
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Imagem de lapso de tempo da arborização neuronal usando rotulagem de vírus associado adeno esparsa de populações de células de retina geneticamente direcionadas
Neste Artigo
Resumo
Aqui, apresentamos um método para investigar morfogênese neurita em explantes de retina de camundongos pós-natal por microscopia confocal de lapso de tempo. Descrevemos uma abordagem para rotulagem e aquisição esparsa de tipos de células da retina e seus processos finos usando vetores de vírus associados a adeno recombinantes que expressam proteínas fluorescentes direcionadas à membrana de forma dependente de Cre.
Resumo
Descobrir mecanismos que padronizam as arboris dendríticas requer métodos para visualizar, imagem e analisar dendritos durante o desenvolvimento. A retina do rato é um poderoso sistema modelo para a investigação de mecanismos específicos do tipo celular de morfogênese neuronal e conectividade. A organização e a composição dos subtipos da retina estão bem definidas, e ferramentas genéticas estão disponíveis para acessar tipos específicos durante o desenvolvimento. Muitos tipos de células de retina também restringem seus dendritos e/ou axônios a camadas estreitas, o que facilita a imagem de lapso de tempo. As culturas de explant da retina do rato são adequadas para imagens de células vivas usando microscopia confocal ou multifotônio, mas métodos otimizados para dinâmica de dendrite de imagem com resolução temporal e estrutural são carentes. Apresentado aqui é um método para rotular e imaginar escassamente o desenvolvimento de populações específicas de retina marcadas pelo sistema Cre-Lox. Vírus associados a adeno (AAVs) comercialmente disponíveis (AAVs) usados aqui expressavam proteínas fluorescentes direcionadas à membrana de forma dependente de Cre. A entrega intraocular de AAVs em camundongos neonatais produz rotulagem fluorescente de tipos de células-alvo por 4-5 dias após a injeção (dpi). Os sinais fluorescentes de membrana são detectáveis por imagens confocal e resolvem estruturas e dinâmicas finas do ramo. Vídeos de alta qualidade que abrangem 2-4 h são adquiridos a partir de imagens de montagem plana de retina perfumadas com fluido cerebrospinal artificial oxigenado (aCSF). Também é fornecido um pipeline de pós-processamento de imagem para desconvolução e correção de deriva tridimensional (3D). Este protocolo pode ser usado para capturar vários comportamentos celulares na retina intacta e identificar novos fatores que controlam a morfogênese neurita. Muitas estratégias de desenvolvimento aprendidas na retina serão relevantes para a compreensão da formação de circuitos neurais em outros lugares do sistema nervoso central.
Introdução
Dendritos de neurônios da retina formam padrões intrincados, mas específicos, que influenciam sua função dentro de circuitos neurais. Na retina vertebrada, diversos tipos de células gânglios da retina (RGCs) e interneurônios de células amacrinas possuem morfologias dendríticas únicas que diferem em tamanho, localização, comprimento do ramo e densidade1. Durante o desenvolvimento pós-natal, RGCs e células amacrinas estendem processos dendráticos exuberantes em um neuropiloto chamado camada plexiforme interna (IPL), onde recebem entradas de células bipolares transmitindo sinais fotorreceptores2. Como capturado por imagens de lapso de tempo de populações de retina fluorescentes rotuladas em larvas de filhotes ou zebrafish, a morfogênese dendrite é altamente dinâmica3,4,5. Em poucos dias, as arbóreas dendríticas expandem, remodelam e ramificam para subcamadas estreitas do IPL, onde sinapse com parceiros selecionados. As arbors apresentam dinâmicas estruturais diferentes sobre o desenvolvimento, com alterações nas taxas relativas de adição de filiais, retração e estabilização. Os dendritos amacrinos e RGC também apresentam diferentes comportamentos de crescimento e remodelação que podem refletir arborização específica do tipo. No entanto, esses estudos acompanharam populações de amacrinas ou RGC amplas e se concentraram na segmentação laminar, que é apenas um aspecto da morfologia.
Os mecanismos que produzem a vasta diversidade morfológica observada entre os subtipos da retina são mal compreendidos. O objetivo deste grupo foi desenvolver um método para capturar a dinâmica dendrite e a remodelação de arbor de subtipos de retina definidos em camundongos. Identificar mecanismos específicos do tipo celular de padronização de dendrite requer métodos para visualizar e medir comportamentos dendrite de células de interesse. Culturas organotípicas de retinas de camundongos são adequadas para estudos de imagem de células vivas usando microscopia confocal ou multifotífera. Retinas em desenvolvimento são dissecadas e montadas em uma explanta plana que pode ser imageda por várias horas em uma câmara de gravação ou cultivada ao longo de alguns dias com efeitos limitados no circuito6,7. Os neurônios de retina ao vivo podem ser rotulados por uma variedade de técnicas, incluindo preenchimento de corante por eletrodos, eletroporação, entrega biolística de partículas revestidas com corantes lipofílicos ou plasmídeos codificando proteínas fluorescentes (por exemplo, Gene Gun), bem como rótulos de células geneticamente codificadas7,8, 9,10 . No entanto, essas abordagens são ineficientes para a dinâmica de dendrite de imagem de subtipos específicos da retina. Por exemplo, os métodos de enchimento de corantes são de baixa produtividade e requerem aparelhos de eletrofisiologia e rótulos genéticos adicionais para direcionar de forma confiável células de interesse. Além disso, os fortes sinais de fluorescência na soma podem obscurecer dendritos próximos.
Métodos de entrega de genes biolísticos podem simultaneamente rotular dezenas de células, mas etapas que envolvem a entrega de partículas de alta pressão e a incubação noturna de retina isolada podem comprometer a fisiologia celular e o crescimento dendrático. Este artigo propõe que ferramentas genéticas recentes podem ser empregadas para capturar dinâmicas dendrita precoces com tipo celular e resolução estrutural, dados os seguintes critérios experimentais. Primeiro, para resolver os ramos finos e filopodia que dominam as árvores em desenvolvimento, o método deve rotular neurônios com proteínas brilhantes e fluorescentes que preenchem processos em toda a arbor. A rotulagem de fluorescência não deve desaparecer devido ao fotobleaching durante o período de imagem. Uma variedade de variantes de proteína fluorescente foram geradas e comparadas para adequação para imagens in vivo/ex vivo11 baseadas em brilho e fotoestabilidade. Em segundo lugar, as proteínas fluorescentes (XFPs) devem ser expressas em níveis suficientemente altos pelo estágio inicial da morfogênese dendrite, de modo que a estreita janela de desenvolvimento não seja perdida. Em análises de pontos de tempo estáticos na retina do camundongo, o desenvolvimento do dendrite ocorre durante a primeira semana pós-natal e inclui fases de crescimento, remodelação e estabilização10,12,13,14,15. Em terceiro lugar, o método deve levar à rotulagem seletiva ou a uma maior probabilidade de rotulagem da subpopulação neuronal de interesse. Em quarto lugar, a rotulagem da subpopulação alvo deve ser suficientemente escassa para que toda a árvore neuronal possa ser identificada e rastreada. Embora os subtipos de RGC e amacrine possam ser distinguidos por suas características morfológicas maduras e padrões de estratificação de IPL16,17,18,19,20, o desafio é identificar subtipos durante o desenvolvimento com base em estruturas imaturas. Essa tarefa é facilitada pela expansão de ferramentas transgênicas para rotular tipos específicos de células da retina durante o desenvolvimento.
Linhas transgênicas e de camundongos em que a expressão celular e temporal de proteínas fluorescentes ou Cre é determinada por elementos regulatórios genéticos são amplamente utilizadas para estudar os tipos de células da retina13,21,22,23. Observações-chave sobre padrões específicos de desenvolvimento de dendrítos de subtipo vieram de estudos de retinas de camundongos transgênicos em momentos estáticos10,14,24,25. O sistema Cre-Lox, em particular, permite a manipulação genética requintada e o monitoramento de subtipos usando uma variedade de repórteres, sensores e ativadores optogenéticos dependentes de recombinase. Essas ferramentas levaram a descobertas de programas moleculares específicos do subtipo e propriedades funcionais que estão por trás do conjunto do circuito de retina26,27,28,29,30. No entanto, eles ainda não foram aproveitados para estudar a dinâmica dendrite específica do subtipo na retina do rato. A rotulagem de baixa densidade pode ser alcançada combinando linhas de mouse Cre com transgênicos introduzidos por eletroporação ou por AAVs recombinantes. Se disponível, linhas cre indutíveis de tamoxifen ou estratégias genéticas interseccionais também podem ser usadas. Finalmente, a célula deve ser rotulada de forma minimamente invasiva e imageada usando parâmetros de aquisição para não comprometer o tecido ou interferir com a função celular necessária para morfogênese dendrita.
Apresentado aqui é um método para aplicar ferramentas transgênicas e microscopia confocal para investigar a dinâmica do dendrite em explants de retina de rato vivo. Linhas de camundongos transgênicos cre foram combinadas com vetores AAV que expressam proteínas fluorescentes após a recombinação de Cre, o que permite rotulagem esparsa de células de interesse da retina. As AAVs disponíveis comercialmente são entregues à retina neonatal por injeções intravitreal. Este artigo demonstra que os AAVs produzem expressões fluorescentes significativamente altas e específicas do tipo celular por 4 dpi, permitindo o acesso aos pontos de tempo pós-natal. Para ilustrar essa abordagem, o interneuron amacrina "starburst" cholinergic "starburst" foi rotulado pela entrega de Brainbow AAV em camundongos neonatais expressando o site de entrada de acetiltransferase de colina (ChAT)-internal ribosome (IRES)-Cre transgene, que está ativo na retina pós-natal inicial31,32. As células amacrinas starburst desenvolvem uma morfologia arbórea estereotipada e radial que é moldada pela auto-evasão dendrite mediada pelas protocadherinas agrupadas33,34. Este artigo mostra que a resolução de dendritos e filopodia de starburst é significativamente melhorada pelos XFPs para a membrana plasmática com a adição do motivo CAAX que sofre farnesylation, como usado para o Brainbow AAVs31. Finalmente, foram determinados protocolos de imagem e pós-processamento de lapso de tempo que produzem imagens de alta qualidade para reconstrução dendrito e quantificação morfométrica. Este protocolo pode ser usado para identificar fatores que controlam a morfogênese dendrita e para capturar vários comportamentos celulares na retina intacta.
Protocolo
NOTA: Este protocolo abrange 2 dias com um período mínimo de 4 a 5 dias para transdução viral entre dias experimentais (Figura 1A). Os experimentos em animais são realizados de acordo com o Conselho Canadense de Diretrizes de Cuidados com Animais para Uso de Animais em Pesquisa e Cuidados Com Animais de Laboratório sob protocolos aprovados pelo Comitê de Uso e Cuidado animal do Laboratório de Serviços Animais do Hospital para Crianças Doentes (Toronto, Canadá).
1. Preparações para as injeções neonatais de AAV e experimentos de imagem
- Selecione uma linha de mouse Cre para rotular as populações de células de retina de interesse. Confirme a expressão de recombinase cre no momento das injeções de AAV cruzando para um repórter transgênico Cre ou através de imunostaining com um anticorpo Cre.
- Criar camundongos cre transgênicos (8-16 semanas de idade) para gerar animais neonatais cre-positivos.
NOTA: Para esta demonstração, os ratos knock-in ChAT-IRES-Cre foram usados para atingir as células amacrinas starburst colinérgicas. - Obtenha vírus AAV dependente de recombinase codificando proteínas fluorescentes. Para uma rotulagem ideal de processos finos, selecione vetores que expressem XFPs modificados visando a membrana plasmática.
NOTA: Este estudo utilizou o vírus Brainbow (BBV), AAV-EF1a-BbTagBY e AAV9-EF1a-BbChT (ver Tabela de Materiais), que fornecem a opção para rotulagem multicolor (Figura 1B). A expressão de proteína fluorescente amarela (eYFP) e a expressão de cereja monomérica (mCherry) produziram os sinais fluorescentes mais fortes para imagens vivas. Um único BBV pode ser usado para imagem de arboris individuais, enquanto a co-injeção de BBVs pode ser usada para rotular mais células ou arboros vizinhos com fluoroforos distintos. Se usar múltiplas proteínas fluorescentes, garanta a sobreposição mínima do espectro de emissões (Figura 1C). Os parâmetros de aquisição de microscópio devem ser ajustados para capturar sinais XFP distintos. - Prepare ∼ 3-5 μL alíquotas de AAVs em tubos individuais de baixa ligação para estoques de uso único para evitar ciclos de congelamento/degelo. Armazém a -80 °C.
- Prepare micropipettos de vidro borossilicatos com pontas muito finas usando um puxador de micropipette.
NOTA: As configurações do puller variam dependendo do filamento e do puxador que estão sendo usados; ver Figura 2A para o tamanho e forma da ponta final. Os diâmetros típicos das gotículas são de 600 μm. - Obtenha um sistema de microinjeção e tubos associados. Conecte o microinjetor a uma porta de ar pressurizada.

Figura 1: Visão geral experimental. (A) Este protocolo abrange 2 dias de experimentos com um período mínimo de infecção de 4 a 5 dias entre os dias experimentais. As injeções intraoculares são realizadas em camundongos neonatais não mais velhos que o pós-natal 3. As retinas são então dissecadas, montadas planas e imagens ao vivo para capturar a janela de desenvolvimento desejada. As células rotuladas podem ser imagens a qualquer momento após os 4-5 dias necessários para a expressão viral, pois não há efeitos aparentes da expressão AAV prolongada na morfologia dendrite. (B) Os vetores Brainbow AAV (BBV) dependentes de Cre são injetados em animais que expressam Cre31. Neste estudo, os camundongos knock-in chat-cre foram usados para impulsionar a recombinação de Cre em células amacrinas starburst. Os dois BBVs codificam eYFP modificados ou tagBFP, ou mCherry e mTFP, que terminam em um motivo CAAX que é sequencialmente farnesylated para localização de membrana. Os locais de lox são representados com triângulos. Os vetores expressam ou XFPs farnesylated de forma estocástica e combinatória dependente da recombinação de Cre. O promotor EF1α inclui elementos regulatórios do gene fator de alongamento 1α. W representa elementos do elemento regulador pós-transcrição pós-transcricional do vírus da hepatite de marmuck, e pA indica a sequência de poliadenilação. (C) Os espectros de excitação e emissão de mCherry e eYFP, os fluoroforos bbv imagemdo neste estudo. Quando a imagem ao vivo de múltiplas proteínas fluorescentes, os parâmetros de detecção devem ser organizados para separar adequadamente os espectros de emissões em canais distintos. Abreviaturas: AAV = vírus associado ao adeno; BBV = Brainbow AAV; ChAT = acetiltransferase de colina; iRES = site interno de entrada ribossosome; eYFP = proteína fluorescente amarela aprimorada; iTR = repetição do terminal invertido; tagBP = Proteína fluorescente azul-tag; mCherry = cereja monomérica; mTFP = proteína fluorescente teal; XFP = qualquer proteína fluorescente; EF1α= fator de alongamento 1 alfa. Clique aqui para ver uma versão maior desta figura.
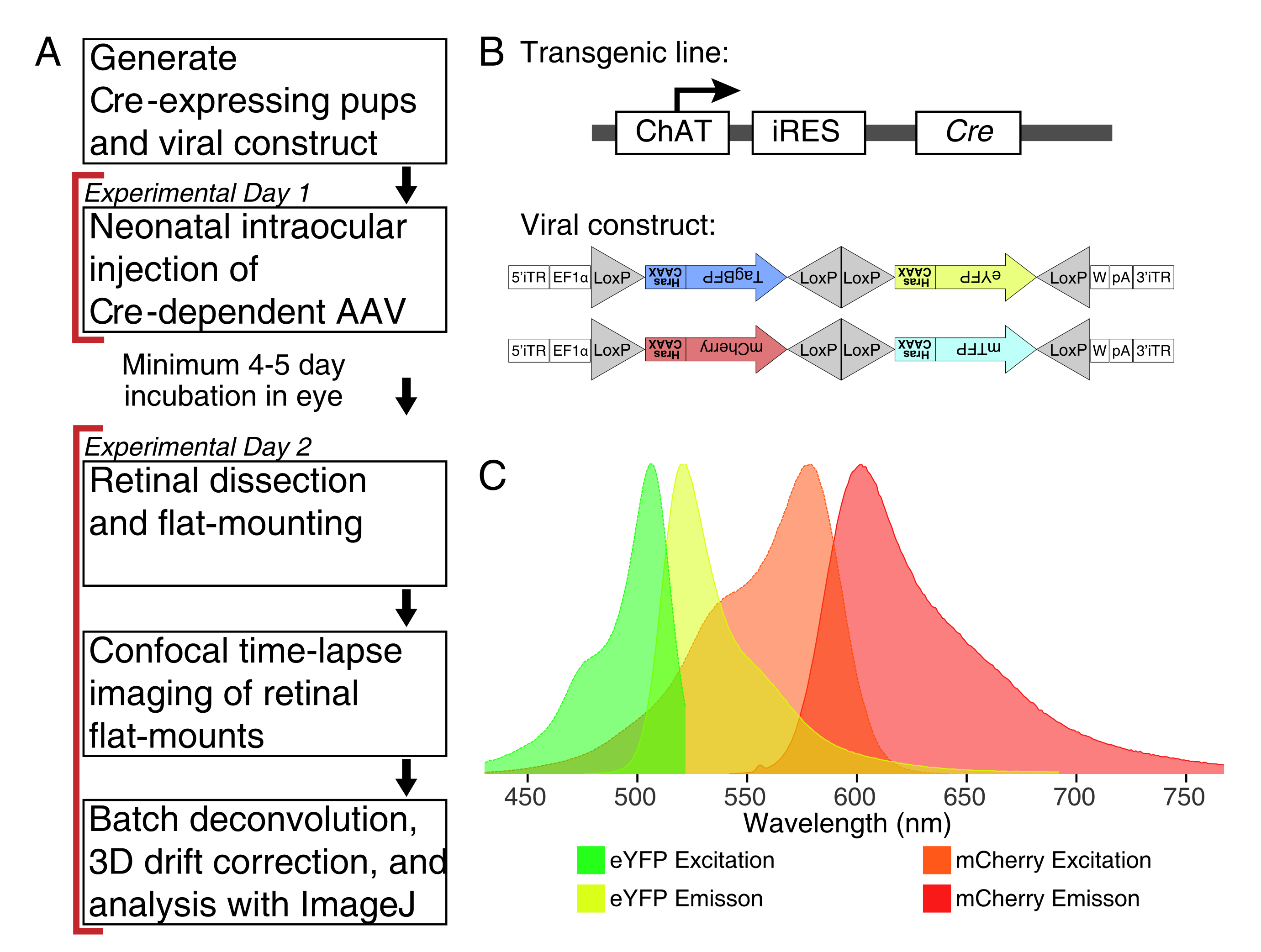{kind=link}
2. Injeções intravitais de AAVs em camundongos neonatais
- Descongele uma alíquota AAV no gelo. Prepare uma diluição de AAV ~1:4 usando soro fisiológico estéril ou salina tamponada com fosfato. Prepare ~0,5 μL de diluição de AAV por animal (~0,25 μL por olho) no caso de a micropipette quebrar, e uma nova pipeta precisa ser preenchida. Armazene o AAV não diluído restante a 4 °C e use dentro de 2 semanas.
NOTA: Neste experimento, a concentração de AAV do fornecedor foi ~1-2 × 1013 teor de genoma (GC)/mL e diluída a uma concentração final de 4 × 1012 GC/mL. Otimize a concentração viral para obter a densidade de rotulagem desejada. - Para visualizar as injeções, adicione aproximadamente 1 μL de solução de corante Fast Green FCF de 0,02% para cada 15 μL de diluição AAV para colorir a solução azul.
- Transfira uma gaiola de rato com barragem e lixo recém-nascido (dia pós-natal (P) 0,5-3) para uma sala de procedimentos com equipamento de microinjeção. Mantenha os animais na mesma gaiola com ninhos e roupas de cama para minimizar o estresse materno e a rejeição dos filhotes.
- Esterilize a área de injeção com 70% de etanol e coloque almofadas de fraldas estéreis em superfícies de banco. Prepare uma plataforma quente (por exemplo, uma almofada de aquecimento) para recuperação da anestesia induzida por hipotermia.
- Enchimento de micropipette com a diluição AAV usando uma microsinga. Sob um estereótipo, quebre a ponta da micropipette com uma agulha de 30 G para desslasar a ponta.
NOTA: A Figura 2A mostra a ponta recheada e selada (superior) e não selada (meio). - Anestesiar camundongos neonatais por hipotermia colocando 1-2 animais no gelo. Coloque os animais em uma luva de látex para proteger a pele do contato direto com o gelo. Uma vez que o animal não se move mais em resposta ao aperto de pata (~2 min), coloque o animal sob um estereóscópio. Se desejar, tatue as pastilhas de pata com tinta de tatuagem e agulha de 30 G, e colete recortes de cauda para isolamento de DNA para identificar animais por genotipagem.
NOTA: Deve ocorrer o devido monitoramento da profundidade da anestesia durante todo o procedimento. - Esfregue a pele sobrepondo os olhos com 70% de etanol. Use uma agulha de 30 G para abrir a pálpebra fundida (Figura 2B). Aplique pressão leve com os dedos para abrir o olho, e faça um pequeno buraco através da córnea na junção córnea-esclera (Figura 2C).
- Insira o micropipette de vidro no orifício e pressione o pedal do pé microinjetor 2-4 vezes para injetar o AAV no espaço intravitreal. Remova lentamente a micropipette e confirme a injeção de AAV visualizando corante azul através da pupila (Figura 2D).
NOTA: Com uma pressão de ejeção de 6-8 psi e tempo de pulso de 600-800 ms, uma gotícula de 600 μm de diâmetro é ejetada (Figura 2A, inferior). Aproximadamente 0,23-0,45 μL de solução AAV é injetado por olho. A solução azul fora do olho indica que o AAV não foi injetado no olho. O vazamento da solução azul do local da injeção indica que o AAV pode ter vazado, reduzindo a eficiência da transfecção. - Pressione suavemente as pálpebras juntas para selar novamente, e coloque o filhote em uma almofada aquecida. Uma vez que os animais recuperem uma cor rosada e sejam responsivos, transfira-os suavemente de volta para a gaiola de habitação.
NOTA: Certifique-se de que as precauções apropriadas sejam tomadas para evitar o aquecimento muito rápido dos filhotes. - Repita o procedimento de injeção para os demais animais da ninhada. Permita um mínimo de 4-5 dias para transdução viral antes de imaginar a retina.

Figura 2: Injeções intraoculares neonatais. (A) Micropipette recheada mostra a forma da ponta da pipeta quando selada (superior) e depois que a ponta é deslalada (inferior, esquerda). A pressão de injeção de pico de 6-8 psi e o tempo de pulso de 600-800 ms produz uma gotícula de 600 μm de diâmetro (inferior, direita). Barra de escala = 1 mm.(B) Filhote P0 anestesiado sob ampliação de 16x. A junção da pálpebra fundida (branca) é cortada usando uma agulha afiada de 30 G. Barra de escala = 2 mm. (C) A pressão leve aplicada ao olho expõe a córnea; barra de escala = 2 mm. A seção ampliada (vermelha) mostra o pequeno orifício na junção córnea-esclera (amarelo) criada com uma agulha de 30 G; barra de escala = 0,5 mm. (D) Retirada da ponta da pipeta após a injeção (esquerda). O corante verde rápido na solução AAV aparece como azul-cinza através da pupila (meio), em comparação com a cor da pupila clara antes da injeção (à direita). Barra de escala = 1 mm. (E) Após 4 dias, a pálpebra se curou e fundiu fechada (esquerda); barra de escala = 2 mm. Após a enucleação, o local de injeção curada é visível (amarelo); barra de escala = 1 mm. Observe a localização do local da injeção na fronteira entre a córnea e a esclera. Clique aqui para ver uma versão maior desta figura.
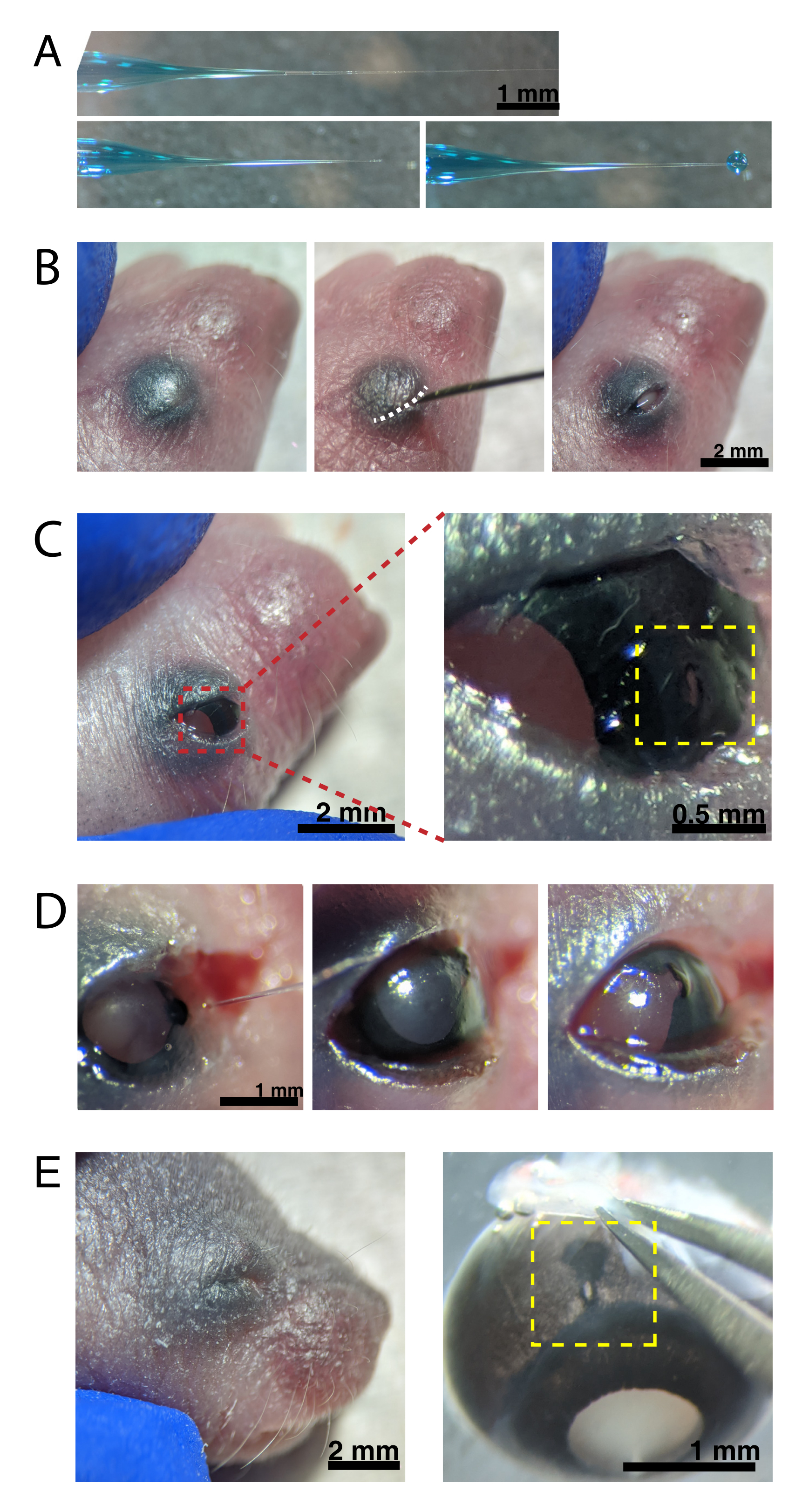{kind=link}
3. Dissecções de retina para experimento de imagem
- Prepare aCSF de retina (119 mM NaCl, 2,5 mM KCl, 1,3 mM MgCl2·6H2O, 2,5 mM CaCl2·2H2O, 1 mM NaH2PO4, 11 mM de glicose e 20 mM 4-(2-hidroxitil)-1-piperazineethanelfonic (hepes livre)ácido 35. Se desejar, prepare e congele a solução 10x; descongelar e diluir para 1x, conforme necessário.
- Oxigene a retina aCSF borbulhando com carbogen (95% O2, 5% CO2) por um mínimo de 15 min. Ajuste o pH para 7,4. Mantenha em um recipiente selado até o uso para garantir que o aCSF permaneça oxigenado.
NOTA: As retinas requerem uma alta concentração de O2. É importante manter oCSF oxigenado durante todo o experimento. - Incorpore uma placa de Petri de 60 mm de diâmetro em uma bandeja de gelo (forre uma bandeja de gelo de plástico de laboratório, por exemplo, uma tampa de caixa de pipeta), e coloque-a sob um estereoscópio. Encha a placa de Petri com aCSF de retina oxigenada.
- Eutanize ratos P9 e mais jovens por decapitação. Eutanize camundongos P10 ou mais velhos por indução de isoflurano, ou um método alternativo aprovado sob o protocolo animal, seguido de decapitação.
- Se as pálpebras estiverem seladas, corte a aba das pálpebras para expor o olho. Use fórceps para enuclear os olhos e transferi-los para a aCSF de retina fria a partir do passo 3.3.
- Para dissecar o copo de retina, sob um estereomicroscópio (ampliação 25x), estabilize o olho apertando o nervo óptico usando uma força Dumont #5 (Figura 3A).
- Faça um buraco no centro da córnea com uma agulha de 30 G e, em seguida, insira uma ponta dos microscissores no orifício para fazer uma incisão do orifício até o fim da córnea. Repita para fazer 4 fatias nas direções cardeais, criando 4 "retalhos" (Figura 3A).
- Com duas fórceps Dumont #5, segure e puxe os dois retalhos adjacentes, descascando suavemente a esclera da retina. Repita com os retalhos restantes da córnea e remova a esclera da retina (Figura 3B).
- Remova a lente do copo de retina usando os fórceps. Com microscisores, faça 4 incisões radiais da borda da retina em direção ao nervo óptico, criando 4 pétalas iguais (Figura 3B). Repita os passos 3.4-3.7 para o segundo olho.
4. Preparação de montagem plana de retina
- Prepare os discos de filtro de membrana de celulose mista cinza (MCE) para montagem. Se usar filtros de membrana MCE de grande diâmetro, corte o disco em quadrantes (aproximadamente 1 cm de diâmetro). Coloque o disco MCE no centro de um papel filtro branco maior (Figura 3D).
NOTA: Os filtros MCE também estão disponíveis em discos de 1 cm de diâmetro. O disco do filtro MCE deve ser grande o suficiente para caber retinas de 1-2, mas pequeno o suficiente para caber na câmara de imagem. Como as membranas MCE possuem uma carga estática, minimize o contato e o manuseio da membrana MCE. - Manuseando retinas usando dois pincéis de tinta tamanho 3/0, vire um copo de retina em um pincel com o lado da célula gânglio da retina para baixo. Retire suavemente a retina do aCSF, certificando-se de que a tensão da água não rasgue a retina (Figura 3C).
- Enquanto ainda segura o pincel com a retina, use uma pipeta de transferência para colocar uma gota de aCSF no centro do papel filtro MCE (Figura 3C, à direita). Flutue a retina para a gota de aCSF criada pela tensão superficial. Use pincéis de tinta para posicionar o lado RGC da retina dentro da gota e desdobrar as quatro pétalas.
- Uma vez posicionado, crie uma ponte de água entre o pincel e o papel filtro branco para quebrar a tensão superficial da gotícula.
NOTA: Quando a ACSF estiver perversa, a retina aderirá ao papel filtro MCE (Figura 3D,E). Retinas planas podem ser manuseadas agarrando o disco MCE com fórceps. Se a gotícula aCSF rapidamente se afastar antes de formar uma ponte de água, isso pode indicar que a membrana MCE não está carregada. Use uma membrana MCE fresca e minimize o tempo entre a enucleação e a imagem dissecando e montando retinas imediatamente antes de mover retinas para a câmara de imagem.

Figura 3: Dissecção de retina e montagem plana em membranas de filtro de éster de celulose mista. (A) À esquerda, um olho enucleado é estabilizado agarrando o nervo óptico com fórceps Dumont #5 (esquerda). Um pequeno buraco é criado no centro da córnea usando uma agulha de 30 G (centro). A tesoura de micro dissecção é usada para cortar a córnea em 4 retalhos iguais (direita). Barra de escala = 1 mm. (B) Retina dissecada com a esclera descascada usando duas fórceps Dumont #5 (esquerda) e com a lente removida (centro). Retina com 4 cortes iguais no meio da retina (direita). Barra de escala = 1 mm. (C) Manuseio da retina com duas pincéis finas (tamanho 3/0, esquerda). Retina virada com células de gânglios de retina de lado para baixo em um pincel (centro) e levantada para fora do ACSF, certificando-se de que a tensão da água não rasga a retina (direita). Barra de escala = 2 mm. (D) Disco de membrana MCE cinza colocado em um papel filtro branco (esquerda). Uma gota de aCSF no disco MCE (à direita). Barra de escala = 1 cm. (E) Depois de flutuar as retinas na gotícula e posicioná-las, crie uma ponte de água com o papel filtro branco para pavio do aCSF, puxando a retina para o papel MCE carregado (esquerda); barra de escala = 1 cm. Imagem ampliada da membrana MCE (vermelha) mostra 2 retinas montadas com células gânglios retinais lado para cima (direita); barra de escala = 2 mm. Uma retina é delineada em branco. Abreviaturas: MCE = éster de celulose mista; aCSF = fluido cerebrospinal artificial. Clique aqui para ver uma versão maior desta figura.
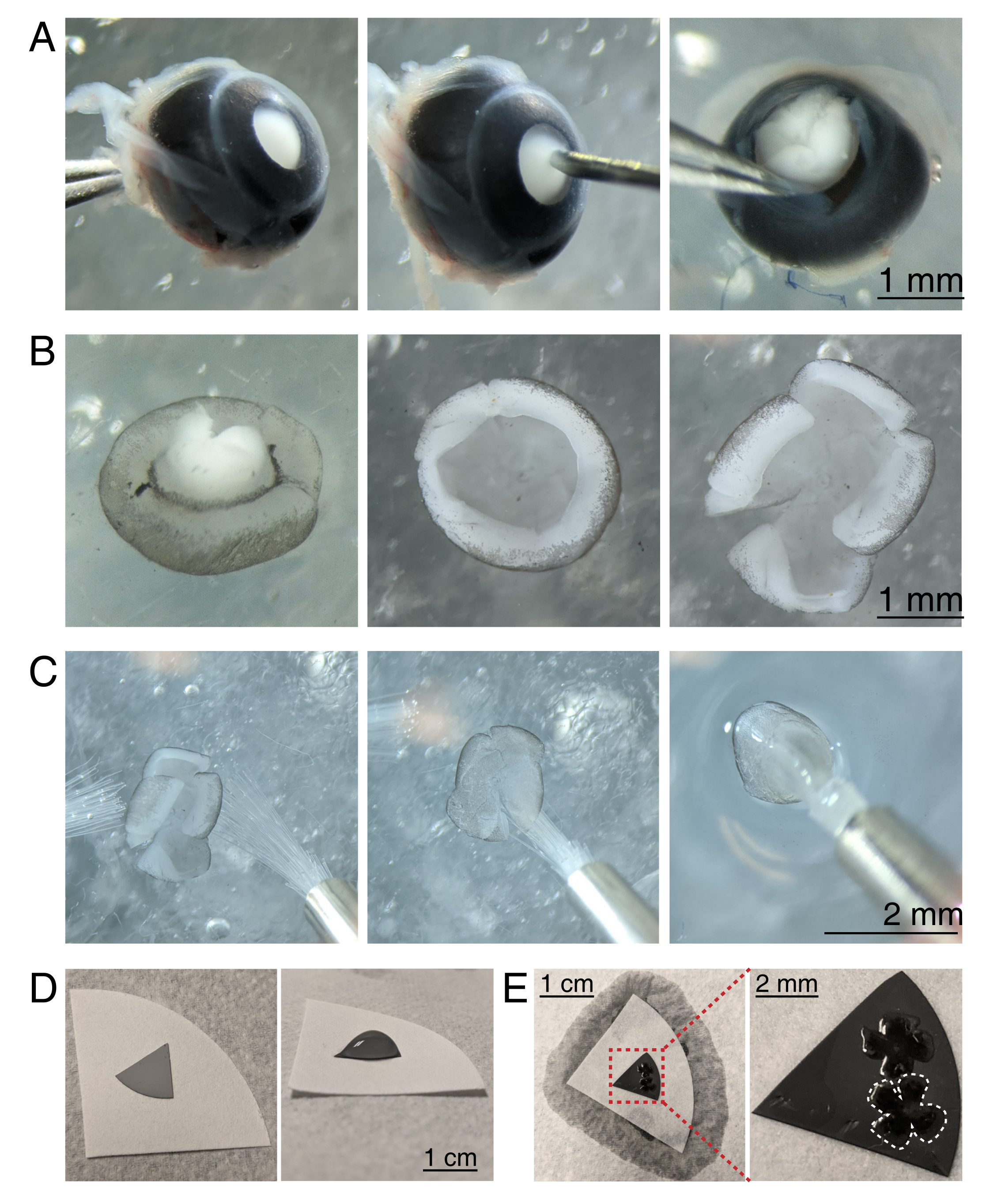{kind=link}
5. Imagem confocal de lapso de tempo de preparações de retina ao vivo
- Monte a câmara de incubação de imagens ao vivo para um microscópio confocal vertical, como visto na Figura 4.
NOTA: Para sistemas confocal invertidos, as montagens planas são colocadas do lado RGC diretamente para baixo na tampa do fundo de vidro da câmara de incubação. Uma vez que as retinas fazem contato com o deslizamento, elas não podem ser movidas. - Encha a câmara com aCSF oxigenado e ligue a bomba e o controlador de temperatura (temperatura de 32-34 °C, taxa de fluxo de 1 mL/min). Não permita que a temperatura suba acima de 34 °C.
- Para transferir o suporte plano da retina para a câmara de perfusão, pare a bomba e remova o aCSF que está na câmara. Coloque o disco MCE com o suporte plano de retina na câmara de incubação (vazia).
- Coloque um peso amostral sobre o montagem plana; pré-molhar o peso para quebrar a tensão superficial. Reabastecer a câmara com o aCSF aquecido e circular aCSF a ~1 mL/min.
- Posicione a peça do nariz com o objetivo de imersão de água de 25x (abertura numérica 0,95) na câmara de imagem. Tela para células rotuladas de interesse usando luz epifluorescente (Figura 4C).
- Ajuste o volume de imagem para capturar características deníticas de interesse.
NOTA: Este estudo capturou a árvore dendrítica completa em 1024 x 1024 pixels por quadro, z-passo 1 μm e uma taxa de quadro de 2 minutos entre cada pilha z. Os tamanhos finais da imagem são ~100 μm x 100 μm x 20 μm. - Para ajustar a potência do laser a uma configuração ideal, use uma tabela de procuração que identifique pixels supersaturados e subsaturados. Durante a varredura, ajuste a potência do laser de modo que nenhum pixel esteja sobresaturado (ou seja, a uma intensidade de 255 ou mais). Continue a imagem pelo tempo necessário, ou até que haja um declínio significativo e detectável no sinal fluorescente e aumente o ruído (tipicamente 2-4 h).
NOTA: A potência a laser reduzida é recomendada, pois os algoritmos de desconvolução funcionam de forma ideal quando os pixels são distribuídos ao longo de todo o alcance dinâmico. A intensidade do pixel não deve exceder 254; análises empíricas de neurites revelaram que os valores dos pixels abaixo de 170 são ideais para a desconvolução. Velocidades de varredura rápidas (400-600 Hz) com média de linha (2-3) são preferíveis a varreduras únicas e mais lentas do mesmo tempo total de habitação de pixels. A área de imagens prolongadas muitas vezes fotobleaches, mas outras seções de explant permanecem viáveis. Várias regiões em um suporte plano podem ser imagens, cada uma por 2-4 h, com um tempo total de incubação de 6 horas. Sessões de imagem além de 6h não foram sistematicamente testadas. A degradação e a brancação da neurita são sinais de que a viabilidade da explant está diminuindo. - Após a imagem, fixe os suportes planos de retina e o filtro de membrana com paraformaldeído frio de 4% por 1-2 h a 4 °C. Amplificar os rótulos fluorescentes nas retinas fixas por imunohistoquímica para análises posteriores.
NOTA: A fixação pós-imagem não é possível ao usar um confocal invertido; retinas não são removíveis sem destruir o tecido.

Figura 4: Configuração da câmara de incubação de imagens de células vivas. (A) Aparelho de incubação de imagens ao vivo mostrando componentes de aquecimento, solução e imagem. O aquecedor duplo inclui uma plataforma de câmara de incubação aquecida (círculo traseiro com eletrodos de conector azul) e um aquecedor de solução em linha. (B) Diagrama esquemático de 4A. O suporte plano de retina (vermelho) na membrana MCE (cinza) é colocado na câmara de incubação. O aquecedor de solução em linha está conectado a uma bomba de solução (não retratado em 4A). As dimensões da câmara de imagem permitem uma boa área de trabalho para o nariz objetivo de mergulho. (C) 25x ver uma explanta de retina injetada com 0,23-0,45 μL de 4 × 1012 GC/mL de diluição de AAV; barra de escala = 100 μm. Rotulagem densa de células muitas vezes envolve a cabeça do nervo óptico (linha tracejada, inferior direita); barra de escala = 50 μm. Abreviaturas: MCE = éster de celulose mista; GC = cópias do genoma; mCherry = cereja monomérica; eYFP = proteína fluorescente amarela aprimorada. Clique aqui para ver uma versão maior desta figura.
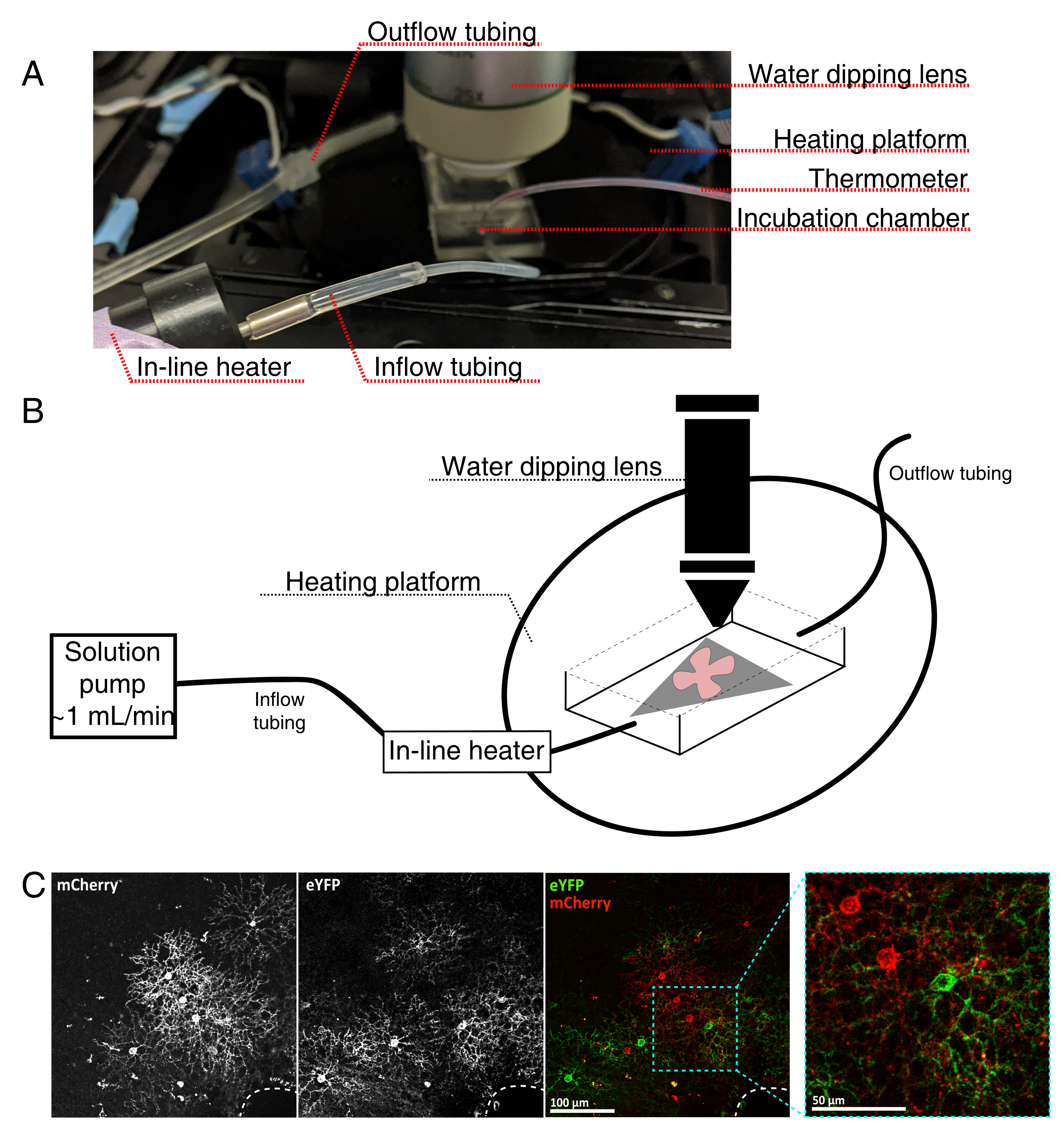{kind=link}
6. Desconvolução de imagem e pós-processamento no ImageJ
- Importe a série de imagens, divida a série por tempo e colora se for uma imagem multicolorida. Certifique-se de que todos os pontos de tempo para uma cor estão contidos na mesma pasta.
NOTA: Importe bioformes se usar ImageJ (Bio-formats é automaticamente incluído no FIJI). - Crie uma função teórica de propagação de ponto (PSF) usando o plugin ImageJ, Diffraction PSF 3D.
NOTA: Cada canal de imagem requer seu próprio PSF, pois o comprimento de onda de emissão de proteína fluorescente impacta o PSF. - Use plugins | Macros | Execute para executar o lote Deconvolução Iterativa Paralela para todos os pontos de tempo usando a macro fornecida (Arquivo Suplementar 1).
NOTA: Esta macro executa 25 iterações do algoritmo iterativo Landweber (WPL) pré-condicionado do filtro Wiener. Cada canal de cores deve ser desconvolvizado separadamente. - Mescle os canais de cores e compile todos os pontos de tempo em um Hyperstack (Image | Hyperstacks | Stacks to Hyperstack). Corrija a deriva 3D usando plugins | | de inscrição Corrija o drift 3D.
- Retorne a imagem para uma pilha regular (| de imagem Hyperstacks | Hyperstack para Stack) e divida os pontos de tempo (Imagem | Pilhas | Ferramentas | Stack Splitter). Use o processamento em lote para criar projeção máxima para todos os pontos de tempo (Processe | | de lote macro | executar ("Projeto Z...", "projeção=[Intensidade Máxima]"); ).
- Importe a sequência de imagem de lapso de tempo (| de arquivos | de importação Sequência de imagem). Use ferramentas convencionais imageJ para análise desejada de vídeo bidimensional (2D) desconvolved e pós-processado.
NOTA: Vídeos desconvolvados e corrigidos à deriva podem ser vistos como um hipersaco. Omitir a etapa anterior para manter os pontos de tempo 3D.
Resultados
Usando o protocolo acima, um vídeo 3D de alta resolução de dendritos de células de explosão estelar em desenvolvimento foi adquirido, desconvolved e corrigido para deriva 3D. Foram produzidas projeções máximas de plano Z para fazer vídeos 2D para análise (Vídeo Suplementar 1, Figura 5A). A desconvolução 3D de cada ponto de tempo aumentou a resolução de projeções finas de filopodia (Figura 5B,C). Saliências fina...
Discussão
Este vídeo demonstra um pipeline experimental que utiliza ferramentas genéticas existentes para a dinâmica dendrite de imagem do desenvolvimento de neurônios da retina com imagens ao vivo confocal. Também são demonstradas injeções intraoculares de AAVs dependentes de Cre codificando proteínas fluorescentes direcionadas à membrana em camundongos neonatais. Células únicas de populações geneticamente direcionadas são brilhantemente rotuladas tão cedo quanto 4-5 dpi. Os suportes planos de retina foram prepara...
Divulgações
Os autores não têm nada a revelar.
Agradecimentos
Agradecemos a Madison Gray por me ajudar quando eu não tinha nenhuma. Esta pesquisa foi apoiada por um NSERC Discovery Grant (RGPIN-2016-06128), uma Sloan Fellowship in Neuroscience e uma Cadeira de Pesquisa do Canadá Nível 2 (para J.L.L). S. Ing-Esteves foi apoiado pelo Vision Science Research Program e NSERC Postgraduate Scholarships-Doctoral.
Materiais
| Name | Company | Catalog Number | Comments |
| Addgene viral prep #45185-AAV9 | |||
| Addgene viral prep #45186-AAV9 | |||
| Dissection tools | |||
| Cellulose filter paper | Whatman | 1001-070 | |
| Dumont #5 fine forceps | FST | 11252-20 | Two Dumont #5 forceps are required for retinal micro-dissection |
| Dumont forceps | VWR | 82027-426 | |
| Fine Scissors | FST | 14058-09 | |
| Mixed cellulose ester membrane (MCE) filter papers, hydrophilic, 0.45 µm pore size | Millipore | HABG01 300 | |
| Petri Dish, 50 × 15 mm | VWR | 470313-352 | |
| Polyethylene disposable transfer pipette | VWR | 470225-034 | |
| Round tip paint brush, size 3/0 | Conventional art supply store | Two size 3/0 paint brushes (or smaller) are required for retinal flat-mounting | |
| Surgical Scissors | FST | 14007-14 | |
| Vannas Spring Scissors - 2.5 mm Cutting Edge | FST | 15000-08 | |
| Live-imaging incubation system | |||
| Chamber polyethylene tubing, PE-160 10' | Warner Instruments | 64-0755 | |
| Dual channel heater controller, Model TC-344C | Warner Instruments | 64-2401 | |
| HC FLUOTAR L 25x/0.95 W VISIR dipping objective | Leica | 15506374 | |
| Heater controller cable | Warner Instruments | CC-28 | |
| Large bath incubation chamber with slice support | Warner Instruments | RC-27L | |
| MPII Mini-Peristaltic Pump | Harvard Apparatus | 70-2027 | |
| PM-6D Magnetic Heated Platform (incubation chamber heater) | Warner Instruments | PM-6D | |
| Pump Head Tubing Pieces For MPII Mini-Peristaltic Pump | Harvard Apparatus | 55-4148 | |
| Sample anchor (Harps) | Warner Instruments | 64-0260 | Sample anchor must be compatible with incubation chamber |
| Sloflo In-line Solution Heater | Warner Instruments | SF-28 | |
| Neonatal Injections | |||
| 10 µL Microliter Syringe Series 700, Removable Needle | Hamilton Company | 80314 | |
| 30 G Hypodermic Needles (0.5 inch) | BD PrecisionGlide | 305106 | |
| 4 inch thinwall glass capillary, no filament (1.0 mm outer diameter/0.75 mm) | WPI World Precision Instruments | TW100-4 | |
| Ethanol 99.8% (to dilute to 70% with double-distilled water [ddH2O]) | Sigma-Aldrich | V001229 | |
| AAV9.hEF1a.lox.TagBFP. lox.eYFP.lox.WPRE.hGH-InvBYF | Penn Vector Core | AV-9-PV2453 | Addgene Plasmid #45185 |
| AAV9.hEF1a.lox.mCherry.lox.mTFP 1.lox.WPRE.hGH-InvCheTF | Penn Vector Core | AV-9-PV2454 | Addgene Plasmid #45186 |
| ChAT-IRES-Cre knock-in transgenic mouse line | The Jackson Laboratory | 6410 | |
| Fast Green FCF Dye content ≥85 % | Sigma-Aldrich | F7252-25G | |
| Flaming/Brown Micropipette Puller, model P-97 | Sutter Instrument Co. | P-97 | |
| Green tattoo paste | Ketchum MFG Co | 329A | |
| Phosphate-Buffered Saline, pH 7.4, liquid, sterile-filtered, suitable for cell culture | Sigma-Aldrich | 806552 | |
| Pneumatic PicoPump | WPI World Precision Instruments | PV-820 | |
| Oxygenated artifiial cerebrospinal fluid (aCSF) Reagents | |||
| Calcium chloride dihydrate (CaCl2·2H2O) | Sigma-Aldrich | C7902 | |
| Carbogen (5% CO2, 95% O2) | AirGas | X02OX95C2003102 | Supplier may vary depending on region |
| D-(+)-Glucose | Sigma-Aldrich | G7021 | |
| HEPES, Free Acid | Bio Basic | HB0264 | |
| Hydrochloric acid solution, 1 N | Sigma-Aldrich | H9892 | |
| Magnesium chloride hexahydrate (MgCl2·6H2O) | Sigma-Aldrich | M2670 | |
| pH-Test strips (6.0-7.7) | VWR | BDH35317.604 | |
| Potassium chloride (KCl) | Sigma-Aldrich | P9541 | |
| Sodium chloride (NaCl) | Bio Basic | DB0483 | |
| Sodium phosphate monobasic (NaH2PO4) | Sigma-Aldrich | RDD007 | |
| Software | |||
| ImageJ | National Institutes of Health (NIH) | Open source |
Referências
- Lefebvre, J. L., Sanes, J. R., Kay, J. N. Development of dendritic form and function. Annual Review of Cell and Developmental Biology. 31, 741-777 (2015).
- Graham, H. K., Duan, X. Molecular mechanisms regulating synaptic specificity and retinal circuit formation. Wiley Interdisciplinary Reviews Developmental biology. 10 (1), 379 (2021).
- Godinho, L., et al. Targeting of amacrine cell neurites to appropriate synaptic laminae in the developing zebrafish retina. Development. 132 (22), 5069-5079 (2005).
- Mumm, J. S., et al. In vivo imaging reveals dendritic targeting of laminated afferents by zebrafish retinal ganglion cells. Neuron. 52 (4), 609-621 (2006).
- Wong, W. T., Faulkner-Jones, B. E., Sanes, J. R., Wong, R. O. Rapid dendritic remodeling in the developing retina: dependence on neurotransmission and reciprocal regulation by Rac and Rho. The Journal of Neuroscience. 20 (13), 5024-5036 (2000).
- Wei, W., Elstrott, J., Feller, M. B. Two-photon targeted recording of GFP-expressing neurons for light responses and live-cell imaging in the mouse retina. Nature Protocols. 5 (7), 1347-1352 (2010).
- Morgan, J. L., Wong, R. O. L. Ballistic labeling with fluorescent dyes and indicators. Current Protocols in Neuroscience. 43 (1), 1-10 (2008).
- Nickerson, P. E. B., et al. Live imaging and analysis of postnatal mouse retinal development. BMC Developmental Biology. 13, 24 (2013).
- Morgan, J. L., Dhingra, A., Vardi, N., Wong, R. O. L. Axons and dendrites originate from neuroepithelial-like processes of retinal bipolar cells. Nature Neuroscience. 9 (1), 85-92 (2006).
- Coombs, J. L., Van Der List, D., Chalupa, L. M. Morphological properties of mouse retinal ganglion cells during postnatal development. The Journal of Comparative Neurology. 503 (6), 803-814 (2007).
- Cranfill, P. J., et al. Quantitative assessment of fluorescent proteins. Nature Methods. 13 (7), 557-562 (2016).
- Stacy, R. C., Wong, R. O. L. Developmental relationship between cholinergic amacrine cell processes and ganglion cell dendrites of the mouse retina. The Journal of Comparative Neurology. 456 (2), 154-166 (2003).
- Kay, J. N., et al. Retinal ganglion cells with distinct directional preferences differ in molecular identity, structure, and central projections. The Journal of Neuroscience. 31 (21), 7753-7762 (2011).
- Liu, J., Sanes, J. R. Cellular and molecular analysis of dendritic morphogenesis in a retinal cell type that senses color contrast and ventral motion. The Journal of Neuroscience. 37 (50), 12247-12262 (2017).
- Diao, L., Sun, W., Deng, Q., He, S. Development of the mouse retina: emerging morphological diversity of the ganglion cells. Journal of Neurobiology. 61 (2), 236-249 (2004).
- Coombs, J., vander List, D., Wang, G. Y., Chalupa, L. M. Morphological properties of mouse retinal ganglion cells. Neuroscience. 140 (1), 123-136 (2006).
- Sanes, J. R., Masland, R. H. The types of retinal ganglion cells: current status and implications for neuronal classification. Annual Review of Neuroscience. 38, 221-246 (2015).
- Sümbül, U., et al. A genetic and computational approach to structurally classify neuronal types. Nature Communications. 5, 3512 (2014).
- Lin, B., Masland, R. H. Populations of wide-field amacrine cells in the mouse retina. The Journal of Comparative Neurology. 499 (5), 797-809 (2006).
- Macneil, M. A., Heussy, J. K., Dacheux, R. F., Raviola, E., Masland, R. H. The shapes and numbers of amacrine cells: Matching of photofilled with Golgi-stained cells in the rabbit retina and comparison with other mammalian species. Journal of Comparative Neurology. 413 (2), 305-326 (1999).
- Ivanova, E., Hwang, G. S., Pan, Z. H. Characterization of transgenic mouse lines expressing Cre recombinase in the retina. Neuroscience. 165 (1), 233-243 (2010).
- Jo, A., Xu, J., Deniz, S., Cherian, S., DeVries, S. H., Zhu, Y. Intersectional strategies for targeting amacrine and ganglion cell types in the mouse retina. Frontiers in Neural Circuits. 12, 66 (2018).
- Siegert, S., et al. Genetic address book for retinal cell types. Nature Neuroscience. 12 (9), 1197-1204 (2009).
- Kim, I. -. J., Zhang, Y., Meister, M., Sanes, J. R. Laminar restriction of retinal ganglion cell dendrites and axons: subtype-specific developmental patterns revealed with transgenic markers. The Journal of Neuroscience. 30 (4), 1452-1462 (2010).
- Peng, Y. -. R., Tran, N. M., Krishnaswamy, A., Kostadinov, D., Martersteck, E. M., Sanes, J. R. Satb1 regulates contactin 5 to pattern dendrites of a mammalian retinal ganglion cell. Neuron. 95 (4), 869-883 (2017).
- Duan, X., Krishnaswamy, A., Dela Huerta, I., Sanes, J. R. Type II cadherins guide assembly of a direction-selective retinal circuit. Cell. 158 (4), 793-807 (2014).
- Ray, T. A., et al. Formation of retinal direction-selective circuitry initiated by starburst amacrine cell homotypic contact. eLife. 7, 34241 (2018).
- Krishnaswamy, A., Yamagata, M., Duan, X., Hong, Y. K., Sanes, J. R. Sidekick 2 directs formation of a retinal circuit that detects differential motion. Nature. 524 (7566), 466-470 (2015).
- Caval-Holme, F., Zhang, Y., Feller, M. B. Gap junction coupling shapes the encoding of light in the developing retina. Current Biology. 29 (23), 4024-4035 (2019).
- Lucas, J. A., Schmidt, T. M. Cellular properties of intrinsically photosensitive retinal ganglion cells during postnatal development. Neural Development. 14 (1), 8 (2019).
- Cai, D., Cohen, K. B., Luo, T., Lichtman, J. W., Sanes, J. R. Improved tools for the Brainbow toolbox. Nature Methods. 10 (6), 540-547 (2013).
- Rossi, J., et al. Melanocortin-4 receptors expressed by cholinergic neurons regulate energy balance and glucose homeostasis. Cell Metabolism. 13 (2), 195-204 (2011).
- Lefebvre, J. L., Kostadinov, D., Chen, W. V., Maniatis, T., Sanes, J. R. Protocadherins mediate dendritic self-avoidance in the mammalian nervous system. Nature. 488 (7412), 517-521 (2012).
- Ing-Esteves, S., et al. Combinatorial effects of alpha- and gamma-protocadherins on neuronal survival and dendritic self-avoidance. The Journal of Neuroscience. 38 (11), 2713-2729 (2018).
- Williams, P. R., Morgan, J. L., Kerschensteiner, D., Wong, R. O. L. In vitro imaging of retinal whole mounts. Cold Spring Harbor Protocols. 2013 (1), (2013).
- Ramoa, A. S., Campbell, G., Shatz, C. J. Transient morphological features of identified ganglion cells in living fetal and neonatal retina. Science. 237 (4814), 522-525 (1987).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 9, 676-682 (2012).
- Peng, H., Ruan, Z., Long, F., Simpson, J. H., Myers, E. W. V3D enables real-time 3D visualization and quantitative analysis of large-scale biological image data sets. Nature Biotechnology. 28 (4), 348-353 (2010).
- Cuntz, H., Forstner, F., Borst, A., Häusser, M. One rule to grow them all: a general theory of neuronal branching and its practical application. PLoS Computational Biology. 6 (8), 1000877 (2010).
- Xiao, H., Peng, H. APP2: automatic tracing of 3D neuron morphology based on hierarchical pruning of a gray-weighted image distance-tree. Bioinformatics. 29 (11), 1448-1454 (2013).
- Nanda, S., et al. Design and implementation of multi-signal and time-varying neural reconstructions. Scientific data. 5, 170207 (2018).
- Sherry, D. M., Wang, M. M., Bates, J., Frishman, L. J. Expression of vesicular glutamate transporter 1 in the mouse retina reveals temporal ordering in development of rod vs. cone and ON vs. OFF circuits. The Journal of Comparative Neurology. 465 (4), 480-498 (2003).
- Johnson, J., et al. Vesicular neurotransmitter transporter expression in developing postnatal rodent retina: GABA and glycine precede glutamate. The Journal of Neuroscience. 23 (2), 518-529 (2003).
- Jüttner, J., et al. Targeting neuronal and glial cell types with synthetic promoter AAVs in mice, non-human primates and humans. Nature Neuroscience. 22 (8), 1345-1356 (2019).
- Zincarelli, C., Soltys, S., Rengo, G., Rabinowitz, J. E. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Molecular Therapy. 16 (6), 1073-1080 (2008).
- Petros, T. J., Rebsam, A., Mason, C. A. In utero and ex vivo electroporation for gene expression in mouse retinal ganglion cells. Journal of Visualized Experiments: JoVE. (31), e1333 (2009).
- Lye, M. H., Jakobs, T. C., Masland, R. H., Koizumi, A. Organotypic culture of adult rabbit retina. Journal of Visualized Experiments: JoVE. (3), e190 (2007).
- Pignatelli, V., Strettoi, E. Bipolar cells of the mouse retina: a gene gun, morphological study. The Journal of Comparative Neurology. 476 (3), 254-266 (2004).
- Huckfeldt, R. M., et al. Transient neurites of retinal horizontal cells exhibit columnar tiling via homotypic interactions. Nature Neuroscience. 12 (1), 35-43 (2009).
- Prahst, C., et al. Mouse retinal cell behaviour in space and time using light sheet fluorescence microscopy. eLife. 9, 49779 (2020).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados