
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Reactivación de células madre neurales en explantes cerebrales de Drosophila cultivados
En este artículo
Resumen
Se ha establecido un método para reactivar las células madre neurales inactivas en explantes cerebrales de Drosophila cultivados. Usando este método, el papel de las señales sistémicas se puede desacoplar de las señales intrínsecas del tejido en la regulación de la inactividad, entrada y salida de las células madre neurales.
Resumen
Las células madre neurales (NSC) tienen la capacidad de proliferar, diferenciarse, sufrir apoptosis e incluso entrar y salir de la inactividad. Muchos de estos procesos están controlados por la compleja interacción entre los programas genéticos intrínsecos del NSC con los factores extrínsecos del NSC, locales y sistémicos. En el organismo modelo genético, Drosophila melanogaster, las NSC, conocidas como neuroblastos (NB), cambian de la inactividad a la proliferación durante la transición embrionaria a larvaria. Durante este tiempo, las larvas emergen de sus cáscaras de huevo y comienzan a gatear, buscando nutrientes dietéticos. En respuesta a la alimentación animal, el cuerpo graso, un órgano endocrino con capacidad de almacenamiento de lípidos, produce una señal, que se libera sistémicamente en la hemolinfa circulante. En respuesta a la señal derivada del cuerpo graso (FBDS), los péptidos similares a la insulina Drosophila (Dilps) se producen y liberan de las neuronas neurosecretoras cerebrales y la glía, lo que lleva a la activación posterior de la señalización de crecimiento de PI3-quinasa en NB y su nicho glial y traqueal. Aunque este es el modelo actual de cómo los NB cambian de la inactividad a la proliferación, la naturaleza de la señal extrínseca FBDS sigue siendo difícil de alcanzar. Para comprender mejor cómo las señales sistémicas extrínsecas NB regulan la salida de la inactividad, se desarrolló un método para cultivar cerebros larvarios tempranos in vitro antes de la alimentación animal. Con este método, se pueden suministrar factores exógenos a los medios de cultivo y se puede ensayar la salida de NB de la inactividad. Encontramos que la insulina exógena es suficiente para reactivar los NB de la inactividad en los explantes de todo el cerebro. Debido a que este método es adecuado para pantallas a gran escala, nuestro objetivo es identificar señales extrínsecas adicionales que regulen la inactividad NB frente a las decisiones de proliferación. Debido a que los genes y las vías que regulan las decisiones de proliferación de NSC se conservan evolutivamente, los resultados de este ensayo podrían proporcionar información sobre la mejora de las terapias regenerativas en la clínica.
Introducción
Las células madre son de gran interés por su potencial de uso en medicina regenerativa 1,2. Muchos animales, especialmente aquellos que son de larga vida, mantienen células madre dentro de sus tejidos adultos. Estas células madre residentes funcionan para mantener la homeostasis tisular y se utilizan para la reparación después de una lesión física o enfermedad 3,4. La mayoría de las células madre en animales adultos están inactivas, un estado relativamente latente caracterizado por la detención del ciclo celular y la inactivación de la señalización del crecimiento5. En respuesta a las señales extrínsecas, las células madre salen de la inactividad, entran en el ciclo celular y comienzan a generar una progenie hija específica para su tipo de tejido. Por ejemplo, para montar una respuesta inmune efectiva, las células presentadoras de antígenos inducen a las células T ingenuas inactivas a entrar en el ciclo celular y expandirse clonalmente6. En respuesta al daño del músculo esquelético, las células madre satélite musculares entran en el ciclo celular y generan mioblastos hijos para reemplazar las miofibrillas dañadas 5,7. Si bien está claro que las células madre inactivas responden a las señales extrínsecas, en muchos casos, la naturaleza de la señal extrínseca sigue sin estar clara, así como el mecanismo de activación de las células madre inducida por la señal. Obtener una mejor comprensión de cómo las células madre inactivas responden a las señales extrínsecas y entran en el ciclo celular ayudará en el desarrollo de mejores terapias con células madre en la clínica y aumentará el conocimiento científico.
Durante décadas, los organismos modelo se han utilizado para descubrir los genes y las vías de señalización celular que regulan la proliferación de células madre durante el desarrollo y en la edad adulta. En Drosophila, las células madre neurales (NSC), conocidas como neuroblastos (NB), se dividen a lo largo del desarrollo para generar todas las neuronas y la glía que finalmente se integran, formando el circuito neuronal requerido para la función cerebral 8,9. Al igual que otras células madre, las NB se dividen asimétricamente para autorrenovarse y, en algunos casos, simétricamente para expandir el grupo de células madre. Los NB se especifican durante la embriogénesis y la mayoría entran en reposo hacia el final, coincidiendo con la disminución de las reservas maternas de nutrientes (Figura 1). Después de que se completa la embriogénesis, las larvas eclosionan y comienzan a alimentarse. En respuesta a la alimentación animal, los NB se reactivan de la inactividad y reanudan las divisiones celulares 10,11,12,13,14,15,16. Debido a que el SNC de Drosophila es relativamente simple y debido a que los NB entran y salen de la inactividad en momentos definidos, el uso de Drosophila para investigar la regulación de la inactividad, la entrada y la salida, resulta ideal.

Figura 1: Proliferación relativa de CB NB (neuroblastos cerebrales centrales, rojo) y MB NB (neuroblastos del cuerpo de hongos, azul) durante el tiempo de desarrollo. Al final de la embriogénesis, la mayoría de los NB (línea roja) cesan la proliferación y entran en reposo. La inactividad continúa hasta que las larvas recién eclosionadas consumen su primera comida completa. Los puntos temporales de enfoque para esta metodología se denotan en círculos rojos (1, inactividad y 2, reactivación). Los NB MB (azules) son un subconjunto de NB cerebrales centrales que se dividen continuamente a lo largo del desarrollo (4 por hemisferio cerebral). Haga clic aquí para ver una versión más grande de esta figura.
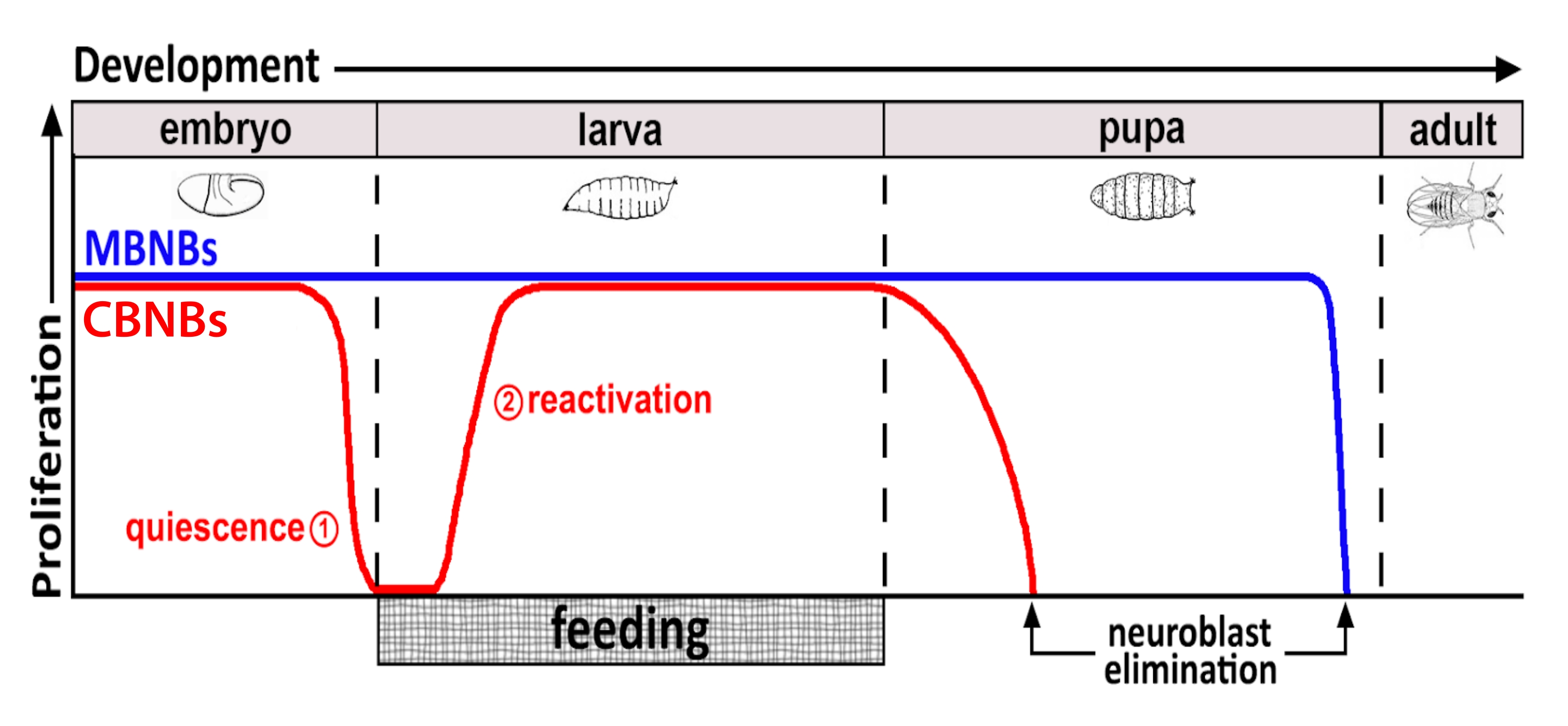{kind=link}
En respuesta a la alimentación animal, las vías de señalización de crecimiento pi3-quinasa y TOR se activan en los NB y en su nicho glial y traqueal 10,11,15,16. Cuando se retiran los nutrientes dietéticos o cuando se reducen los niveles de PI3-quinasa, los NB no se reactivan y el crecimiento de la glía y la tráquea también se reduce 10,11,15,16. El modelo actual postula que la reactivación de NB se acopla al crecimiento larvario por parte del cuerpo graso, que libera una señal sistémica en respuesta a la alimentación animal 12,17,18. Esta señal, que sigue siendo esquiva, probablemente promueve la expresión y liberación del péptido similar a la insulina Drosophila (Dilps) en el cerebro, lo que conduce a la activación aguas abajo de la PI3-quinasa en nbes y su nicho glial y traqueal. Para comprender mejor la naturaleza de las señales sistémicas, desarrollamos un método para reactivar los NB quiescentes en explantes cerebrales cultivados. Con este método, la reactivación de los NB se puede ensayar en ausencia de señales sistémicas animales completas. Los factores exógenos pueden ser reabastecidos a los medios de cultivo y la reactivación de NB ensayada en base a la incorporación del análogo de timidina, EdU. Usando este método, determinamos que la insulina exógena es suficiente para reactivar los NB quiescentes en los explantes cerebrales. El trabajo futuro estará dirigido a identificar factores adicionales que, cuando se agregan de nuevo, regulan positiva o negativamente la inactividad NB en los explantes cerebrales.
Protocolo
1. Recolección de larvas de Drosophila
NOTA: Prepare el plato de levadura, la pasta de uva y el condominio Fly antes de comenzar:
- Pasta de levadura: En un recipiente pequeño, mezcle 5 g de levadura seca activa con 10 ml de agua para formar una pasta que tenga la consistencia de la mantequilla de maní. Cubra la pasta de levadura con una envoltura de plástico y use una banda elástica para unirla firmemente al recipiente.
NOTA: La pasta de levadura fresca se expandirá en su recipiente y saldrá de la tapa a menos que esté firmemente unida. La pasta de levadura durará varios días a temperatura ambiente (RT). - Platos de uva: Siga la receta para hacer platos de uva (Tabla 1). Si usa placas almacenadas a 4 ° C, asegúrese de precalentar las placas antes de usarlas colocándolas en RT durante 1 h.
- Mezcle agua (750 ml) y agar (18,75 g) en un matraz de 4 L, remolino y autoclave durante 20 min (ciclo líquido).
- Mezcle el jugo de uva (250 ml) y la sacarosa (25 g) en un matraz de 1 L con una barra de agitación grande en un plato calentado (a fuego lento). Cuando la sacarosa se disuelva, apague el fuego, espere hasta que se pueda tocar el matraz antes de agregar Tegosept (10%, 4 ml) y ácido propiónico (5 ml). Mantenga la barra de agitación encendida.
- Cuando se complete el autoclave, deje que se enfríe hasta que se pueda tocar el matraz (~ 60 ° C), luego mezcle la mezcla de jugo de uva.
- Combine todas las soluciones en un matraz y deje remover en la placa.
- Pipetear la solución en tapas de placas de Petri de tamaño pequeño (35 mm). Pipetear aproximadamente 9 ml por tapa o hasta obtener una cúpula convexa.
- OPCIONAL: Flamear las tapas para deshacerse de cualquier burbuja.
- Cuando las placas se solidifiquen, apile las placas de uva en una caja con una tapa hermética y coloque la caja a 4 ° C. Las placas se pueden almacenar hasta por 1 mes.
- Condominio Fly: Perfore ~ 20 agujeros en una botella de polipropileno Drosophila de fondo cuadrado de 6 onzas con una aguja de 18 G.
- Transfiera moscas adultas (~ 100 OregonR o cualquier genotipo) a un condominio de moscas y cubra el condominio con un plato de agar de uva cubierto con una pizca de pasta de levadura. Coloque el dab hacia el centro de la placa y fije la placa al condominio con cinta de laboratorio.
- Invierta el recipiente para que el plato de agar de uva esté en el fondo y colóquelo en una incubadora de 25 °C durante 24 h (Figura 2).

Figura 2: Representación visual de la botella de mosca invertida (condominio) con adultos machos y hembras de Drosophila . La botella de plástico tiene pequeños pinchazos, generados con una aguja de 18 G, para el intercambio de oxígeno. La boca de la botella está sellada con una tapa de jugo de uva de agar y se invierte y se almacena en una incubadora de 25 ° C. Haga clic aquí para ver una versión más grande de esta figura.
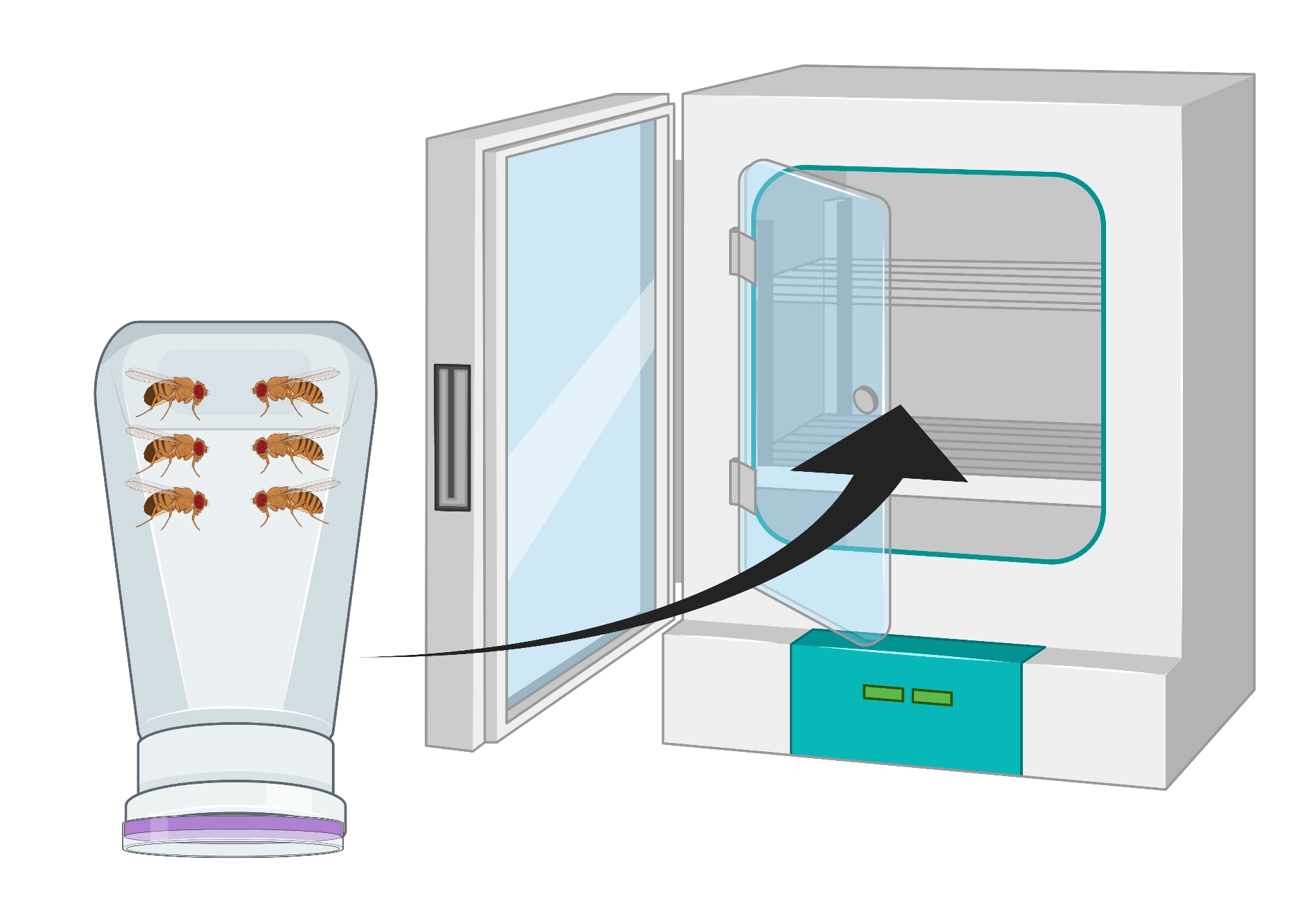{kind=link}
- Después de 24 h, cambie el plato de agar de uva y reemplácelo con un nuevo plato cubierto con pasta de levadura. Cambie rápidamente las dos placas mientras golpea continuamente el condominio ligeramente en el banco para que las moscas adultas no escapen.
- Examine la placa a ojo y evalúe el número de embriones en la placa. Los embriones de Drosophila son oblongos y blancos con dos apéndices en forma de cuerda.
- Si hay muy pocos embriones en el plato (menos de 100), deseche el plato (raspe el agar en la basura de moscas y guarde la tapa de plástico para su reutilización). En muchos casos, las hembras adultas no pondrán muchos embriones la primera noche en un nuevo condominio. Si este es el caso, dé a las moscas adultas otras 24 h para aclimatarse.
- Si hay una gran cantidad de embriones en la placa (al menos 100), manténgala y retire cuidadosamente la pasta de levadura con una espátula de fondo plano.
- Una vez que se retira la pasta de levadura, use una púa de metal para eliminar manualmente todas las larvas de la placa de uva bajo un microscopio de disección. Al observar la placa bajo un microscopio de disección, se deben observar larvas que se arrastran, así como embriones.
- Retire todas las larvas cepillando la púa de metal hacia el lado de una larva. Las larvas son pegajosas y se pegarán a la púa. Una vez que una larva está en la selección, las larvas adicionales se pueden recoger fácilmente usando la larva en la herramienta para unir más.
NOTA: A las larvas les gusta pegarse entre sí. En este punto, no importa si las larvas se dañan. Estas larvas serán descartadas. - Después de recoger y eliminar todas las larvas, coloque la placa de nuevo en la incubadora de 25 ° C. Asegúrese de colocar la placa en un recipiente más grande que pueda sellarse. Coloque toallas de papel mojadas en el fondo del recipiente más grande para mantener la humedad.
- Después de 30-60 min, lleve la placa de vuelta al microscopio de disección y ahora, recoja cuidadosamente ~ 20-25 larvas de la misma placa de agar de uva para asegurarse de que las larvas recolectadas estén recién eclosionadas dentro de un período de tiempo de 30-60 minutos.
- Sumerja la punta de la herramienta con las 20-25 larvas recién eclosionadas en una placa de Petri (60 mm) llena de 1-2 ml de solución salina tamponada con fosfato (PBS) durante 2 minutos.
- Después de 2 min, incline el plato en ángulo para acumular el líquido en la parte inferior. Usando un pincel pequeño, cepille las larvas del líquido hasta el fondo de la placa de Petri.
- Recolecte todas las larvas en el pincel y transfiera las larvas a una nueva placa de Petri (60 mm) que contenga 1-2 ml de etanol al 70%. Repita los pasos 1.15 para recolectar larvas con un pincel y transfiéralas a una nueva placa de Petri con 1-2 ml de 1x PBS.
2. Medios de cultivo y preparación de herramientas
- Rocíe el banco y el área de trabajo con etanol al 70% y deje secar.
- Rocíe las herramientas de disección, las pinzas y dos platos de reloj de vidrio, con etanol al 70% y déjelos secar en el banco.
- Haga los medios de Schneider suplementados (SSM, Tabla 2) y colóquelos sobre hielo.
- Pipete 1 ml de SSM en cada uno de los platos de reloj de vidrio.
- Usando una micropipeta con una punta estéril, transfiera las larvas recién eclosionadas de la placa de PBS al SSM en la primera placa de reloj de vidrio. Usando una micropipeta con una punta estéril, transfiera las larvas recién eclosionadas al SSM en el segundo plato de reloj de vidrio.
3. Disecciones y cultivos cerebrales
- Una vez que las larvas estén en el segundo plato de reloj de vidrio con SSM, diseccione los cerebros de las larvas usando fórceps y un microscopio de disección. Ajuste la ampliación según sea necesario.
- Use un fórceps para agarrar los ganchos de la boca y con el otro, agarre suavemente el cuerpo hasta la mitad hacia abajo y tire en la dirección opuesta (Figura 3) para dividir la larva en dos pedazos.
NOTA: El cerebro se ubicará justo detrás de los ganchos bucales. Tenga en cuenta que puede haber otros tejidos que rodean el cerebro. Tenga mucho cuidado al eliminar estos tejidos, ya que puede dañar el cerebro

Figura 3: Larvas de Drosophila en una placa de reloj de vidrio con SSM. Las pinzas están colocadas correctamente para la disección. La ubicación del cerebro larvario (gris oscuro) es posterior a los ganchos bucales (negro), y ambos se muestran dentro de la larva. Haga clic aquí para ver una versión más grande de esta figura.
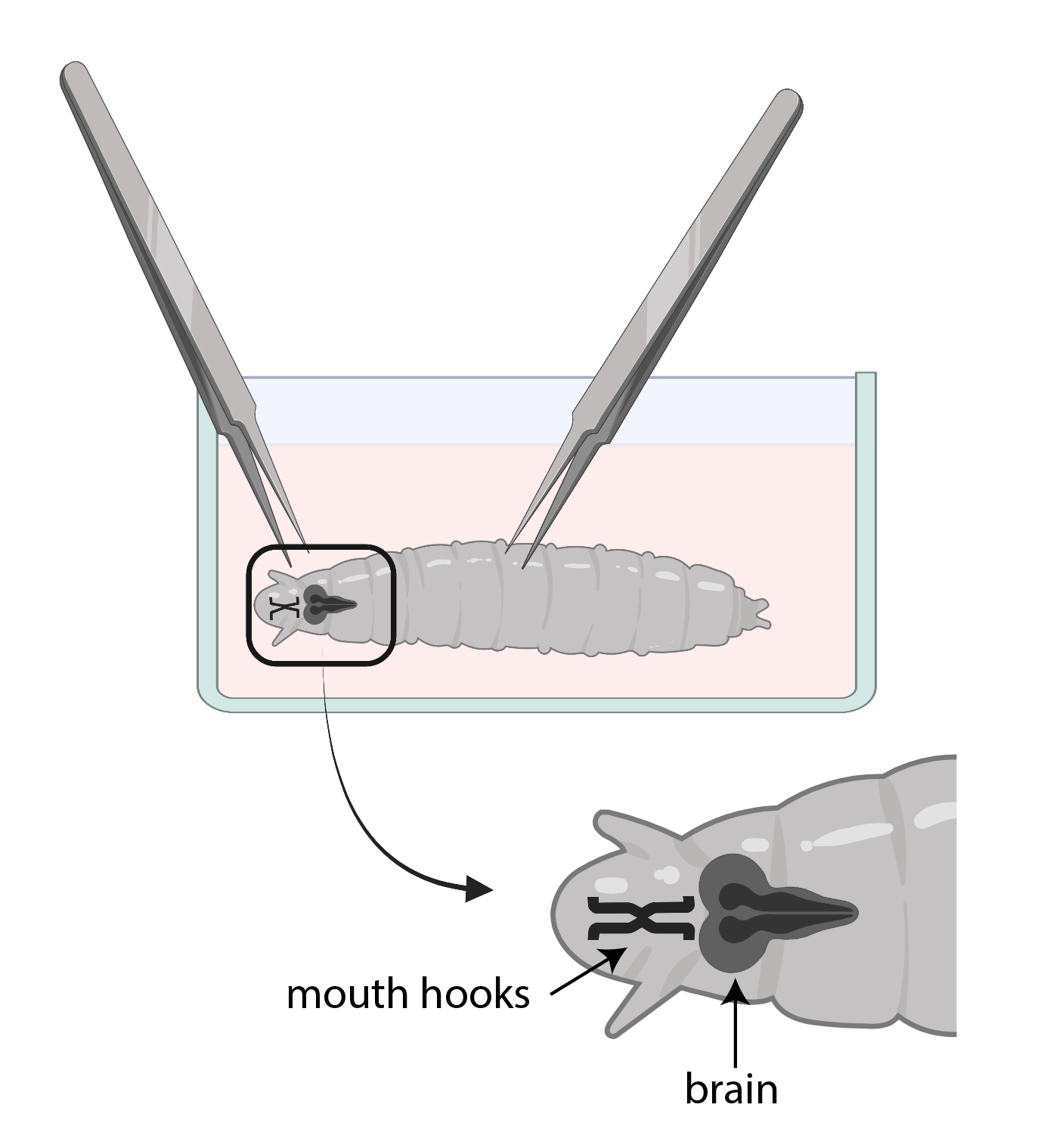{kind=link}
- Una vez que se hayan diseccionado 15-20 cerebros, agregue 1 ml de SSM en un pozo de una bandeja de cultivo estéril de 12 pocillos. Transfiera los cerebros recién diseccionados al SSM utilizando una micropipeta y una punta estéril (Figura 4A).
- Coloque los cerebros en el medio SSM en la bandeja de cultivo de 12 pocillos en una incubadora a 25 °C durante 24 h (Figura 4A).

Figura 4: Cultivo cerebral e inmunotinción. (A) Cerebros enteros en un plato de cultivo de 12 pocillos que contiene 1 ml de SSM. El plato de cultivo se coloca en una incubadora de 25 ° C durante 24 h. (B) Mini bandeja de 72 pocillos que contiene explantes cerebrales durante la inmunotinción. Los cerebros se lavan y las soluciones se transfieren utilizando una micropipeta P20 ajustada a 10 μL. Haga clic aquí para ver una versión más grande de esta figura.
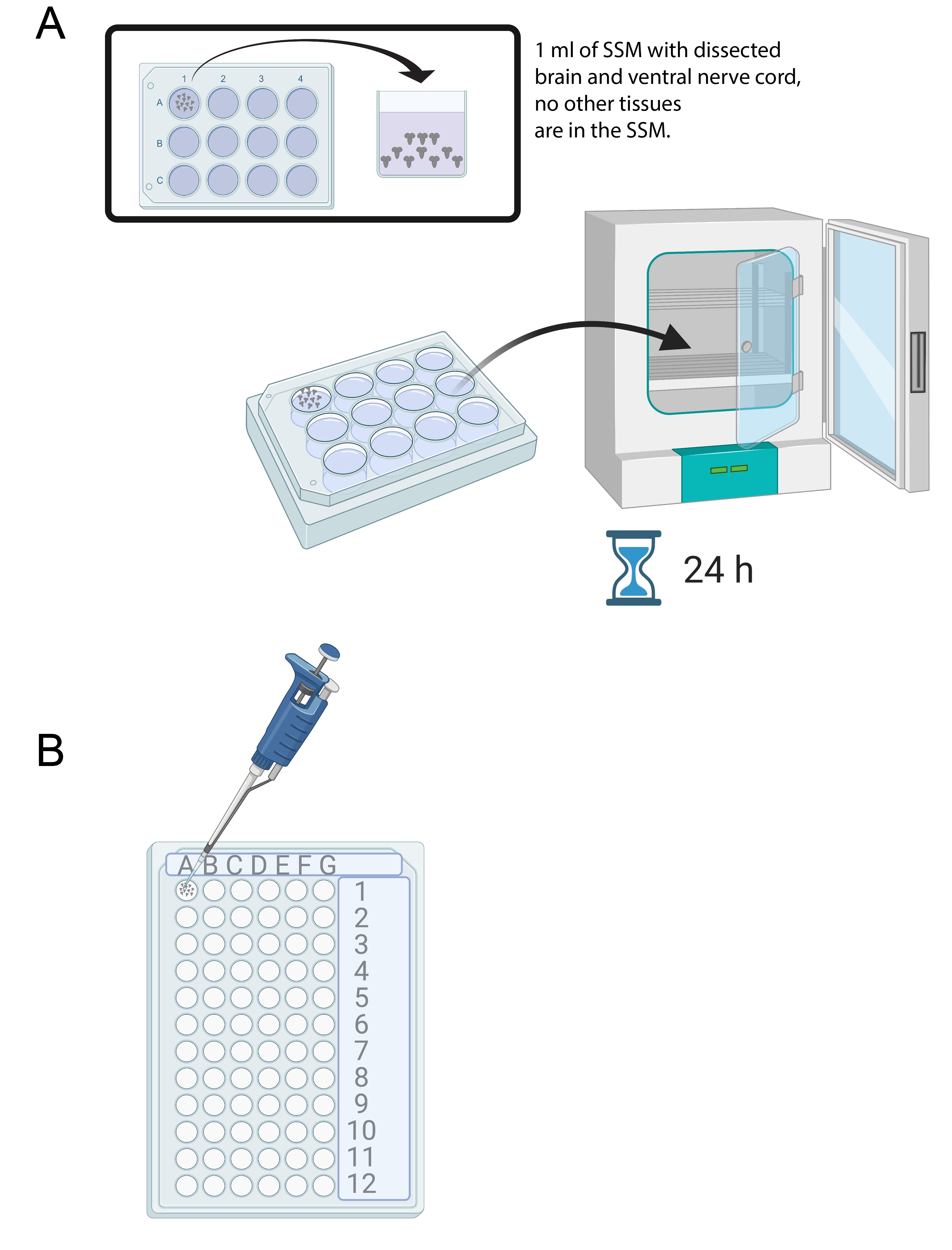{kind=link}
4. Ensayo de proliferación, fijación cerebral y tinción de anticuerpos
- Al día siguiente, haga 1 ml de solución EdU SSM. Pipetear 10 μL de una cepa de 10 mM de 5-etinil-2′-desoxiuridina (EdU) con 990 μL de SSM (la concentración final de EdU es igual a 0,1 mM) en un tubo de microcentrífuga estéril y mezclar. Después de completar la incubación de 24 h, pipetee el 1 ml de EdU SSM en un pozo de la bandeja de cultivo estéril de 12 pocillos.
- Transfiera los cerebros usando una micropipeta con una punta estéril del pozo que contiene SSM solo al nuevo pozo que contiene la solución EdU SSM. Incubar durante 1 h a 25 °C.
- A continuación, transfiera los cerebros marcados con EdU a otro pozo en la misma bandeja de cultivo que contenga 1 ml de fijador (4% de paraformaldehído, consulte la Tabla 3 para la receta) durante 20 min.
PRECAUCIÓN: El paraformaldehído es un peligro biológico y debe eliminarse adecuadamente. - Después de la fijación, transfiera rápidamente los cerebros a los pozos de una mini bandeja de 72 pocillos usando una micropipeta. Cada pozo puede contener 10 cerebros y 10-15 μL de líquido (Figura 4B). Una vez que los cerebros se transfieren a la mini bandeja (no más de 10 cerebros por pozo), retire la solución y enjuague los cerebros 3 veces en 10 μL de 1x PBT (tampón de fosfato, pH 7.4 que contiene 0.1% Triton-X 100).
NOTA: Enjuagar significa pipetear 10 μL de 1x PBT en los cerebros, eliminar y repetir 3 veces. - A continuación, lave los cerebros 3 veces durante 10 minutos cada uno, nuevamente en 10 μL 1x PBT. Asegúrese de que los cerebros estén cubiertos con un poco de líquido en todo momento.
- Después de completar los lavados, pipetee 10 μL de solución bloqueadora (1x PBT con 10% de suero de cabra normal) en el cerebro. Cubra la bandeja y séllela con una tira de parafilm alrededor del borde.
- Una vez sellada, coloque la mini bandeja en una caja sellada con toallas húmedas para proporcionar un ambiente húmedo para evitar la evaporación. Coloque la caja que contiene la bandeja a 4 °C durante la noche.
- Al día siguiente, haga una solución primaria de anticuerpos.
NOTA: En este protocolo, se utilizó anti-garabato de conejo para etiquetar las membranas celulares y anti-deadpan de rata para etiquetar neuroblastos, aunque se podría usar cualquier número de otros anticuerpos primarios.- Para hacer la solución primaria de anticuerpos, primero, haga diluciones de anticuerpos primarios en la solución de bloqueo. Por ejemplo, el anti-garabato de conejo se usa en una concentración final de 1: 1000. Por lo tanto, primero, diluya el anticuerpo anti-garabato de conejo a 1:100 (1 μL de anticuerpo más 99 μL de solución de bloqueo). Rat-deadpan se utiliza en una concentración final de 1:100. Por lo tanto, primero, diluya el anticuerpo de rata muerta a 1:10 (1 μL de anticuerpo más 9 μL de solución de bloqueo).
NOTA: Estas diluciones se pueden almacenar a largo plazo a 4 °C si también se agrega azida de sodio (0,05%) para inhibir el crecimiento bacteriano. - A continuación, cuente el número de pozos que contienen cerebros. El número de pocillos determina el volumen de solución de anticuerpos primarios a producir. Por ejemplo, si hay 2 pocillos de cerebro, prepare 20 μL de solución primaria de anticuerpos (para 10 pocillos, 100 μL, etc.). Para hacer una solución de anticuerpo primario de 20 μL, agregue 2 μL de cada dilución de anticuerpo primario y 16 μL de la solución de bloqueo.
NOTA: La concentración final de cada uno de los anticuerpos primarios es de 1:1000 y 1:100, respectivamente. En resumen, hacer la primera dilución de anticuerpos primarios a una concentración para que la segunda dilución sea siempre 1:10 para llegar a las respectivas concentraciones finales. En este caso, 1:1000 para conejo anti-garabato y 1:100 para rata anti-deadpan.
- Para hacer la solución primaria de anticuerpos, primero, haga diluciones de anticuerpos primarios en la solución de bloqueo. Por ejemplo, el anti-garabato de conejo se usa en una concentración final de 1: 1000. Por lo tanto, primero, diluya el anticuerpo anti-garabato de conejo a 1:100 (1 μL de anticuerpo más 99 μL de solución de bloqueo). Rat-deadpan se utiliza en una concentración final de 1:100. Por lo tanto, primero, diluya el anticuerpo de rata muerta a 1:10 (1 μL de anticuerpo más 9 μL de solución de bloqueo).
- Retire la solución de bloqueo con la micropipeta ajustada a 10 μL y pipetee 10 μL de solución de anticuerpos primarios en cada pocillo.
- Cubra y selle la bandeja con parafilm y colóquela de nuevo en la caja sellada con toallas húmedas. Incubar durante la noche a 4 °C.
NOTA: No se requiere agitar y se desaconseja encarecidamente. Los anticuerpos penetrarán en los cerebros sin temblar ni mezclarse. - Al día siguiente, retire la solución primaria de anticuerpos con una micropipeta y enjuague los cerebros 3 veces con 10 μL de 1x PBT. A continuación, lave los cerebros 4 veces con 10 μL de 1x PBT durante 10 min cada uno. Durante los lavados de 10 minutos, prepare la solución secundaria de anticuerpos.
- Para hacer la solución de anticuerpos secundarios, elija anticuerpos secundarios que reconozcan los anticuerpos primarios. En este protocolo, se utilizaron alexa Fluor 488 anti-conejo de cabra y alexa 555 antirata de cabra.
- Pipetear 1 μL de cada uno de los anticuerpos secundarios en un tubo de microcentrífuga con 298 μL de la solución bloqueadora para hacer la concentración final 1:300 para cada anticuerpo secundario.
- Después de los últimos 10 minutos de lavado, retire el PBT 1x y pipetee 10 μL de la solución secundaria de anticuerpos en cada pocillo. Selle la bandeja con parafilm y colóquela de nuevo en la caja con toallas húmedas. Incubar durante la noche a 4 °C.
NOTA: No se preocupe por eliminar hasta el último μL en los pocillos entre enjuagues, lavados o al agregar soluciones de anticuerpos primarios y secundarios. Los cerebros siempre permanecerán sumergidos en unos pocos μL de líquido, lo cual está bien. - Al día siguiente, retire la solución secundaria de anticuerpos con una micropipeta y enjuague los cerebros 3 veces con 10 μL cada uno de 1x PBT. A continuación, lave los cerebros 4 veces con 10 μL cada uno de 1x PBT durante 10 min cada uno.
- Durante los lavados de 10 minutos, prepare la mezcla de reacción de EdU para detectar la incorporación de EdU. Prepare la mezcla de reacción EdU de acuerdo con las directrices de los fabricantes.
- Después del lavado final, retire el 1x PBT y la pipeta 10 μL de la mezcla de reacción EdU en cada pozo con cerebros. Selle la placa del micropocillo con parafilm y cúbrala con papel de aluminio. Dejar el plato en el banco durante 30 min.
- Después de 30 min, enjuague los cerebros 3 veces con 10 μL cada uno de 1x PBT y lave los cerebros 3 veces con 10 μL cada uno de 1x PBT durante 5 min cada uno.
- Después del último lavado, retire el 1x PBT y la pipeta 10 μL de una solución de medios de montaje a base de glicerol. Selle la placa y colóquela a 4 °C durante la noche.
5. Montaje e imágenes de los cerebros
- Al día siguiente, prepare portaobjetos de microscopio (25 mm x 75 mm x 1 mm): Pegar (por ejemplo, superpegamento) un vidrio de cubierta cuadrada de 22 mm x 22 mm x 1 mm en cada extremo del portaobjetos de microscopio para crear un "puente" sobre el cual se colocará el lata de cubierta más grande de 22 mm x 50 mm x 1 mm para hacer un espacio entre la diapositiva y la funda más grande (Figura 5A). Este espacio permitirá que el cerebro tenga el movimiento justo para estar correctamente orientado mientras evita que sea aplastado.

Figura 5: Esquema que muestra el portaobjetos del microscopio, la orientación y los tipos de células en el cerebro larvario. (A) Representación visual del portaobjetos del microscopio sobre el cual se monta un cerebro larvario y está listo para ser fotografiado. (B) También se ha demostrado que una guía se usa para la orientación del tejido. (C) Portaobjetos de microscopio listo para imágenes en un microscopio confocal. (D) Dibujos animados que muestran algunos de los tipos de células en el cerebro larvario. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
- Después de pegar las gafas de cubierta de 22 mm x 22 mm x 1 mm al portaobjetos del microscopio, pipetee 9,3 μL de la solución de medios de montaje a base de glicerol que contiene un cerebro de un pozo de la placa de micropocillos y colóquelo en el centro de la diapositiva (Figura 5A).
NOTA: Los cerebros larvales pueden adherirse a la punta de la pipeta, así que tenga cuidado. Para evitar la adhesión, comience aspirando el antifade y luego aspirando el cerebro único hacia el final del volumen de 9.3 μL. - Una vez que el cerebro esté en la diapositiva, coloque suavemente el capa de cubierta de 22 mm x 50 mm x 1 mm en la parte superior. Coloque el cerebro como se ve en la Figura 5B. Mueva suavemente el cubrehojas para orientar el cerebro. La muestra está lista para la obtención de imágenes.
- Utilice un microscopio confocal equipado con un alto aumento y un objetivo de alta apertura numérica (Figura 5C) para adquirir las mejores imágenes. Por ejemplo: lente de inmersión en aceite de 60x o 63x, 1.4 NA.
- Imagen de los cerebros con la superficie dorsal más cercana a la cubierta (y objetivo). Adquiera las pilas Z a través de todo el hemisferio cerebral comenzando en la superficie ventral (más alejada del objetivo) a intervalos de 1 μm o tamaño de paso Z.
NOTA: Los láseres utilizados dependen de los anticuerpos secundarios. En este protocolo, las líneas láser utilizadas fueron de 488 nm para detectar tinción de Scribble, 555 nm para detectar Deadpan y 633 nm para detectar EdU.
- Imagen de los cerebros con la superficie dorsal más cercana a la cubierta (y objetivo). Adquiera las pilas Z a través de todo el hemisferio cerebral comenzando en la superficie ventral (más alejada del objetivo) a intervalos de 1 μm o tamaño de paso Z.
6. Análisis de datos
- Utilice el software de código abierto de Fiji para analizar los hemisferios cerebrales y use el complemento de contador de células de Fiji para contar las células.
Resultados
Los cerebros de tipo salvaje OregonR recién nacidos fueron diseccionados y cultivados durante 24 horas en medios de Schneider suplementados (SSM) con insulina. Los tejidos se fijaron y teñieron de acuerdo con el protocolo. Se utilizaron anticuerpos primarios generados contra Deadpan (Dpn) para detectar NB y Scribble para etiquetar las membranas celulares. Se agregó el análogo de timidina 5-Etinil-2′-desoxiuridina (Edu) para detectar la entrada en fase S y la reactivación de NB. Se encontraron NB Edu positivos y Dp...
Discusión
El método descrito aquí para cultivar explantes cerebrales se puede llevar a cabo en la mayoría de los entornos de laboratorio. Las herramientas requeridas, así como el procedimiento y la recopilación de datos, son simples y directas. Con este método, se pueden probar una variedad de hipótesis, incluidas las relacionadas con las cascadas de señalización celular y los factores extrínsecos que regulan la reactivación y proliferación de NB. Aquí, utilizando animales OregonR de tipo salvaje, encontramos que la i...
Divulgaciones
Los autores no tienen intereses contrapuestos.
Agradecimientos
Reconocemos el programa LSAMP Bridges to Doctorate para la financiación (CNK), así como NIH / NIGMS (R01-GM120421 y R35-GM141886). Agradecemos al Dr. Conor Sipe por la Figura 1. También agradecemos a todos los miembros del laboratorio Siegrist por su continuo apoyo y tutoría. Agradecemos especialmente a Chhavi Sood y Gary Teeters por su cuidadosa lectura del manuscrito y por proporcionar comentarios.
Materiales
| Name | Company | Catalog Number | Comments |
| 10 µL Pipette tips | Denville Sci | P2102 | |
| 1000 µL Pipette tips | Denville Sci | P2103-N | |
| 1000 µL Pipettor | Gilson | P1000 | |
| 16% paraformaldehyde (10 x 10 mL) | Electron Microscopy Sciences | 2912.60.0000 | Used for Fixation of Larval Brains |
| 20 µL Pipette | Gilson | P20 | |
| 200 µL Pipette tips | Denville Sci | 1158U56 | |
| 24-well multiwell culture plates | Fisher Scientific | 50-197-4477 | |
| 35 mm Petri dishes | Fisher Scientific | 08-757-100A | Grape Plate Ingredients |
| 4 °C refrigerator | Fisher Scientific | Provides an ideal temperature for >24 h incubations in antibody solution | |
| 63x Objective | Lecia | ||
| Active dry yeast | Most supermarkets | ||
| Agarose | Fisher Scientific | 214010 | Grape Plate Ingredients |
| Click-iT EdU Cell Proliferation Kit for Imaging, Alexa Fluor 647 dye | Thermo Fisher Scientific | C10340 | to label proliferating cells |
| Confocal Microscope | Leica | SP8 | |
| Coverslips 22 mm x 22 mm x 1 mm , 10 pack of 4 oz | Fisher Scientific | 12-544-10 | Two Coverslips are super glued to the ends of the microscope slide. This creates a space that allows for the brains to float in antifade while being imaged. |
| Coverslips, 22 mm x 50 mm x 1 mm | Fisher Scientific | 12-545E | The coverslip is placed on two square coverslips on the microscope slide ensuring that the brain in the antifade does not move while imaging. |
| Dissecting microscope | Zeiss | Stemi 2000 | |
| Ethanol 200 proof (100%), Decon Labs, 1 gallon bottle | Fisher Scientific | 2701 | Used to wash off the larvae before the 24 hr hold in culture medium |
| Fetal Bovine Serum (10%) | Sigma | F4135-100ML | Supplement for cell culture media. |
| Fine forceps for dissection | Fine Science Tools | 11295-20 | Forcepts used in disections. They work best when sharpened. |
| Fly Bottles for Crossing | Genessee Scientific | 32-130 | This bottle is used as a container that lets the flies lay eggs on the grape plate. |
| Glass Dissection Dish (3 well) | These are no longer available | ||
| Glutathione | Sigma | G6013 | Provides oxidative protection during cell culture. |
| Goat Serum | Sigma | G9023- 10ML | Blocking Agent |
| Grape Plates | Made in house | Made in house | Grape juice/agarose plates for collecting freshly hatched eggs |
| Image J | Imagej.net/fiji/downloads | Free Download: https://fiji.sc | Imaging platform that is used to count cells and Edu reactivation |
| Incubator | Thermo Fisher Scientific | Ensures that the temperature, humidity, and light exposure is exactly the same throughout experiment. | |
| Insulin | Sigma | I0516 | Independant variable of the experiment |
| Laminar flow hood | For aliquoting culture media | ||
| L-Glutamine | Sigma | G7513 | Provides support during cell culture |
| Nunc 72-well Microwell Mini Trays | Fisher Scientific | 12-565-154 | Immunostaining steps are performed in this tray |
| Parafilm | Fisher Scientific | S37440 | Film used to seal plates in order to prevent evaporation |
| Pen-Strep | Sigma | P4458-100ml | Antibiodics used to prevent bacterial contamination of cells during culture. |
| Phosphate Buffer, pH7.4 | Made in house | Made in house | Solvent used to wash the brains after fixing and staining steps |
| Pick | Fine Science Tools | 10140-01 | Used to pick larvae off of the grape plate |
| Propionic acid | Fisher Scientific | A-258 | Grape Plate Ingredients |
| Rabbit 405 | Abcam | ab175653 | Antibodies used for immunostaining |
| Rat 555 | Abcam | ab150166 | Antibodies used for immunostaining |
| Rb Scribble | A Gift from Chris Doe | Antibodies used for immunostaining | |
| Rt Deadpan | Abcam | ab195173 | Antibodies used for immunostaining |
| Schneiders Culture Medium | Life Tech | 21720024 | Contains nutrients that help the cells grow and proliferate |
| SlowFade Diamond Antifade (5 x 2 mL) | Life Tech | S36963 | Reagent that provides protection against fading fluorophores |
| Sterile Water | Autoclave Milli-Q water made in house | Needed for Solutions | |
| Sucrose | Fisher | S2-12 | Grape Plate Ingredients |
| Superfrost Microscope Slides | Fisher Scientific | 12-544-7 | |
| Superglue | Most supermarkets | ||
| Tegosept | Genesee Scientific | 20-259 | Grape Plate Ingredients |
| Triton-X 100 | Sigma | T9284-100ML | PBT |
| Welch's 100% grape grape juice | Most supermarkets | Grape Plate Ingredients |
Referencias
- Suman, S., Domingues, A., Ratajczak, J., Ratajczak, M. Z. Potential clinical applications of stem cells in regenerative medicine. Advances in Experimental Medicine and Biology. 1201, 1-22 (2019).
- Tabar, V., Studer, L. Pluripotent stem cells in regenerative medicine: challenges and recent progress. Nature Reviews Genetics. 15, 82-92 (2014).
- Daley, G. Q. Stem cells and the evolving notion of cellular identity. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 370, 20140376 (2015).
- Rodrigues, M., Kosaric, N., Bonham, C. A., Gurtner, G. C. Wound healing: A cellular perspective. Physiological Reviews. 99, 665-706 (2019).
- van Velthoven, C. T. J., Rando, T. A. Stem cell quiescence: Dynamism, restraint, and cellular idling. Cell Stem Cell. 24, 213-225 (2019).
- Chapman, N. M., Boothby, M. R., Chi, H. Metabolic coordination of T cell quiescence and activation. Nature Reviews Immunology. 20, 55-70 (2020).
- Wosczyna, M. N., Rando, T. A. A muscle stem cell support group: Coordinated cellular responses in muscle regeneration. Developmental Cell. 46, 135-143 (2018).
- Homem, C. C., Knoblich, J. A. Drosophila neuroblasts: a model for stem cell biology. Development. 139, 4297-4310 (2012).
- Kang, K. H., Reichert, H. Control of neural stem cell self-renewal and differentiation in Drosophila. Cell and Tissue Research. 359, 33-45 (2015).
- Chell, J. M., Brand, A. H. Nutrition-responsive glia control exit of neural stem cells from quiescence. Cell. 143, 1161-1173 (2010).
- Sousa-Nunes, R., Yee, L. L., Gould, A. P. Fat cells reactivate quiescent neuroblasts via TOR and glial insulin relays in Drosophila. Nature. 471, 508-512 (2011).
- Britton, J. S., Edgar, B. A. Environmental control of the cell cycle in Drosophila: nutrition activates mitotic and endoreplicative cells by distinct mechanisms. Development. 125, 2149-2158 (1998).
- Lin, S., et al. Extremes of lineage plasticity in the Drosophila brain. Current biology : CB. 23, 1908-1913 (2013).
- Sipe, C. W., Siegrist, S. E. Eyeless uncouples mushroom body neuroblast proliferation from dietary amino acids in Drosophila. Elife. 6, 26343 (2017).
- Speder, P., Brand, A. H. Systemic and local cues drive neural stem cell niche remodelling during neurogenesis in Drosophila. Elife. 7, 30413 (2018).
- Yuan, X., Sipe, C. W., Suzawa, M., Bland, M. L., Siegrist, S. E. Dilp-2-mediated PI3-kinase activation coordinates reactivation of quiescent neuroblasts with growth of their glial stem cell niche. PLoS Biology. 18, 3000721 (2020).
- Colombani, J., et al. A nutrient sensor mechanism controls Drosophila growth. Cell. 114, 739-749 (2003).
- Geminard, C., Rulifson, E. J., Leopold, P. Remote control of insulin secretion by fat cells in Drosophila. Cell Metabolism. 10, 199-207 (2009).
- Siller, K. H., Serr, M., Steward, R., Hays, T. S., Doe, C. Q. Live imaging of Drosophila brain neuroblasts reveals a role for Lis1/dynactin in spindle assembly and mitotic checkpoint control. Molecular Biology of the Cell. 16, 5127-5140 (2005).
- Prithviraj, R., Trunova, S., Giniger, E. Ex vivo culturing of whole, developing Drosophila brains. Journal of Visualized Experiments: JoVE. (65), e4270 (2012).
- Bostock, M. P., et al. An immobilization technique for long-term time-lapse imaging of explanted drosophila tissues. Frontiers in Cell and Developmental Biology. 8, 590094 (2020).
- Datta, S. Activation of neuroblast proliferation in explant culture of the Drosophila larval CNS. Brain Research. 818, 77-83 (1999).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados