
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Imágenes de lapso de tiempo de arborización neuronal utilizando el etiquetado de virus adenoasociados dispersos de poblaciones de células retinianas genéticamente dirigidas
En este artículo
Resumen
Aquí, presentamos un método para investigar la morfogénesis de neuritas en explantes retinianos de ratón postnatales mediante microscopía confocal de lapso de tiempo. Describimos un enfoque para el etiquetado disperso y la adquisición de tipos de células retinianas y sus procesos finos utilizando vectores de virus adenoasociados recombinantes que expresan proteínas fluorescentes dirigidas a la membrana de una manera dependiente de Cre.
Resumen
Descubrir los mecanismos que modelan los cenadores dendríticos requiere métodos para visualizar, visualizar y analizar las dendritas durante el desarrollo. La retina de ratón es un potente sistema modelo para la investigación de mecanismos específicos del tipo celular de morfogénesis neuronal y conectividad. La organización y composición de los subtipos de retina están bien definidas, y las herramientas genéticas están disponibles para acceder a tipos específicos durante el desarrollo. Muchos tipos de células de la retina también restringen sus dendritas y / o axones a capas estrechas, lo que facilita las imágenes de lapso de tiempo. Los cultivos de explantes de retina de ratón son adecuados para la obtención de imágenes de células vivas mediante microscopía confocal o multifotónica, pero faltan métodos optimizados para la dinámica de las dendritas de imágenes con resolución temporal y estructural. Aquí se presenta un método para etiquetar e visualizar escasamente el desarrollo de poblaciones específicas de retina marcadas por el sistema Cre-Lox. Los virus adenoasociados (AVA) disponibles comercialmente utilizados aquí expresaron proteínas fluorescentes dirigidas a la membrana de una manera dependiente de Cre. La administración intraocular de AAV en ratones neonatales produce un marcado fluorescente de tipos de células objetivo a los 4-5 días después de la inyección (dpi). Las señales fluorescentes de membrana son detectables por imágenes confocales y resuelven estructuras y dinámicas de ramas finas. Los videos de alta calidad que abarcan de 2 a 4 h se adquieren a partir de imágenes de montajes planos de retina perfundidos con líquido cefalorraquídeo artificial oxigenado (aCSF). También se proporciona una tubería de postprocesamiento de imágenes para la deconvolución y la corrección de deriva tridimensional (3D). Este protocolo se puede utilizar para capturar varios comportamientos celulares en la retina intacta y para identificar nuevos factores que controlan la morfogénesis de la neurita. Muchas estrategias de desarrollo aprendidas en la retina serán relevantes para comprender la formación de circuitos neuronales en otras partes del sistema nervioso central.
Introducción
Las dendritas de las neuronas de la retina forman patrones intrincados, pero específicos, que influyen en su función dentro de los circuitos neuronales. En la retina de los vertebrados, diversos tipos de células ganglionares retinianas (RGC) e interneuronas de células amacrinas tienen morfologías dendríticas únicas que difieren en tamaño del cenador, ubicación, longitud de la rama y densidad1. Durante el desarrollo postnatal, las RGC y las células amacrinas extienden procesos dendríticos exuberantes en un neuropil llamado capa plexiforme interna (IPL), donde reciben entradas de células bipolares que transmiten señales fotorreceptoras2. Según lo capturado por imágenes de lapso de tiempo de poblaciones retinianas marcadas fluorescentemente en larvas de pollitos o peces cebra, la morfogénesis de dendritas es altamente dinámica3,4,5. En cuestión de días, los cenadores dendríticos se expanden, remodelan y ramifican a subcapas estrechas de la IPL, donde hacen sinapsis con socios seleccionados. Los cenadores exhiben diferentes dinámicas estructurales a lo largo del desarrollo, con cambios en las tasas relativas de adición, retracción y estabilización de ramas. Las dendritas amacrinas y RGC también exhiben diferentes comportamientos de crecimiento y remodelación que podrían reflejar la arborización específica del tipo. Sin embargo, estos estudios rastrearon amplias poblaciones de amacrina o RGC y se centraron en la orientación laminar, que es solo un aspecto de la morfología.
Los mecanismos que producen la vasta diversidad morfológica observada a través de los subtipos de retina son poco conocidos. El objetivo de este grupo fue desarrollar un método para capturar la dinámica de las dendritas y la remodelación del cenador de subtipos retinianos definidos en ratones. La identificación de mecanismos específicos del tipo de célula de patrón de dendritas requiere métodos para visualizar y medir el comportamiento de las dendritas de las células de interés. Los cultivos organotípicos de retinas de ratón son adecuados para estudios de imágenes de células vivas que utilizan microscopía confocal o multifotónica. Las retinas en desarrollo se diseccionan y se montan en un explante plano que puede ser fotografiado durante varias horas en una cámara de grabación o cultivado durante unos días con efectos limitados en el circuito6,7. Las neuronas retinianas vivas pueden ser marcadas por una variedad de técnicas, incluyendo el llenado de tinte por electrodos, electroporación, entrega biolística de partículas recubiertas con colorantes lipofílicos o plásmidos que codifican proteínas fluorescentes (por ejemplo, Gene Gun), así como etiquetas celulares codificadas genéticamente7,8,9,10 . Sin embargo, estos enfoques son ineficientes para la dinámica de las dendritas por imágenes de subtipos específicos de retina. Por ejemplo, los métodos de llenado de colorantes son de bajo rendimiento y requieren aparatos de electrofisiología y etiquetas genéticas adicionales para dirigirse de manera confiable a las células de interés. Además, las fuertes señales de fluorescencia en el soma pueden oscurecer las dendritas cercanas.
Los métodos de administración de genes biolísticos pueden etiquetar simultáneamente docenas de células, pero los pasos que involucran la entrega de partículas a alta presión y la incubación nocturna de retina aislada pueden comprometer la fisiología celular y el crecimiento dendrítico. Este artículo propone que se pueden emplear herramientas genéticas recientes para capturar la dinámica temprana de las dendritas con el tipo de célula y la resolución estructural, dados los siguientes criterios experimentales. Primero, para resolver las ramas finas y los filopodios que dominan los cenadores en desarrollo, el método debe etiquetar las neuronas con proteínas brillantes y fluorescentes que llenan los procesos en todo el cenador. El etiquetado de fluorescencia no debe desvanecerse debido al fotoblanqueo durante el período de obtención de imágenes. Se han generado y comparado una variedad de variantes de proteínas fluorescentes para la idoneidad de las imágenes in vivo/ex vivo11 basadas en el brillo y la fotoestabilidad. En segundo lugar, las proteínas fluorescentes (PFX) deben expresarse en niveles suficientemente altos en la etapa más temprana de la morfogénesis de la dendrita, de modo que no se pierda la estrecha ventana de desarrollo. En los análisis de puntos de tiempo estáticos en la retina del ratón, el desarrollo de dendritas ocurre durante la primera semana postnatal e incluye fases de crecimiento, remodelación y estabilización10,12,13,14,15. En tercer lugar, el método debe conducir a un etiquetado selectivo o a una mayor probabilidad de etiquetado de la subpoblación neuronal de interés. En cuarto lugar, el etiquetado de la subpoblación objetivo debe ser lo suficientemente escaso como para que se pueda identificar y rastrear todo el cenador neuronal. Aunque los subtipos RGC y amacrina pueden distinguirse por sus características morfológicas maduras y patrones de estratificación de IPL16,17,18,19,20, el desafío es identificar subtipos durante el desarrollo basados en estructuras inmaduras. Esta tarea se ve facilitada por la expansión de herramientas transgénicas para etiquetar tipos específicos de células de la retina durante el desarrollo.
Las líneas de ratón transgénicos y knock-in en las que la expresión celular y temporal de proteínas fluorescentes o Cre está determinada por elementos reguladores de genes son ampliamente utilizadas para estudiar tipos de células retinianas13,21,22,23. Las observaciones clave sobre los patrones específicos de subtipo de desarrollo de dendritas provienen de estudios de retinas de ratones transgénicos en puntos de tiempo estáticos10,14,24,25. El sistema Cre-Lox, en particular, permite una exquisita manipulación genética y monitoreo de subtipos utilizando una variedad de reporteros, sensores y activadores optogenéticos dependientes de la recombinasa. Estas herramientas han llevado a descubrimientos de programas moleculares específicos de subtipos y propiedades funcionales que subyacen al ensamblaje del circuito retiniano26,27,28,29,30. Sin embargo, aún no se han aprovechado para estudiar la dinámica de las dendritas específicas del subtipo en la retina del ratón. El etiquetado de baja densidad se puede lograr combinando líneas de ratón Cre con transgenes introducidos por electroporación o por AAV recombinantes. Si está disponible, también se pueden usar líneas de Cre inducibles por tamoxifeno o estrategias genéticas interseccionales. Finalmente, la célula debe ser etiquetada de una manera mínimamente invasiva y fotografiada utilizando parámetros de adquisición para no comprometer el tejido o interferir con la función celular requerida para la morfogénesis de la dendrita.
Aquí se presenta un método para aplicar herramientas transgénicas y microscopía confocal para investigar la dinámica de las dendritas en explantes de retina de ratones vivos. Las líneas de ratón transgénico Cre se han combinado con vectores AAV que expresan proteínas fluorescentes tras la recombinación de Cre, lo que permite un etiquetado escaso de las células de la retina de interés. Los AAV disponibles comercialmente se administran a la retina neonatal mediante inyecciones intravítreas. Este artículo demuestra que los AAV producen una expresión fluorescente significativamente alta y específica del tipo celular en 4 dpi, lo que permite el acceso a los puntos de tiempo postnatales. Para ilustrar este enfoque, la interneurona colinérgica "starburst" amacrina fue marcada mediante la administración de Brainbow AAV en ratones neonatales que expresan el transgén de colina acetiltransferasa (ChAT)-ribosoma interno (IRES)-Cre, que es activo en la retina postnatal temprana31,32. Las células amacrinas starburst desarrollan una morfología estereotipada y radial del cenador que está formada por la autoevitabilidad de las dendritas mediada por las protocadherinas agrupadas33,34. Este artículo muestra que la resolución de las dendritas y filopodias de estallido estelar se mejora significativamente mediante XFP a la membrana plasmática con la adición del motivo CAAX que sufre farnesilación, como se utiliza para los AAV de Brainbow31. Finalmente, se han determinado los protocolos de imágenes de lapso de tiempo y post-procesamiento que producen imágenes de alta calidad susceptibles de reconstrucción de dendritas y cuantificación morfométrica. Este protocolo se puede utilizar para identificar factores que controlan la morfogénesis de las dendritas y para capturar varios comportamientos celulares en la retina intacta.
Protocolo
NOTA: Este protocolo abarca 2 días con un período mínimo de 4-5 días para la transducción viral entre días experimentales (Figura 1A). Los experimentos con animales se realizan de acuerdo con las Directrices del Consejo Canadiense de Cuidado animal para el uso de animales en investigación y cuidado de animales de laboratorio bajo protocolos aprobados por el Comité de Uso y Cuidado de Animales del Laboratorio de Servicios para Animales en el Hospital para Niños Enfermos (Toronto, Canadá).
1. Preparativos para las inyecciones neonatales de AAV y experimentos de imagen
- Seleccione una línea de ratón Cre para etiquetar las poblaciones de células de la retina de interés. Confirme la expresión de la recombinasa de Cre en el momento de las inyecciones de AAV cruzando a un reportero transgénico de Cre o mediante inmunotinción con un anticuerpo Cre.
- Criar ratones Cre transgénicos (8-16 semanas de edad) para generar animales neonatales Cre positivos.
NOTA: Para esta demostración, se utilizaron ratones ChAT-IRES-Cre knock-in para atacar las células colinérgicas de Starburst amacrine. - Obtener el virus AAV dependiente de la recombinasa que codifica proteínas fluorescentes. Para un etiquetado óptimo de procesos finos, seleccione vectores que expresen XFP modificados dirigidos a la membrana plasmática.
NOTA: Este estudio utilizó el virus Brainbow (BBV), AAV-EF1a-BbTagBY y AAV9-EF1a-BbChT (ver Tabla de Materiales), que proporcionan la opción de etiquetado multicolor (Figura 1B). La proteína fluorescente amarilla mejorada farnesilada (eYFP) y la expresión monomérica de cereza (mCherry) produjeron las señales fluorescentes más fuertes para imágenes en vivo. Se puede usar un solo BBV para obtener imágenes de cenadores individuales, mientras que la coinyección de BBV se puede usar para etiquetar más células o cenadores vecinos con fluoróforos distintos. Si utiliza múltiples proteínas fluorescentes, asegúrese de una superposición mínima del espectro de emisión (Figura 1C). Los parámetros de adquisición del microscopio deben ajustarse para capturar distintas señales XFP. - Preparar alícuotas de ∼3-5 μL de AAV en tubos individuales de baja unión para existencias de un solo uso para evitar ciclos de congelación/descongelación. Conservar a -80 °C.
- Prepare micropipetas de vidrio de borosilicato con puntas muy finas usando un extractor de micropipetas.
NOTA: La configuración del extractor varía según el filamento y el extractor que se utilicen; consulte la Figura 2A para conocer el tamaño y la forma finales de la punta. Los diámetros típicos de las gotas son de 600 μm. - Obtener un sistema de microinyección y tubos asociados. Conecte el microinyector a un puerto de aire presurizado.

Figura 1: Visión general experimental. (A) Este protocolo abarca 2 días de experimentos con un período mínimo de infección de 4-5 días entre días experimentales. Las inyecciones intraoculares se realizan en ratones neonatales no mayores del día 3 postnatal. Las retinas se diseccionan, se montan en plano y se visualizan en vivo para capturar la ventana de desarrollo deseada. Las células marcadas se pueden obtener imágenes en cualquier momento después de los 4-5 días necesarios para la expresión viral, ya que no hay efectos aparentes de la expresión prolongada de AAV en la morfología de la dendrita. (B) Los vectores Brainbow AAV (BBV) dependientes de Cre se inyectan en animales que expresan Cre31. En este estudio, se utilizaron ratones Knock-in ChAT-Cre para impulsar la recombinación de Cre en células amacrinas de estallido estelar. Los dos BBV codifican eYFP o tagBFP modificados, o mCherry y mTFP, que terminan en un motivo CAAX que se farnesila secuencialmente para la localización de la membrana. Los sitios lox se representan con triángulos. Los vectores expresan XFP farnesilados de manera estocástica y combinatoria dependiente de la recombinación de Cre. El promotor EF1α incluye elementos reguladores del gen del factor de elongación 1α. W representa elementos del elemento regulador posttranscripcional del virus de la hepatitis de la carpintería, y pA indica la secuencia de poliadenilación. (C) Los espectros de excitación y emisión de mCherry y eYFP, los fluoróforos BBV fotografiados en este estudio. Cuando se toman imágenes en vivo de múltiples proteínas fluorescentes, los parámetros de detección deben organizarse para separar adecuadamente los espectros de emisión en canales distintos. Abreviaturas: AAV = virus adenoasociado; BBV = Brainbow AAV; ChAT = colina acetiltransferasa; iRES = sitio de entrada del ribosoma interno; eYFP = proteína fluorescente amarilla mejorada; iTR = repetición terminal invertida; tagBP = Proteína fluorescente tag-blue; mCherry = cereza monomérica; mTFP = proteína fluorescente verde azulado; XFP = cualquier proteína fluorescente; EF1α= factor de elongación 1 alfa. Haga clic aquí para ver una versión más grande de esta figura.
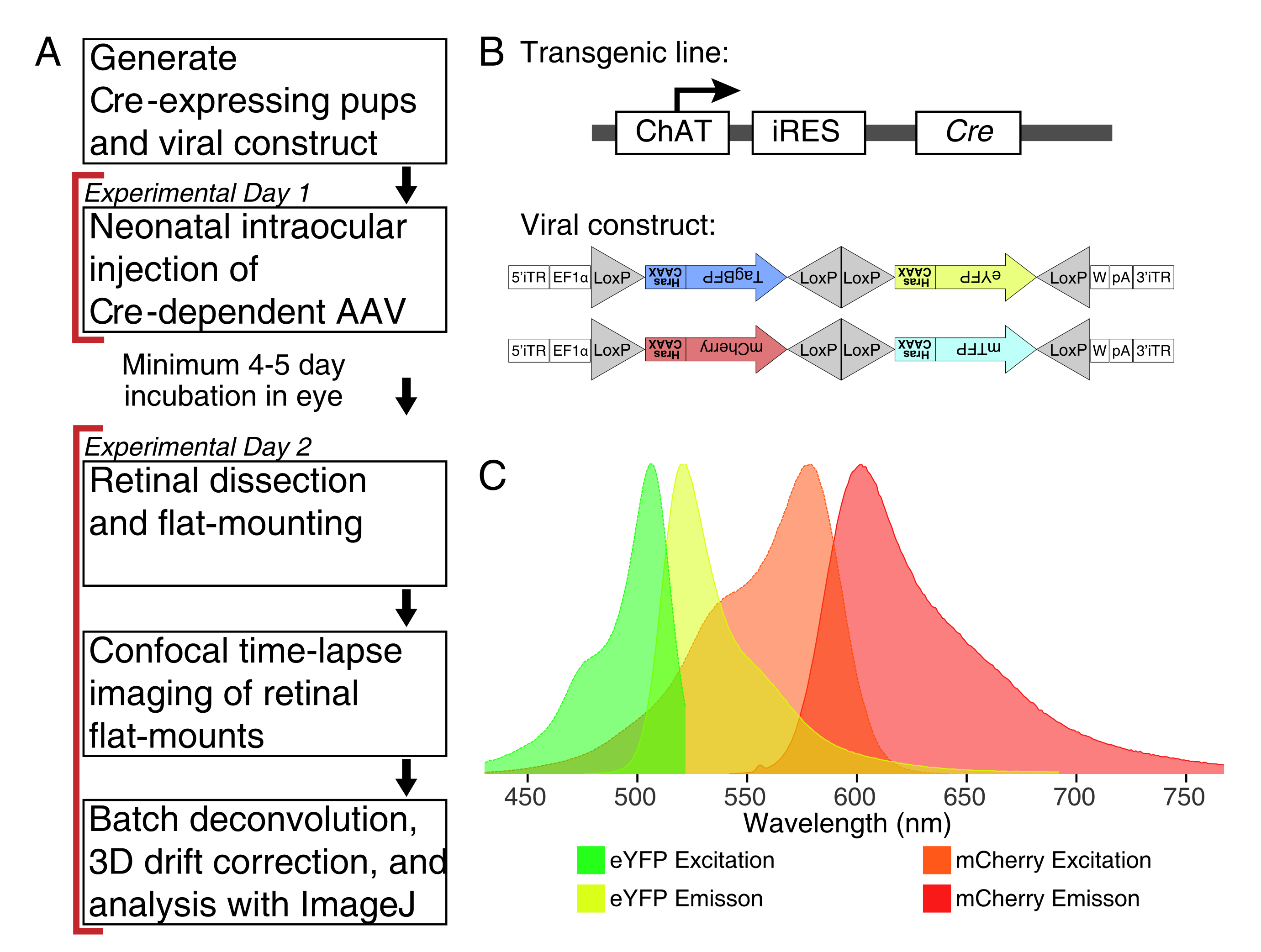{kind=link}
2. Inyecciones intravítreas de AAV en ratones neonatales
- Descongelar una alícuota AAV sobre hielo. Prepare una dilución de ~1:4 AAV usando solución salina estéril o solución salina tamponada con fosfato. Prepare ~ 0.5 μL de dilución de AAV por animal (~ 0.25 μL por ojo) en caso de que la micropipeta se rompa y se deba llenar una nueva pipeta. Guarde el AAV restante sin diluir a 4 °C y úselo dentro de las 2 semanas.
NOTA: En este experimento, la concentración de AAV del proveedor fue de ~ 1-2 × contenido del genoma (GC) / ml de 1013 y se diluyó a una concentración final de 4 × 1012 GC / ml. Optimizar la concentración viral para obtener la densidad de etiquetado deseada. - Para visualizar las inyecciones, agregue aproximadamente 1 μL de solución de colorante Fast Green FCF al 0,02% por cada 15 μL de dilución de AAV para colorear la solución de azul.
- Transfiera una jaula de ratón con camada de presa y recién nacido (día postnatal (P) 0.5-3) a una sala de procedimientos con equipo de microinyección. Mantenga a los animales en la misma jaula con nidos y ropa de cama para minimizar el estrés materno y el rechazo de los cachorros.
- Esterilice el área de inyección con etanol al 70% y coloque almohadillas de pañales estériles en las superficies del banco. Prepare una plataforma caliente (por ejemplo, una almohadilla térmica) para la recuperación de la anestesia inducida por hipotermia.
- Rellene la micropipeta con la dilución AAV con una microjeringa. Debajo de un estereomicroscopio, rompa la punta de la micropipeta con una aguja de 30 G para desprecintar la punta.
NOTA: La Figura 2A muestra la punta rellena, sellada (arriba) y sin sellar (centro). - Anestesiar ratones neonatales por hipotermia colocando 1-2 animales en hielo. Coloque a los animales en un guante de látex para proteger la piel del contacto directo con el hielo. Una vez que el animal ya no se mueva en respuesta al pellizco de la pata (~ 2 min), coloque al animal debajo de un estereomicroscopio. Si lo desea, tatúe almohadillas para las patas con tinta para tatuajes y aguja de 30 G, y recoja recortes de cola para el aislamiento del ADN para identificar a los animales mediante genotipado.
NOTA: El monitoreo apropiado de la profundidad de la anestesia debe ocurrir durante todo el procedimiento. - Frote la piel que recubre los ojos con etanol al 70%. Use una aguja de 30 G para abrir el párpado fusionado (Figura 2B). Aplique una ligera presión con los dedos para abrir el ojo y haga un pequeño orificio a través de la córnea en la unión córnea-esclerótica (Figura 2C).
- Inserte la micropipeta de vidrio en el orificio y presione el pedal del microinyector 2-4 veces para inyectar el AAV en el espacio intravítreo. Retire lentamente la micropipeta y confirme la inyección de AAV visualizando el tinte azul a través de la pupila (Figura 2D).
NOTA: Con una presión de eyección de 6-8 psi y un tiempo de pulso de 600-800 ms, se expulsa una gota de 600 μm de diámetro (Figura 2A, abajo). Se inyecta aproximadamente 0.23-0.45 μL de solución de AAV por ojo. La solución azul fuera del ojo indica que el AAV no se inyectó en el ojo. La fuga de solución azul del sitio de inyección indica que el AAV puede haberse filtrado, lo que reduce la eficiencia de la transfección. - Presione suavemente los párpados juntos para volver a sellar y coloque al cachorro en una almohadilla calentada. Una vez que los animales recuperen un color rosado y respondan, transfiéralos suavemente a la jaula de la vivienda.
NOTA: Asegúrese de que se tomen las precauciones adecuadas para evitar un recalentamiento demasiado rápido de los cachorros. - Repita el procedimiento de inyección para los animales restantes en la camada. Espere un mínimo de 4-5 días para la transducción viral antes de obtener imágenes de la retina.

Figura 2: Inyecciones intraoculares neonatales. (A) La micropipeta rellena muestra la forma de la punta de la pipeta cuando está sellada (arriba) y después de que la punta se desprecinta (abajo, izquierda). La presión de picoinyección de 6-8 psi y el tiempo de pulso de 600-800 ms producen una gota de 600 μm de diámetro (abajo, derecha). Barra de escala = 1 mm.(B) Cachorro P0 anestesiado con un aumento de 16x. La unión del párpado fusionado (blanco) se abre con una aguja afilada de 30 G. Barra de escala = 2 mm. (C) La presión ligera aplicada al ojo expone la córnea; barra de escala = 2 mm. La sección ampliada (rojo) muestra el pequeño orificio en la unión córnea-esclerótica (amarillo) creado con una aguja de 30 G; barra de escala = 0,5 mm. (D) Retirada de la punta de la pipeta después de la inyección (izquierda). El tinte verde rápido en la solución AAV aparece como gris azulado a través de la pupila (centro), en comparación con el color claro de la pupila antes de la inyección (derecha). Barra de escamas = 1 mm. (E) Después de 4 días, el párpado se ha curado y se ha fusionado cerrado (izquierda); barra de escala = 2 mm. Tras la enucleación, el sitio de inyección curado es visible (amarillo); barra de escala = 1 mm. Tenga en cuenta la ubicación del sitio de inyección en el borde entre la córnea y la esclerótica. Haga clic aquí para ver una versión más grande de esta figura.
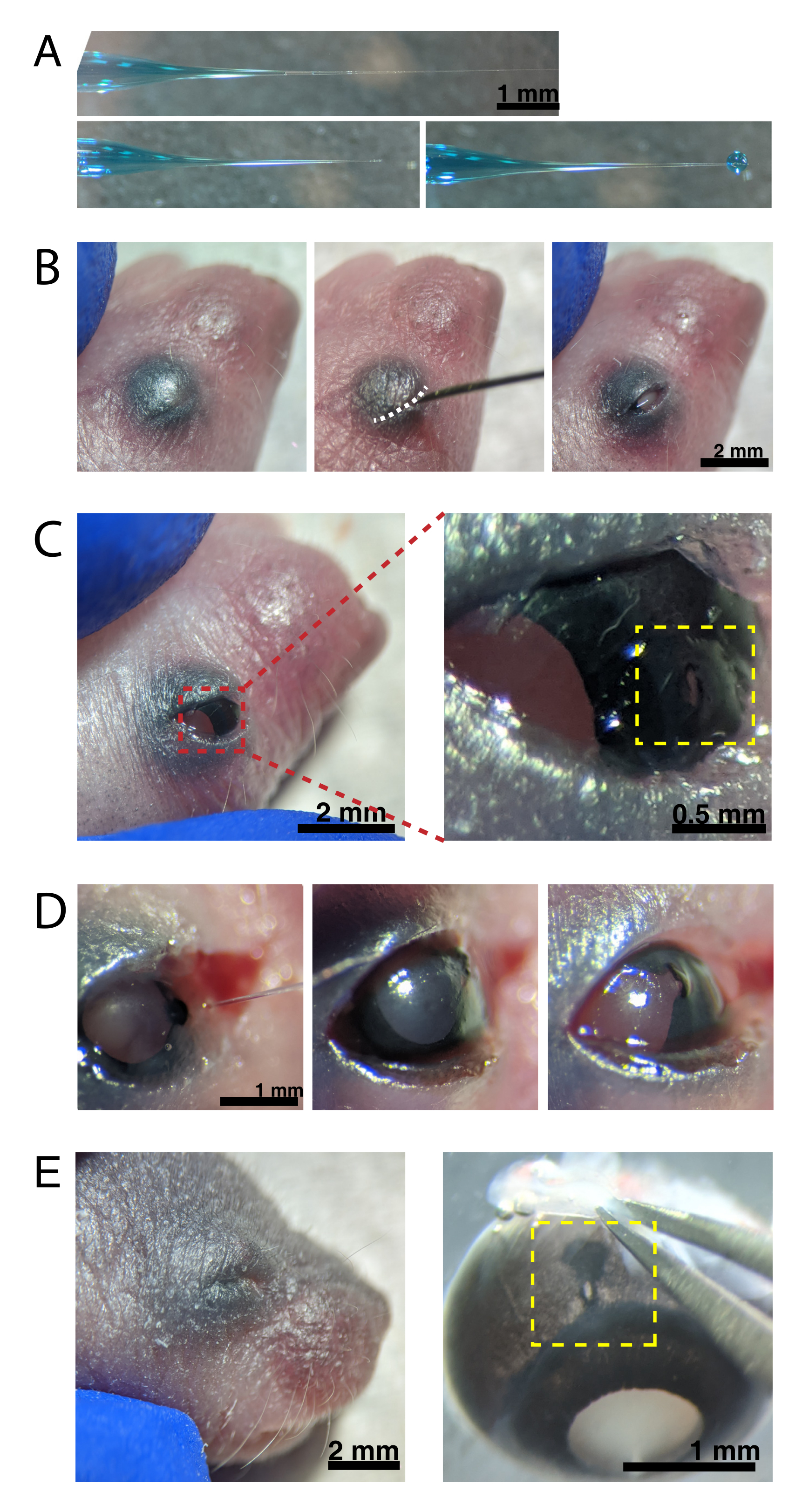{kind=link}
3. Disecciones retinianas para experimentos de imagen
- Preparar aCSF retiniano (119 mM NaCl, 2.5 mM KCl, 1.3 mM MgCl2·6H2O, 2.5 mM CaCl2·2H2O, 1 mM NaH2PO4, 11 mM glucosa y 20 mM 4-(2-hidroxietil)-1-piperazinetanosulfónico ácido (ácido libre HEPES)35. Si lo desea, prepare y congele la solución 10x; descongelar y diluir a 1x según sea necesario.
- Oxigenar el aCSF retiniano burbujeando con carbógeno (95% O2, 5% CO2) durante un mínimo de 15 min. Ajuste el pH a 7.4. Mantener en un recipiente sellado hasta su uso para asegurarse de que el aCSF permanezca oxigenado.
NOTA: Las retinas requieren una alta concentración de O2. Es importante mantener el aCSF oxigenado durante todo el experimento. - Incruste una placa de Petri de 60 mm de diámetro en una bandeja de hielo (coloque una bandeja de hielo con plástico de laboratorio, por ejemplo, la tapa de una caja de pipetas) y colóquela debajo de un estereomicroscopio. Llene la placa de Petri con aCSF retiniana oxigenada.
- Sacrificar ratones P9 y menores por decapitación. Sacrificar ratones P10 y mayores por inducción de isoflurano, o un método alternativo aprobado bajo el protocolo animal, seguido de decapitación.
- Si los párpados están sellados, corte el colgajo del párpado para exponer el ojo. Use fórceps para enuclear los ojos y transfiéralos a la aCSF retiniana fría del paso 3.3.
- Para diseccionar la copa de la retina, bajo un estereomicroscopio (aumento de 25x), estabilice el ojo apretando el nervio óptico usando un forcep de Dumont #5 (Figura 3A).
- Haga un agujero en el centro de la córnea con una aguja de 30 G y luego inserte una punta de las microescisoras en el orificio para hacer una incisión desde el orificio hasta el final de la córnea. Repita para hacer 4 cortes en las direcciones cardinales, creando 4 "solapas" (Figura 3A).
- Con dos fórceps Dumont #5, agarre y separe los dos colgajos adyacentes, pelando suavemente la esclerótica de la retina. Repetir con los colgajos de córnea restantes y extraer la esclerótica de la retina (Figura 3B).
- Retire el cristalino de la copa de la retina con los fórceps. Con microscisores, realizar 4 incisiones radiales desde el borde de la retina hacia el nervio óptico, creando 4 pétalos iguales (Figura 3B). Repita los pasos 3.4-3.7 para el segundo ojo.
4. Preparación de montaje plano retinal
- Prepare discos filtrantes de membrana de éster de celulosa mixta gris (MCE) para su montaje. Si usa filtros de membrana MCE de gran diámetro, corte el disco en cuadrantes (aproximadamente 1 cm de ancho). Coloque el disco MCE en el centro de un papel de filtro blanco más grande (Figura 3D).
NOTA: Los filtros MCE también están disponibles en discos de 1 cm de diámetro. El disco del filtro MCE debe ser lo suficientemente grande como para caber 1-2 retinas, pero lo suficientemente pequeño como para caber en la cámara de imágenes. Como las membranas MCE mantienen una carga estática, minimice el contacto y el manejo de la membrana MCE. - Manejando retinas usando dos pinceles de tamaño 3/0, voltee una taza de retina sobre un pincel con la célula ganglionar retiniana hacia abajo. Levante suavemente la retina fuera del aCSF, asegurándose de que la tensión del agua no desgarre la retina (Figura 3C).
- Mientras mantiene el pincel con la retina, use una pipeta de transferencia para colocar una gota de aCSF en el centro del papel de filtro MCE (Figura 3C, derecha). Flote la retina en la gota de aCSF creada por la tensión superficial. Use pinceles para colocar el lado RGC de la retina hacia arriba dentro de la gota y para desplegar los cuatro pétalos.
- Una vez colocado, cree un puente de agua entre el pincel y el papel de filtro blanco para romper la tensión superficial de la gota.
NOTA: Cuando el aCSF desaparece, la retina se adherirá al papel de filtro MCE (Figura 3D, E). Las retinas de montaje plano se pueden manejar agarrando el disco MCE con fórceps. Si la gota de aCSF se absorbe rápidamente antes de formar un puente de agua, esto puede indicar que la membrana MCE no está cargada. Use una membrana MCE fresca y minimice el tiempo entre la enucleación y la obtención de imágenes diseccionando y montando las retinas inmediatamente antes de mover las retinas a la cámara de imágenes.

Figura 3: Disección retiniana y montaje plano en membranas mixtas de filtro de éster de celulosa. (A) A la izquierda, un ojo enucleado se estabiliza agarrando el nervio óptico con fórceps Dumont #5 (izquierda). Se crea un pequeño orificio en el centro de la córnea utilizando una aguja de 30 G (centro). Las tijeras de microdisección se utilizan para cortar la córnea en 4 colgajos iguales (derecha). Barra de escala = 1 mm. (B) Retina diseccionada con la esclerótica despegada usando dos pinzas Dumont #5 (izquierda) y con la lente removida (centro). Retina con 4 cortes iguales a la mitad de la retina (derecha). Barra de escala = 1 mm. (C) Manejo de retina con dos pinceles finos (tamaño 3/0, izquierda). Retina volteada con células ganglionares de la retina hacia abajo en un pincel (centro) y levantada de aCSF, asegurándose de que la tensión del agua no desgarre la retina (derecha). Barra de escala = 2 mm. (D) Disco de membrana MCE gris colocado sobre un papel de filtro blanco (izquierda). Una gota de aCSF en el disco MCE (derecha). Barra de escala = 1 cm. (E) Después de flotar las retinas en la gota y posicionarlas, cree un puente de agua con el papel de filtro blanco para absorber el aCSF, tirando de la retina sobre el papel MCE cargado (izquierda); barra de escala = 1 cm. La imagen ampliada de la membrana MCE (rojo) muestra 2 retinas montadas con células ganglionares retinianas hacia arriba (derecha); barra de escala = 2 mm. Una retina está delineada en blanco. Abreviaturas: MCE = éster de celulosa mixta; aCSF = líquido cefalorraquídeo artificial. Haga clic aquí para ver una versión más grande de esta figura.
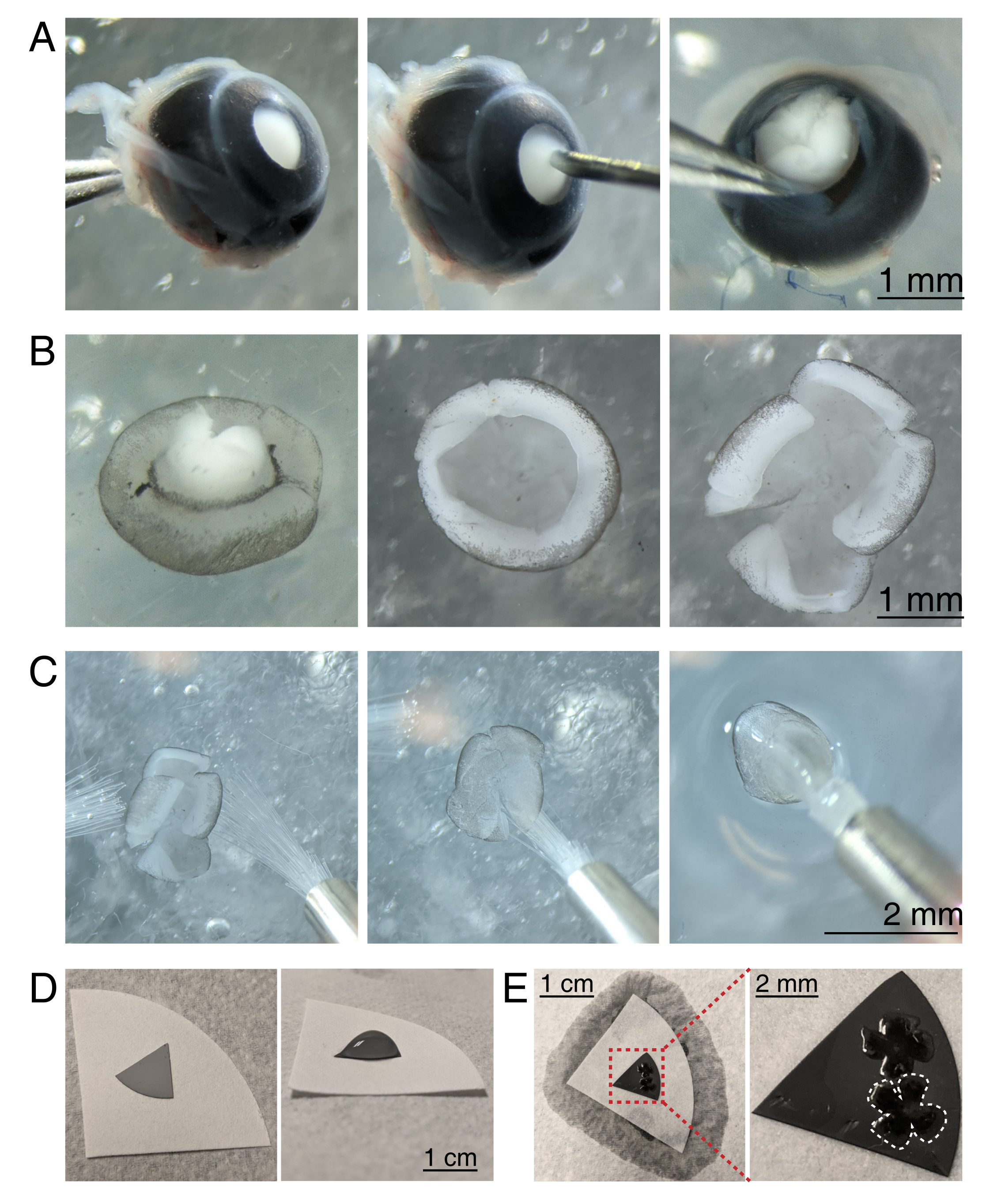{kind=link}
5. Imágenes confocales de lapso de tiempo de preparaciones de retina de montaje completo en vivo
- Ensamble la cámara de incubación de imágenes en vivo para un microscopio confocal vertical como se ve en la Figura 4.
NOTA: Para los sistemas confocales invertidos, los soportes planos se colocan RGC de lado hacia abajo directamente sobre la cubierta inferior de vidrio de la cámara de incubación. Una vez que las retinas hacen contacto con el coverslip, no se pueden mover. - Llene la cámara con aCSF oxigenado y encienda la bomba y el controlador de temperatura (temperatura 32-34 °C, caudal de 1 ml/min). No permita que la temperatura aumente por encima de 34 °C.
- Para transferir el soporte plano de retina a la cámara de perfusión, detenga la bomba y retire el aCSF que se encuentra en la cámara. Coloque el disco MCE con el soporte plano de la retina en la cámara de incubación (vacía).
- Coloque un peso de muestra sobre el soporte plano; mojar previamente el peso para romper la tensión superficial. Rellene la cámara con el aCSF calentado y haga circular el aCSF a ~1 mL/min.
- Coloque la pieza nasal con el objetivo de inmersión de agua 25x (apertura numérica 0.95) en la cámara de imagen. Detección de células marcadas de interés utilizando luz epifluorescente (Figura 4C).
- Ajuste el volumen de la imagen para capturar las características dendríticas de interés.
NOTA: Este estudio capturó el cenador dendrítico completo a 1024 x 1024 píxeles por fotograma, z-step 1 μm y una velocidad de fotogramas de 2 min entre cada z-stack. Los tamaños finales de imagen son ~100 μm x 100 μm x 20 μm. - Para ajustar la potencia del láser a un ajuste óptimo, utilice una tabla de búsqueda que identifique los píxeles sobresaturados y subsaturados. Durante el escaneo, ajuste la potencia del láser de tal manera que no haya píxeles sobresaturados (es decir, a una intensidad de 255 o superior). Continúe con las imágenes el tiempo que sea necesario, o hasta que haya una disminución significativa y detectable en la señal fluorescente y un aumento en el ruido (generalmente 2-4 h).
NOTA: Se recomienda una potencia láser reducida ya que los algoritmos de deconvolución funcionan de manera óptima cuando los píxeles se distribuyen en todo el rango dinámico. La intensidad de píxeles no debe exceder de 254; Los análisis empíricos de neuritas revelaron que los valores de píxeles por debajo de 170 son ideales para la deconvolución. Las velocidades de escaneo rápidas (400-600 Hz) con promedio de línea (2-3) son preferibles a los escaneos únicos y más lentos del mismo tiempo total de permanencia de píxeles. El área de imágenes prolongadas a menudo se fotobleaches, pero otras secciones de explante siguen siendo viables. Se pueden obtener imágenes de múltiples regiones en un montaje plano, cada una durante 2-4 h, con un tiempo total de incubación de 6 horas. Las sesiones de imágenes más allá de las 6 h no se han probado sistemáticamente. La degradación de la neurita y el sangrado son signos de que la viabilidad del explante está disminuyendo. - Después de la toma de imágenes, fije los soportes planos de la retina y el filtro de membrana con paraformaldehído frío al 4% durante 1-2 h a 4 °C. Amplificar las etiquetas fluorescentes en las retinas fijas mediante inmunohistoquímica para análisis adicionales.
NOTA: La fijación posterior a la obtención de imágenes no es posible cuando se utiliza un confocal invertido; las retinas no son removibles sin destruir el tejido.

Figura 4: Configuración de la cámara de incubación de imágenes de células vivas. (A) Aparato de incubación de imágenes en vivo que muestra los componentes de calefacción, solución e imágenes. El calentador dual incluye una plataforma de cámara de incubación calentada (círculo posterior con electrodos de conector azul) y un calentador de solución en línea. (B) Diagrama esquemático de 4A. La montura plana de la retina (rojo) en la membrana MCE (gris) se coloca en la cámara de incubación. El calentador de solución en línea está conectado a una bomba de solución (no se muestra en 4A). Las dimensiones de la cámara de imágenes permiten una buena área de trabajo para la nariz del objetivo de inmersión. (C) vista 25x un explante de retina inyectado con 0.23-0.45 μL de 4 × 1012 GC/mL de dilución de AAV; barra de escala = 100 μm. El marcado denso de las células a menudo rodea la cabeza del nervio óptico (línea discontinua, abajo a la derecha); barra de escala = 50 μm. Abreviaturas: MCE = éster de celulosa mixta; GC = copias del genoma; mCherry = cereza monomérica; eYFP = proteína fluorescente amarilla mejorada. Haga clic aquí para ver una versión más grande de esta figura.
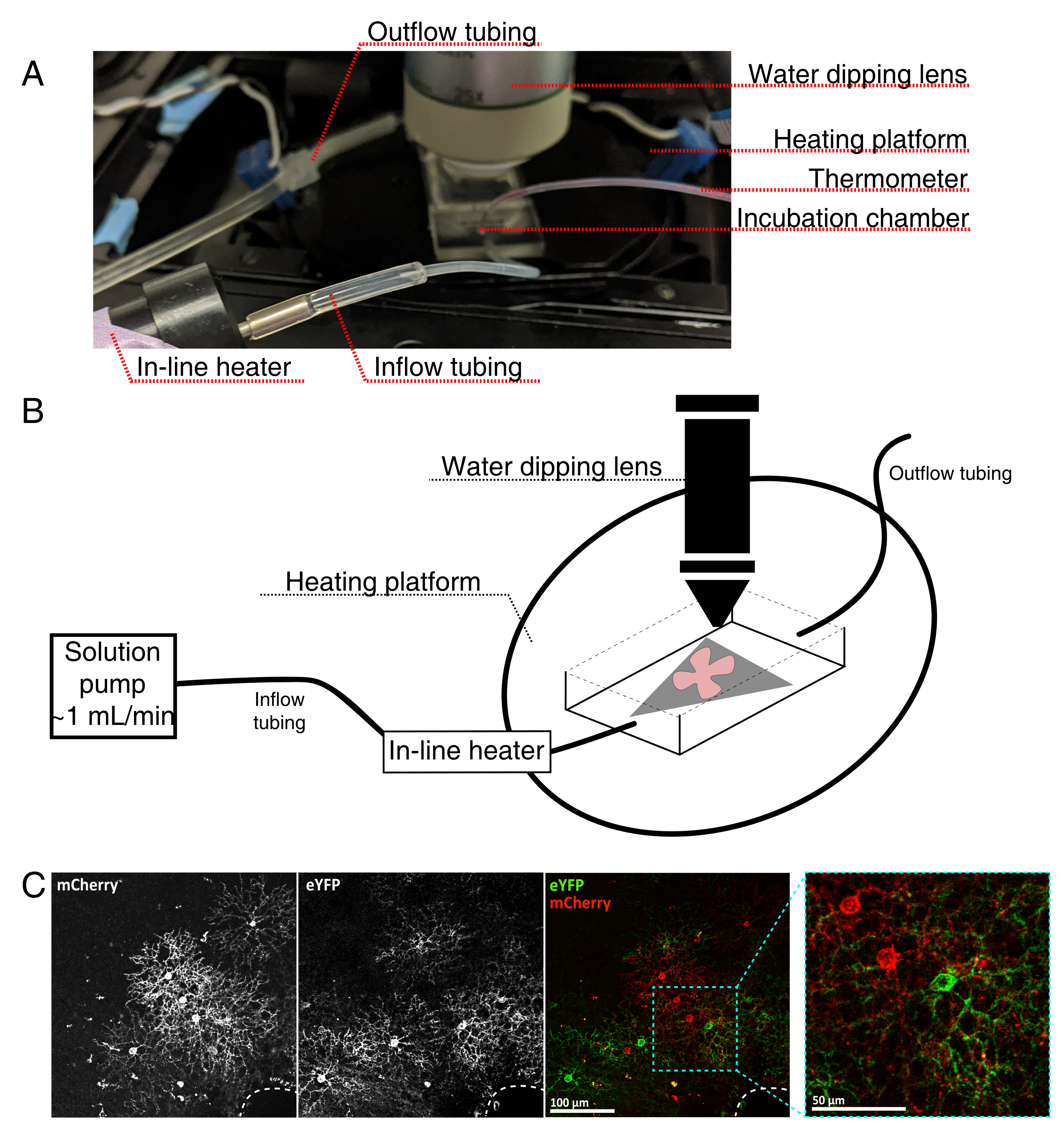{kind=link}
6. Deconvolución de imágenes y post-procesamiento en ImageJ
- Importe la serie de imágenes, divida la serie por tiempo y coloree si se trata de una imagen multicolor. Asegúrese de que todos los puntos de tiempo de un color estén contenidos en la misma carpeta.
NOTA: Importe bioformatos si utiliza ImageJ (los bioformatos se incluyen automáticamente en FIJI). - Cree una función teórica de dispersión de puntos (PSF) utilizando el complemento ImageJ, Diffraction PSF 3D.
NOTA: Cada canal de imagen requiere su propio PSF ya que la longitud de onda de emisión de proteínas fluorescentes afecta al PSF. - Usar plugins | | macros Ejecutar para realizar la deconvolución iterativa paralela por lotes para todos los puntos de tiempo mediante la macro proporcionada (archivo complementario 1).
Nota : esta macro ejecuta 25 iteraciones del algoritmo iterativo Wiener Filter Preconditioned Landweber (WPL). Cada canal de color debe ser desconvolucrado por separado. - Combinar los canales de color y compilar todos los puntos de tiempo en una Hyperstack (Imagen | Hyperstacks | Pilas a Hyperstack). Corrige la deriva 3D usando Plugins | | de registro Deriva 3D correcta.
- Devolver la imagen a una pila normal (Imagen | Hyperstacks | Hyperstack to Stack), y divida los puntos de tiempo (Image | Pilas | Herramientas | Divisor de pila). Utilice el procesamiento por lotes para crear la proyección máxima para todos los puntos de tiempo (Process | | por lotes | de macros run("Proyecto Z...", "projection=[Intensidad máxima]"); ).
- Importar la secuencia de imágenes time-lapse (Archivo | Importar | Secuencia de imágenes). Utilice las herramientas convencionales de ImageJ para el análisis deseado de vídeo bidimensional (2D) descontornado y postprocesado.
NOTA: Los videos descontorneados en cuatro dimensiones (3D + tiempo) y corregidos por deriva se pueden ver como un hyperstack. Omita el paso anterior para mantener los puntos de tiempo 3D.
Resultados
Usando el protocolo anterior, se adquirió, descontornó y corrigió un video 3D de alta resolución del desarrollo de dendritas celulares starburst para la deriva 3D. Se produjeron proyecciones máximas en plano Z para realizar videos 2D para su análisis (Video Complementario 1, Figura 5A). La deconvolución 3D de cada punto temporal aumentó la resolución de las proyecciones de filopodia fina (Figura 5B, C). Las protuberanci...
Discusión
Este video muestra una tubería experimental que utiliza las herramientas genéticas existentes para obtener imágenes de la dinámica de las dendritas del desarrollo de las neuronas de la retina con imágenes en vivo confocales. También se han demostrado las inyecciones intraoculares de AAV dependientes de Cre que codifican proteínas fluorescentes dirigidas a la membrana en ratones neonatales. Las células individuales de poblaciones genéticamente dirigidas están brillantemente etiquetadas tan pronto como 4-5 dpi. S...
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Agradecemos a Madison Gray por darme una mano cuando no tenía ninguna. Esta investigación fue apoyada por una beca de descubrimiento de NSERC (RGPIN-2016-06128), una beca Sloan en neurociencia y una cátedra de investigación de Canadá Nivel 2 (a J.L.L). S. Ing-Esteves fue apoyado por el Programa de Investigación de Ciencias de la Visión y las Becas de Postgrado-Doctorado de NSERC.
Materiales
| Name | Company | Catalog Number | Comments |
| Addgene viral prep #45185-AAV9 | |||
| Addgene viral prep #45186-AAV9 | |||
| Dissection tools | |||
| Cellulose filter paper | Whatman | 1001-070 | |
| Dumont #5 fine forceps | FST | 11252-20 | Two Dumont #5 forceps are required for retinal micro-dissection |
| Dumont forceps | VWR | 82027-426 | |
| Fine Scissors | FST | 14058-09 | |
| Mixed cellulose ester membrane (MCE) filter papers, hydrophilic, 0.45 µm pore size | Millipore | HABG01 300 | |
| Petri Dish, 50 × 15 mm | VWR | 470313-352 | |
| Polyethylene disposable transfer pipette | VWR | 470225-034 | |
| Round tip paint brush, size 3/0 | Conventional art supply store | Two size 3/0 paint brushes (or smaller) are required for retinal flat-mounting | |
| Surgical Scissors | FST | 14007-14 | |
| Vannas Spring Scissors - 2.5 mm Cutting Edge | FST | 15000-08 | |
| Live-imaging incubation system | |||
| Chamber polyethylene tubing, PE-160 10' | Warner Instruments | 64-0755 | |
| Dual channel heater controller, Model TC-344C | Warner Instruments | 64-2401 | |
| HC FLUOTAR L 25x/0.95 W VISIR dipping objective | Leica | 15506374 | |
| Heater controller cable | Warner Instruments | CC-28 | |
| Large bath incubation chamber with slice support | Warner Instruments | RC-27L | |
| MPII Mini-Peristaltic Pump | Harvard Apparatus | 70-2027 | |
| PM-6D Magnetic Heated Platform (incubation chamber heater) | Warner Instruments | PM-6D | |
| Pump Head Tubing Pieces For MPII Mini-Peristaltic Pump | Harvard Apparatus | 55-4148 | |
| Sample anchor (Harps) | Warner Instruments | 64-0260 | Sample anchor must be compatible with incubation chamber |
| Sloflo In-line Solution Heater | Warner Instruments | SF-28 | |
| Neonatal Injections | |||
| 10 µL Microliter Syringe Series 700, Removable Needle | Hamilton Company | 80314 | |
| 30 G Hypodermic Needles (0.5 inch) | BD PrecisionGlide | 305106 | |
| 4 inch thinwall glass capillary, no filament (1.0 mm outer diameter/0.75 mm) | WPI World Precision Instruments | TW100-4 | |
| Ethanol 99.8% (to dilute to 70% with double-distilled water [ddH2O]) | Sigma-Aldrich | V001229 | |
| AAV9.hEF1a.lox.TagBFP. lox.eYFP.lox.WPRE.hGH-InvBYF | Penn Vector Core | AV-9-PV2453 | Addgene Plasmid #45185 |
| AAV9.hEF1a.lox.mCherry.lox.mTFP 1.lox.WPRE.hGH-InvCheTF | Penn Vector Core | AV-9-PV2454 | Addgene Plasmid #45186 |
| ChAT-IRES-Cre knock-in transgenic mouse line | The Jackson Laboratory | 6410 | |
| Fast Green FCF Dye content ≥85 % | Sigma-Aldrich | F7252-25G | |
| Flaming/Brown Micropipette Puller, model P-97 | Sutter Instrument Co. | P-97 | |
| Green tattoo paste | Ketchum MFG Co | 329A | |
| Phosphate-Buffered Saline, pH 7.4, liquid, sterile-filtered, suitable for cell culture | Sigma-Aldrich | 806552 | |
| Pneumatic PicoPump | WPI World Precision Instruments | PV-820 | |
| Oxygenated artifiial cerebrospinal fluid (aCSF) Reagents | |||
| Calcium chloride dihydrate (CaCl2·2H2O) | Sigma-Aldrich | C7902 | |
| Carbogen (5% CO2, 95% O2) | AirGas | X02OX95C2003102 | Supplier may vary depending on region |
| D-(+)-Glucose | Sigma-Aldrich | G7021 | |
| HEPES, Free Acid | Bio Basic | HB0264 | |
| Hydrochloric acid solution, 1 N | Sigma-Aldrich | H9892 | |
| Magnesium chloride hexahydrate (MgCl2·6H2O) | Sigma-Aldrich | M2670 | |
| pH-Test strips (6.0-7.7) | VWR | BDH35317.604 | |
| Potassium chloride (KCl) | Sigma-Aldrich | P9541 | |
| Sodium chloride (NaCl) | Bio Basic | DB0483 | |
| Sodium phosphate monobasic (NaH2PO4) | Sigma-Aldrich | RDD007 | |
| Software | |||
| ImageJ | National Institutes of Health (NIH) | Open source |
Referencias
- Lefebvre, J. L., Sanes, J. R., Kay, J. N. Development of dendritic form and function. Annual Review of Cell and Developmental Biology. 31, 741-777 (2015).
- Graham, H. K., Duan, X. Molecular mechanisms regulating synaptic specificity and retinal circuit formation. Wiley Interdisciplinary Reviews Developmental biology. 10 (1), 379 (2021).
- Godinho, L., et al. Targeting of amacrine cell neurites to appropriate synaptic laminae in the developing zebrafish retina. Development. 132 (22), 5069-5079 (2005).
- Mumm, J. S., et al. In vivo imaging reveals dendritic targeting of laminated afferents by zebrafish retinal ganglion cells. Neuron. 52 (4), 609-621 (2006).
- Wong, W. T., Faulkner-Jones, B. E., Sanes, J. R., Wong, R. O. Rapid dendritic remodeling in the developing retina: dependence on neurotransmission and reciprocal regulation by Rac and Rho. The Journal of Neuroscience. 20 (13), 5024-5036 (2000).
- Wei, W., Elstrott, J., Feller, M. B. Two-photon targeted recording of GFP-expressing neurons for light responses and live-cell imaging in the mouse retina. Nature Protocols. 5 (7), 1347-1352 (2010).
- Morgan, J. L., Wong, R. O. L. Ballistic labeling with fluorescent dyes and indicators. Current Protocols in Neuroscience. 43 (1), 1-10 (2008).
- Nickerson, P. E. B., et al. Live imaging and analysis of postnatal mouse retinal development. BMC Developmental Biology. 13, 24 (2013).
- Morgan, J. L., Dhingra, A., Vardi, N., Wong, R. O. L. Axons and dendrites originate from neuroepithelial-like processes of retinal bipolar cells. Nature Neuroscience. 9 (1), 85-92 (2006).
- Coombs, J. L., Van Der List, D., Chalupa, L. M. Morphological properties of mouse retinal ganglion cells during postnatal development. The Journal of Comparative Neurology. 503 (6), 803-814 (2007).
- Cranfill, P. J., et al. Quantitative assessment of fluorescent proteins. Nature Methods. 13 (7), 557-562 (2016).
- Stacy, R. C., Wong, R. O. L. Developmental relationship between cholinergic amacrine cell processes and ganglion cell dendrites of the mouse retina. The Journal of Comparative Neurology. 456 (2), 154-166 (2003).
- Kay, J. N., et al. Retinal ganglion cells with distinct directional preferences differ in molecular identity, structure, and central projections. The Journal of Neuroscience. 31 (21), 7753-7762 (2011).
- Liu, J., Sanes, J. R. Cellular and molecular analysis of dendritic morphogenesis in a retinal cell type that senses color contrast and ventral motion. The Journal of Neuroscience. 37 (50), 12247-12262 (2017).
- Diao, L., Sun, W., Deng, Q., He, S. Development of the mouse retina: emerging morphological diversity of the ganglion cells. Journal of Neurobiology. 61 (2), 236-249 (2004).
- Coombs, J., vander List, D., Wang, G. Y., Chalupa, L. M. Morphological properties of mouse retinal ganglion cells. Neuroscience. 140 (1), 123-136 (2006).
- Sanes, J. R., Masland, R. H. The types of retinal ganglion cells: current status and implications for neuronal classification. Annual Review of Neuroscience. 38, 221-246 (2015).
- Sümbül, U., et al. A genetic and computational approach to structurally classify neuronal types. Nature Communications. 5, 3512 (2014).
- Lin, B., Masland, R. H. Populations of wide-field amacrine cells in the mouse retina. The Journal of Comparative Neurology. 499 (5), 797-809 (2006).
- Macneil, M. A., Heussy, J. K., Dacheux, R. F., Raviola, E., Masland, R. H. The shapes and numbers of amacrine cells: Matching of photofilled with Golgi-stained cells in the rabbit retina and comparison with other mammalian species. Journal of Comparative Neurology. 413 (2), 305-326 (1999).
- Ivanova, E., Hwang, G. S., Pan, Z. H. Characterization of transgenic mouse lines expressing Cre recombinase in the retina. Neuroscience. 165 (1), 233-243 (2010).
- Jo, A., Xu, J., Deniz, S., Cherian, S., DeVries, S. H., Zhu, Y. Intersectional strategies for targeting amacrine and ganglion cell types in the mouse retina. Frontiers in Neural Circuits. 12, 66 (2018).
- Siegert, S., et al. Genetic address book for retinal cell types. Nature Neuroscience. 12 (9), 1197-1204 (2009).
- Kim, I. -. J., Zhang, Y., Meister, M., Sanes, J. R. Laminar restriction of retinal ganglion cell dendrites and axons: subtype-specific developmental patterns revealed with transgenic markers. The Journal of Neuroscience. 30 (4), 1452-1462 (2010).
- Peng, Y. -. R., Tran, N. M., Krishnaswamy, A., Kostadinov, D., Martersteck, E. M., Sanes, J. R. Satb1 regulates contactin 5 to pattern dendrites of a mammalian retinal ganglion cell. Neuron. 95 (4), 869-883 (2017).
- Duan, X., Krishnaswamy, A., Dela Huerta, I., Sanes, J. R. Type II cadherins guide assembly of a direction-selective retinal circuit. Cell. 158 (4), 793-807 (2014).
- Ray, T. A., et al. Formation of retinal direction-selective circuitry initiated by starburst amacrine cell homotypic contact. eLife. 7, 34241 (2018).
- Krishnaswamy, A., Yamagata, M., Duan, X., Hong, Y. K., Sanes, J. R. Sidekick 2 directs formation of a retinal circuit that detects differential motion. Nature. 524 (7566), 466-470 (2015).
- Caval-Holme, F., Zhang, Y., Feller, M. B. Gap junction coupling shapes the encoding of light in the developing retina. Current Biology. 29 (23), 4024-4035 (2019).
- Lucas, J. A., Schmidt, T. M. Cellular properties of intrinsically photosensitive retinal ganglion cells during postnatal development. Neural Development. 14 (1), 8 (2019).
- Cai, D., Cohen, K. B., Luo, T., Lichtman, J. W., Sanes, J. R. Improved tools for the Brainbow toolbox. Nature Methods. 10 (6), 540-547 (2013).
- Rossi, J., et al. Melanocortin-4 receptors expressed by cholinergic neurons regulate energy balance and glucose homeostasis. Cell Metabolism. 13 (2), 195-204 (2011).
- Lefebvre, J. L., Kostadinov, D., Chen, W. V., Maniatis, T., Sanes, J. R. Protocadherins mediate dendritic self-avoidance in the mammalian nervous system. Nature. 488 (7412), 517-521 (2012).
- Ing-Esteves, S., et al. Combinatorial effects of alpha- and gamma-protocadherins on neuronal survival and dendritic self-avoidance. The Journal of Neuroscience. 38 (11), 2713-2729 (2018).
- Williams, P. R., Morgan, J. L., Kerschensteiner, D., Wong, R. O. L. In vitro imaging of retinal whole mounts. Cold Spring Harbor Protocols. 2013 (1), (2013).
- Ramoa, A. S., Campbell, G., Shatz, C. J. Transient morphological features of identified ganglion cells in living fetal and neonatal retina. Science. 237 (4814), 522-525 (1987).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 9, 676-682 (2012).
- Peng, H., Ruan, Z., Long, F., Simpson, J. H., Myers, E. W. V3D enables real-time 3D visualization and quantitative analysis of large-scale biological image data sets. Nature Biotechnology. 28 (4), 348-353 (2010).
- Cuntz, H., Forstner, F., Borst, A., Häusser, M. One rule to grow them all: a general theory of neuronal branching and its practical application. PLoS Computational Biology. 6 (8), 1000877 (2010).
- Xiao, H., Peng, H. APP2: automatic tracing of 3D neuron morphology based on hierarchical pruning of a gray-weighted image distance-tree. Bioinformatics. 29 (11), 1448-1454 (2013).
- Nanda, S., et al. Design and implementation of multi-signal and time-varying neural reconstructions. Scientific data. 5, 170207 (2018).
- Sherry, D. M., Wang, M. M., Bates, J., Frishman, L. J. Expression of vesicular glutamate transporter 1 in the mouse retina reveals temporal ordering in development of rod vs. cone and ON vs. OFF circuits. The Journal of Comparative Neurology. 465 (4), 480-498 (2003).
- Johnson, J., et al. Vesicular neurotransmitter transporter expression in developing postnatal rodent retina: GABA and glycine precede glutamate. The Journal of Neuroscience. 23 (2), 518-529 (2003).
- Jüttner, J., et al. Targeting neuronal and glial cell types with synthetic promoter AAVs in mice, non-human primates and humans. Nature Neuroscience. 22 (8), 1345-1356 (2019).
- Zincarelli, C., Soltys, S., Rengo, G., Rabinowitz, J. E. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Molecular Therapy. 16 (6), 1073-1080 (2008).
- Petros, T. J., Rebsam, A., Mason, C. A. In utero and ex vivo electroporation for gene expression in mouse retinal ganglion cells. Journal of Visualized Experiments: JoVE. (31), e1333 (2009).
- Lye, M. H., Jakobs, T. C., Masland, R. H., Koizumi, A. Organotypic culture of adult rabbit retina. Journal of Visualized Experiments: JoVE. (3), e190 (2007).
- Pignatelli, V., Strettoi, E. Bipolar cells of the mouse retina: a gene gun, morphological study. The Journal of Comparative Neurology. 476 (3), 254-266 (2004).
- Huckfeldt, R. M., et al. Transient neurites of retinal horizontal cells exhibit columnar tiling via homotypic interactions. Nature Neuroscience. 12 (1), 35-43 (2009).
- Prahst, C., et al. Mouse retinal cell behaviour in space and time using light sheet fluorescence microscopy. eLife. 9, 49779 (2020).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados