
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Zeitraffer-Bildgebung neuronaler Arborisation mittels spärlicher adenoassoziierter Virusmarkierung genetisch zielgerichteter Netzhautzellpopulationen
In diesem Artikel
Zusammenfassung
Hier stellen wir eine Methode zur Untersuchung der Neuritenmorphogenese in postnatalen Maus-Netzhautexplantationen mittels konfokaler Zeitraffinalmikroskopie vor. Wir beschreiben einen Ansatz zur spärlichen Markierung und Erfassung von retinalen Zelltypen und deren Feinprozessen unter Verwendung rekombinanter adenoassoziierter Virusvektoren, die membrangerichtete fluoreszierende Proteine in einer Cre-abhängigen Weise exprimieren.
Zusammenfassung
Die Entdeckung von Mechanismen, die dendritische Dornen strukturieren, erfordert Methoden zur Visualisierung, Abbildung und Analyse von Dendriten während der Entwicklung. Die Mausnetzhaut ist ein leistungsfähiges Modellsystem zur Untersuchung zelltypspezifischer Mechanismen der neuronalen Morphogenese und Konnektivität. Die Organisation und Zusammensetzung der retinalen Subtypen ist klar definiert, und genetische Werkzeuge stehen zur Verfügung, um während der Entwicklung auf bestimmte Typen zuzugreifen. Viele retinale Zelltypen beschränken ihre Dendriten und / oder Axone auch auf schmale Schichten, was die Zeitrafferbildgebung erleichtert. Maus-Retina-Explantationskulturen eignen sich gut für die Lebendzellbildgebung mittels konfokaler oder Multiphotonenmikroskopie, aber es fehlen Methoden, die für die Bildgebung der Dendritendynamik mit zeitlicher und struktureller Auflösung optimiert sind. Hier wird eine Methode vorgestellt, um die Entwicklung spezifischer Netzhautpopulationen, die durch das Cre-Lox-System gekennzeichnet sind, spärlich zu markieren und abzubilden. Kommerziell erhältliche Adeno-assoziierte Viren (AAVs), die hier verwendet werden, exprimierten membranzielgerichtete fluoreszierende Proteine in einer Cre-abhängigen Weise. Die intraokulare Verabreichung von AAVs bei neonatalen Mäusen führt zu einer fluoreszierenden Markierung der Zielzelltypen durch 4-5 Tage nach der Injektion (dpi). Die Membranfluoreszenzsignale sind durch konfokale Bildgebung detektierbar und lösen feine Aststrukturen und -dynamiken auf. Hochwertige Videos von 2-4 h werden von bildgebenden retinalen Flatmounts aufgenommen, die mit sauerstoffreicher künstlicher Zerebrospinalflüssigkeit (aCSF) durchblutet sind. Ebenfalls enthalten ist eine Bildnachbearbeitungspipeline zur Dekonvolution und dreidimensionalen (3D) Driftkorrektur. Dieses Protokoll kann verwendet werden, um mehrere zelluläre Verhaltensweisen in der intakten Netzhaut zu erfassen und neue Faktoren zu identifizieren, die die Morphogenese der Neuriten steuern. Viele Entwicklungsstrategien, die in der Netzhaut erlernt werden, werden für das Verständnis der Bildung neuronaler Schaltkreise an anderer Stelle im zentralen Nervensystem relevant sein.
Einleitung
Dendriten von Netzhautneuronen bilden komplizierte, aber spezifische Muster, die ihre Funktion innerhalb neuronaler Schaltkreise beeinflussen. In der Netzhaut von Wirbeltieren tragen verschiedene Arten von retinalen Ganglienzellen (RGCs) und Amacrinzell-Interneuronen einzigartige dendritische Morphologien, die sich in Baumgröße, Lage, Astlänge und Dichte unterscheiden1. Während der postnatalen Entwicklung verlängern RGCs und Amacrinzellen überschwängliche dendritische Prozesse in einen Neuropil, der als innere plexiforme Schicht (IPL) bezeichnet wird, wo sie bipolare Zelleingänge empfangen, die Photorezeptorsignale übertragen2. Wie durch Zeitraffer-Bildgebung fluoreszierend markierter Netzhautpopulationen in Küken- oder Zebrafischlarven erfasst, ist die Dendritenmorphogenese hochdynamisch3,4,5. Innerhalb weniger Tage dehnen sich dendritische Lauben aus, remodellieren und verzweigen sich zu engen Unterschichten des IPL, wo sie mit ausgewählten Partnern synapsieren. Die Dornen weisen im Laufe der Entwicklung unterschiedliche strukturelle Dynamiken auf, mit Änderungen der relativen Raten der Zweigaddition, des Rückzugs und der Stabilisierung. Amacrin- und RGC-Dendriten zeigen auch unterschiedliche Auswuchs- und Umbauverhaltensweisen, die eine typspezifische Arborisierung widerspiegeln können. Diese Studien verfolgten jedoch breite amakrine oder RGC-Populationen und konzentrierten sich auf laminares Targeting, das nur ein Aspekt der Morphologie ist.
Die Mechanismen, die die enorme morphologische Vielfalt erzeugen, die bei retinalen Subtypen beobachtet wird, sind kaum verstanden. Ziel dieser Gruppe war es, eine Methode zur Erfassung der Dendritendynamik und des Dornumbaus definierter retinaler Subtypen in Mäusen zu entwickeln. Die Identifizierung zelltypspezifischer Mechanismen der Dendritenmusterung erfordert Methoden zur Visualisierung und Messung des Dendritenverhaltens von Zellen von Interesse. Organotypische Kulturen von Mausretinas eignen sich gut für Lebendzellbildgebungsstudien mit konfokaler oder Multiphotonenmikroskopie. Sich entwickelnde Netzhäute werden seziert und zu einem flachen Explant montiert, das für mehrere Stunden in einer Aufnahmekammer abgebildet oder über einige Tage mit begrenzten Auswirkungen auf die Schaltung kultiviert werden kann6,7. Lebende retinale Neuronen können durch eine Vielzahl von Techniken markiert werden, einschließlich Farbstofffüllung durch Elektroden, Elektroporation, biolistische Abgabe von Partikeln, die mit lipophilen Farbstoffen oder Plasmiden, die für fluoreszierende Proteine (z. B. Gene Gun) beschichtet sind, sowie genetisch kodierte Zellmarkierungen7,8,9,10 . Diese Ansätze sind jedoch ineffizient für die Bildgebung der Dendritendynamik bestimmter retinaler Subtypen. Zum Beispiel sind Farbstofffüllmethoden mit geringem Durchsatz und erfordern elektrophysiologische Geräte und zusätzliche genetische Markierungen, um zuverlässig auf Zellen von Interesse abzuzielen. Darüber hinaus können die starken Fluoreszenzsignale im Soma benachbarte Dendriten verdecken.
Biolistische Genabgabemethoden können gleichzeitig Dutzende von Zellen markieren, aber Schritte, die die Hochdruckpartikelabgabe und die Inkubation isolierter Netzhaut über Nacht beinhalten, können die Zellphysiologie und das dendritische Auswachsen beeinträchtigen. Dieses Papier schlägt vor, dass neuere genetische Werkzeuge eingesetzt werden können, um die frühe Dendritendynamik mit Zelltyp und struktureller Auflösung zu erfassen, wobei die folgenden experimentellen Kriterien berücksichtigt werden. Erstens, um die feinen Zweige und Filopodien aufzulösen, die sich entwickelnde Lauben dominieren, sollte die Methode Neuronen mit hellen, fluoreszierenden Proteinen markieren, die Prozesse in der gesamten Laube füllen. Die Fluoreszenzmarkierung sollte aufgrund von Photobleichen während der Bildgebungsphase nicht verblassen. Eine Vielzahl von fluoreszierenden Proteinvarianten wurde generiert und auf Eignung für in vivo/ex vivo imaging11 basierend auf Helligkeit und Photostabilität verglichen. Zweitens müssen die fluoreszierenden Proteine (XFPs) im frühesten Stadium der Dendritenmorphogenese in ausreichend hohen Mengen exprimiert werden, damit das enge Entwicklungsfenster nicht übersehen wird. Bei Analysen statischer Zeitpunkte in der Netzhaut der Maus tritt die Entwicklung der Dendriten während der ersten postnatalen Woche auf und umfasst Phasen des Auswachsens, des Umbaus und der Stabilisierung10,12,13,14,15. Drittens sollte die Methode zu einer selektiven Markierung oder zu einer erhöhten Wahrscheinlichkeit der Markierung der interessierenden neuronalen Subpopulation führen. Viertens muss die Markierung der Zielsubpopulation ausreichend spärlich sein, damit die gesamte neuronale Laube identifiziert und verfolgt werden kann. Obwohl RGC- und amakrine Subtypen durch ihre reifen morphologischen Eigenschaften und IPL-Schichtungsmuster unterschieden werden können16,17,18,19,20, besteht die Herausforderung darin, Subtypen während der Entwicklung basierend auf unreifen Strukturen zu identifizieren. Diese Aufgabe wird durch die Erweiterung transgener Werkzeuge erleichtert, um bestimmte retinale Zelltypen während der Entwicklung zu markieren.
Transgene und Knock-in-Mauslinien, in denen die zelluläre und zeitliche Expression fluoreszierender Proteine oder Cre durch genregulatorische Elemente bestimmt wird, werden häufig zur Untersuchung von Netzhautzelltypen verwendet13,21,22,23. Wichtige Beobachtungen zu subtypspezifischen Mustern der Dendritenentwicklung stammen aus Studien an transgenen Mausnetzhäuten zu statischen Zeitpunkten10,14,24,25. Insbesondere das Cre-Lox-System ermöglicht eine exquisite Genmanipulation und Überwachung von Subtypen mit einer Vielzahl von rekombinaseabhängigen Reportern, Sensoren und optogenetischen Aktivatoren. Diese Werkzeuge haben zur Entdeckung subtypspezifischer molekularer Programme und funktioneller Eigenschaften geführt, die der retinalen Schaltkreismontage zugrunde liegen26,27,28,29,30. Sie müssen jedoch noch genutzt werden, um die subtypspezifische Dendritendynamik in der Netzhaut der Maus zu untersuchen. Eine Markierung mit geringer Dichte kann durch die Kombination von Cre-Mauslinien mit Transgenen erreicht werden, die durch Elektroporation oder durch rekombinante AAVs eingeführt werden. Falls verfügbar, können auch Tamoxifen-induzierbare Cre-Linien oder intersektionale genetische Strategien verwendet werden. Schließlich sollte die Zelle minimal-invasiv markiert und unter Verwendung von Erfassungsparametern abgebildet werden, um das Gewebe nicht zu beeinträchtigen oder die für die Dendritenmorphogenese erforderliche Zellfunktion zu beeinträchtigen.
Hier wird eine Methode zur Anwendung transgener Werkzeuge und konfokaler Mikroskopie vorgestellt, um die Dendritendynamik in lebenden Netzhautexplantationen von Mäusen zu untersuchen. Cre transgene Mauslinien wurden mit AAV-Vektoren kombiniert, die fluoreszierende Proteine bei Cre-Rekombination exprimieren, was eine spärliche Markierung von Netzhautzellen von Interesse ermöglicht. Kommerziell erhältliche AAVs werden durch intravitreale Injektionen an die neonatale Netzhaut abgegeben. Diese Arbeit zeigt, dass AAVs eine signifikant hohe und zelltypspezifische Fluoreszenzexpression von 4 dpi erzeugen, was den Zugang zu postnatalen Zeitpunkten ermöglicht. Um diesen Ansatz zu veranschaulichen, wurde das cholinerge "Starburst" amacrine Interneuron markiert, indem Brainbow AAV in neonatalen Mäusen verabreicht wurde, die das Cholin-Acetyltransferase (ChAT)-interne Ribosom-Entry-Site (IRES)-Cre-Transgen exprimierten, das in der frühen postnatalen Netzhaut aktiv ist31,32. Starburst-Amakrinzellen entwickeln eine stereotype und radiale Dornmorphologie, die durch die Selbstvermeidung von Dendriten geformt wird, die durch die geclusterten Protocadherine vermittelt wird33,34. Diese Arbeit zeigt, dass die Auflösung von Starburst-Dendriten und Filopodien durch XFPs zur Plasmamembran durch Zugabe des CAAX-Motivs, das einer Farnesylierung unterzogen wird, wie es für den Brainbow AAVs31 verwendet wird, signifikant verbessert wird. Schließlich wurden Zeitraffer-Bildgebungs- und Nachbearbeitungsprotokolle bestimmt, die qualitativ hochwertige Bilder erzeugen, die für die Dendritenrekonstruktion und morphometrische Quantifizierung geeignet sind. Dieses Protokoll kann verwendet werden, um Faktoren zu identifizieren, die die Dendritenmorphogenese kontrollieren, und um mehrere zelluläre Verhaltensweisen in der intakten Netzhaut zu erfassen.
Protokoll
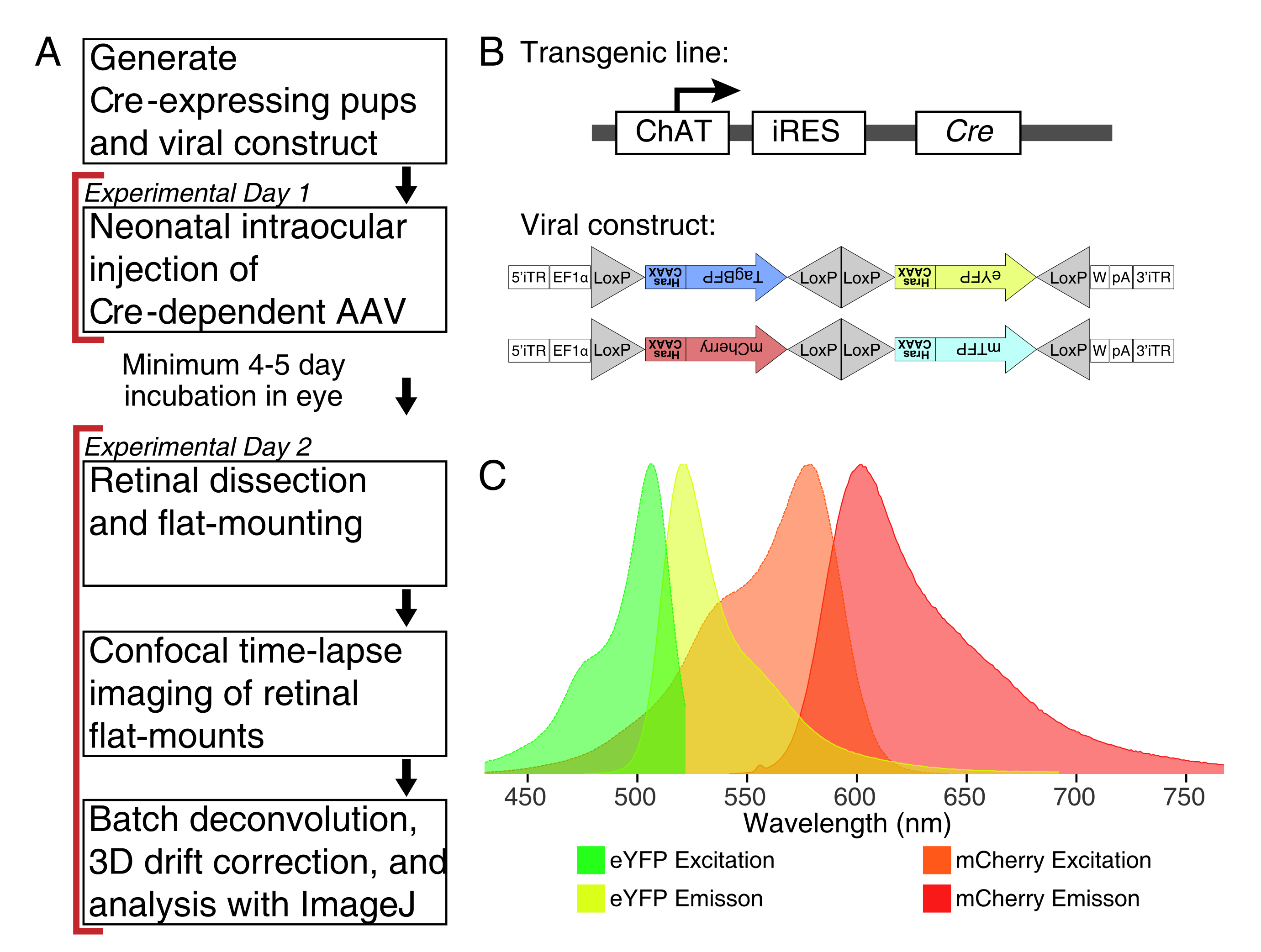
HINWEIS: Dieses Protokoll erstreckt sich über 2 Tage mit einem Mindestzeitraum von 4-5 Tagen für die Virustransduktion zwischen den Versuchstagen (Abbildung 1A). Tierversuche werden in Übereinstimmung mit den Richtlinien des Canadian Council on Animal Care für die Verwendung von Tieren in der Forschung und Labortierpflege gemäß Protokollen durchgeführt, die vom Laboratory of Animal Services Animal Use and Care Committee am Hospital for Sick Children (Toronto, Kanada) genehmigt wurden.
1. Vorbereitungen für die neugeborenen AAV-Injektionen und bildgebenden Versuche
- Wählen Sie eine Cre-Mauslinie aus, um die interessierenden retinalen Zellpopulationen zu kennzeichnen. Bestätigen Sie die Cre-Rekombinase-Expression zum Zeitpunkt der AAV-Injektionen, indem Sie zu einem transgenen Cre-Reporter übergehen oder durch Immunfärbung mit einem Cre-Antikörper.
- Züchten Sie transgene Cre-Mäuse (8-16 Wochen alt), um Cre-positive Neugeborenentiere zu erzeugen.
HINWEIS: Für diese Demonstration wurden ChAT-IRES-Cre-Knock-in-Mäuse verwendet, um die cholinergen Starburst-Amakrinzellen anzugreifen. - Erhalten Sie ein rekombinaseabhängiges AAV-Virus, das für fluoreszierende Proteine kodiert. Für eine optimale Markierung von Feinprozessen wählen Sie Vektoren, die modifizierte XFPs ausdrücken, die auf die Plasmamembran abzielen.
HINWEIS: Diese Studie verwendete das Brainbow-Virus (BBV), AAV-EF1a-BbTagBY und AAV9-EF1a-BbChT (siehe Materialtabelle), die die Möglichkeit zur mehrfarbigen Kennzeichnung bieten (Abbildung 1B). Farnesylierte Enhanced Yellow Fluorescent Protein (eYFP) und monomere Kirsche (mCherry) Expression erzeugten die stärksten fluoreszierenden Signale für die Live-Bildgebung. Ein einzelnes BBV kann verwendet werden, um einzelne Lauben abzubilden, während die Co-Injektion von BBVs verwendet werden kann, um mehr Zellen oder benachbarte Lauben mit unterschiedlichen Fluorophoren zu markieren. Wenn Sie mehrere fluoreszierende Proteine verwenden, achten Sie auf eine minimale Überlappung des Emissionsspektrums (Abbildung 1C). Die Parameter der Mikroskoperfassung müssen angepasst werden, um unterschiedliche XFP-Signale zu erfassen. - Bereiten Sie ∼3-5 μL Aliquots von AAVs in einzelnen niedrig bindenden Röhrchen für Einwegvorräte vor, um Frost-/Tauzyklen zu vermeiden. Bei -80 °C lagern.
- Borosilikatglas-Mikropipetten mit sehr feinen Spitzen mit einem Mikropipettenabzieher zubereiten.
HINWEIS: Die Puller-Einstellungen variieren je nach verwendetem Filament und Puller. Siehe Abbildung 2A für die endgültige Größe und Form der Spitze. Typische Tröpfchendurchmesser sind 600 μm. - Erhalten Sie ein Mikroinjektionssystem und den zugehörigen Schlauch. Schließen Sie den Mikroinjektor an einen unter Druck stehenden Luftanschluss an.

Abbildung 1: Experimentelle Übersicht . (A) Dieses Protokoll umfasst 2 Tage Experimente mit einer Mindestinfektionsdauer von 4-5 Tagen zwischen den Versuchstagen. Intraokulare Injektionen werden an neonatalen Mäusen durchgeführt, die nicht älter als der postnatale Tag 3 sind. Netzhäute werden dann seziert, flach montiert und live abgebildet, um das gewünschte Entwicklungsfenster einzufangen. Markierte Zellen können jederzeit nach den für die virale Expression erforderlichen 4-5 Tagen abgebildet werden, da es keine offensichtlichen Auswirkungen einer längeren AAV-Expression auf die Dendritenmorphologie gibt. (B) Cre-abhängige Brainbow-AAV-Vektoren (BBV) werden in Tiere injiziert, die Cre31 exprimieren. In dieser Studie wurden ChAT-Cre-Knock-in-Mäuse verwendet, um die Cre-Rekombination in Starburst-Amakrinzellen voranzutreiben. Die beiden BBVs kodieren modifiziertes eYFP oder tagBFP oder mCherry und mTFP, die in einem CAAX-Motiv enden, das für die Membranlokalisierung sequentiell farnesyliert wird. Lox-Standorte werden mit Dreiecken dargestellt. Die Vektoren exprimieren entweder farnesylierte XFPs in einer stochastischen und kombinatorischen Weise, abhängig von der Cre-Rekombination. Der EF1α-Promotor enthält regulatorische Elemente aus dem Dehnungsfaktor-1α-Gen. W stellt Elemente aus dem posttranskriptionellen regulatorischen Element des Woodchuck-Hepatitis-Virus dar, und pA gibt die Polyadenylierungssequenz an. (C) Die Anregungs- und Emissionsspektren von mCherry und eYFP, den in dieser Studie abgebildeten BBV-Fluorophoren. Bei der Live-Bildgebung mehrerer fluoreszierender Proteine müssen die Detektionsparameter so angeordnet werden, dass Emissionsspektren angemessen in verschiedene Kanäle getrennt werden. Abkürzungen: AAV = Adeno-assoziiertes Virus; BBV = Brainbow AAV; ChAT = Cholin-Acetyltransferase; iRES = interne Ribosomen-Eintrittsstelle; eYFP = Enhanced Yellow Fluorescent Protein; iTR = invertierte Klemmenwiederholung; tagBP = Tag-blau fluoreszierendes Protein; mCherry = monomere Kirsche; mTFP = türkisfarbenes fluoreszierendes Protein; XFP = jedes fluoreszierende Protein; EF1α= Dehnungsfaktor 1 alpha. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
2. Intravitreale Injektionen von AAVs bei neonatalen Mäusen
- Tauen Sie ein AAV-Aliquot auf Eis auf. Bereiten Sie eine ~ 1: 4 AAV-Verdünnung mit steriler Kochsalzlösung oder phosphatgepufferter Kochsalzlösung vor. Bereiten Sie ~ 0,5 μL AAV-Verdünnung pro Tier (~ 0,25 μL pro Auge) vor, falls die Mikropipette bricht und eine neue Pipette gefüllt werden muss. Lagern Sie das verbleibende unverdünnte AAV bei 4 °C und verwenden Sie es innerhalb von 2 Wochen.
HINWEIS: In diesem Experiment betrug die AAV-Konzentration des Lieferanten ~ 1-2 × 1013 Genomgehalt (GC) / ml und wurde auf eine Endkonzentration von 4 × 1012 GC / ml verdünnt. Optimieren Sie die Viruskonzentration, um die gewünschte Markierungsdichte zu erhalten. - Um die Injektionen zu visualisieren, fügen Sie etwa 1 μL 0,02% Fast Green FCF-Farbstofflösung für jede 15 μL AAV-Verdünnung hinzu, um die Lösung blau zu färben.
- Transfer eines Mauskäfigs mit Mutter- und Neugeborenenstreu (postnataler Tag (P) 0,5-3) in einen Behandlungsraum mit Mikroinjektionsgeräten. Halten Sie die Tiere im selben Käfig mit Nestern und Einstreu, um den mütterlichen Stress und die Ablehnung von Welpen zu minimieren.
- Sterilisieren Sie den Injektionsbereich mit 70% Ethanol und legen Sie sterile Windelkissen auf die Tischflächen. Bereiten Sie eine warme Plattform (z. B. ein Heizkissen) für die Erholung von der durch Unterkühlung induzierten Anästhesie vor.
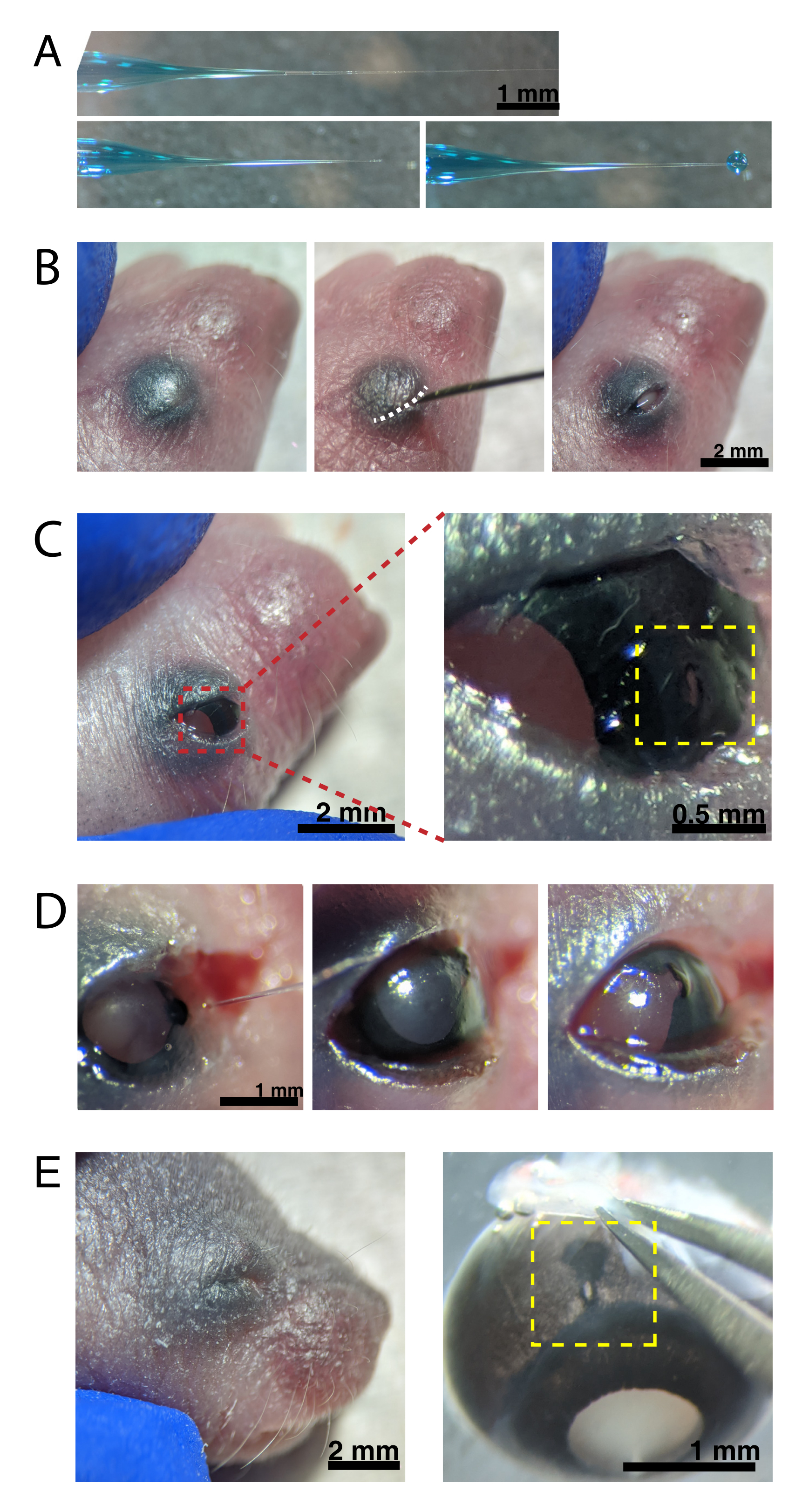
- Füllen Sie die Mikropipette mit einer Mikrospritze mit der AAV-Verdünnung ab. Unter einem Stereomikroskop die Mikropipettenspitze mit einer 30-G-Nadel brechen, um die Spitze zu entsiegeln.
HINWEIS: Abbildung 2A zeigt die verfüllte Spitze - sowohl versiegelt (oben) als auch unversiegelt (Mitte). - Betäuben Sie neonatale Mäuse durch Unterkühlung, indem Sie 1-2 Tiere auf Eis legen. Legen Sie Tiere auf einen Latexhandschuh, um die Haut vor direktem Kontakt mit Eis zu schützen. Sobald sich das Tier als Reaktion auf das Pfotenkneifen (~2 min) nicht mehr bewegt, legen Sie das Tier unter ein Stereomikroskop. Wenn gewünscht, tätowieren Sie Pfotenpolster mit Tätowierfarbe und 30 G Nadel und sammeln Sie Schwanzschnitte für die DNA-Isolierung, um Tiere durch Genotypisierung zu identifizieren.
HINWEIS: Eine angemessene Überwachung der Anästhesietiefe muss während des gesamten Eingriffs erfolgen. - Tupfen Sie die Haut über den Augen mit 70% Ethanol ab. Verwenden Sie eine 30-G-Nadel, um das verschmolzene Augenlid zu öffnen (Abbildung 2B). Üben Sie leichten Druck mit den Fingern aus, um das Auge zu öffnen, und stechen Sie ein kleines Loch durch die Hornhaut an der Hornhaut-Sklera-Verbindung (Abbildung 2C).
- Setzen Sie die Glasmikropipette in das Loch ein und drücken Sie das Mikroinjektor-Fußpedal 2-4 Mal, um das AAV in den intravitrealen Raum zu injizieren. Entfernen Sie langsam die Mikropipette und bestätigen Sie die AAV-Injektion, indem Sie blauen Farbstoff durch die Pupille visualisieren (Abbildung 2D).
HINWEIS: Bei einem Auswurfdruck von 6-8 psi und einer Pulszeit von 600-800 ms wird ein Tröpfchen mit einem Durchmesser von 600 μm ausgestoßen (Abbildung 2A, unten). Pro Auge werden ca. 0,23-0,45 μL AAV-Lösung injiziert. Blaue Lösung außerhalb des Auges zeigt an, dass das AAV nicht in das Auge injiziert wurde. Blaue Lösung, die von der Injektionsstelle austritt, deutet darauf hin, dass das AAV möglicherweise ausgetreten ist, wodurch die Transfektionseffizienz verringert wird. - Drücken Sie die Augenlider vorsichtig zusammen, um sie wieder zu verschließen, und legen Sie den Welpen auf ein beheiztes Pad. Sobald die Tiere eine rosa Farbe wiedererlangt haben und ansprechbar sind, bringen Sie sie vorsichtig zurück in den Haltungskäfig.
HINWEIS: Stellen Sie sicher, dass geeignete Vorsichtsmaßnahmen getroffen werden, um eine zu schnelle Wiedererwärmung der Welpen zu vermeiden. - Wiederholen Sie das Injektionsverfahren für die verbleibenden Tiere in der Einstreu. Erlauben Sie mindestens 4-5 Tage für die virale Transduktion, bevor Sie die Netzhaut abbilden.

Abbildung 2: Neugeborene intraokulare Injektionen. (A) Die hinterlegte Mikropipette zeigt die Form der Pipettenspitze an, wenn sie versiegelt ist (oben) und nachdem die Spitze entsiegelt wurde (unten, links). Der Pico-Injektionsdruck von 6-8 psi und die Pulszeit von 600-800 ms erzeugen einen Tröpfchen mit einem Durchmesser von 600 μm (unten, rechts). Maßstabsleiste = 1 mm.(B) Anästhesierter P0-Welpe unter 16-facher Vergrößerung. Die verschmolzene Augenlidverbindung (weiß) wird mit einer scharfen 30 G Nadel aufgeschlitzt. Maßstabsleiste = 2 mm. (C) Leichter Druck, der auf das Auge ausgeübt wird, legt die Hornhaut frei; Maßstabsleiste = 2 mm. Der vergrößerte Abschnitt (rot) zeigt das kleine Loch an der Hornhaut-Sklera-Verbindung (gelb), das mit einer 30-G-Nadel erzeugt wurde; Maßstabsleiste = 0,5 mm. (D) Entzug der Pipettenspitze nach der Injektion (links). Fast Green-Farbstoff in der AAV-Lösung erscheint als blaugrau durch die Pupille (Mitte), verglichen mit der hellen Pupillenfarbe vor der Injektion (rechts). Maßstabsleiste = 1 mm. (E) Nach 4 Tagen ist das Augenlid verheilt und verschmolzen (links); Maßstabsleiste = 2 mm. Bei der Enukleation ist die verheilte Injektionsstelle sichtbar (gelb); Maßstabsleiste = 1 mm. Beachten Sie die Lage der Injektionsstelle an der Grenze zwischen Hornhaut und Sklera. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
3. Netzhautdissektionen für bildgebende Versuche
- Es wird retinales aCSF (119 mM NaCl, 2,5 mM KCl, 1,3 mM MgCl2·6H2O, 2,5 mM CaCl2·2H2O, 1 mM NaH2PO4, 11 mM Glukose und 20 mM 4-(2-hydroxyethyl)-1-piperazinethansulfonsäure (HEPES-freie Säure)35 hergestellt. Falls gewünscht, 10-fache Lösung vorbereiten und einfrieren; Nach Bedarf auftauen und auf 1x verdünnen.
- Oxygenieren Sie das retinale aCSF, indem Sie mit Carbogen (95% O2, 5% CO2) für mindestens 15 min sprudeln. Stellen Sie den pH-Wert auf 7,4 ein. Bis zur Verwendung in einem versiegelten Behälter aufbewahren, um sicherzustellen, dass das aCSF mit Sauerstoff versorgt bleibt.
HINWEIS: Netzhäute erfordern eine hohe Konzentration von O2. Es ist wichtig, aCSF während des gesamten Experiments mit Sauerstoff zu versorgen. - Betten Sie eine Petrischale mit einem Durchmesser von 60 mm in eine Eisschale ein (machen Sie eine Eisschale aus Laborplastik, z. B. einem Pipettenkastendeckel) und legen Sie sie unter ein Stereomikroskop. Füllen Sie die Petrischale mit sauerstoffreichem retinalem aCSF.
- Euthanasie Mäuse P9 und jünger durch Enthauptung. Euthanasie von Mäusen P10 und älter durch Isofluran-Induktion oder eine alternative Methode, die nach dem Tierprotokoll zugelassen ist, gefolgt von Enthauptung.
- Wenn die Augenlider versiegelt sind, schneiden Sie die Augenlidklappe ab, um das Auge freizulegen. Verwenden Sie eine Pinzette, um die Augen zu enukleieren, und übertragen Sie sie ab Schritt 3.3 in das kalte retinale aCSF.
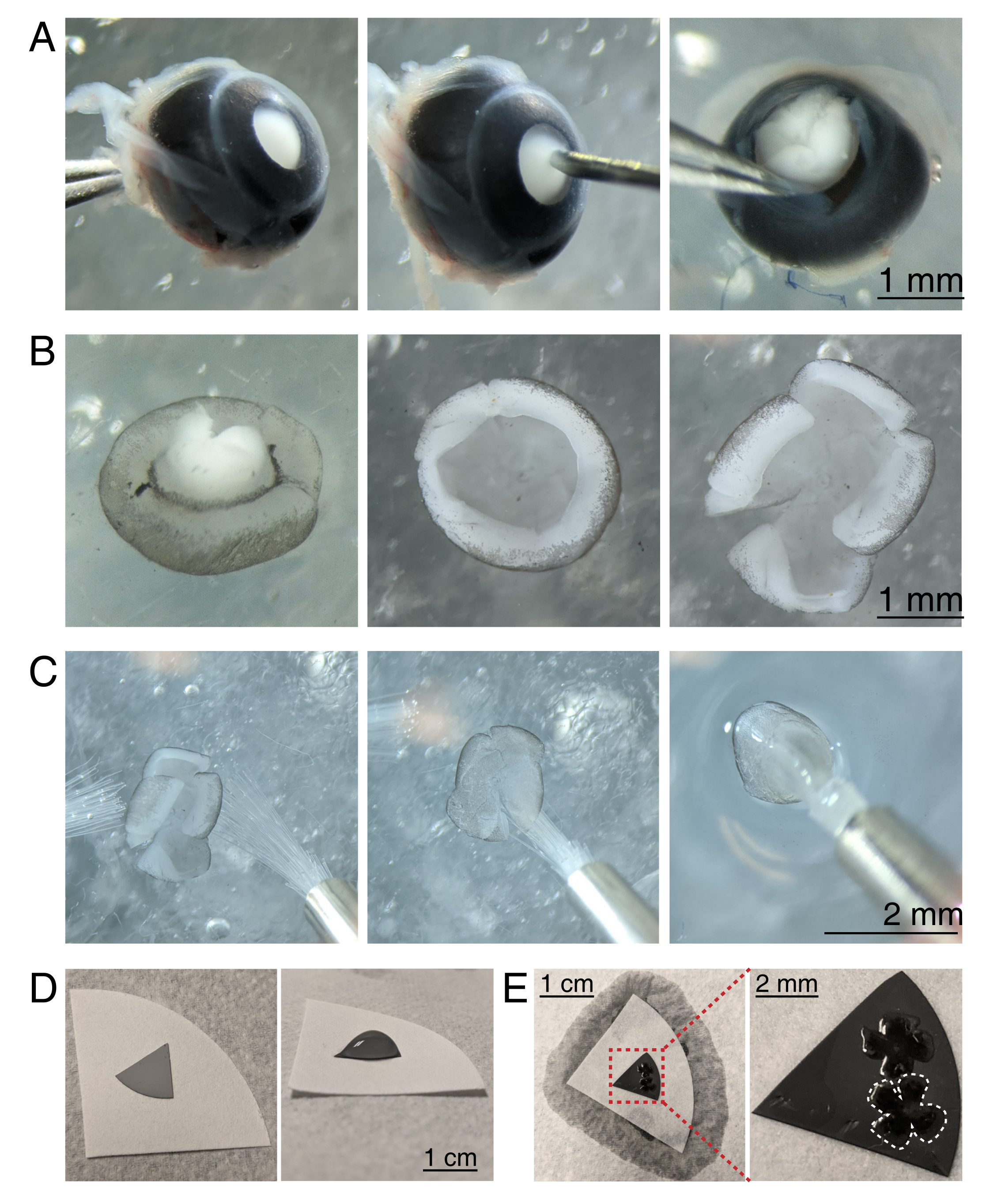
- Um die Netzhautschale zu sezieren, stabilisieren Sie das Auge unter einem Stereomikroskop (Vergrößerung 25x), indem Sie den Sehnerv mit einer Dumont #5-Pinzette umklammern (Abbildung 3A).
- Stecken Sie ein Loch mit einer 30-G-Nadel in die Mitte der Hornhaut und führen Sie dann eine Spitze der Mikroschere in das Loch ein, um einen Schnitt vom Loch bis zum Ende der Hornhaut zu machen. Wiederholen Sie den Vorgang, um 4 Scheiben in die Himmelsrichtungen zu erstellen und 4 "Klappen" zu erzeugen (Abbildung 3A).
- Greifen und ziehen Sie mit zwei Dumont #5 Pinzetten die beiden benachbarten Klappen auseinander und schälen Sie die Sklera sanft von der Netzhaut. Wiederholen Sie dies mit den verbleibenden Hornhautklappen und entfernen Sie die Sklera von der Netzhaut (Abbildung 3B).
- Entfernen Sie die Linse mit der Pinzette aus der Netzhauttasse. Machen Sie mit einer Mikroschere 4 radiale Schnitte vom Rand der Netzhaut zum Sehnerv, wodurch 4 gleiche Blütenblätter entstehen (Abbildung 3B). Wiederholen Sie die Schritte 3.4-3.7 für das zweite Auge.
4. Netzhaut-Flat-Mount-Vorbereitung
- Bereiten Sie graue Mischzelluloseester (MCE) Membranfilterscheiben für die Montage vor. Wenn Sie MCE-Membranfilter mit großem Durchmesser verwenden, schneiden Sie die Scheibe in Quadranten (ca. 1 cm Durchmesser). Legen Sie die MCE-Scheibe in die Mitte eines größeren weißen Filterpapiers (Abbildung 3D).
HINWEIS: MCE-Filter sind auch in Scheiben mit einem Durchmesser von 1 cm erhältlich. Die MCE-Filterscheibe muss groß genug sein, um 1-2 Netzhäute aufzunehmen, aber klein genug, um in die Bildgebungskammer zu passen. Da MCE-Membranen eine statische Aufladung halten, minimieren Sie den Kontakt und die Handhabung der MCE-Membran. - Behandeln Sie Netzhäute mit zwei Pinseln der Größe 3/0 und drehen Sie eine Netzhauttasse auf einen Pinsel, wobei die retinale Ganglienzelle mit der Seite nach unten zeigt. Heben Sie die Netzhaut vorsichtig aus dem aCSF heraus und stellen Sie sicher, dass die Wasserspannung die Netzhaut nicht reißt (Abbildung 3C).
- Während Sie den Pinsel noch mit der Netzhaut halten, platzieren Sie mit einer Transferpipette einen ACSF-Tröpfchen in der Mitte des MCE-Filterpapiers (Abbildung 3C, rechts). Schweben Sie die Netzhaut in den Tröpfchen von aCSF, der durch die Oberflächenspannung erzeugt wird. Verwenden Sie Pinsel, um die Retina-RGC-Seite im Tröpfchen nach oben zu positionieren und die vier Blütenblätter zu entfalten.
- Sobald Sie positioniert sind, erstellen Sie eine Wasserbrücke zwischen dem Pinsel und dem weißen Filterpapier, um die Oberflächenspannung des Tröpfchens zu brechen.
HINWEIS: Wenn aCSF böse ist, haftet die Netzhaut am MCE-Filterpapier (Abbildung 3D,E). Flach montierte Netzhäute können durch Greifen der MCE-Disc mit einer Pinzette gehandhabt werden. Wenn sich der aCSF-Tröpfchen schnell ableitet, bevor er eine Wasserbrücke bildet, kann dies darauf hindeuten, dass die MCE-Membran nicht aufgeladen ist. Verwenden Sie eine frische MCE-Membran und minimieren Sie die Zeit zwischen Enukleation und Bildgebung, indem Sie Netzhäute unmittelbar vor dem Bewegen der Netzhaut in die Bildgebungskammer sezieren und flach montieren.

Abbildung 3: Netzhautdissektion und flache Montage auf Filtermembranen mit gemischtem Cellulosester . (A) Links wird ein enukleiertes Auge stabilisiert, indem der Sehnerv mit einer Dumont #5 Pinzette (links) gegriffen wird. Ein kleines Loch wird in der Mitte der Hornhaut mit einer 30 G Nadel (Mitte) erzeugt. Mikrodissektionsscheren werden verwendet, um die Hornhaut in 4 gleiche Klappen zu schneiden (rechts). Maßstabsleiste = 1 mm. (B) Sezierte Netzhaut mit abgeschälter Sklera mit zwei Dumont #5 Pinzetten (links) und mit entfernter Linse (Mitte). Netzhaut mit 4 gleichen Schnitten auf halbem Weg in die Netzhaut (rechts). Maßstabsleiste = 1 mm. (C) Handhabung der Netzhaut mit zwei feinen Pinseln (Größe 3/0, links). Die Netzhaut drehte sich mit retinalen Ganglienzellen auf einen Pinsel (Mitte) und hob sie aus einem CSF heraus, um sicherzustellen, dass die Wasserspannung die Netzhaut nicht zerreißt (rechts). Maßstabsleiste = 2 mm. (D) Graue MCE-Membranscheibe auf einem weißen Filterpapier (links). Ein Tropfen aCSF auf der MCE-Disc (rechts). Maßstabsleiste = 1 cm. (E) Nachdem Sie die Netzhaut in das Tröpfchen geschwommen und positioniert haben, erstellen Sie eine Wasserbrücke mit dem weißen Filterpapier, um das aCSF abzuleiten, und ziehen Sie die Netzhaut auf das geladene MCE-Papier (links); Maßstabsleiste = 1 cm. Vergrößertes Bild der MCE-Membran (rot) zeigt 2 Netzhäute, die mit retinalen Ganglienzellen montiert sind, die nach oben zeigen (rechts); Maßstabsleiste = 2 mm. Eine Netzhaut ist weiß umrandet. Abkürzungen: MCE = gemischter Celluloseester; aCSF = künstliche Zerebrospinalflüssigkeit. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
5. Konfokale Zeitraffer-Bildgebung von lebenden Retina-Präparaten für die gesamte Montierung
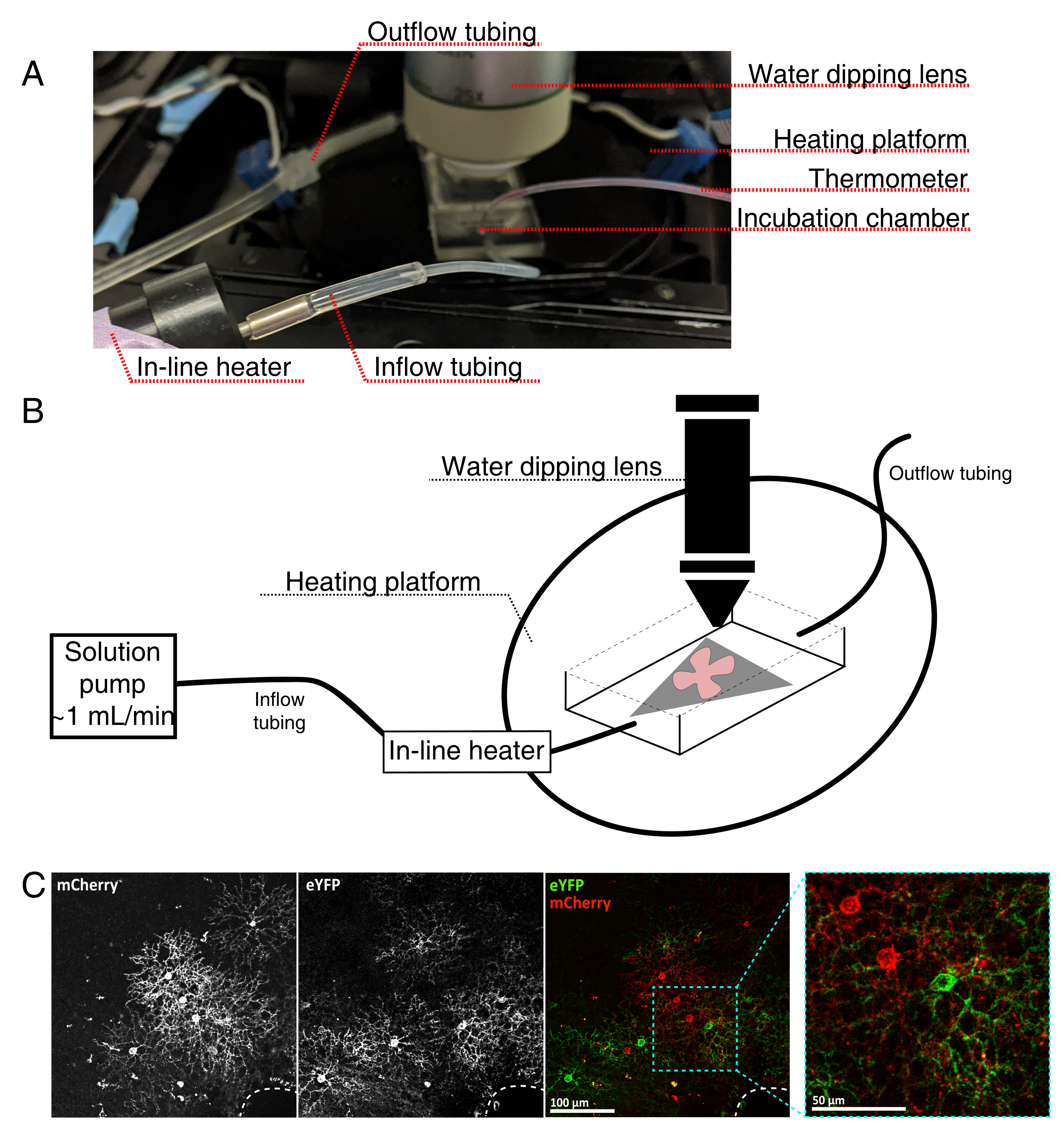
- Montieren Sie die Live-Imaging-Inkubationskammer für ein aufrechtes konfokales Mikroskop wie in Abbildung 4 dargestellt.
HINWEIS: Bei invertierten konfokalen Systemen werden flache Halterungen RGC mit der Seite nach unten direkt auf den Glasbodendeckel der Inkubationskammer gelegt. Sobald die Netzhaut mit dem Deckglas in Kontakt kommt, können sie nicht mehr bewegt werden. - Füllen Sie die Kammer mit oxygeniertem aCSF und schalten Sie die Pumpe und den Temperaturregler ein (Temperatur 32-34 °C, Durchfluss 1 ml/min). Lassen Sie nicht zu, dass die Temperatur über 34 °C steigt.
- Um die retinale flache Montierung in die Perfusionskammer zu übertragen, stoppen Sie die Pumpe und entfernen Sie den aCSF, der sich in der Kammer befindet. Legen Sie die MCE-Disc mit der retinalen Flat-Mount in die (leere) Inkubationskammer.
- Legen Sie ein Probengewicht auf die flache Halterung; Befeuchten Sie das Gewicht vor, um die Oberflächenspannung zu brechen. Füllen Sie die Kammer mit dem erwärmten aCSF auf und zirkulieren Sie aCSF mit ~ 1 ml / min.
- Positionieren Sie den Revolver mit dem 25-fachen Wassertauchobjektiv (numerische Apertur 0,95) in der Abbildungskammer. Screening auf markierte Zellen von Interesse mit epifluoreszierendem Licht (Abbildung 4C).
- Passen Sie das Bildvolumen an, um dendritische Merkmale von Interesse zu erfassen.
HINWEIS: Diese Studie erfasste die komplette dendritische Laube mit 1024 x 1024 Pixeln pro Bild, Z-Schritt 1 μm und einer Bildrate von 2 Minuten zwischen jedem Z-Stack. Die endgültigen Bildgrößen betragen ~100 μm x 100 μm x 20 μm. - Um die Laserleistung auf eine optimale Einstellung einzustellen, verwenden Sie eine Nachschlagetabelle, die sowohl übersättigte als auch untersättigte Pixel identifiziert. Stellen Sie während des Scannens die Laserleistung so ein, dass keine Pixel übersättigt sind (d. h. bei einer Intensität von 255 oder höher). Setzen Sie die Bildgebung so lange wie nötig fort oder bis es zu einem signifikanten und nachweisbaren Rückgang des Fluoreszenzsignals und einer Zunahme des Rauschens (typischerweise 2-4 h) kommt.
HINWEIS: Eine reduzierte Laserleistung wird empfohlen, da Dekonvolutionsalgorithmen optimal funktionieren, wenn Pixel über den gesamten Dynamikbereich verteilt sind. Die Pixelintensität sollte 254 nicht überschreiten; Empirische Analysen von Neuriten ergaben, dass Pixelwerte unter 170 ideal für die Dekonvolution sind. Schnelle Scangeschwindigkeiten (400-600 Hz) mit Zeilenmittelung (2-3) sind einzelnen, langsameren Scans der gleichen Gesamtpixelverweilzeit vorzuziehen. Der Bereich der verlängerten Bildgebung bleicht oft, aber andere Explantationsabschnitte bleiben lebensfähig. Mehrere Bereiche in einer flachen Montierung können für jeweils 2-4 h mit einer Gesamtinkubationszeit von 6 Stunden abgebildet werden. Bildgebungssitzungen über 6 h hinaus wurden nicht systematisch getestet. Neuritenabbau und Blebbing sind Anzeichen dafür, dass die Explantierbarkeit abnimmt. - Fixieren Sie nach der Bildgebung die retinalen Flatmounts und den Membranfilter mit kaltem 4% Paraformaldehyd für 1-2 h bei 4 °C. Amplifizieren Sie die fluoreszierenden Markierungen in der fixierten Netzhaut durch Immunhistochemie für weitere Analysen.
HINWEIS: Eine post-bildgebende Fixierung ist nicht möglich, wenn ein invertierter Konfokal verwendet wird. Netzhaut ist nicht entfernbar, ohne das Gewebe zu zerstören.

Abbildung 4: Aufbau der Inkubationskammer für die Live-Cell-Bildgebung. (A) Inkubationsapparat für Live-Imaging mit Erwärmungs-, Lösungs- und Bildgebungskomponenten. Die Doppelheizung umfasst eine beheizte Inkubationskammerplattform (hinterer Kreis mit blauen Anschlusselektroden) und eine Inline-Lösungsheizung. (B) Schematische Darstellung von 4A. Retinal flat-mount (rot) auf MCE-Membran (grau) wird in der Inkubationskammer platziert. Die Inline-Lösungsheizung ist an eine Lösungspumpe angeschlossen (nicht in 4A abgebildet). Die Abmessungen der Bildkammer ermöglichen einen guten Arbeitsbereich für die Tauchobjektivnase. (C) 25x Ansicht eines retinalen Explantas, das mit 0,23-0,45 μL von 4 × 1012 GC/ml AAV-Verdünnung injiziert wurde; Maßstabsstab = 100 μm. Eine dichte Markierung von Zellen umgibt oft den Sehnervenkopf (gestrichelte Linie, unten rechts); Maßstabsleiste = 50 μm. Abkürzungen: MCE = gemischter Celluloseester; GC = Genomkopien; mCherry = monomere Kirsche; eYFP = Enhanced Yellow Fluorescent Protein. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
6. Bildentfaltung und Nachbearbeitung in ImageJ
- Importieren Sie die Bildserie, teilen Sie die Serie nach Zeit und Farbe, wenn es sich um ein mehrfarbiges Bild handelt. Stellen Sie sicher, dass alle Zeitpunkte für eine Farbe im selben Ordner enthalten sind.
HINWEIS: Importieren Sie Bioformate, wenn Sie ImageJ verwenden (Bioformate sind automatisch in FIDSCHI enthalten). - Erstellen Sie eine theoretische Point-Spread-Funktion (PSF) mit dem ImageJ-Plugin Diffraction PSF 3D.
HINWEIS: Jeder Bildgebungskanal benötigt sein eigenes PSF, da die Fluoreszenzprotein-Emissionswellenlänge die PSF beeinflusst. - Verwenden Sie Plugins | Makros | Ausführen zum Ausführen der parallelen iterativen Dekonvolution im Batch für alle Zeitpunkte mithilfe des bereitgestellten Makros (Ergänzende Datei 1).
HINWEIS: Dieses Makro führt 25 Iterationen des iterativen Algorithmus Wiener Filter Preconditioned Landweber (WPL) aus. Jeder Farbkanal muss separat entwickelt werden. - Zusammenführen der Farbkanäle und Kompilieren aller Zeitpunkte zu einem Hyperstack (Image | Hyperstacks-| Stapelt zu Hyperstack). Korrigieren Sie die 3D-Drift mit Plugins | Registrierung | Korrekte 3D-Drift.
- Zurücksetzen des Bilds auf einen regulären Stapel (Bild | Hyperstacks-| Hyperstack zu Stack) und teilen Sie die Zeitpunkte auf (Abbildung | Stapel | Tools | B. Stack Splitter). Verwenden Sie die Stapelverarbeitung, um eine maximale Projektion für alle Zeitpunkte zu erstellen (Process | Batch-| Makro-| run("Z-Projekt...", "projection=[Max Intensity]"); ).
- Importieren der Zeitraffer-Bildsequenz (Datei | | importieren B. Bildsequenz). Verwenden Sie herkömmliche ImageJ-Tools für die gewünschte Analyse von dekonvolviertem und nachbearbeitetem zweidimensionalem (2D) Video.
HINWEIS: Vierdimensionale (3D + Zeit) dekonvolvierte und driftkorrigierte Videos können als Hyperstack angesehen werden. Lassen Sie den vorherigen Schritt weg, um 3D-Zeitpunkte beizubehalten.
Ergebnisse
Mit dem obigen Protokoll wurde ein hochauflösendes 3D-Video der Entwicklung von Starburst-Zelldendriten aufgenommen, dekonvolviert und für die 3D-Drift korrigiert. Es wurden maximale Projektionen der Z-Ebene erstellt, um 2D-Videos für die Analyse zu erstellen (Ergänzendes Video 1, Abbildung 5A). Die 3D-Dekonvolution jedes Zeitpunkts erhöhte die Auflösung von feinen Filopodienprojektionen (Abbildung 5B,C). Feine Filopodienv...
Diskussion
Dieses Video zeigt eine experimentelle Pipeline, die vorhandene genetische Werkzeuge nutzt, um die Dendritendynamik der sich entwickelnden Netzhautneuronen mit konfokaler Live-Bildgebung abzubilden. Gezeigt werden auch intraokulare Injektionen von Cre-abhängigen AAVs, die membrangerichtete fluoreszierende Proteine in neonatale Mäuse kodieren. Einzelzellen genetisch zielgerichteter Populationen sind bereits mit 4-5 dpi hell markiert. Retinale flache Montierungen wurden für Standard-Bildgebungskammern vorbereitet, um ko...
Offenlegungen
Die Autoren haben nichts offenzulegen.
Danksagungen
Wir danken Madison Gray, dass sie mir geholfen hat, als ich keine hatte. Diese Forschung wurde durch einen NSERC Discovery Grant (RGPIN-2016-06128), ein Sloan Fellowship in Neuroscience und einen Canada Research Chair Tier 2 (an J.L.L.) unterstützt. S. Ing-Esteves wurde durch das Vision Science Research Program und NSERC Postgraduate Scholarships-Doctoral unterstützt.
Materialien
| Name | Company | Catalog Number | Comments |
| Addgene viral prep #45185-AAV9 | |||
| Addgene viral prep #45186-AAV9 | |||
| Dissection tools | |||
| Cellulose filter paper | Whatman | 1001-070 | |
| Dumont #5 fine forceps | FST | 11252-20 | Two Dumont #5 forceps are required for retinal micro-dissection |
| Dumont forceps | VWR | 82027-426 | |
| Fine Scissors | FST | 14058-09 | |
| Mixed cellulose ester membrane (MCE) filter papers, hydrophilic, 0.45 µm pore size | Millipore | HABG01 300 | |
| Petri Dish, 50 × 15 mm | VWR | 470313-352 | |
| Polyethylene disposable transfer pipette | VWR | 470225-034 | |
| Round tip paint brush, size 3/0 | Conventional art supply store | Two size 3/0 paint brushes (or smaller) are required for retinal flat-mounting | |
| Surgical Scissors | FST | 14007-14 | |
| Vannas Spring Scissors - 2.5 mm Cutting Edge | FST | 15000-08 | |
| Live-imaging incubation system | |||
| Chamber polyethylene tubing, PE-160 10' | Warner Instruments | 64-0755 | |
| Dual channel heater controller, Model TC-344C | Warner Instruments | 64-2401 | |
| HC FLUOTAR L 25x/0.95 W VISIR dipping objective | Leica | 15506374 | |
| Heater controller cable | Warner Instruments | CC-28 | |
| Large bath incubation chamber with slice support | Warner Instruments | RC-27L | |
| MPII Mini-Peristaltic Pump | Harvard Apparatus | 70-2027 | |
| PM-6D Magnetic Heated Platform (incubation chamber heater) | Warner Instruments | PM-6D | |
| Pump Head Tubing Pieces For MPII Mini-Peristaltic Pump | Harvard Apparatus | 55-4148 | |
| Sample anchor (Harps) | Warner Instruments | 64-0260 | Sample anchor must be compatible with incubation chamber |
| Sloflo In-line Solution Heater | Warner Instruments | SF-28 | |
| Neonatal Injections | |||
| 10 µL Microliter Syringe Series 700, Removable Needle | Hamilton Company | 80314 | |
| 30 G Hypodermic Needles (0.5 inch) | BD PrecisionGlide | 305106 | |
| 4 inch thinwall glass capillary, no filament (1.0 mm outer diameter/0.75 mm) | WPI World Precision Instruments | TW100-4 | |
| Ethanol 99.8% (to dilute to 70% with double-distilled water [ddH2O]) | Sigma-Aldrich | V001229 | |
| AAV9.hEF1a.lox.TagBFP. lox.eYFP.lox.WPRE.hGH-InvBYF | Penn Vector Core | AV-9-PV2453 | Addgene Plasmid #45185 |
| AAV9.hEF1a.lox.mCherry.lox.mTFP 1.lox.WPRE.hGH-InvCheTF | Penn Vector Core | AV-9-PV2454 | Addgene Plasmid #45186 |
| ChAT-IRES-Cre knock-in transgenic mouse line | The Jackson Laboratory | 6410 | |
| Fast Green FCF Dye content ≥85 % | Sigma-Aldrich | F7252-25G | |
| Flaming/Brown Micropipette Puller, model P-97 | Sutter Instrument Co. | P-97 | |
| Green tattoo paste | Ketchum MFG Co | 329A | |
| Phosphate-Buffered Saline, pH 7.4, liquid, sterile-filtered, suitable for cell culture | Sigma-Aldrich | 806552 | |
| Pneumatic PicoPump | WPI World Precision Instruments | PV-820 | |
| Oxygenated artifiial cerebrospinal fluid (aCSF) Reagents | |||
| Calcium chloride dihydrate (CaCl2·2H2O) | Sigma-Aldrich | C7902 | |
| Carbogen (5% CO2, 95% O2) | AirGas | X02OX95C2003102 | Supplier may vary depending on region |
| D-(+)-Glucose | Sigma-Aldrich | G7021 | |
| HEPES, Free Acid | Bio Basic | HB0264 | |
| Hydrochloric acid solution, 1 N | Sigma-Aldrich | H9892 | |
| Magnesium chloride hexahydrate (MgCl2·6H2O) | Sigma-Aldrich | M2670 | |
| pH-Test strips (6.0-7.7) | VWR | BDH35317.604 | |
| Potassium chloride (KCl) | Sigma-Aldrich | P9541 | |
| Sodium chloride (NaCl) | Bio Basic | DB0483 | |
| Sodium phosphate monobasic (NaH2PO4) | Sigma-Aldrich | RDD007 | |
| Software | |||
| ImageJ | National Institutes of Health (NIH) | Open source |
Referenzen
- Lefebvre, J. L., Sanes, J. R., Kay, J. N. Development of dendritic form and function. Annual Review of Cell and Developmental Biology. 31, 741-777 (2015).
- Graham, H. K., Duan, X. Molecular mechanisms regulating synaptic specificity and retinal circuit formation. Wiley Interdisciplinary Reviews Developmental biology. 10 (1), 379 (2021).
- Godinho, L., et al. Targeting of amacrine cell neurites to appropriate synaptic laminae in the developing zebrafish retina. Development. 132 (22), 5069-5079 (2005).
- Mumm, J. S., et al. In vivo imaging reveals dendritic targeting of laminated afferents by zebrafish retinal ganglion cells. Neuron. 52 (4), 609-621 (2006).
- Wong, W. T., Faulkner-Jones, B. E., Sanes, J. R., Wong, R. O. Rapid dendritic remodeling in the developing retina: dependence on neurotransmission and reciprocal regulation by Rac and Rho. The Journal of Neuroscience. 20 (13), 5024-5036 (2000).
- Wei, W., Elstrott, J., Feller, M. B. Two-photon targeted recording of GFP-expressing neurons for light responses and live-cell imaging in the mouse retina. Nature Protocols. 5 (7), 1347-1352 (2010).
- Morgan, J. L., Wong, R. O. L. Ballistic labeling with fluorescent dyes and indicators. Current Protocols in Neuroscience. 43 (1), 1-10 (2008).
- Nickerson, P. E. B., et al. Live imaging and analysis of postnatal mouse retinal development. BMC Developmental Biology. 13, 24 (2013).
- Morgan, J. L., Dhingra, A., Vardi, N., Wong, R. O. L. Axons and dendrites originate from neuroepithelial-like processes of retinal bipolar cells. Nature Neuroscience. 9 (1), 85-92 (2006).
- Coombs, J. L., Van Der List, D., Chalupa, L. M. Morphological properties of mouse retinal ganglion cells during postnatal development. The Journal of Comparative Neurology. 503 (6), 803-814 (2007).
- Cranfill, P. J., et al. Quantitative assessment of fluorescent proteins. Nature Methods. 13 (7), 557-562 (2016).
- Stacy, R. C., Wong, R. O. L. Developmental relationship between cholinergic amacrine cell processes and ganglion cell dendrites of the mouse retina. The Journal of Comparative Neurology. 456 (2), 154-166 (2003).
- Kay, J. N., et al. Retinal ganglion cells with distinct directional preferences differ in molecular identity, structure, and central projections. The Journal of Neuroscience. 31 (21), 7753-7762 (2011).
- Liu, J., Sanes, J. R. Cellular and molecular analysis of dendritic morphogenesis in a retinal cell type that senses color contrast and ventral motion. The Journal of Neuroscience. 37 (50), 12247-12262 (2017).
- Diao, L., Sun, W., Deng, Q., He, S. Development of the mouse retina: emerging morphological diversity of the ganglion cells. Journal of Neurobiology. 61 (2), 236-249 (2004).
- Coombs, J., vander List, D., Wang, G. Y., Chalupa, L. M. Morphological properties of mouse retinal ganglion cells. Neuroscience. 140 (1), 123-136 (2006).
- Sanes, J. R., Masland, R. H. The types of retinal ganglion cells: current status and implications for neuronal classification. Annual Review of Neuroscience. 38, 221-246 (2015).
- Sümbül, U., et al. A genetic and computational approach to structurally classify neuronal types. Nature Communications. 5, 3512 (2014).
- Lin, B., Masland, R. H. Populations of wide-field amacrine cells in the mouse retina. The Journal of Comparative Neurology. 499 (5), 797-809 (2006).
- Macneil, M. A., Heussy, J. K., Dacheux, R. F., Raviola, E., Masland, R. H. The shapes and numbers of amacrine cells: Matching of photofilled with Golgi-stained cells in the rabbit retina and comparison with other mammalian species. Journal of Comparative Neurology. 413 (2), 305-326 (1999).
- Ivanova, E., Hwang, G. S., Pan, Z. H. Characterization of transgenic mouse lines expressing Cre recombinase in the retina. Neuroscience. 165 (1), 233-243 (2010).
- Jo, A., Xu, J., Deniz, S., Cherian, S., DeVries, S. H., Zhu, Y. Intersectional strategies for targeting amacrine and ganglion cell types in the mouse retina. Frontiers in Neural Circuits. 12, 66 (2018).
- Siegert, S., et al. Genetic address book for retinal cell types. Nature Neuroscience. 12 (9), 1197-1204 (2009).
- Kim, I. -. J., Zhang, Y., Meister, M., Sanes, J. R. Laminar restriction of retinal ganglion cell dendrites and axons: subtype-specific developmental patterns revealed with transgenic markers. The Journal of Neuroscience. 30 (4), 1452-1462 (2010).
- Peng, Y. -. R., Tran, N. M., Krishnaswamy, A., Kostadinov, D., Martersteck, E. M., Sanes, J. R. Satb1 regulates contactin 5 to pattern dendrites of a mammalian retinal ganglion cell. Neuron. 95 (4), 869-883 (2017).
- Duan, X., Krishnaswamy, A., Dela Huerta, I., Sanes, J. R. Type II cadherins guide assembly of a direction-selective retinal circuit. Cell. 158 (4), 793-807 (2014).
- Ray, T. A., et al. Formation of retinal direction-selective circuitry initiated by starburst amacrine cell homotypic contact. eLife. 7, 34241 (2018).
- Krishnaswamy, A., Yamagata, M., Duan, X., Hong, Y. K., Sanes, J. R. Sidekick 2 directs formation of a retinal circuit that detects differential motion. Nature. 524 (7566), 466-470 (2015).
- Caval-Holme, F., Zhang, Y., Feller, M. B. Gap junction coupling shapes the encoding of light in the developing retina. Current Biology. 29 (23), 4024-4035 (2019).
- Lucas, J. A., Schmidt, T. M. Cellular properties of intrinsically photosensitive retinal ganglion cells during postnatal development. Neural Development. 14 (1), 8 (2019).
- Cai, D., Cohen, K. B., Luo, T., Lichtman, J. W., Sanes, J. R. Improved tools for the Brainbow toolbox. Nature Methods. 10 (6), 540-547 (2013).
- Rossi, J., et al. Melanocortin-4 receptors expressed by cholinergic neurons regulate energy balance and glucose homeostasis. Cell Metabolism. 13 (2), 195-204 (2011).
- Lefebvre, J. L., Kostadinov, D., Chen, W. V., Maniatis, T., Sanes, J. R. Protocadherins mediate dendritic self-avoidance in the mammalian nervous system. Nature. 488 (7412), 517-521 (2012).
- Ing-Esteves, S., et al. Combinatorial effects of alpha- and gamma-protocadherins on neuronal survival and dendritic self-avoidance. The Journal of Neuroscience. 38 (11), 2713-2729 (2018).
- Williams, P. R., Morgan, J. L., Kerschensteiner, D., Wong, R. O. L. In vitro imaging of retinal whole mounts. Cold Spring Harbor Protocols. 2013 (1), (2013).
- Ramoa, A. S., Campbell, G., Shatz, C. J. Transient morphological features of identified ganglion cells in living fetal and neonatal retina. Science. 237 (4814), 522-525 (1987).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 9, 676-682 (2012).
- Peng, H., Ruan, Z., Long, F., Simpson, J. H., Myers, E. W. V3D enables real-time 3D visualization and quantitative analysis of large-scale biological image data sets. Nature Biotechnology. 28 (4), 348-353 (2010).
- Cuntz, H., Forstner, F., Borst, A., Häusser, M. One rule to grow them all: a general theory of neuronal branching and its practical application. PLoS Computational Biology. 6 (8), 1000877 (2010).
- Xiao, H., Peng, H. APP2: automatic tracing of 3D neuron morphology based on hierarchical pruning of a gray-weighted image distance-tree. Bioinformatics. 29 (11), 1448-1454 (2013).
- Nanda, S., et al. Design and implementation of multi-signal and time-varying neural reconstructions. Scientific data. 5, 170207 (2018).
- Sherry, D. M., Wang, M. M., Bates, J., Frishman, L. J. Expression of vesicular glutamate transporter 1 in the mouse retina reveals temporal ordering in development of rod vs. cone and ON vs. OFF circuits. The Journal of Comparative Neurology. 465 (4), 480-498 (2003).
- Johnson, J., et al. Vesicular neurotransmitter transporter expression in developing postnatal rodent retina: GABA and glycine precede glutamate. The Journal of Neuroscience. 23 (2), 518-529 (2003).
- Jüttner, J., et al. Targeting neuronal and glial cell types with synthetic promoter AAVs in mice, non-human primates and humans. Nature Neuroscience. 22 (8), 1345-1356 (2019).
- Zincarelli, C., Soltys, S., Rengo, G., Rabinowitz, J. E. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Molecular Therapy. 16 (6), 1073-1080 (2008).
- Petros, T. J., Rebsam, A., Mason, C. A. In utero and ex vivo electroporation for gene expression in mouse retinal ganglion cells. Journal of Visualized Experiments: JoVE. (31), e1333 (2009).
- Lye, M. H., Jakobs, T. C., Masland, R. H., Koizumi, A. Organotypic culture of adult rabbit retina. Journal of Visualized Experiments: JoVE. (3), e190 (2007).
- Pignatelli, V., Strettoi, E. Bipolar cells of the mouse retina: a gene gun, morphological study. The Journal of Comparative Neurology. 476 (3), 254-266 (2004).
- Huckfeldt, R. M., et al. Transient neurites of retinal horizontal cells exhibit columnar tiling via homotypic interactions. Nature Neuroscience. 12 (1), 35-43 (2009).
- Prahst, C., et al. Mouse retinal cell behaviour in space and time using light sheet fluorescence microscopy. eLife. 9, 49779 (2020).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten