
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Imaging time-lapse dell'arborizzazione neuronale utilizzando l'etichettatura del virus adeno-associato sparso di popolazioni di cellule retiniche geneticamente mirate
In questo articolo
Riepilogo
Qui, presentiamo un metodo per studiare la morfogenesi dei neuriti negli espianti retinici postnatali di topo mediante microscopia confocale time-lapse. Descriviamo un approccio per l'etichettatura sparsa e l'acquisizione di tipi di cellule retiniche e dei loro processi fini utilizzando vettori virali adeno-associati ricombinanti che esprimono proteine fluorescenti mirate alla membrana in modo Cre-dipendente.
Abstract
La scoperta di meccanismi che modellano i pergole dendritiche richiede metodi per visualizzare, immaginare e analizzare i dendriti durante lo sviluppo. La retina del topo è un potente sistema modello per lo studio dei meccanismi specifici del tipo cellulare della morfogenesi neuronale e della connettività. L'organizzazione e la composizione dei sottotipi di retina sono ben definite e sono disponibili strumenti genetici per accedere a tipi specifici durante lo sviluppo. Molti tipi di cellule retiniche vincolano anche i loro dendriti e / o assoni a strati stretti, il che facilita l'imaging time-lapse. Le colture di espianto della retina del topo sono adatte per l'imaging di cellule vive utilizzando la microscopia confocale o multifotonica, ma mancano metodi ottimizzati per l'imaging della dinamica della dendrite con risoluzione temporale e strutturale. Qui viene presentato un metodo per etichettare e visualizzare scarsamente lo sviluppo di specifiche popolazioni retiniche caratterizzate dal sistema Cre-Lox. I virus adeno-associati disponibili in commercio (AAV) qui utilizzati hanno espresso proteine fluorescenti mirate alla membrana in modo Cre-dipendente. La somministrazione intraoculare di AAV nei topi neonatali produce l'etichettatura fluorescente dei tipi di cellule mirate entro 4-5 giorni dall'iniezione (dpi). I segnali fluorescenti a membrana sono rilevabili mediante imaging confocale e risolvono strutture e dinamiche di rami fini. Video di alta qualità della durata di 2-4 ore vengono acquisiti dall'imaging di supporti piatti retinici perfusi con liquido cerebrospinale artificiale ossigenato (aCSF). Viene inoltre fornita una pipeline di post-elaborazione delle immagini per la deconvoluzione e la correzione tridimensionale (3D) della deriva. Questo protocollo può essere utilizzato per catturare diversi comportamenti cellulari nella retina intatta e per identificare nuovi fattori che controllano la morfogenesi dei neuriti. Molte strategie di sviluppo apprese nella retina saranno rilevanti per comprendere la formazione di circuiti neurali altrove nel sistema nervoso centrale.
Introduzione
I dendriti dei neuroni retinici formano schemi intricati, ma specifici, che influenzano la loro funzione all'interno dei circuiti neurali. Nella retina dei vertebrati, diversi tipi di cellule gangliari retiniche (RGC) e interneuroni a cellule amacrine portano morfologie dendritiche uniche che differiscono per dimensioni del pergolato, posizione, lunghezza del ramo e densità1. Durante lo sviluppo postnatale, gli RGC e le cellule amacrine estendono i processi dendritici esuberanti in un neuropil chiamato strato plessiforme interno (IPL), dove ricevono input di cellule bipolari che trasmettono segnali fotorecettori2. Come catturato dall'imaging time-lapse di popolazioni retiniche marcate fluorescentemente in larve di pulcino o zebrafish, la morfogenesi dei dendrite è altamente dinamica3,4,5. In pochi giorni, i pergole dendritiche si espandono, rimodellano e si ramificano in stretti sottostrati dell'IPL, dove si sinapsino con partner selezionati. I pergole mostrano diverse dinamiche strutturali rispetto allo sviluppo, con cambiamenti nei tassi relativi di aggiunta di rami, retrazione e stabilizzazione. I dendriti di amacrina e RGC mostrano anche diversi comportamenti di crescita e rimodellamento che potrebbero riflettere l'arborizzazione specifica del tipo. Tuttavia, questi studi hanno monitorato ampie popolazioni di amacrine o RGC e si sono concentrati sul targeting laminare, che è solo un aspetto della morfologia.
I meccanismi che producono la vasta diversità morfologica osservata tra i sottotipi di retina sono poco conosciuti. L'obiettivo di questo gruppo era quello di sviluppare un metodo per catturare la dinamica dei dendriti e il rimodellamento del pergolato di sottotipi retinici definiti nei topi. L'identificazione dei meccanismi specifici del tipo di cellula del pattern dendrite richiede metodi per visualizzare e misurare i comportamenti dei dendrite delle cellule di interesse. Le colture organotipiche di retine di topo sono adatte per studi di imaging di cellule vive utilizzando la microscopia confocale o multifotonica. Le retine in via di sviluppo vengono sezionate e montate in un impianto piatto che può essere ripreso per diverse ore in una camera di registrazione o coltivato in pochi giorni con effetti limitati sul circuito6,7. I neuroni retinici vivi possono essere etichettati con una varietà di tecniche, tra cui il riempimento del colorante mediante elettrodi, l'elettroporazione, la somministrazione biolistica di particelle rivestite con coloranti lipofili o plasmidi che codificano proteine fluorescenti (ad esempio, Gene Gun), nonché etichette cellulari geneticamente codificate7,8,9,10 . Tuttavia, questi approcci sono inefficienti per l'imaging della dinamica dei dendriti di specifici sottotipi di retina. Ad esempio, i metodi di riempimento dei coloranti sono a bassa produttività e richiedono apparecchiature di elettrofisiologia ed etichette genetiche aggiuntive per indirizzare in modo affidabile le cellule di interesse. Inoltre, i forti segnali di fluorescenza nel soma possono oscurare i dendriti vicini.
I metodi di consegna dei geni biolistici possono etichettare simultaneamente dozzine di cellule, ma i passaggi che coinvolgono la consegna di particelle ad alta pressione e l'incubazione notturna della retina isolata possono compromettere la fisiologia cellulare e la crescita dendritica. Questo documento propone che i recenti strumenti genetici possano essere impiegati per catturare le prime dinamiche dei dendriti con il tipo di cellula e la risoluzione strutturale, dati i seguenti criteri sperimentali. In primo luogo, per risolvere i rami sottili e i filopodi che dominano i pergolati in via di sviluppo, il metodo dovrebbe etichettare i neuroni con proteine luminose e fluorescenti che riempiono i processi nell'intero pergolato. L'etichettatura a fluorescenza non deve sbiadire a causa del fotosbiancamento durante il periodo di imaging. È stata generata una varietà di varianti proteiche fluorescenti e confrontate per l'idoneità per l'imaging in vivo/ex vivo11 in base alla luminosità e alla fotostabilità. In secondo luogo, le proteine fluorescenti (XFP) devono essere espresse a livelli sufficientemente elevati dal primo stadio della morfogenesi della dendrite, in modo che non si perda la stretta finestra di sviluppo. Nelle analisi dei timepoint statici nella retina del topo, lo sviluppo di dendrite avviene durante la prima settimana postnatale e comprende fasi di crescita, rimodellamento e stabilizzazione10,12,13,14,15. In terzo luogo, il metodo dovrebbe portare a un'etichettatura selettiva o ad una maggiore probabilità di etichettatura della sottopopolazione neuronale di interesse. In quarto luogo, l'etichettatura della sottopopolazione bersaglio deve essere sufficientemente scarsa in modo che l'intero pergolato neuronale possa essere identificato e rintracciato. Sebbene i sottotipi di RGC e amacrina possano essere distinti per le loro caratteristiche morfologiche mature e i modelli di stratificazione IPL16,17,18,19,20, la sfida è identificare i sottotipi durante lo sviluppo basati su strutture immature. Questo compito è facilitato dall'espansione di strumenti transgenici per etichettare specifici tipi di cellule retiniche durante lo sviluppo.
Le linee di topo transgeniche e knock-in in cui l'espressione cellulare e temporale di proteine fluorescenti o Cre è determinata da elementi regolatori genici sono ampiamente utilizzate per studiare i tipi di cellule retiniche13,21,22,23. Osservazioni chiave sui modelli specifici del sottotipo di sviluppo di dendrite provengono da studi su retine di topi transgenici a timepoint statici10,14,24,25. Il sistema Cre-Lox, in particolare, consente una squisita manipolazione genica e il monitoraggio dei sottotipi utilizzando una varietà di reporter, sensori e attivatori optogenetici dipendenti dalla ricombinasi. Questi strumenti hanno portato alla scoperta di programmi molecolari specifici del sottotipo e proprietà funzionali che sono alla base dell'assemblaggio del circuito retinico26,27,28,29,30. Tuttavia, devono ancora essere sfruttati per studiare la dinamica dei dendriti specifici del sottotipo nella retina del topo. L'etichettatura a bassa densità può essere ottenuta combinando le linee di topo Cre con i transgeni introdotti dall'elettroporazione o dagli AAV ricombinanti. Se disponibili, possono essere utilizzate anche linee Cre inducibili dal tamoxifene o strategie genetiche intersezionali. Infine, la cellula deve essere marcata in modo minimamente invasivo e fotografata utilizzando parametri di acquisizione in modo da non compromettere il tessuto o interferire con la funzione cellulare richiesta per la morfogenesi dei dendriti.
Qui viene presentato un metodo per applicare strumenti transgenici e microscopia confocale per studiare la dinamica dei dendriti negli espianti retinici di topo vivo. Le linee di topo transgenico Cre sono state combinate con vettori AAV che esprimono proteine fluorescenti sulla ricombinazione cre, che consente l'etichettatura sparsa delle cellule retiniche di interesse. Gli AAV disponibili in commercio vengono consegnati alla retina neonatale mediante iniezioni intravitreali. Questo documento dimostra che gli AAV producono un'espressione fluorescente significativamente elevata e specifica del tipo di cellula di 4 dpi, consentendo l'accesso ai punti temporali postnatali. Per illustrare questo approccio, l'interneurone colinergico "starburst" amacrina è stato etichettato consegnando Brainbow AAV in topi neonatali che esprimono il sito di ingresso del ribosoma interno alla colina acetilsi (ChAT) (IRES)-Cre transgene, che è attivo nella retina postnatale precoce31,32. Le cellule amacrine Starburst sviluppano una morfologia stereotipata e radiale del pergolato che è modellata dall'auto-evitamento della dendrite mediata dalle protocadherine raggruppate33,34. Questo documento mostra che la risoluzione dei dendriti starburst e dei filopodi è significativamente migliorata dagli XFP alla membrana plasmatica con l'aggiunta del motivo CAAX che subisce la farnesilazione, come usato per gli AAV Brainbow31. Infine, sono stati determinati protocolli di imaging e post-elaborazione time-lapse che producono immagini di alta qualità suscettibili di ricostruzione di dendrite e quantificazione morfometrica. Questo protocollo può essere utilizzato per identificare i fattori che controllano la morfogenesi dei dendriti e per catturare diversi comportamenti cellulari nella retina intatta.
Protocollo
NOTA: Questo protocollo si estende su 2 giorni con un periodo minimo di 4-5 giorni per la trasduzione virale tra i giorni sperimentali (Figura 1A). Gli esperimenti sugli animali vengono eseguiti in conformità con le linee guida del Canadian Council on Animal Care per l'uso degli animali nella ricerca e nella cura degli animali da laboratorio secondo protocolli approvati dal Laboratory of Animal Services Animal Use and Care Committee presso l'Hospital for Sick Children (Toronto, Canada).
1. Preparazioni per le iniezioni neonatali di AAV ed esperimenti di imaging
- Selezionare una linea del mouse Cre per etichettare le popolazioni di cellule retiniche di interesse. Confermare l'espressione di Cre ricombinasi al momento delle iniezioni di AAV incrociando un reporter transgenico Cre o attraverso l'immunocolorazione con un anticorpo Cre.
- Alleva topi Cre transgenici (8-16 settimane) per generare animali neonatali Cre-positivi.
NOTA: Per questa dimostrazione, i topi knock-in ChAT-IRES-Cre sono stati utilizzati per colpire le cellule colinergiche di amacrina Starburst. - Ottenere il virus AAV ricombinasi-dipendente che codifica per le proteine fluorescenti. Per un'etichettatura ottimale dei processi fini, selezionare i vettori che esprimono XFP modificati mirati alla membrana plasmatica.
NOTA: Questo studio ha utilizzato il virus Brainbow (BBV), AAV-EF1a-BbTagBY e AAV9-EF1a-BbChT (vedi Tabella dei materiali), che forniscono l'opzione per l'etichettatura multicolore (Figura 1B). L'espressione della proteina fluorescente gialla potenziata farnesilata (eYFP) e della ciliegia monomerica (mCherry) ha prodotto i segnali fluorescenti più forti per l'imaging dal vivo. Un singolo BBV può essere utilizzato per visualizzare singoli pergole, mentre la co-iniezione di BBV può essere utilizzata per etichettare più cellule o pergole vicine con fluorofori distinti. Se si utilizzano più proteine fluorescenti, assicurarsi una sovrapposizione minima dello spettro di emissione (Figura 1C). I parametri di acquisizione del microscopio devono essere regolati per catturare segnali XFP distinti. - Preparare aliquote ∼3-5 μL di AAV in singoli tubi a bassa legatura per scorte monouso per evitare cicli di congelamento/scongelamento. Conservare a -80 °C.
- Preparare micropipette di vetro borosilicato con punte molto fini utilizzando un estrattore di micropipette.
NOTA: le impostazioni dell'estrattore variano a seconda del filamento e dell'estrattore utilizzato; vedere la Figura 2A per le dimensioni e la forma della punta finale. I diametri tipici delle goccioline sono 600 μm. - Ottenere un sistema di microiniezione e tubi associati. Collegare il microiniettore a una porta d'aria pressurizzata.

Figura 1: Panoramica sperimentale. (A) Questo protocollo copre 2 giorni di esperimenti con un periodo di infezione minimo di 4-5 giorni tra i giorni sperimentali. Le iniezioni intraoculari vengono eseguite su topi neonatali non più vecchi del giorno 3 postnatale. Le retine vengono quindi sezionate, montate in piano e fotografate dal vivo per catturare la finestra di sviluppo desiderata. Le cellule marcate possono essere fotografate in qualsiasi momento dopo i 4-5 giorni necessari per l'espressione virale, poiché non vi sono effetti apparenti dell'espressione prolungata di AAV sulla morfologia dei dendriti. (B) I vettori AAV Cre-dipendenti brainbow (BBV) vengono iniettati in animali che esprimono Cre31. In questo studio, i topi knock-in ChAT-Cre sono stati utilizzati per guidare la ricombinazione cre nelle cellule amacrine starburst. I due BBV codificano eYFP o tagBFP modificati, o mCherry e mTFP, che terminano in un motivo CAAX che viene farnesilato sequenzialmente per la localizzazione della membrana. I siti Lox sono raffigurati con triangoli. I vettori esprimono XFP farnesilati in modo stocastico e combinatorio dipendente dalla ricombinazione di Cre. Il promotore EF1α include elementi regolatori del gene del fattore di allungamento 1α. W rappresenta gli elementi dell'elemento regolatore post-trascrizionale del virus dell'epatite woodchuck e pA indica la sequenza di poliadenilazione. (C) Gli spettri di eccitazione ed emissione di mCherry ed eYFP, i fluorofori BBV ripresi in questo studio. Quando si esegue l'imaging dal vivo di più proteine fluorescenti, i parametri di rilevamento devono essere disposti in modo da separare adeguatamente gli spettri di emissione in canali distinti. Abbreviazioni: AAV = virus adeno-associato; BBV = Brainbow AAV; ChAT = colina acetiltransferasi; iRES = sito di ingresso interno del ribosoma; eYFP = proteina fluorescente gialla potenziata; iTR = ripetizione terminale invertita; tagBP = Tag-blue proteina fluorescente; mCherry = ciliegia monomerica; mTFP = proteina fluorescente verde acqua; XFP = qualsiasi proteina fluorescente; EF1α= fattore di allungamento 1 alfa. Fare clic qui per visualizzare una versione più grande di questa figura.
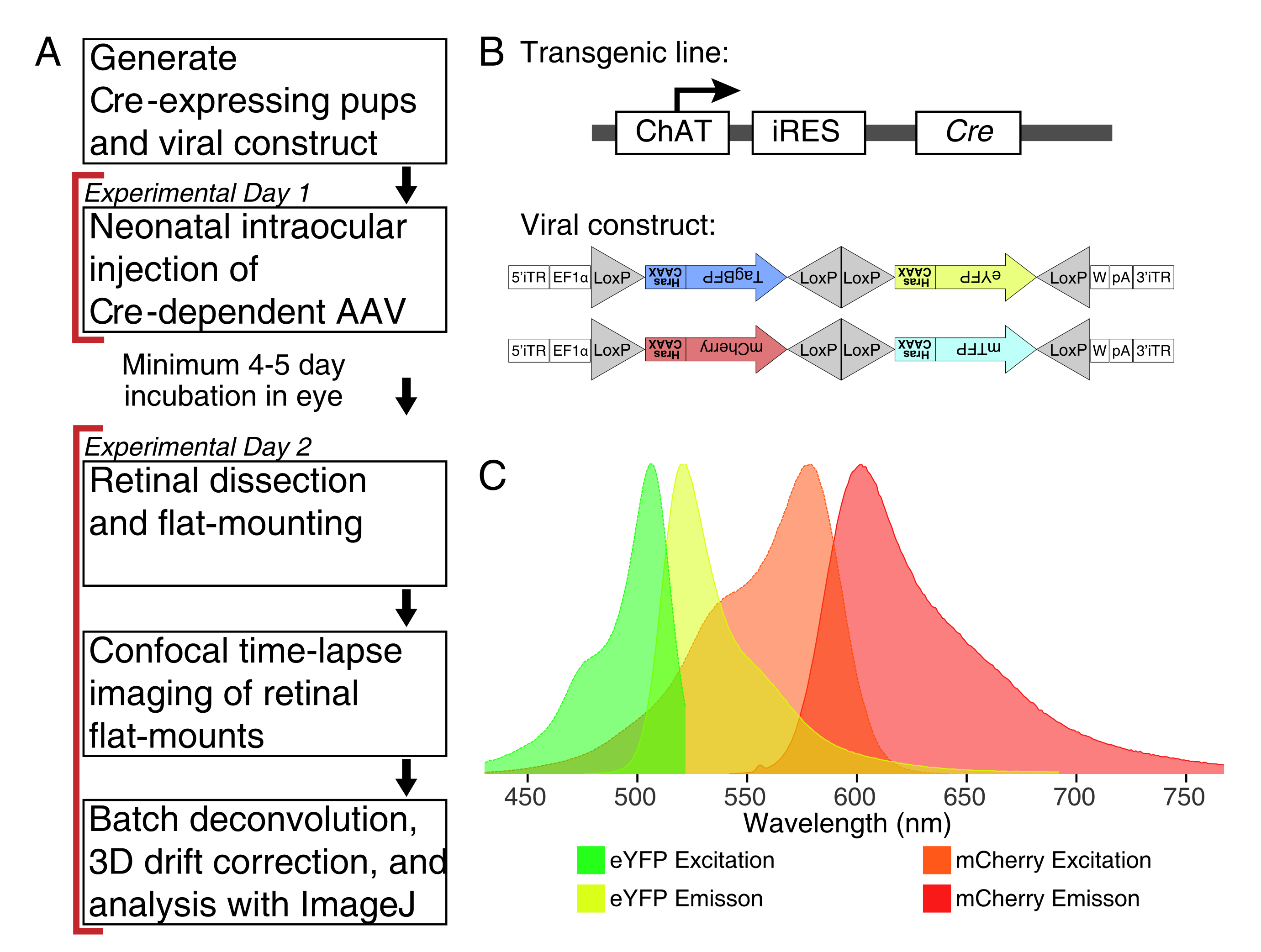{kind=link}
2. Iniezioni intravitreali di AAV in topi neonatali
- Scongelare un'aliquota AAV sul ghiaccio. Preparare una diluizione AAV ~1:4 utilizzando soluzione salina sterile o soluzione salina tamponata con fosfato. Preparare ~ 0,5 μL di diluizione AAV per animale (~ 0,25 μL per occhio) nel caso in cui la micropipetta si rompa e una nuova pipetta debba essere riempita. Conservare il restante AAV non diluito a 4 °C e utilizzare entro 2 settimane.
NOTA: In questo esperimento, la concentrazione di AAV del fornitore era ~ 1-2 × contenuto del genoma 1013 (GC) / mL e diluita a una concentrazione finale di 4 × 1012 GC / mL. Ottimizzare la concentrazione virale per ottenere la densità di etichettatura desiderata. - Per visualizzare le iniezioni, aggiungere circa 1 μL di soluzione colorante Fast Green FCF allo 0,02% per ogni 15 μL di diluizione AAV per colorare la soluzione di blu.
- Trasferire una gabbia per topi con diga e lettiera neonatale (giorno postnatale (P) 0,5-3) in una sala operatoria con apparecchiature di microiniezione. Tieni gli animali nella stessa gabbia con nidi e lettiere per ridurre al minimo lo stress materno e il rifiuto dei cuccioli.
- Sterilizzare l'area di iniezione con il 70% di etanolo e posizionare i cuscinetti per pannolini sterili sulle superfici del banco. Preparare una piattaforma calda (ad esempio, una piastra riscaldante) per il recupero dall'anestesia indotta da ipotermia.
- Riempire la micropipetta con la diluizione AAV utilizzando una microsiringa. Sotto uno stereomicroscopio, rompere la punta della micropipetta con un ago da 30 G per aprire la punta.
NOTA: la Figura 2A mostra la punta riempita, sia sigillata (in alto) che non sigillata (al centro). - Anestetizzare i topi neonatali per ipotermia mettendo 1-2 animali sul ghiaccio. Metti gli animali su un guanto di lattice per proteggere la pelle dal contatto diretto con il ghiaccio. Una volta che l'animale non si muove più in risposta al pizzico della zampa (~ 2 min), posiziona l'animale sotto uno stereomicroscopio. Se lo si desidera, tatuare i cuscinetti delle zampe con inchiostro per tatuaggi e ago da 30 G e raccogliere ritagli di coda per l'isolamento del DNA per identificare gli animali mediante genotipizzazione.
NOTA: durante tutta la procedura deve essere effettuato un adeguato monitoraggio della profondità dell'anestesia. - Tamponare la pelle sovrastante gli occhi con il 70% di etanolo. Utilizzare un ago da 30 G per aprire la palpebra fusa (Figura 2B). Applicare una leggera pressione con le dita per aprire l'occhio e praticare un piccolo foro attraverso la cornea alla giunzione cornea-sclera (Figura 2C).
- Inserire la micropipetta di vetro nel foro e premere il pedale del microiniettore 2-4 volte per iniettare l'AAV nello spazio intravitreale. Rimuovere lentamente la micropipetta e confermare l'iniezione di AAV visualizzando il colorante blu attraverso la pupilla (Figura 2D).
NOTA: con una pressione di espulsione di 6-8 psi e un tempo di impulso di 600-800 ms, viene espulsa una goccia di diametro 600 μm (Figura 2A, in basso). Circa 0,23-0,45 μL di soluzione AAV vengono iniettati per occhio. La soluzione blu all'esterno dell'occhio indica che l'AAV non è stato iniettato nell'occhio. La soluzione blu che fuoriesce dal sito di iniezione indica che l'AAV potrebbe essere fuoriuscito, riducendo l'efficienza di trasfezione. - Premere delicatamente le palpebre insieme per sigillare nuovamente e posizionare il cucciolo su un cuscinetto riscaldato. Una volta che gli animali recuperano un colore rosato e sono reattivi, trasferiscili delicatamente nella gabbia di stabulazione.
NOTA: Assicurarsi che vengano prese le precauzioni appropriate per evitare un riscaldamento troppo rapido dei cuccioli. - Ripetere la procedura di iniezione per gli animali rimanenti nella lettiera. Lasciare un minimo di 4-5 giorni per la trasduzione virale prima di visualizzare la retina.

Figura 2: Iniezioni intraoculari neonatali. (A) La micropipetta riempita mostra la forma della punta della pipetta quando è sigillata (in alto) e dopo che la punta è stata aperta (in basso, a sinistra). La pressione di pico-iniezione di 6-8 psi e il tempo di impulso di 600-800 ms producono una goccia di 600 μm di diametro (in basso, a destra). Barra della scala = 1 mm.(B) Cucciolo P0 anestetizzato con ingrandimento 16x. La giunzione palpebrale fusa (bianca) viene aperta a fessura utilizzando un ago affilato da 30 G. Scala bar = 2 mm. (C) La leggera pressione applicata all'occhio espone la cornea; barra della scala = 2 mm. La sezione ingrandita (rossa) mostra il piccolo foro alla giunzione cornea-sclera (giallo) creato con un ago da 30 G; barra della scala = 0,5 mm. (D) Ritiro della punta della pipetta dopo l'iniezione (a sinistra). Il colorante Fast Green nella soluzione AAV appare come blu-grigio attraverso la pupilla (al centro), rispetto al colore della pupilla chiara prima dell'iniezione (a destra). Barra della scala = 1 mm. (E) Dopo 4 giorni, la palpebra è guarita e si è fusa chiusa (a sinistra); barra della scala = 2 mm. Al momento dell'enucleazione, il sito di iniezione guarito è visibile (giallo); barra della scala = 1 mm. Si noti la posizione del sito di iniezione al confine tra la cornea e la sclera. Fare clic qui per visualizzare una versione più grande di questa figura.
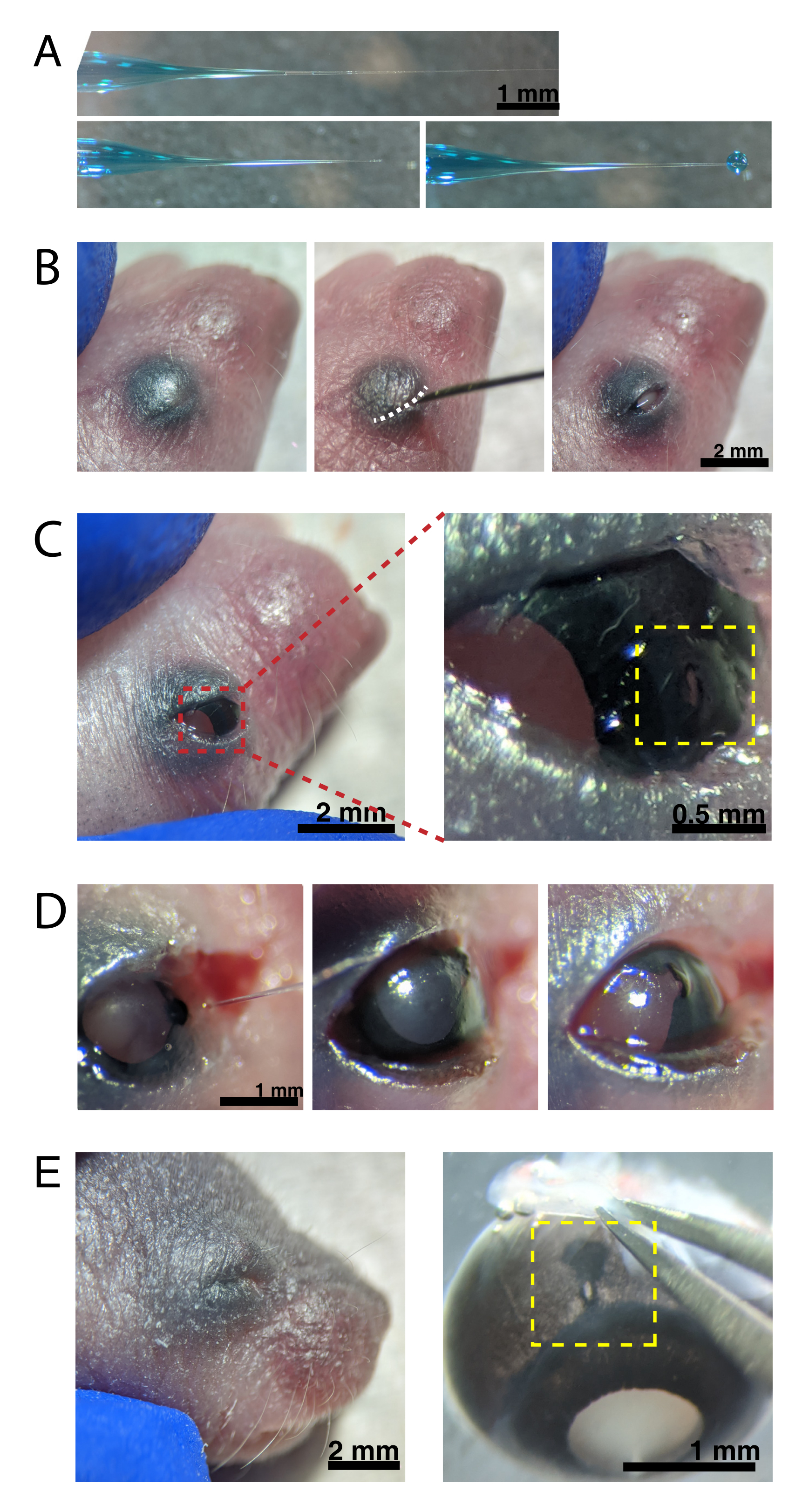{kind=link}
3. Dissezioni retiniche per esperimenti di imaging
- Preparare l'aCSF retinico (119 mM NaCl, 2,5 mM KCl, 1,3 mM MgCl2·6H2O, 2,5 mM CaCl2·2H2O, 1 mM NaH2PO4, 11 mM glucosio e 20 mM 4-(2-idrossietil)-1-piperazineetanosolfonico (acido libero HEPES)35. Se lo si desidera, preparare e congelare la soluzione 10x; scongelare e diluire a 1x secondo necessità.
- Ossigenare l'aCSF retinico gorgogliando con carbogeno (95% O2, 5% CO2) per un minimo di 15 min. Regolare il pH a 7,4. Conservare in un contenitore sigillato fino all'uso per assicurarsi che l'aCSF rimanga ossigenato.
NOTA: le retine richiedono un'alta concentrazione di O2. È importante mantenere l'aCSF ossigenato durante l'esperimento. - Incorporare una capsula di Petri di 60 mm di diametro in un vassoio del ghiaccio (creare un vassoio del ghiaccio in plastica da laboratorio, ad esempio un coperchio della scatola della pipetta) e posizionarlo sotto uno stereomicroscopio. Riempire la capsula di Petri con aCSF retinico ossigenato.
- Eutanasia topi P9 e più giovani per decapitazione. Eutanasia topi P10 e più anziani per induzione di isoflurano, o un metodo alternativo approvato secondo il protocollo animale, seguito da decapitazione.
- Se le palpebre sono sigillate, tagliare il lembo palpebrale per esporre l'occhio. Utilizzare una pinza per enucleare gli occhi e trasferirli nell'aCSF retinico freddo dal passaggio 3.3.
- Per sezionare la coppa retinica, sotto uno stereomicroscopio (ingrandimento 25x), stabilizzare l'occhio stringendo il nervo ottico usando un forcepe Dumont #5 (Figura 3A).
- Fai un buco al centro della cornea con un ago da 30 G, quindi inserisci una punta dei microscissori nel foro per fare un'incisione dal foro all'estremità della cornea. Ripetere l'operazione per fare 4 fette nelle direzioni cardinali, creando 4 "lembi" (Figura 3A).
- Con due pinze Dumont #5, afferrare e tirare i due lembi adiacenti, staccando delicatamente la sclera dalla retina. Ripetere con i lembi corneali rimanenti e rimuovere la sclera dalla retina (Figura 3B).
- Rimuovere la lente dalla coppa retinica usando la pinza. Con i microscissori, fare 4 incisioni radiali dal bordo della retina verso il nervo ottico, creando 4 petali uguali (Figura 3B). Ripetere i passaggi 3.4-3.7 per il secondo occhio.
4. Preparazione retinica a montaggio piatto
- Preparare dischi filtranti a membrana a membrana MCE (Grey Mixed Cellulose Estere) per il montaggio. Se si utilizzano filtri a membrana MCE di grande diametro, tagliare il disco in quadranti (circa 1 cm di diametro). Posizionare il disco MCE al centro di una carta da filtro bianca più grande (Figura 3D).
NOTA: i filtri MCE sono disponibili anche in dischi da 1 cm di diametro. Il disco filtrante MCE deve essere abbastanza grande da contenere 1-2 retine, ma abbastanza piccolo da adattarsi alla camera di imaging. Poiché le membrane MCE mantengono una carica statica, ridurre al minimo il contatto e la gestione della membrana MCE. - Maneggiando le retine usando due pennelli 3/0 di dimensioni, capovolgere una tazza retinica su un pennello con la cellula gangliare retinica rivolta verso il basso. Sollevare delicatamente la retina dall'aCSF, assicurandosi che la tensione dell'acqua non strappi la retina (Figura 3C).
- Mentre si tiene ancora il pennello con la retina, utilizzare una pipetta di trasferimento per posizionare una goccia di aCSF al centro della carta da filtro MCE (Figura 3C, a destra). Far galleggiare la retina nella goccia di aCSF creata dalla tensione superficiale. Utilizzare pennelli per posizionare il lato RGC della retina verso l'alto all'interno della goccia e per aprire i quattro petali.
- Una volta posizionato, creare un ponte d'acqua tra il pennello e la carta da filtro bianca per rompere la tensione superficiale della goccia.
NOTA: quando aCSF è lontano, la retina aderirà alla carta da filtro MCE (Figura 3D,E). Le retine piatte possono essere maneggiate afferrando il disco MCE con una pinza. Se la goccia aCSF si allontana rapidamente prima di formare un ponte d'acqua, ciò potrebbe indicare che la membrana MCE non è carica. Utilizzare una nuova membrana MCE e ridurre al minimo il tempo tra l'enucleazione e l'imaging sezionando e montando le retine immediatamente prima di spostare le retine nella camera di imaging.

Figura 3: Dissezione retinica e montaggio piatto su membrane filtranti miste estere di cellulosa. (A) A sinistra, un occhio enucleato viene stabilizzato afferrando il nervo ottico con una pinza Dumont #5 (a sinistra). Un piccolo foro viene creato al centro della cornea usando un ago da 30 G (al centro). Le forbici da micro-dissezione vengono utilizzate per tagliare la cornea in 4 lembi uguali (a destra). Barra della scala = 1 mm. (B) Retina sezionata con la sclera staccata usando due pinze Dumont #5 (a sinistra) e con la lente rimossa (al centro). Retina con 4 tagli uguali a metà della retina (a destra). Barra della scala = 1 mm. (C) Manipolazione della retina con due pennelli fini (dimensione 3/0, a sinistra). Retina capovolta con cellule gangliari retiniche rivolte verso il basso su un pennello (al centro) e sollevata da aCSF, assicurandosi che la tensione dell'acqua non strappi la retina (a destra). Barra della scala = 2 mm. (D) Disco a membrana MCE grigia posto su una carta da filtro bianca (a sinistra). Una goccia di aCSF sul disco MCE (a destra). Barra della scala = 1 cm. (E) Dopo aver fatto fluttuare le retine nella goccia e averle posizionate, creare un ponte d'acqua con la carta da filtro bianca per aspirare l'aCSF, tirando la retina sulla carta MCE carica (a sinistra); barra della scala = 1 cm. L'immagine ingrandita della membrana MCE (rossa) mostra 2 retine montate con cellule gangliari retiniche rivolte verso l'alto (a destra); barra della scala = 2 mm. Una retina è delineata in bianco. Abbreviazioni: MCE = estere misto di cellulosa; aCSF = liquido cerebrospinale artificiale. Fare clic qui per visualizzare una versione più grande di questa figura.
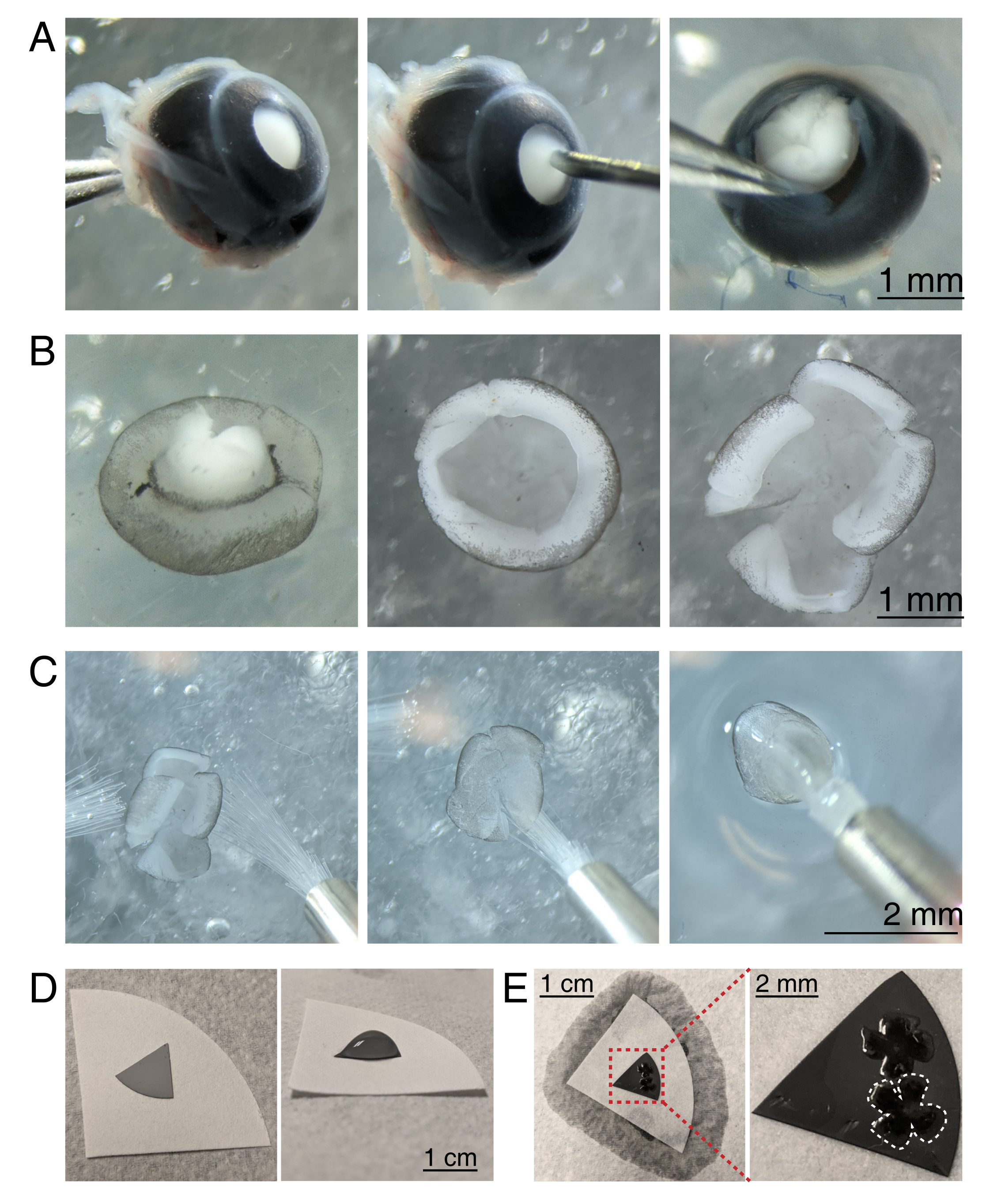{kind=link}
5. Imaging confocale time-lapse di preparati di retina a montaggio intero dal vivo
- Assemblare la camera di incubazione live-imaging per un microscopio confocale verticale come mostrato nella Figura 4.
NOTA: per i sistemi confocali invertiti, i supporti piatti sono posizionati RGC lateralmente verso il basso direttamente sul coperchio del fondo di vetro della camera di incubazione. Una volta che le retine entrano in contatto con il coverslip, non possono essere spostate. - Riempire la camera con aCSF ossigenato e accendere la pompa e il regolatore di temperatura (temperatura 32-34 °C, portata 1 mL/min). Non lasciare che la temperatura superi i 34 °C.
- Per trasferire il supporto piatto retinico nella camera di perfusione, arrestare la pompa e rimuovere l'aCSF che si trova nella camera. Posizionare il disco MCE con il supporto piatto retinico nella camera di incubazione (vuota).
- Posizionare un peso del campione sul supporto piatto; pre-bagnare il peso per rompere la tensione superficiale. Riempire la camera con l'aCSF riscaldato e far circolare aCSF a ~1 mL/min.
- Posizionare il nasello con l'obiettivo di immersione in acqua 25x (apertura numerica 0,95) nella camera di imaging. Screening per le cellule di interesse marcate utilizzando luce epifluorescente (Figura 4C).
- Regolare il volume di imaging per acquisire le caratteristiche dendritiche di interesse.
NOTA: questo studio ha catturato il pergolato dendritico completo a 1024 x 1024 pixel per fotogramma, z-step 1 μm e un frame rate di 2 min tra ogni z-stack. Le dimensioni finali dell'immagine sono ~ 100 μm x 100 μm x 20 μm. - Per regolare la potenza del laser su un'impostazione ottimale, utilizzare una tabella di ricerca che identifichi sia i pixel sovrasaturi che quelli sottosaturi. Durante la scansione, regolare la potenza del laser in modo tale che nessun pixel sia sovrasaturato (cioè ad un'intensità di 255 o superiore). Continuare l'imaging per tutto il tempo necessario o fino a quando non si verifica un calo significativo e rilevabile del segnale fluorescente e un aumento del rumore (in genere 2-4 ore).
NOTA: si consiglia una potenza laser ridotta in quanto gli algoritmi di deconvoluzione funzionano in modo ottimale quando i pixel sono distribuiti sull'intera gamma dinamica. L'intensità dei pixel non deve superare 254; le analisi empiriche dei neuriti hanno rivelato che valori di pixel inferiori a 170 sono ideali per la deconvoluzione. Velocità di scansione elevate (400-600 Hz) con media di linea (2-3) sono preferibili a scansioni singole e più lente dello stesso tempo di permanenza totale dei pixel. L'area di imaging prolungato spesso fotosbianche, ma altre sezioni di espianto rimangono vitali. È possibile visualizzare più regioni in un montaggio piatto, ciascuna per 2-4 ore, con un tempo di incubazione totale di 6 ore. Le sessioni di imaging oltre le 6 ore non sono state testate sistematicamente. La degradazione dei neuriti e il blebbing sono segni che la vitalità dell'espianto è in declino. - Dopo l'imaging, fissare i supporti piatti retinici e il filtro a membrana con paraformaldeide fredda al 4% per 1-2 ore a 4 °C. Amplificare le etichette fluorescenti nelle retine fisse mediante immunoistochimica per ulteriori analisi.
NOTA: la fissazione post-imaging non è possibile quando si utilizza una confocale invertita; le retine non sono rimovibili senza distruggere il tessuto.

Figura 4: Configurazione della camera di incubazione per l'imaging a cellule vive. (A) Apparecchiature di incubazione per immagini dal vivo che mostrano componenti di riscaldamento, soluzione e imaging. Il doppio riscaldatore include una piattaforma a camera di incubazione riscaldata (cerchio posteriore con elettrodi connettore blu) e un riscaldatore di soluzione in linea. (B) Schema schematico di 4A. Il montaggio piatto retinico (rosso) sulla membrana MCE (grigio) viene posizionato nella camera di incubazione. Il riscaldatore di soluzione in linea è collegato a una pompa di soluzione (non raffigurata in 4A). Le dimensioni della camera di imaging consentono una buona area di lavoro per il naso dell'obiettivo di immersione. (C) 25x vista un espianto retinico iniettato con 0,23-0,45 μL di 4 × 1012 GC/mL di diluizione AAV; barra della scala = 100 μm. L'etichettatura densa delle cellule spesso circonda la testa del nervo ottico (linea tratteggiata, in basso a destra); barra della scala = 50 μm. Abbreviazioni: MCE = estere misto di cellulosa; GC = copie del genoma; mCherry = ciliegia monomerica; eYFP = proteina fluorescente gialla potenziata. Fare clic qui per visualizzare una versione più grande di questa figura.
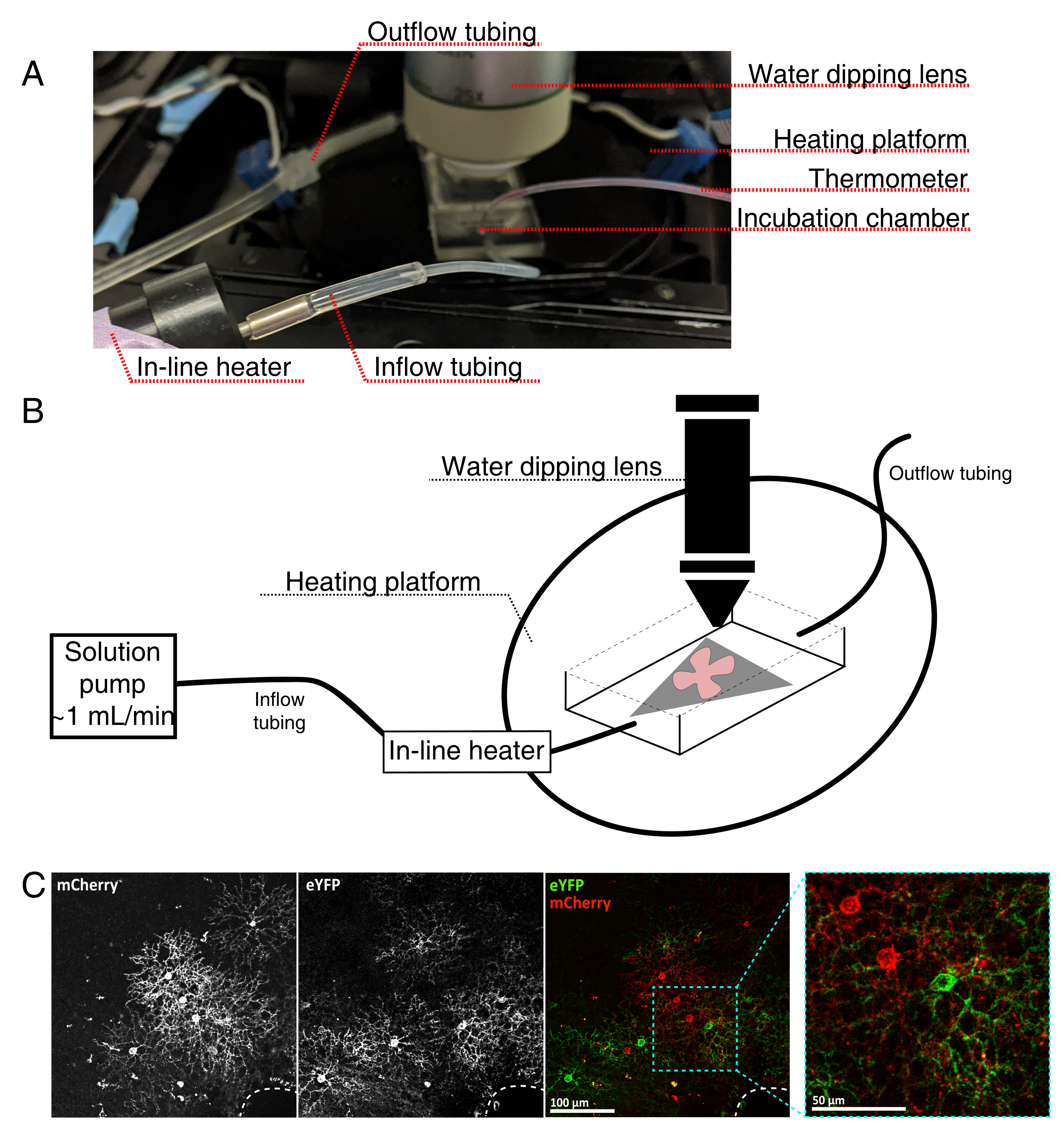{kind=link}
6. Deconvoluzione e post-elaborazione delle immagini in ImageJ
- Importare la serie di immagini, dividere la serie per tempo e colore se si tratta di un'immagine multicolore. Assicurarsi che tutti i punti temporali per un colore siano contenuti nella stessa cartella.
NOTA: Importare i formati bio se si utilizza ImageJ (i formati bio sono automaticamente inclusi in FIJI). - Creare una funzione teorica di diffusione puntuale (PSF) utilizzando il plugin ImageJ, Diffraction PSF 3D.
NOTA: ogni canale di imaging richiede la propria PSF poiché la lunghezza d'onda di emissione della proteina fluorescente influisce sulla PSF. - Usa i plugin | macro | Eseguire per eseguire la deconvoluzione iterativa parallela batch per tutti i punti temporali utilizzando la macro fornita (file supplementare 1).
Nota : questa macro esegue 25 iterazioni dell'algoritmo iterativo WpL (Node Filter Preconditioned Landweber) del filtro di Wiener. Ogni canale di colore deve essere deconvolto separatamente. - Unisci i canali di colore e compila tutti i punti temporali in un Hyperstack (Image | Hyperstacks | Stacks to Hyperstack). Correggi la deriva 3D usando Plugins | | di registrazione Deriva 3D corretta.
- Riportare l'immagine a una pila normale (Image | Hyperstacks | Hyperstack to Stack) e dividere i punti temporali (Immagine | Stack | Strumenti | Stack Splitter). Utilizzare l'elaborazione batch per creare la proiezione massima per tutti i punti temporali (Process | | batch macro | run("Z Project...", "projection=[Max Intensity]"); ).
- Importare la sequenza di immagini time-lapse (File | Importare | Sequenza di immagini). Utilizzare gli strumenti ImageJ convenzionali per l'analisi desiderata di video bidimensionali (2D) deconvoluti e post-elaborati.
NOTA: i video deconvolti e corretti alla deriva quadridimensionali (3D + tempo) possono essere visualizzati come un hyperstack. Ometti il passaggio precedente per mantenere i punti temporali 3D.
Risultati
Utilizzando il protocollo di cui sopra, è stato acquisito, deconvoluto e corretto un video 3D ad alta risoluzione dello sviluppo di dendriti di cellule starburst. Sono state prodotte proiezioni massime del piano Z per realizzare video 2D per l'analisi (Video supplementare 1, Figura 5A). La deconvoluzione 3D di ogni punto temporale ha aumentato la risoluzione delle proiezioni di filopodi fini (Figura 5B, C). Le protrusioni sotti...
Discussione
Questo video dimostra una pipeline sperimentale che utilizza gli strumenti genetici esistenti per visualizzare le dinamiche dendrite dello sviluppo di neuroni retinici con imaging vivo confocale. Sono state dimostrate anche iniezioni intraoculari di AAV Cre-dipendenti che codificano proteine fluorescenti mirate alla membrana nei topi neonatali. Le singole cellule di popolazioni geneticamente mirate sono marcate brillantemente già a 4-5 dpi. I supporti piatti retinici sono stati preparati per le camere di imaging standar...
Divulgazioni
Gli autori non hanno nulla da rivelare.
Riconoscimenti
Ringraziamo Madison Gray per avermi dato una mano quando non ne avevo. Questa ricerca è stata supportata da un NSERC Discovery Grant (RGPIN-2016-06128), una Sloan Fellowship in Neuroscience e una Canada Research Chair Tier 2 (a J.L.L). S. Ing-Esteves è stato supportato dal Vision Science Research Program e dal NSERC Postgraduate Scholarships-Doctorals.
Materiali
| Name | Company | Catalog Number | Comments |
| Addgene viral prep #45185-AAV9 | |||
| Addgene viral prep #45186-AAV9 | |||
| Dissection tools | |||
| Cellulose filter paper | Whatman | 1001-070 | |
| Dumont #5 fine forceps | FST | 11252-20 | Two Dumont #5 forceps are required for retinal micro-dissection |
| Dumont forceps | VWR | 82027-426 | |
| Fine Scissors | FST | 14058-09 | |
| Mixed cellulose ester membrane (MCE) filter papers, hydrophilic, 0.45 µm pore size | Millipore | HABG01 300 | |
| Petri Dish, 50 × 15 mm | VWR | 470313-352 | |
| Polyethylene disposable transfer pipette | VWR | 470225-034 | |
| Round tip paint brush, size 3/0 | Conventional art supply store | Two size 3/0 paint brushes (or smaller) are required for retinal flat-mounting | |
| Surgical Scissors | FST | 14007-14 | |
| Vannas Spring Scissors - 2.5 mm Cutting Edge | FST | 15000-08 | |
| Live-imaging incubation system | |||
| Chamber polyethylene tubing, PE-160 10' | Warner Instruments | 64-0755 | |
| Dual channel heater controller, Model TC-344C | Warner Instruments | 64-2401 | |
| HC FLUOTAR L 25x/0.95 W VISIR dipping objective | Leica | 15506374 | |
| Heater controller cable | Warner Instruments | CC-28 | |
| Large bath incubation chamber with slice support | Warner Instruments | RC-27L | |
| MPII Mini-Peristaltic Pump | Harvard Apparatus | 70-2027 | |
| PM-6D Magnetic Heated Platform (incubation chamber heater) | Warner Instruments | PM-6D | |
| Pump Head Tubing Pieces For MPII Mini-Peristaltic Pump | Harvard Apparatus | 55-4148 | |
| Sample anchor (Harps) | Warner Instruments | 64-0260 | Sample anchor must be compatible with incubation chamber |
| Sloflo In-line Solution Heater | Warner Instruments | SF-28 | |
| Neonatal Injections | |||
| 10 µL Microliter Syringe Series 700, Removable Needle | Hamilton Company | 80314 | |
| 30 G Hypodermic Needles (0.5 inch) | BD PrecisionGlide | 305106 | |
| 4 inch thinwall glass capillary, no filament (1.0 mm outer diameter/0.75 mm) | WPI World Precision Instruments | TW100-4 | |
| Ethanol 99.8% (to dilute to 70% with double-distilled water [ddH2O]) | Sigma-Aldrich | V001229 | |
| AAV9.hEF1a.lox.TagBFP. lox.eYFP.lox.WPRE.hGH-InvBYF | Penn Vector Core | AV-9-PV2453 | Addgene Plasmid #45185 |
| AAV9.hEF1a.lox.mCherry.lox.mTFP 1.lox.WPRE.hGH-InvCheTF | Penn Vector Core | AV-9-PV2454 | Addgene Plasmid #45186 |
| ChAT-IRES-Cre knock-in transgenic mouse line | The Jackson Laboratory | 6410 | |
| Fast Green FCF Dye content ≥85 % | Sigma-Aldrich | F7252-25G | |
| Flaming/Brown Micropipette Puller, model P-97 | Sutter Instrument Co. | P-97 | |
| Green tattoo paste | Ketchum MFG Co | 329A | |
| Phosphate-Buffered Saline, pH 7.4, liquid, sterile-filtered, suitable for cell culture | Sigma-Aldrich | 806552 | |
| Pneumatic PicoPump | WPI World Precision Instruments | PV-820 | |
| Oxygenated artifiial cerebrospinal fluid (aCSF) Reagents | |||
| Calcium chloride dihydrate (CaCl2·2H2O) | Sigma-Aldrich | C7902 | |
| Carbogen (5% CO2, 95% O2) | AirGas | X02OX95C2003102 | Supplier may vary depending on region |
| D-(+)-Glucose | Sigma-Aldrich | G7021 | |
| HEPES, Free Acid | Bio Basic | HB0264 | |
| Hydrochloric acid solution, 1 N | Sigma-Aldrich | H9892 | |
| Magnesium chloride hexahydrate (MgCl2·6H2O) | Sigma-Aldrich | M2670 | |
| pH-Test strips (6.0-7.7) | VWR | BDH35317.604 | |
| Potassium chloride (KCl) | Sigma-Aldrich | P9541 | |
| Sodium chloride (NaCl) | Bio Basic | DB0483 | |
| Sodium phosphate monobasic (NaH2PO4) | Sigma-Aldrich | RDD007 | |
| Software | |||
| ImageJ | National Institutes of Health (NIH) | Open source |
Riferimenti
- Lefebvre, J. L., Sanes, J. R., Kay, J. N. Development of dendritic form and function. Annual Review of Cell and Developmental Biology. 31, 741-777 (2015).
- Graham, H. K., Duan, X. Molecular mechanisms regulating synaptic specificity and retinal circuit formation. Wiley Interdisciplinary Reviews Developmental biology. 10 (1), 379 (2021).
- Godinho, L., et al. Targeting of amacrine cell neurites to appropriate synaptic laminae in the developing zebrafish retina. Development. 132 (22), 5069-5079 (2005).
- Mumm, J. S., et al. In vivo imaging reveals dendritic targeting of laminated afferents by zebrafish retinal ganglion cells. Neuron. 52 (4), 609-621 (2006).
- Wong, W. T., Faulkner-Jones, B. E., Sanes, J. R., Wong, R. O. Rapid dendritic remodeling in the developing retina: dependence on neurotransmission and reciprocal regulation by Rac and Rho. The Journal of Neuroscience. 20 (13), 5024-5036 (2000).
- Wei, W., Elstrott, J., Feller, M. B. Two-photon targeted recording of GFP-expressing neurons for light responses and live-cell imaging in the mouse retina. Nature Protocols. 5 (7), 1347-1352 (2010).
- Morgan, J. L., Wong, R. O. L. Ballistic labeling with fluorescent dyes and indicators. Current Protocols in Neuroscience. 43 (1), 1-10 (2008).
- Nickerson, P. E. B., et al. Live imaging and analysis of postnatal mouse retinal development. BMC Developmental Biology. 13, 24 (2013).
- Morgan, J. L., Dhingra, A., Vardi, N., Wong, R. O. L. Axons and dendrites originate from neuroepithelial-like processes of retinal bipolar cells. Nature Neuroscience. 9 (1), 85-92 (2006).
- Coombs, J. L., Van Der List, D., Chalupa, L. M. Morphological properties of mouse retinal ganglion cells during postnatal development. The Journal of Comparative Neurology. 503 (6), 803-814 (2007).
- Cranfill, P. J., et al. Quantitative assessment of fluorescent proteins. Nature Methods. 13 (7), 557-562 (2016).
- Stacy, R. C., Wong, R. O. L. Developmental relationship between cholinergic amacrine cell processes and ganglion cell dendrites of the mouse retina. The Journal of Comparative Neurology. 456 (2), 154-166 (2003).
- Kay, J. N., et al. Retinal ganglion cells with distinct directional preferences differ in molecular identity, structure, and central projections. The Journal of Neuroscience. 31 (21), 7753-7762 (2011).
- Liu, J., Sanes, J. R. Cellular and molecular analysis of dendritic morphogenesis in a retinal cell type that senses color contrast and ventral motion. The Journal of Neuroscience. 37 (50), 12247-12262 (2017).
- Diao, L., Sun, W., Deng, Q., He, S. Development of the mouse retina: emerging morphological diversity of the ganglion cells. Journal of Neurobiology. 61 (2), 236-249 (2004).
- Coombs, J., vander List, D., Wang, G. Y., Chalupa, L. M. Morphological properties of mouse retinal ganglion cells. Neuroscience. 140 (1), 123-136 (2006).
- Sanes, J. R., Masland, R. H. The types of retinal ganglion cells: current status and implications for neuronal classification. Annual Review of Neuroscience. 38, 221-246 (2015).
- Sümbül, U., et al. A genetic and computational approach to structurally classify neuronal types. Nature Communications. 5, 3512 (2014).
- Lin, B., Masland, R. H. Populations of wide-field amacrine cells in the mouse retina. The Journal of Comparative Neurology. 499 (5), 797-809 (2006).
- Macneil, M. A., Heussy, J. K., Dacheux, R. F., Raviola, E., Masland, R. H. The shapes and numbers of amacrine cells: Matching of photofilled with Golgi-stained cells in the rabbit retina and comparison with other mammalian species. Journal of Comparative Neurology. 413 (2), 305-326 (1999).
- Ivanova, E., Hwang, G. S., Pan, Z. H. Characterization of transgenic mouse lines expressing Cre recombinase in the retina. Neuroscience. 165 (1), 233-243 (2010).
- Jo, A., Xu, J., Deniz, S., Cherian, S., DeVries, S. H., Zhu, Y. Intersectional strategies for targeting amacrine and ganglion cell types in the mouse retina. Frontiers in Neural Circuits. 12, 66 (2018).
- Siegert, S., et al. Genetic address book for retinal cell types. Nature Neuroscience. 12 (9), 1197-1204 (2009).
- Kim, I. -. J., Zhang, Y., Meister, M., Sanes, J. R. Laminar restriction of retinal ganglion cell dendrites and axons: subtype-specific developmental patterns revealed with transgenic markers. The Journal of Neuroscience. 30 (4), 1452-1462 (2010).
- Peng, Y. -. R., Tran, N. M., Krishnaswamy, A., Kostadinov, D., Martersteck, E. M., Sanes, J. R. Satb1 regulates contactin 5 to pattern dendrites of a mammalian retinal ganglion cell. Neuron. 95 (4), 869-883 (2017).
- Duan, X., Krishnaswamy, A., Dela Huerta, I., Sanes, J. R. Type II cadherins guide assembly of a direction-selective retinal circuit. Cell. 158 (4), 793-807 (2014).
- Ray, T. A., et al. Formation of retinal direction-selective circuitry initiated by starburst amacrine cell homotypic contact. eLife. 7, 34241 (2018).
- Krishnaswamy, A., Yamagata, M., Duan, X., Hong, Y. K., Sanes, J. R. Sidekick 2 directs formation of a retinal circuit that detects differential motion. Nature. 524 (7566), 466-470 (2015).
- Caval-Holme, F., Zhang, Y., Feller, M. B. Gap junction coupling shapes the encoding of light in the developing retina. Current Biology. 29 (23), 4024-4035 (2019).
- Lucas, J. A., Schmidt, T. M. Cellular properties of intrinsically photosensitive retinal ganglion cells during postnatal development. Neural Development. 14 (1), 8 (2019).
- Cai, D., Cohen, K. B., Luo, T., Lichtman, J. W., Sanes, J. R. Improved tools for the Brainbow toolbox. Nature Methods. 10 (6), 540-547 (2013).
- Rossi, J., et al. Melanocortin-4 receptors expressed by cholinergic neurons regulate energy balance and glucose homeostasis. Cell Metabolism. 13 (2), 195-204 (2011).
- Lefebvre, J. L., Kostadinov, D., Chen, W. V., Maniatis, T., Sanes, J. R. Protocadherins mediate dendritic self-avoidance in the mammalian nervous system. Nature. 488 (7412), 517-521 (2012).
- Ing-Esteves, S., et al. Combinatorial effects of alpha- and gamma-protocadherins on neuronal survival and dendritic self-avoidance. The Journal of Neuroscience. 38 (11), 2713-2729 (2018).
- Williams, P. R., Morgan, J. L., Kerschensteiner, D., Wong, R. O. L. In vitro imaging of retinal whole mounts. Cold Spring Harbor Protocols. 2013 (1), (2013).
- Ramoa, A. S., Campbell, G., Shatz, C. J. Transient morphological features of identified ganglion cells in living fetal and neonatal retina. Science. 237 (4814), 522-525 (1987).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 9, 676-682 (2012).
- Peng, H., Ruan, Z., Long, F., Simpson, J. H., Myers, E. W. V3D enables real-time 3D visualization and quantitative analysis of large-scale biological image data sets. Nature Biotechnology. 28 (4), 348-353 (2010).
- Cuntz, H., Forstner, F., Borst, A., Häusser, M. One rule to grow them all: a general theory of neuronal branching and its practical application. PLoS Computational Biology. 6 (8), 1000877 (2010).
- Xiao, H., Peng, H. APP2: automatic tracing of 3D neuron morphology based on hierarchical pruning of a gray-weighted image distance-tree. Bioinformatics. 29 (11), 1448-1454 (2013).
- Nanda, S., et al. Design and implementation of multi-signal and time-varying neural reconstructions. Scientific data. 5, 170207 (2018).
- Sherry, D. M., Wang, M. M., Bates, J., Frishman, L. J. Expression of vesicular glutamate transporter 1 in the mouse retina reveals temporal ordering in development of rod vs. cone and ON vs. OFF circuits. The Journal of Comparative Neurology. 465 (4), 480-498 (2003).
- Johnson, J., et al. Vesicular neurotransmitter transporter expression in developing postnatal rodent retina: GABA and glycine precede glutamate. The Journal of Neuroscience. 23 (2), 518-529 (2003).
- Jüttner, J., et al. Targeting neuronal and glial cell types with synthetic promoter AAVs in mice, non-human primates and humans. Nature Neuroscience. 22 (8), 1345-1356 (2019).
- Zincarelli, C., Soltys, S., Rengo, G., Rabinowitz, J. E. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Molecular Therapy. 16 (6), 1073-1080 (2008).
- Petros, T. J., Rebsam, A., Mason, C. A. In utero and ex vivo electroporation for gene expression in mouse retinal ganglion cells. Journal of Visualized Experiments: JoVE. (31), e1333 (2009).
- Lye, M. H., Jakobs, T. C., Masland, R. H., Koizumi, A. Organotypic culture of adult rabbit retina. Journal of Visualized Experiments: JoVE. (3), e190 (2007).
- Pignatelli, V., Strettoi, E. Bipolar cells of the mouse retina: a gene gun, morphological study. The Journal of Comparative Neurology. 476 (3), 254-266 (2004).
- Huckfeldt, R. M., et al. Transient neurites of retinal horizontal cells exhibit columnar tiling via homotypic interactions. Nature Neuroscience. 12 (1), 35-43 (2009).
- Prahst, C., et al. Mouse retinal cell behaviour in space and time using light sheet fluorescence microscopy. eLife. 9, 49779 (2020).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati