
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Riattivazione delle cellule staminali neurali negli espianti cerebrali di Drosophila in coltura
In questo articolo
Riepilogo
È stato stabilito un metodo per riattivare le cellule staminali neurali quiescenti negli espianti cerebrali di Drosophila in coltura. Utilizzando questo metodo, il ruolo dei segnali sistemici può essere disaccoppiato dai segnali intrinseci del tessuto nella regolazione della quiescenza, dell'ingresso e dell'uscita delle cellule staminali neurali.
Abstract
Le cellule staminali neurali (NSC) hanno la capacità di proliferare, differenziarsi, subire apoptosi e persino entrare e uscire dalla quiescenza. Molti di questi processi sono controllati dalla complessa interazione tra i programmi genetici intrinseci NSC con i fattori estrinseci NSC, locali e sistemici. Nell'organismo modello genetico, Drosophila melanogaster, le NSC, note come neuroblasti (NB), passano dalla quiescenza alla proliferazione durante la transizione embrionale a quella larvale. Durante questo periodo, le larve emergono dai loro gusci d'uovo e iniziano a gattonare, alla ricerca di nutrienti dietetici. In risposta all'alimentazione animale, il corpo grasso, un organo endocrino con capacità di accumulo lipidico, produce un segnale, che viene rilasciato sistemicamente nell'emolinfa circolante. In risposta al segnale derivato dal corpo grasso (FBDS), i peptidi insulino-simili a Drosophila (Dilps) vengono prodotti e rilasciati dai neuroni neurosecretori del cervello e dalla glia, portando all'attivazione a valle della segnalazione della crescita della PI3-chinasi nei NB e nella loro nicchia gliale e tracheale. Sebbene questo sia il modello attuale di come i NB passano dalla quiescenza alla proliferazione, la natura del segnale estrinseco FBDS rimane sfuggente. Per capire meglio come i segnali sistemici estrinseci NB regolano l'uscita dalla quiescenza, è stato sviluppato un metodo per coltivare i primi cervelli larvali in vitro prima dell'alimentazione animale. Con questo metodo, i fattori esogeni possono essere forniti ai terreni di coltura e NB uscita dalla quiescenza analizzata. Abbiamo scoperto che l'insulina esogena è sufficiente per riattivare i NB dalla quiescenza negli espianti di tutto il cervello. Poiché questo metodo è adatto per schermi su larga scala, miriamo a identificare ulteriori segnali estrinseci che regolano la quiescenza NB rispetto alle decisioni di proliferazione. Poiché i geni e i percorsi che regolano le decisioni di proliferazione NSC sono conservati evolutivamente, i risultati di questo test potrebbero fornire informazioni sul miglioramento delle terapie rigenerative nella clinica.
Introduzione
Le cellule staminali sono di grande interesse per il loro potenziale di utilizzo nella medicina rigenerativa 1,2. Molti animali, specialmente quelli longevi, mantengono le cellule staminali all'interno dei loro tessuti adulti. Queste cellule staminali residenti funzionano per mantenere l'omeostasi tissutale e sono utilizzate per la riparazione a seguito di lesioni fisiche o malattie 3,4. La maggior parte delle cellule staminali negli animali adulti sono quiescenti, uno stato relativamente dormiente caratterizzato dall'arresto del ciclo cellulare e dall'inattivazione della segnalazione della crescita5. In risposta a segnali estrinseci, le cellule staminali escono dalla quiescenza, entrano nel ciclo cellulare e iniziano a generare progenie figlia specifica per il loro tipo di tessuto. Ad esempio, al fine di montare una risposta immunitaria efficace, le cellule che presentano l'antigene inducono cellule T ingenue quiescenti ad entrare nel ciclo cellulare e ad espandersi clonalmente6. In risposta al danno muscolare scheletrico, le cellule staminali satelliti muscolari entrano nel ciclo cellulare e generano mioblasti figli per sostituire le miofibrille danneggiate 5,7. Mentre è chiaro che le cellule staminali quiescenti rispondono ai segnali estrinseci, in molti casi, la natura del segnale estrinseco rimane poco chiara, così come il meccanismo di attivazione delle cellule staminali indotte dal cue. Ottenere una migliore comprensione di come le cellule staminali quiescenti rispondono ai segnali estrinseci ed entrano nel ciclo cellulare aiuterà nello sviluppo di migliori terapie con cellule staminali nella clinica e aumenterà le conoscenze scientifiche.
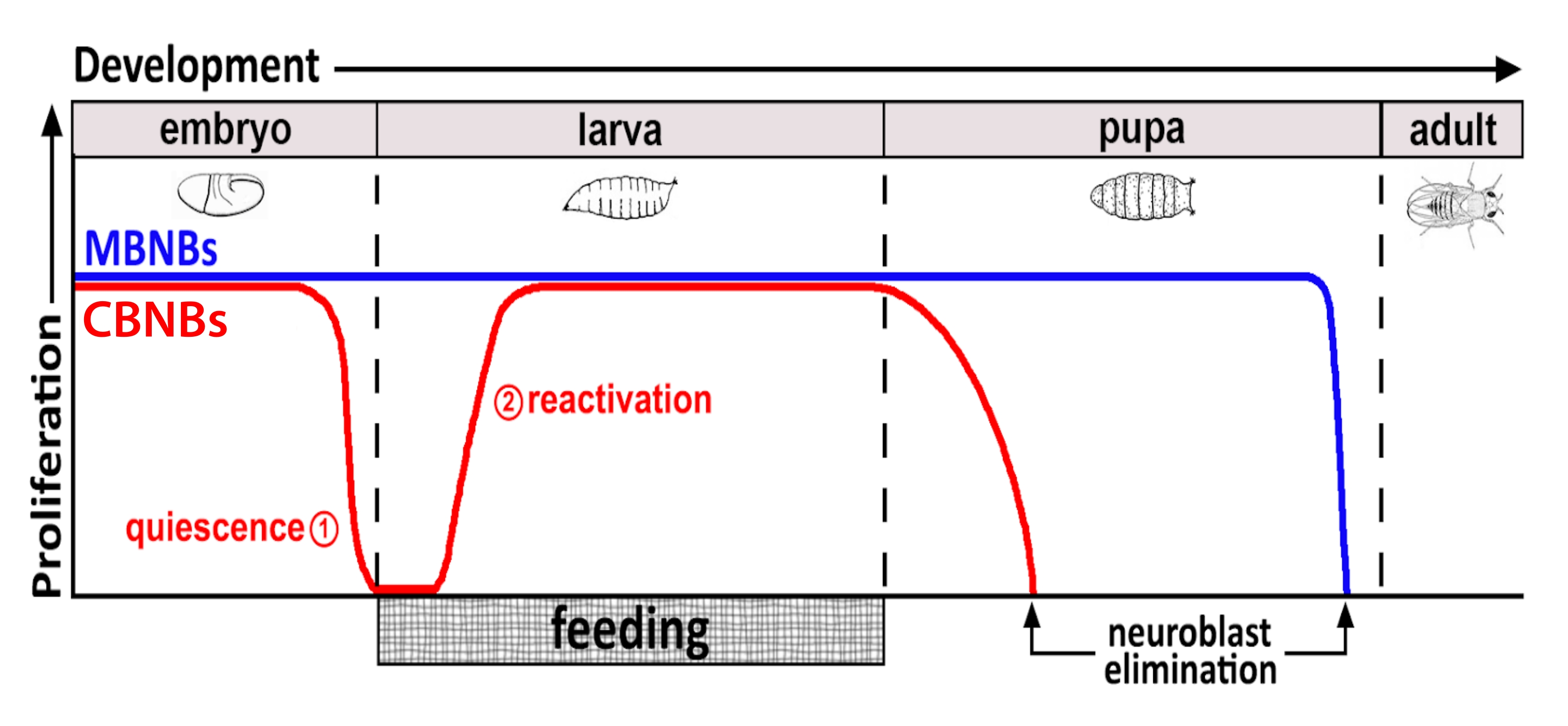
Per decenni, gli organismi modello sono stati utilizzati per scoprire i geni e le vie di segnalazione cellulare che regolano la proliferazione delle cellule staminali durante lo sviluppo e in età adulta. In Drosophila, le cellule staminali neurali (NSC), note come neuroblasti (NB), si dividono durante lo sviluppo per generare tutti i neuroni e glia che alla fine si integrano, formando il circuito neurale richiesto per la funzione cerebrale 8,9. Come altre cellule staminali, le NB si dividono asimmetricamente per auto-rinnovarsi e, in alcuni casi, simmetricamente per espandere il pool di cellule staminali. I NB sono specificati durante l'embriogenesi e la maggior parte entra in quiescenza verso la fine, in coincidenza con il declino delle riserve di nutrienti materni (Figura 1). Dopo che l'embriogenesi è completa, le larve si schiudono e iniziano a nutrirsi. In risposta all'alimentazione animale, i NB si riattivano dalla quiescenza e riprendono le divisioni cellulari 10,11,12,13,14,15,16. Poiché il SNC Di Drosophila è relativamente semplice e poiché i NB entrano ed escono dalla quiescenza in momenti definiti, l'uso di Drosophila per studiare la regolazione della quiescenza, dell'entrata e dell'uscita, si rivela ideale.

Figura 1: Proliferazione relativa di CB NB (neuroblasti cerebrali centrali, rosso) e MB NB (neuroblasti del corpo a fungo, blu) nel corso dello sviluppo. Alla fine dell'embriogenesi, la maggior parte dei NB (linea rossa) cessa la proliferazione ed entra nella quiescenza. La quiescenza continua fino a quando le larve appena schiuse consumano il loro primo pasto completo. I punti temporali di messa a fuoco per questa metodologia sono indicati in cerchi rossi (1, quiescenza e 2, riattivazione). Gli MB NB (blu) sono un sottoinsieme di NB del cervello centrale che si dividono continuamente durante lo sviluppo (4 per emisfero cerebrale). Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
In risposta all'alimentazione animale, le vie di segnalazione della crescita PI3-chinasi e TOR diventano attive nei NB e nella loro nicchia gliale e tracheale 10,11,15,16. Quando i nutrienti dietetici vengono ritirati o quando i livelli di PI3-chinasi sono ridotti, i NB non riescono a riattivarsi e anche la crescita di glia e trachea viene ridottadi 10,11,15,16. Il modello attuale postula che la riattivazione nb sia accoppiata alla crescita larvale da parte del corpo grasso, che rilascia un segnale sistemico in risposta all'alimentazione animale 12,17,18. Questo segnale, che rimane sfuggente, probabilmente promuove l'espressione e il rilascio del peptide insulino-simile alla Drosophila (Dilps) nel cervello, che porta all'attivazione a valle della PI3-chinasi nei NB e nella loro nicchia gliale e tracheale. Per comprendere meglio la natura dei segnali sistemici, abbiamo sviluppato un metodo per riattivare i NB quiescenti negli espianti cerebrali in coltura. Con questo metodo, la riattivazione dei NB può essere analizzata in assenza di segnali sistemici di animali interi. I fattori esogeni possono essere riforniti ai terreni di coltura e la riattivazione NB è stata analizzata in base all'incorporazione dell'analogo della timidina, EdU. Utilizzando questo metodo, abbiamo determinato che l'insulina esogena è sufficiente per riattivare i NB quiescenti negli espianti cerebrali. Il lavoro futuro sarà finalizzato all'identificazione di ulteriori fattori che, una volta aggiunti, regolano positivamente o negativamente la quiescenza NB negli espianti cerebrali.
Protocollo
1. Raccolta delle larve di Drosophila
NOTA: Preparare il piatto di lievito, la pasta d'uva e il condominio Fly prima di iniziare:
- Pasta di lievito: In un piccolo contenitore, mescolare 5 g di lievito secco attivo con 10 ml di acqua per formare una pasta che abbia la consistenza del burro di arachidi. Coprire la pasta di lievito con pellicola trasparente e utilizzare un elastico per fissarlo saldamente al contenitore.
NOTA: la pasta di lievito fresco si espanderà nel suo contenitore e si staccherà dal coperchio a meno che non sia saldamente attaccata. La pasta di lievito durerà per diversi giorni a temperatura ambiente (RT). - Piatti d'uva: Segui la ricetta per fare piatti d'uva (Tabella 1). Se si utilizzano piastre conservate a 4°C, assicurarsi di preriscaldare le piastre prima dell'uso posizionandole a RT per 1 ora.
- Mescolare acqua (750 ml) e agar (18,75 g) in un matraccio da 4 L, vortice e autoclave per 20 minuti (ciclo liquido).
- Mescolare il succo d'uva (250 ml) e il saccarosio (25 g) in un matraccio da 1 L con una grande barra di agitazione su una piastra riscaldata (a fuoco basso). Quando il saccarosio è sciolto, spegnere il fuoco, attendere che il pallone possa essere toccato prima di aggiungere Tegosept (10%, 4 ml) e acido propionico (5 ml). Mantieni la barra di agitazione accesa.
- Quando l'autoclave è completa, lasciarla raffreddare fino a quando il pallone non può essere toccato (~ 60 ° C), quindi mescolare nel mix di succo d'uva.
- Unire tutte le soluzioni in un unico pallone e lasciare mescolare sul piatto.
- Pipettare la soluzione in coperchi di piastre di Petri di piccole dimensioni (35 mm). Pipetta circa 9 ml per coperchio o fino ad ottenere una cupola convessa.
- OPZIONALE: Fiammare i coperchi per eliminare eventuali bolle.
- Quando le piastre si solidificano, impilare le piastre dell'uva in una scatola con un coperchio ermetico e posizionare la scatola a 4 °C. Le piastre possono essere conservate fino a 1 mese.
- Fly condo: Punch ~ 20 fori in una bottiglia di Drosophila in polipropilene quadrato da 6 once usando un ago da 18 G.
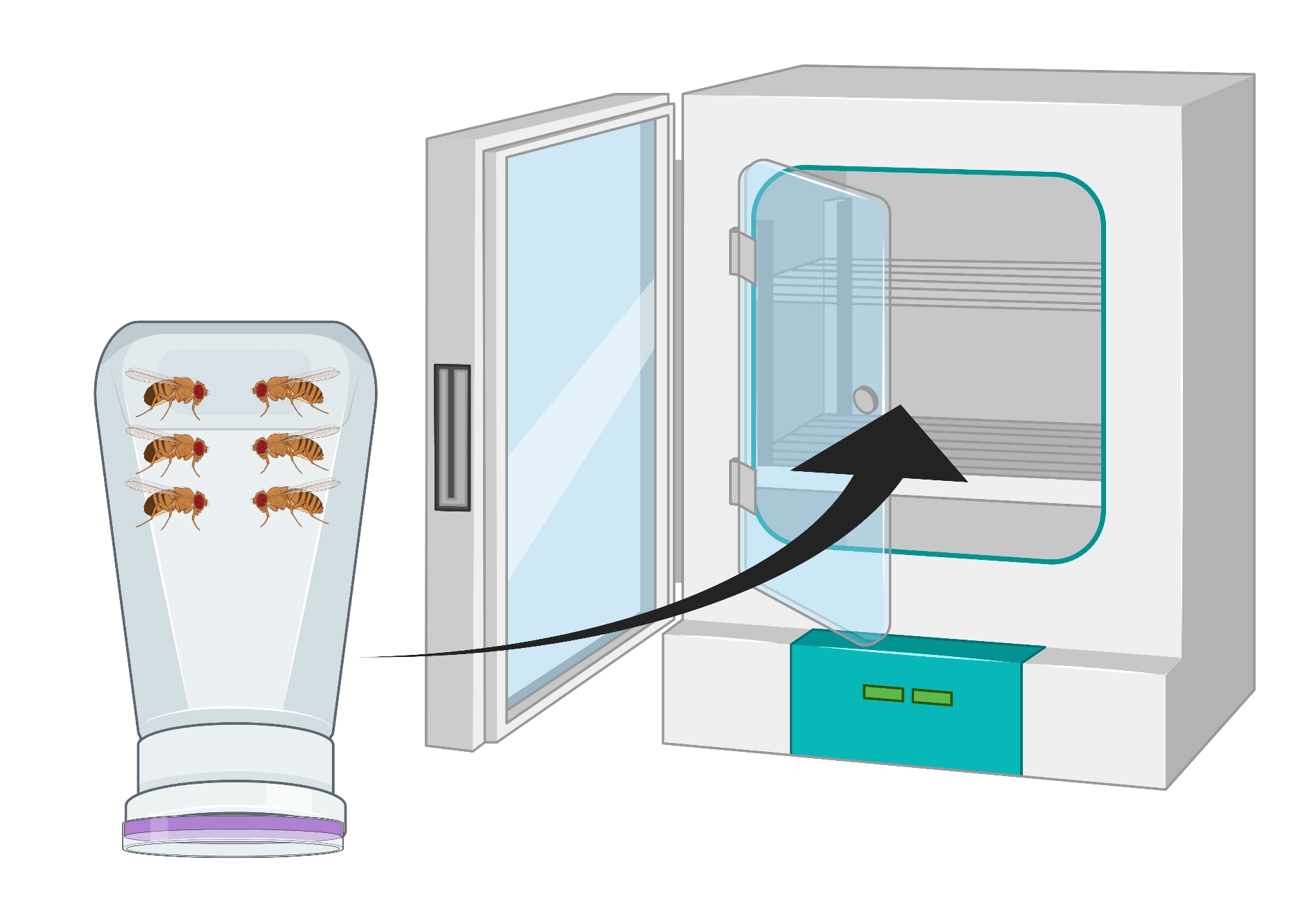
- Trasferisci le mosche adulte (~ 100 OregonR o qualsiasi genotipo) in un condominio di mosche e copri il condominio con un piatto di agar d'uva sormontato da un po 'di pasta di lievito. Posizionare il dab verso il centro della piastra e apporre la piastra al condominio con nastro da laboratorio.
- Capovolgere il contenitore in modo che la piastra di agar d'uva sia sul fondo e metterla in un'incubatrice a 25 °C per 24 ore (Figura 2).

Figura 2: Rappresentazione visiva della bottiglia di mosca invertita (condominio) con adulti Drosophila maschi e femmine. La bottiglia di plastica ha piccole forature, generate con un ago da 18 G, per lo scambio di ossigeno. La bocca della bottiglia è sigillata con un tappo di succo d'uva agar e viene invertita e conservata in un incubatore a 25 °C. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
- Dopo 24 ore, cambiare il piatto di agar d'uva e sostituirlo con un nuovo piatto condito con pasta di lievito. Cambia rapidamente le due piastre mentre tocchi continuamente il condominio leggermente sulla panchina in modo che le mosche adulte non sfuggano.
- Esaminare la piastra ad occhio e valutare il numero di embrioni sulla piastra. Gli embrioni di Drosophila sono oblunghi e bianchi con due appendici simili a stringhe.
- Se ci sono pochissimi embrioni sul piatto (meno di 100), scartare il piatto (raschiare l'agar nella spazzatura della mosca e salvare il coperchio di plastica per il riutilizzo). In molti casi, le femmine adulte non deporranno molti embrioni la prima notte in un nuovo condominio. Se questo è il caso, dare alle mosche adulte altre 24 ore per acclimatarsi.
- Se c'è un gran numero di embrioni sul piatto (almeno 100), conservalo e rimuovi con cura la pasta di lievito usando una spatola a fondo piatto.
- Una volta rimossa la pasta di lievito, utilizzare un plettro metallico per rimuovere manualmente tutte le larve dal piatto dell'uva al microscopio di dissezione. Quando si guarda la piastra al microscopio sezionante, si dovrebbero osservare larve striscianti ed embrioni.
- Rimuovere tutte le larve spazzolando il piccone metallico verso il lato di una larva. Le larve sono appiccicose e si attaccheranno al plettro. Una volta che una larva è sul plettro, le larve aggiuntive possono essere facilmente raccolte usando la larva sullo strumento per attaccarne di più.
NOTA: le larve amano attaccarsi l'una all'altra. A questo punto, non importa se le larve vengono danneggiate. Queste larve saranno scartate. - Dopo aver raccolto e rimosso tutte le larve, riposizionare la piastra nell'incubatrice a 25 °C. Assicurarsi di posizionare la piastra in un contenitore più grande che può essere sigillato. Posizionare asciugamani di carta bagnati sul fondo del contenitore più grande per mantenere l'umidità.
- Dopo 30-60 minuti, riporta la piastra al microscopio di dissezione e ora, scegli con cura ~ 20-25 larve dallo stesso piatto di agar d'uva per assicurarti che le larve raccolte siano appena nate entro una finestra di tempo di 30-60 minuti.
- Immergere la punta dello strumento con le 20-25 larve appena nate in una capsula di Petri (60 mm) riempita con 1-2 ml di 1x soluzione salina tamponata con fosfato (PBS) per 2 minuti.
- Dopo 2 minuti, rovesciare il piatto ad angolo per raggruppare il liquido sul fondo. Usando un piccolo pennello, spazzolare le larve dal liquido sul fondo della capsula di Petri.
- Raccogliere tutte le larve sul pennello e trasferire le larve in una nuova capsula di Petri (60 mm) contenente 1-2 ml di etanolo al 70%. Ripetere i passaggi 1.15 per raccogliere le larve con un pennello e trasferirle in una nuova capsula di Petri con 1-2 ml di 1x PBS.
2. Preparazione di terreni di coltura e strumenti
- Spruzzare il banco e l'area di lavoro con etanolo al 70% e lasciare asciugare.
- Spruzzare gli strumenti di dissezione, la pinza e due piatti in vetro, con etanolo al 70% e lasciarli asciugare sul banco.
- Preparare il supporto integrato di Schneider (SSM, Tabella 2) e metterlo sul ghiaccio.
- Pipettare 1 mL di SSM in ciascuna delle piastre di vetro.
- Utilizzando una micropipetta con una punta sterile, trasferire le larve appena schiuse dalla piastra di PBS all'SSM nel primo piatto di vetro. Utilizzando una micropipetta con una punta sterile, trasferire le larve appena schiuse all'SSM nella seconda parabola di vetro.
3. Dissezioni e colture cerebrali
- Una volta che le larve sono nel secondo piatto di vetro con SSM, seziona il cervello dalle larve usando una pinza e un microscopio di dissezione. Regolare l'ingrandimento in base alle esigenze.
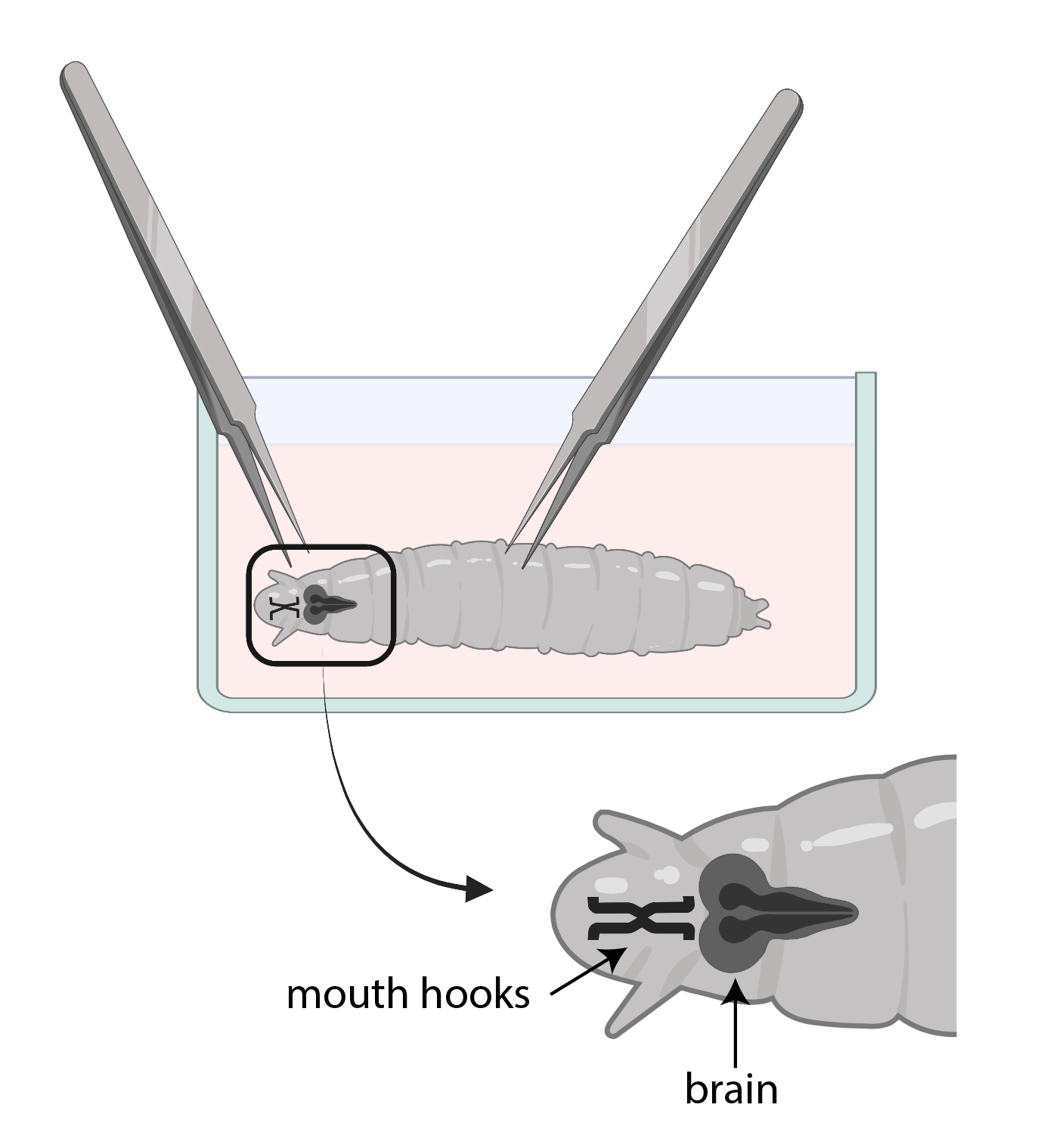
- Usa una pinza per afferrare i ganci della bocca e con l'altra, afferra delicatamente il corpo a metà strada e tira nella direzione opposta (Figura 3) per dividere la larva in due pezzi.
NOTA: Il cervello si troverà proprio dietro i ganci della bocca. Si noti che ci possono essere altri tessuti che circondano il cervello. Fai molta attenzione quando rimuovi questi tessuti in quanto può causare danni al cervello

Figura 3: Larve di Drosophila in un piatto di vetro con SSM. Le pinze sono posizionate correttamente per la dissezione. La posizione del cervello larvale (grigio scuro) è posteriore ai ganci della bocca (nero), ed entrambi sono mostrati all'interno della larva. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
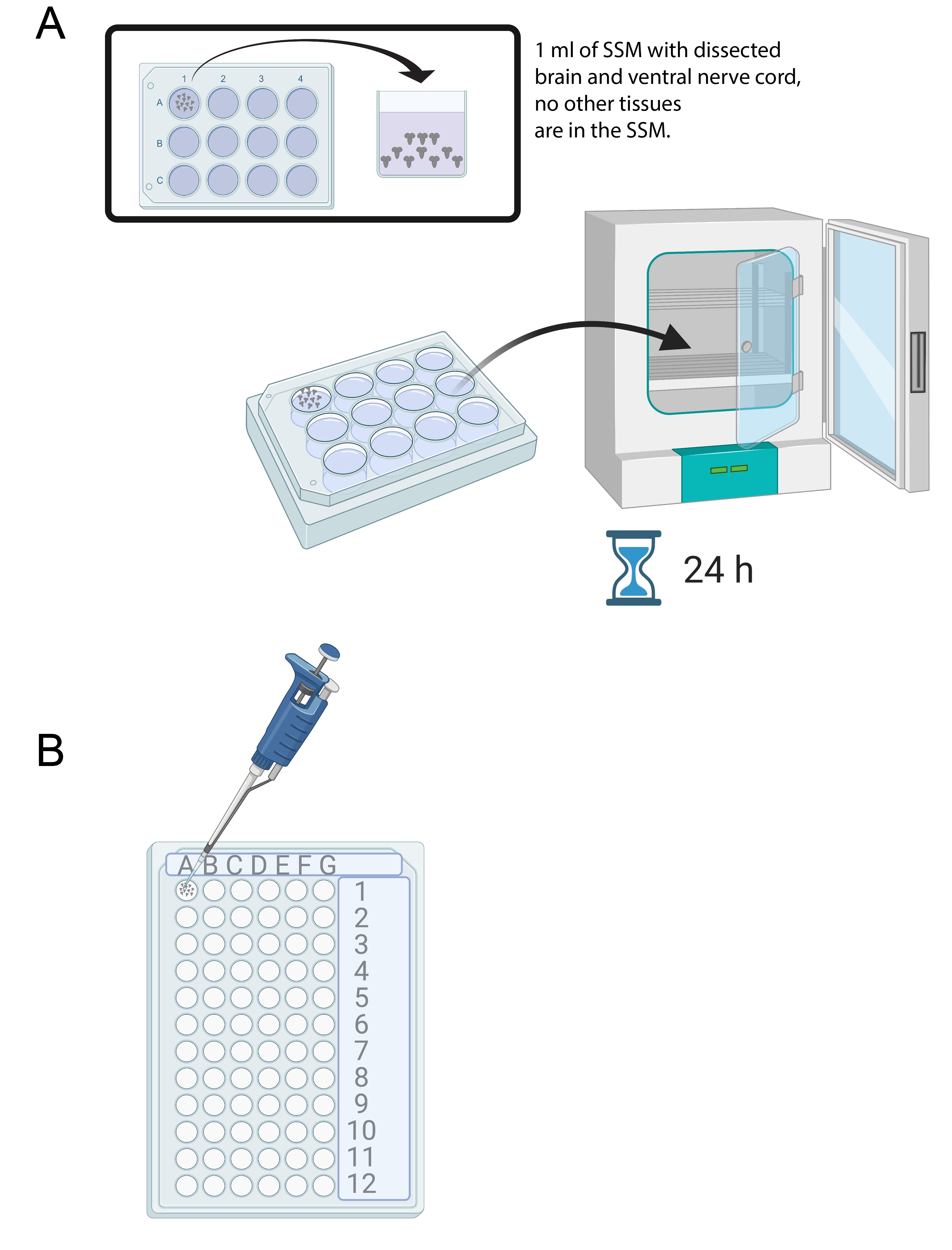
- Una volta che 15-20 cervelli sono stati sezionati, aggiungere 1 ml di SSM in un pozzetto di un vassoio sterile di coltura a 12 pozzetti. Trasferire i cervelli appena sezionati nell'SSM usando una micropipetta e una punta sterile (Figura 4A).
- Posizionare i cervelli nei mezzi SSM nel vassoio di coltura a 12 pozzetti in un'incubatrice a 25 °C per 24 ore (Figura 4A).

Figura 4: Coltura cerebrale e immunocolorazione. (A) Cervello intero in un piatto di coltura a 12 pozzetti contenente 1 mL di SSM. Il piatto di coltura viene quindi posto in un incubatore a 25 °C per 24 ore. (B) Mini vassoio a 72 pozzetti che contiene gli espianti cerebrali durante l'immunocolorazione. I cervelli vengono lavati e le soluzioni trasferite utilizzando una micropipetta P20 impostata su 10 μL. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
4. Test di proliferazione, fissazione del cervello e colorazione degli anticorpi
- Il giorno seguente, effettuare 1 mL di soluzione EdU SSM. Pipettare 10 μL di uno stock di 10 mM di 5-etinile-2′-deossiuridina (EdU) con 990 μL di SSM (concentrazione finale di EdU uguale a 0,1 mM) in un tubo e miscela di microcentrifuga sterile. Al termine dell'incubazione di 24 ore, pipettare 1 mL di EdU SSM in un pozzetto del vassoio di coltura sterile a 12 pozzetti.
- Trasferire il cervello utilizzando una micropipetta con una punta sterile dal pozzo contenente solo SSM al nuovo pozzo contenente la soluzione EdU SSM. Incubare per 1 ora a 25 °C.
- Quindi, trasferire i cervelli marcati con EdU in un altro pozzo nello stesso vassoio di coltura contenente 1 mL di fissativo (4% paraformaldeide, vedere Tabella 3 per la ricetta) per 20 minuti.
ATTENZIONE: La paraformaldeide è un pericolo biologico e deve essere smaltita correttamente. - Dopo la fissazione, trasferire rapidamente il cervello ai pozzetti di un mini vassoio a 72 pozzetti usando una micropipetta. Ogni pozzo può contenere 10 cervelli e 10-15 μL di liquido (Figura 4B). Una volta che i cervelli vengono trasferiti nel mini vassoio (non più di 10 cervelli per pozzetto), rimuovere la correzione e risciacquare il cervello 3 volte in 10 μL di 1x PBT (tampone fosfato, pH 7,4 contenente 0,1% Triton-X 100).
NOTA: risciacquo significa pipettare 10 μL di 1x PBT sul cervello, rimuovere e ripetere 3 volte. - Quindi, lavare il cervello 3 volte per 10 minuti ciascuno, di nuovo in 10 μL 1x PBT. Assicurarsi che il cervello sia coperto con un po 'di liquido in ogni momento.
- Dopo che i lavaggi sono stati completati, pipettare 10 μL di soluzione bloccante (1x PBT con siero di capra normale al 10%) sul cervello. Coprire il vassoio e sigillarlo con una striscia di parafilm attorno al bordo.
- Una volta sigillato, posizionare il mini vassoio in una scatola sigillata con asciugamani bagnati per fornire un ambiente umido per evitare l'evaporazione. Posizionare la scatola contenente il vassoio a 4 °C durante la notte.
- Il giorno seguente, fare una soluzione anticorpale primaria.
NOTA: In questo protocollo, l'anti-scarabocchio di coniglio è stato utilizzato per etichettare le membrane cellulari e l'anti-deadpan di ratto per etichettare i neuroblasti, anche se è stato possibile utilizzare un numero qualsiasi di altri anticorpi primari.- Per produrre la soluzione anticorpale primaria, in primo luogo, effettuare diluizioni di anticorpi primari nella soluzione bloccante. Ad esempio, l'anti-scarabocchio di coniglio viene utilizzato a una concentrazione finale di 1:1000. Pertanto, in primo luogo, diluire l'anticorpo anti-scarabocchio del coniglio a 1:100 (1 μL di anticorpo più 99 μL di soluzione bloccante). Rat-deadpan viene utilizzato ad una concentrazione finale di 1:100. Pertanto, in primo luogo, diluire l'anticorpo rat-deadpan a 1:10 (1 μL di anticorpo più 9 μL di soluzione bloccante).
NOTA: Queste diluizioni possono essere conservate a lungo termine a 4 °C se viene aggiunto anche azide di sodio (0,05%) per inibire la crescita batterica. - Quindi, conta il numero di pozzi che contengono cervelli. Il numero di pozzetti determina il volume della soluzione anticorpale primaria da produrre. Ad esempio, se ci sono 2 pozzetti di cervello, preparare 20 μL di soluzione anticorpale primaria (per 10 pozzetti, 100 μL, ecc.). Per produrre una soluzione anticorpale primaria da 20 μL, aggiungere 2 μL di ciascuna diluizione anticorpale primaria e 16 μL della soluzione bloccante.
NOTA: La concentrazione finale di ciascuno degli anticorpi primari è rispettivamente 1:1000 e 1:100. In breve, effettuare la prima diluizione di anticorpi primari ad una concentrazione in modo che la seconda diluizione sia sempre 1:10 per arrivare alle rispettive concentrazioni finali. In questo caso, 1:1000 per l'anti-scarabocchio di coniglio e 1:100 per l'anti-deadpan di ratto.
- Per produrre la soluzione anticorpale primaria, in primo luogo, effettuare diluizioni di anticorpi primari nella soluzione bloccante. Ad esempio, l'anti-scarabocchio di coniglio viene utilizzato a una concentrazione finale di 1:1000. Pertanto, in primo luogo, diluire l'anticorpo anti-scarabocchio del coniglio a 1:100 (1 μL di anticorpo più 99 μL di soluzione bloccante). Rat-deadpan viene utilizzato ad una concentrazione finale di 1:100. Pertanto, in primo luogo, diluire l'anticorpo rat-deadpan a 1:10 (1 μL di anticorpo più 9 μL di soluzione bloccante).
- Rimuovere la soluzione bloccante con la micropipetta impostata su 10 μL e la pipetta a 10 μL di soluzione anticorpale primaria in ciascun pozzetto.
- Coprire e sigillare il vassoio con parafilm e riposizionarlo nella scatola sigillata con asciugamani bagnati. Incubare durante la notte a 4 °C.
NOTA: L'agitazione non è necessaria ed è fortemente sconsigliata. Gli anticorpi penetreranno nel cervello senza scuotere o mescolare. - Il giorno seguente, rimuovere la soluzione anticorpale primaria utilizzando una micropipetta e risciacquare il cervello 3 volte con 10 μL di 1x PBT. Quindi, lavare il cervello 4 volte con 10 μL di 1x PBT per 10 minuti ciascuno. Durante i lavaggi di 10 minuti, preparare la soluzione anticorpale secondaria.
- Per produrre la soluzione anticorpale secondaria, scegliere anticorpi secondari che riconoscano gli anticorpi primari. In questo protocollo sono stati utilizzati l'anti-coniglio di capra Alexa Fluor 488 e l'anti-ratto di capra Alexa 555.
- Pipettare 1 μL di ciascuno degli anticorpi secondari in un tubo microcentrifuga con 298 μL della soluzione bloccante per rendere la concentrazione finale 1:300 per ciascun anticorpo secondario.
- Dopo gli ultimi 10 minuti di lavaggio, rimuovere 1x PBT e pipettare 10 μL della soluzione anticorpale secondaria in ciascun pozzetto. Sigillare il vassoio con parafilm e rimetterlo nella scatola con asciugamani umidi. Incubare durante la notte a 4 °C.
NOTA: Non preoccuparsi di rimuovere ogni ultimo μL nei pozzetti tra risciacqui, lavaggi o quando si aggiungono soluzioni anticorpali primarie e secondarie. Il cervello rimarrà sempre immerso in pochi μL di liquido, il che va bene. - Il giorno seguente, rimuovere la soluzione anticorpale secondaria usando una micropipetta e risciacquare il cervello 3 volte con 10 μL ciascuno di 1x PBT. Quindi, lavare il cervello 4 volte con 10 μL ciascuno di 1x PBT per 10 minuti ciascuno.
- Durante i lavaggi di 10 minuti, preparare la miscela di reazione EdU per rilevare l'incorporazione di EdU. Preparare la miscela di reazione EdU secondo le linee guida dei produttori.
- Dopo il lavaggio finale, rimuovere il PBT 1x e la pipetta 10 μL della miscela di reazione EdU in ciascun pozzetto con il cervello. Sigillare la piastra microwell con parafilm e coprire con foglio di alluminio. Lasciare il piatto sul banco per 30 minuti.
- Dopo 30 minuti, sciacquare il cervello 3 volte con 10 μL ciascuno di 1x PBT e lavare il cervello 3 volte con 10 μL ciascuno di 1x PBT per 5 minuti ciascuno.
- Dopo l'ultimo lavaggio, rimuovere 1x PBT e pipetta 10 μL di una soluzione di supporto a base di glicerolo. Sigillare la piastra e posizionarla a 4 °C durante la notte.
5. Montare e immaginare il cervello
- Il giorno seguente, preparare vetrini per microscopio (25 mm x 75 mm x 1 mm): colla (ad esempio, supercolla) un vetro di copertura quadrato da 22 mm x 22 mm x 1 mm a ciascuna estremità del vetrino del microscopio per creare un "ponte" su cui verrà posizionato il coperchio più grande di 22 mm x 50 mm x 1 mm per creare uno spazio tra il vetrino e il coperchio più grande (Figura 5A). Questo spazio consentirà al cervello di muoversi abbastanza per essere orientato correttamente impedendogli di essere schiacciato.

Figura 5: Schema che mostra il vetrino del microscopio, l'orientamento e i tipi di cellule nel cervello larvale. (A) Rappresentazione visiva del vetrino del microscopio su cui è montato un cervello larvale ed è pronto per essere ripreso. (B) Viene anche mostrato un orientamento da utilizzare per l'orientamento dei tessuti. (C) Vetrino per microscopio pronto per l'imaging su un microscopio confocale. (D) Cartone animato che mostra alcuni dei tipi di cellule nel cervello larvale. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
- Dopo aver incollato gli occhiali di copertura da 22 mm x 22 mm x 1 mm al vetrino del microscopio, pipettare 9,3 μL della soluzione di supporto di montaggio a base di glicerolo contenente un cervello da un pozzetto della piastra del microwell e posizionarlo al centro del vetrino (Figura 5A).
NOTA: i cervelli larvali possono aderire alla punta della pipetta, quindi fai attenzione. Per evitare l'adesione, iniziare aspirando l'antifade e quindi aspirando il singolo cervello verso la fine del volume di 9,3 μL. - Una volta che il cervello è sulla diapositiva, posizionare delicatamente il coperchio di 22 mm x 50 mm x 1 mm sulla parte superiore. Posizionare il cervello come si vede nella Figura 5B. Spostare delicatamente il coverslip per orientare il cervello. Il campione è quindi pronto per l'imaging.
- Utilizzare un microscopio confocale dotato di un elevato ingrandimento e di un obiettivo ad alta apertura numerica (Figura 5C) per acquisire le migliori immagini. Ad esempio: 60x o 63x, obiettivo ad immersione in olio da 1,4 NA.
- Immagina il cervello con la superficie dorsale più vicina al coverslip (e obiettivo). Acquisire le pile Z attraverso l'intero emisfero cerebrale a partire dalla superficie ventrale (più lontana dall'obiettivo) a intervalli di 1 μm o dimensione del passo Z.
NOTA: I laser utilizzati dipendono dagli anticorpi secondari. In questo protocollo, le linee laser utilizzate erano 488 nm per rilevare la colorazione Scribble, 555 nm per rilevare Deadpan e 633 nm per rilevare EdU.
- Immagina il cervello con la superficie dorsale più vicina al coverslip (e obiettivo). Acquisire le pile Z attraverso l'intero emisfero cerebrale a partire dalla superficie ventrale (più lontana dall'obiettivo) a intervalli di 1 μm o dimensione del passo Z.
6. Analisi dei dati
- Usa il software open source Fiji per analizzare gli emisferi cerebrali e usa il plug-in del contatore di cellule delle Fiji per contare le cellule.
Risultati
I cervelli selvatici dell'OregonR appena nati sono stati sezionati e coltivati per 24 ore nei media di Schneider integrati (SSM) con insulina. I tessuti sono stati fissati e macchiati secondo il protocollo. Sono stati utilizzati anticorpi primari generati contro Deadpan (Dpn) per rilevare NB e Scribble per etichettare le membrane cellulari. L'analogo della timidina 5-etinil-2′-deossiuridina (Edu) è stato aggiunto per rilevare l'ingresso nella fase S e la riattivazione NB. Abbiamo trovato NB Edu positivi e Dpn positivi...
Discussione
Il metodo qui descritto per coltivare gli espianti cerebrali può essere eseguito nella maggior parte degli ambienti di laboratorio. Gli strumenti richiesti, così come la procedura e la raccolta dei dati, sono semplici e diretti. Con questo metodo, si può testare una varietà di ipotesi, comprese quelle relative alle cascate di segnalazione cellulare e ai fattori estrinseci che regolano la riattivazione e la proliferazione nb. Qui, utilizzando animali OregonR selvatici, abbiamo scoperto che l'insulina esogena era suffi...
Divulgazioni
Gli autori non hanno interessi in competizione.
Riconoscimenti
Riconosciamo il programma LSAMP Bridges to Doctorate for funding (CNK) e NIH / NIGMS (R01-GM120421 e R35-GM141886). Siamo grati al Dr. Conor Sipe per la Figura 1. Ringraziamo anche tutti i membri del laboratorio Siegrist per il loro continuo supporto e tutoraggio. Ringraziamo in particolare Chhavi Sood e Gary Teeters per l'attenta lettura del manoscritto e per aver fornito commenti.
Materiali
| Name | Company | Catalog Number | Comments |
| 10 µL Pipette tips | Denville Sci | P2102 | |
| 1000 µL Pipette tips | Denville Sci | P2103-N | |
| 1000 µL Pipettor | Gilson | P1000 | |
| 16% paraformaldehyde (10 x 10 mL) | Electron Microscopy Sciences | 2912.60.0000 | Used for Fixation of Larval Brains |
| 20 µL Pipette | Gilson | P20 | |
| 200 µL Pipette tips | Denville Sci | 1158U56 | |
| 24-well multiwell culture plates | Fisher Scientific | 50-197-4477 | |
| 35 mm Petri dishes | Fisher Scientific | 08-757-100A | Grape Plate Ingredients |
| 4 °C refrigerator | Fisher Scientific | Provides an ideal temperature for >24 h incubations in antibody solution | |
| 63x Objective | Lecia | ||
| Active dry yeast | Most supermarkets | ||
| Agarose | Fisher Scientific | 214010 | Grape Plate Ingredients |
| Click-iT EdU Cell Proliferation Kit for Imaging, Alexa Fluor 647 dye | Thermo Fisher Scientific | C10340 | to label proliferating cells |
| Confocal Microscope | Leica | SP8 | |
| Coverslips 22 mm x 22 mm x 1 mm , 10 pack of 4 oz | Fisher Scientific | 12-544-10 | Two Coverslips are super glued to the ends of the microscope slide. This creates a space that allows for the brains to float in antifade while being imaged. |
| Coverslips, 22 mm x 50 mm x 1 mm | Fisher Scientific | 12-545E | The coverslip is placed on two square coverslips on the microscope slide ensuring that the brain in the antifade does not move while imaging. |
| Dissecting microscope | Zeiss | Stemi 2000 | |
| Ethanol 200 proof (100%), Decon Labs, 1 gallon bottle | Fisher Scientific | 2701 | Used to wash off the larvae before the 24 hr hold in culture medium |
| Fetal Bovine Serum (10%) | Sigma | F4135-100ML | Supplement for cell culture media. |
| Fine forceps for dissection | Fine Science Tools | 11295-20 | Forcepts used in disections. They work best when sharpened. |
| Fly Bottles for Crossing | Genessee Scientific | 32-130 | This bottle is used as a container that lets the flies lay eggs on the grape plate. |
| Glass Dissection Dish (3 well) | These are no longer available | ||
| Glutathione | Sigma | G6013 | Provides oxidative protection during cell culture. |
| Goat Serum | Sigma | G9023- 10ML | Blocking Agent |
| Grape Plates | Made in house | Made in house | Grape juice/agarose plates for collecting freshly hatched eggs |
| Image J | Imagej.net/fiji/downloads | Free Download: https://fiji.sc | Imaging platform that is used to count cells and Edu reactivation |
| Incubator | Thermo Fisher Scientific | Ensures that the temperature, humidity, and light exposure is exactly the same throughout experiment. | |
| Insulin | Sigma | I0516 | Independant variable of the experiment |
| Laminar flow hood | For aliquoting culture media | ||
| L-Glutamine | Sigma | G7513 | Provides support during cell culture |
| Nunc 72-well Microwell Mini Trays | Fisher Scientific | 12-565-154 | Immunostaining steps are performed in this tray |
| Parafilm | Fisher Scientific | S37440 | Film used to seal plates in order to prevent evaporation |
| Pen-Strep | Sigma | P4458-100ml | Antibiodics used to prevent bacterial contamination of cells during culture. |
| Phosphate Buffer, pH7.4 | Made in house | Made in house | Solvent used to wash the brains after fixing and staining steps |
| Pick | Fine Science Tools | 10140-01 | Used to pick larvae off of the grape plate |
| Propionic acid | Fisher Scientific | A-258 | Grape Plate Ingredients |
| Rabbit 405 | Abcam | ab175653 | Antibodies used for immunostaining |
| Rat 555 | Abcam | ab150166 | Antibodies used for immunostaining |
| Rb Scribble | A Gift from Chris Doe | Antibodies used for immunostaining | |
| Rt Deadpan | Abcam | ab195173 | Antibodies used for immunostaining |
| Schneiders Culture Medium | Life Tech | 21720024 | Contains nutrients that help the cells grow and proliferate |
| SlowFade Diamond Antifade (5 x 2 mL) | Life Tech | S36963 | Reagent that provides protection against fading fluorophores |
| Sterile Water | Autoclave Milli-Q water made in house | Needed for Solutions | |
| Sucrose | Fisher | S2-12 | Grape Plate Ingredients |
| Superfrost Microscope Slides | Fisher Scientific | 12-544-7 | |
| Superglue | Most supermarkets | ||
| Tegosept | Genesee Scientific | 20-259 | Grape Plate Ingredients |
| Triton-X 100 | Sigma | T9284-100ML | PBT |
| Welch's 100% grape grape juice | Most supermarkets | Grape Plate Ingredients |
Riferimenti
- Suman, S., Domingues, A., Ratajczak, J., Ratajczak, M. Z. Potential clinical applications of stem cells in regenerative medicine. Advances in Experimental Medicine and Biology. 1201, 1-22 (2019).
- Tabar, V., Studer, L. Pluripotent stem cells in regenerative medicine: challenges and recent progress. Nature Reviews Genetics. 15, 82-92 (2014).
- Daley, G. Q. Stem cells and the evolving notion of cellular identity. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 370, 20140376 (2015).
- Rodrigues, M., Kosaric, N., Bonham, C. A., Gurtner, G. C. Wound healing: A cellular perspective. Physiological Reviews. 99, 665-706 (2019).
- van Velthoven, C. T. J., Rando, T. A. Stem cell quiescence: Dynamism, restraint, and cellular idling. Cell Stem Cell. 24, 213-225 (2019).
- Chapman, N. M., Boothby, M. R., Chi, H. Metabolic coordination of T cell quiescence and activation. Nature Reviews Immunology. 20, 55-70 (2020).
- Wosczyna, M. N., Rando, T. A. A muscle stem cell support group: Coordinated cellular responses in muscle regeneration. Developmental Cell. 46, 135-143 (2018).
- Homem, C. C., Knoblich, J. A. Drosophila neuroblasts: a model for stem cell biology. Development. 139, 4297-4310 (2012).
- Kang, K. H., Reichert, H. Control of neural stem cell self-renewal and differentiation in Drosophila. Cell and Tissue Research. 359, 33-45 (2015).
- Chell, J. M., Brand, A. H. Nutrition-responsive glia control exit of neural stem cells from quiescence. Cell. 143, 1161-1173 (2010).
- Sousa-Nunes, R., Yee, L. L., Gould, A. P. Fat cells reactivate quiescent neuroblasts via TOR and glial insulin relays in Drosophila. Nature. 471, 508-512 (2011).
- Britton, J. S., Edgar, B. A. Environmental control of the cell cycle in Drosophila: nutrition activates mitotic and endoreplicative cells by distinct mechanisms. Development. 125, 2149-2158 (1998).
- Lin, S., et al. Extremes of lineage plasticity in the Drosophila brain. Current biology : CB. 23, 1908-1913 (2013).
- Sipe, C. W., Siegrist, S. E. Eyeless uncouples mushroom body neuroblast proliferation from dietary amino acids in Drosophila. Elife. 6, 26343 (2017).
- Speder, P., Brand, A. H. Systemic and local cues drive neural stem cell niche remodelling during neurogenesis in Drosophila. Elife. 7, 30413 (2018).
- Yuan, X., Sipe, C. W., Suzawa, M., Bland, M. L., Siegrist, S. E. Dilp-2-mediated PI3-kinase activation coordinates reactivation of quiescent neuroblasts with growth of their glial stem cell niche. PLoS Biology. 18, 3000721 (2020).
- Colombani, J., et al. A nutrient sensor mechanism controls Drosophila growth. Cell. 114, 739-749 (2003).
- Geminard, C., Rulifson, E. J., Leopold, P. Remote control of insulin secretion by fat cells in Drosophila. Cell Metabolism. 10, 199-207 (2009).
- Siller, K. H., Serr, M., Steward, R., Hays, T. S., Doe, C. Q. Live imaging of Drosophila brain neuroblasts reveals a role for Lis1/dynactin in spindle assembly and mitotic checkpoint control. Molecular Biology of the Cell. 16, 5127-5140 (2005).
- Prithviraj, R., Trunova, S., Giniger, E. Ex vivo culturing of whole, developing Drosophila brains. Journal of Visualized Experiments: JoVE. (65), e4270 (2012).
- Bostock, M. P., et al. An immobilization technique for long-term time-lapse imaging of explanted drosophila tissues. Frontiers in Cell and Developmental Biology. 8, 590094 (2020).
- Datta, S. Activation of neuroblast proliferation in explant culture of the Drosophila larval CNS. Brain Research. 818, 77-83 (1999).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati