעיניים (לדוגמה, HDAC1 או היסטון H3) בשברים גרעיניים. ניתן להעריך את תרומת הזיהום המיטוכונדריאלי במקטע הגרעיני על ידי ניתוח qPCR עם בדיקות ספציפיות ל-DNA המיטוכונדריאלי. |
| 2.1 | המדגם צמיג מדי | מספר התא גבוה מדי. | צמצם את ה- DNA בחצי והמשך בשלב הסוניקציה 2.1 |
| 2.12 | אין גלולה גלויה | חומר התחלתי לא מספיק או אובדן במהלך החילוץ. | התחל מההתחלה באמצעות תאים נוספים. |
| 2.13 | ריכוז DNA נמוך | ||
| 3.1 | אין מספיק DNA לדילולים | ||
| 3.4 | הדגימה לא תרווי לתוך הממברנה | משרים את הממברנה ב -1x TBST. הניחו לעודפי החיץ להתייבש והתחילו בשלב 3.4 | |
| 4.6 | זוהה דפוס טלאי או מנומר | הוסף 0.1% נתרן דודציל סולפט (SDS) לדגימה והמשך בשלב 3.1 | |
| 4.6 | לנקודות יש מראה של "טבעת קפה" | הוסף 0.01% סרקוזיל לדגימה והמשך בשלב 3.1 | |
| 5.2 | פקד RNaseH אינו מראה ירידה באות | עיכול הריבונוקלאז אינו שלם. | הגדל את הדגירה או הגדל את ריכוז האנזימים. |
| 6.5 | אין אות לפקדי האוליגו | דופלקס לא נוצר. אוליגונוקלאוטידים לא חוסלו כראוי. | ודא את היחסים בין אוליגונוקלאוטידים ומאגר חישול. |
| 6.5 | אות S9.6 אינו ספציפי להיברידיות | לאצוות נוגדנים S9.6 יש קשירה לא ספציפית | אמת את הרגישות והספציפיות של קבוצות חדשות של נוגדן S9.6 באמצעות טיפולי אנזימים RNase או ניתוח אוליגונוקלאוטידים סינתטיים. |
טבלה 2: פתרון בעיות
| ssRNA, גדיל עליון | 5'-UGGGGGCUCGUCCGGGAUAUGGGAACCACUGAUCCC-3' |
| ssDNA, גדיל עליון | 5'-TGGGGGCTCGTCCGGGATATGGACCACTGATCCC-3' |
| ssRNA, גדיל תחתון | 5'-GGGAUCAGUGGUUCCCAUAUCCGGACGAGCCCCCA-3' |
| ssDNA, גדיל תחתון | 5'-GGGATCAGTGGTTCCCATATCCCGGACGAGCCCCCA-3' |
טבלה 3. בקרה על רצפי אוליגונוקלאוטידים
Discussion
חומצות הגרעין התלת-גדיליות, לולאות R, נוצרות בשלבים שונים במהלך מחזור החיים של RNA ונמצא יותר ויותר כמווסתות תהליכים תאיים. כדי להבין באופן מלא את לולאות R, יש צורך בטכניקות אמינות לזיהוי לולאת R. כאן, אנו מתארים גישה לחקירת השפע של לולאות R באמצעות נוגדן S9.6 8,23,24. שיטה זו מאפשרת הערכה מהירה של שפע לולאת R מתאים ודגימות תרבית רקמות. זה לא דורש ציוד מיוחד, או כמות גדולה של חומר מוצא. זה מבטיח תוצאות ספציפיות וניתנות לשחזור באמצעות שילוב של טיפולי RNase.
חלקם דיווחו על חששות לגבי הספציפיות של נוגדן S9.6. כמו בכל מגיב, תיתכן שונות אצווה לאצווה עם נוגדן S9.6. הפרוטוקול שלנו כולל RNase H, RNase T1 ו-RNase III כדי לבדוק את ספציפיות האות. בנוסף, אנו משתמשים באוליגונוקלאוטידים סינתטיים כדי להבטיח את הספציפיות של כל אצווה של נוגדן S9.6.
ביולוגיה של לולאת R היא תחום צומח; פיתוח שיטות זיהוי וכימות אמינות, כמו זו המוצגת כאן, יאפשר מחקרים מכניסטיים להבהיר מתי נוצרות לולאות R, כיצד הן מווסתות ומה הן מווסתות. עם בקרות מתאימות, בדיקת כתמי נקודות זו היא שיטה פשוטה לסינון שפע לולאת R במסגרות קליניות ומחקריות.
Disclosures
למחברים אין מה לחשוף.
Acknowledgements
עבודה זו נתמכה על ידי המכון הרפואי הווארד יוז ומחקר תוך-כיתתי במכון הלאומי להפרעות נוירולוגיות ושבץ מוחי.
אנו מודים לד"ר סטיבן לפלה על מתן קבוצות של נוגדן S9.6 לניתוח. אנו מודים גם לד"ר דונג-ג'ון לי על עזרתו בטיפולי ריבונוקלאז.
Materials
| Name | Company | Catalog Number | Comments |
| Anti-dsDNA antibody | Abcam | ab27156 | |
| Anti-RNA-DNA hybrid antibody (S9.6) | Kerafast | ENH001 | |
| Biorupter sonicator | Diagenode | UCD-200 | |
| EB Buffer | Qiagen | 19086 | |
| EDTA (0.5M) | Invitrogen | AM9261 | |
| Hybond N+ nylon membrane | GE healthcare Life Sciences | RPN203B | |
| KCl (2M) | Invitrogen | AM9640G | |
| NP-40 (Igepal CA-630) | Sigma | I8896 | |
| PBS | Invitrogen | 10010-023 | |
| Phenol:chloroform:isoamyl alcohol | Invitrogen | 15593031 | |
| PIPES (0.5M, pH 8.0) | VWR | AAJ61406-AE | |
| Proteinase K | Qiagen | 19131 | |
| RNase III | Invitrogen | AM2290 | |
| RNase H | New England Biolabs | M0297 | |
| RNase T1 | ThermoFisher Sci. | EN0541 | |
| SDS (10%) | Invitrogen | 15553027 | |
| sodium acetate (3M, pH 5.2) | Invitrogen | AM9740 | |
| Tris-buffered saline (10X) | Corning | 46-012-CM | |
| Tris-HCl (1M, pH 8.0) | KD Medical | RGF-3360 | |
| TrypLE | Invitrogen | 12605010 | |
| Tween-20 | Sigma | P9416 | |
| UV Stratalinker 2400 | Stratagene | Stratalinker 2400 | |
| Whatman marking pen | Sigma | WHA10499001 |
References
- Daube, S. S., von Hippel, P. H. RNA displacement pathways during transcription from synthetic RNA-DNA bubble duplexes. Biochemistry. 33 (1), 340-347 (1994).
- Westover, K. D., Buschnell, D. A., Kornberg, R. D. Structural basis of transcription: separation of RNA from DNA by RNA polymerase II. Science. 303 (5660), 1014-1016 (2004).
- Itoh, T., Tomizawa, J. Formation of an RNA primer for initiation of replication of ColE1 DNA by ribonuclease H. Proceedings of the National Academy of Science USA. 77 (5), 2450-2454 (1980).
- Hamperl, S., Bocek, M. J., Saldivar, J. C., Swigut, T., Cimprich, K. A. Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell. 170 (4), 774-786 (2017).
- Skourti-Stathaki, K., Kamieniarz-Gdula, K., Proudfoot, N. J. R-loops induce repressive chromatin marks over mammalian gene terminators. Nature. 516 (7531), 436-439 (2014).
- Sanz, L. A., et al. conserved R-loop structures associate with specific epigenomic signatures in mammals. Molecular Cell. 63 (1), 167-178 (2016).
- Chen, L., et al. R-ChIP using inactive RNase H reveals dynamic coupling of R-loops with transcriptional pausing at gene promoters. Molecular Cell. 68 (4), 745-757 (2017).
- El Hage, A., French, S. L., Beyer, A. L., Tollervey, D. Loss of topoisomerase I leads to R-loop mediated transcriptional blocks during ribosomal RNA synthesis. Genes and Development. 24 (14), 1546-1558 (2010).
- Tran, P., et al. PIF1 family DNA helicases suppress R-loop mediated genome instability at tRNA genes. Nature Communication. 8, 15025(2017).
- Ginno, P. A., Lott, P. L., Christensen, H. C., Korf, I., Chedin, F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Molecular Cell. 45 (6), 814-825 (2012).
- Colak, D., et al. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science. 343 (6174), 1002-1005 (2014).
- Yu, K., Chedin, F., Hsieh, C., Wilson, T. E., Lieber, M. R. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nature Immunology. 4 (5), 442-451 (2003).
- Gasiunas, G., Barrangou, R., Horvath, P., Siksnys, V. Cas9-crRNA ribonucleotide complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proceedings of the National Academy of Science USA. 109 (39), 2579-2586 (2012).
- Westra, E. R., et al. CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by Cascade and Cas3. Molecular Cell. 46 (5), 595-605 (2012).
- Jiang, F., et al. Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science. 351 (6275), 867-871 (2016).
- Huertas, P., Aguilera, A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Molecular Cell. 12 (3), 711-721 (2003).
- Grunseich, C., et al. Senataxin mutation reveals how R-loops promote transcription by blocking DNA methylation at gene promoters. Molecular Cell. 69 (3), 426-437 (2018).
- Kim, H. D., Choe, J., Seo, Y. S. The sen1(+) gene of Schizosaccharomyces pombe, a homologue of budding yeast SEN1, encodes an RNA and DNA helicase. Biochemistry. 38 (44), 14697-14710 (1999).
- Stein, H., Hausen, P. Enzyme from calf thymus degrading the RNA moiety of DNA-RNA hybrids: effect on DNA-dependent RNA polymerase. Science. 166 (3903), 393-395 (1969).
- Cerritelli, S. M., Crouch, R. J. Ribonuclease H: the enzymes in eukaryotes. FEBS Journal. 276 (6), 1494-1505 (2009).
- Hyjek, M., Figiel, M., Nowotny, M. RNases H: Structure and mechanism. DNA Repair. 84, 102672(2019).
- Pohl, T. J., Zakian, V. A. Pif1 family DNA helicases: A helpmate to RNase H. DNA Repair. 84, 102633(2019).
- Pohjoismaki, J. L., et al. Mammalian mitochondrial DNA replication intermediates are essentially duplex but contain extensive tracts of RNA/DNA hybrid. Journal of Molecular Biology. 397 (5), 1144-1155 (2010).
- Sanz, L. A., Chedin, F. High-resolution, strand-specific R-loop mapping via S9.6-based DNA-RNA immunoprecipitation and high-throughput sequencing. Nature Protocols. 14 (6), 1734-1755 (2019).
- Wahba, L., Costantino, L., Tan, F. J., Zimmer, A., Koshland, D. S1-DRIP-seq identifies high expression and polyA tracts as major contributors to R-loop formation. Genes and Development. 30 (11), 1327-1338 (2016).
- Yan, Q., Shields, E. J., Bonasio, R., Sarma, K. Mapping native r-loops genome-wide using a targeted nuclease approach. Cell Reports. 29 (5), 1369-1380 (2019).
- Hu, Z., Zhang, A., Storz, G., Gottesman, S., Leppla, S. H. An antibody-based microarray assay for small RNA detection. Nucleic Acids Research. 34 (7), 52(2006).
- Nowotny, M., Gaidamakov, S. A., Crouch, R. J., Yang, W. Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell. 121 (7), 1005-1016 (2005).
- Sato, K., Egami, F. Studies on ribonucleases in takadiastase. Journal of Biochemistry. 44 (11), Tokyo. 753-767 (1957).
- Takahashi, K., Moore, S. Ribonuclease T1. Enzymes. 15, 435-468 (1982).
- Dunn, J. J. RNase III cleavage of single-stranded RNA: Effect of ionic strength in the fidelity of cleavage. Journal of Biological Chemistry. 251 (12), 3807-3814 (1976).
- Pertzev, A., Nicholson, A. W. Characterization of RNA sequence determinants and antideterminants of processing reactivity for a minimal substrate of Escherichia coli ribonuclease III. Nucleic Acids Research. 34 (13), 3708-3721 (2006).
- Phillips, D. D., et al. The sub-nanomolar binding of DNA-RNA hybrids by the single chain Fv fragment of antibody S9.6. Journal of Molecular Recognition. 26 (8), 376-381 (2013).
- Haruki, M., Noguchi, E., Kanaya, S., Crouch, R. J. Kinetic and stoichiometric analysis for the binding of Escherichia coli ribonuclease H1 to RNA-DNA hybrids using surface plasmon resonance. Journal of Biological Chemistry. 272 (35), 22015-22022 (1997).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved