
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Reativação de células-tronco neurais em explantes cerebrais de Drosophila cultivadas
Neste Artigo
Resumo
Um método para reativar células-tronco neurais quiescentes em explantas cerebrais de Drosophila cultivadas foi estabelecido. Usando este método, o papel dos sinais sistêmicos pode ser desacoplado a partir de sinais intrínsecos teciduais na regulação da quiescência de células-tronco neurais, entrada e saída.
Resumo
As células-tronco neurais (NSCs) têm a capacidade de proliferar, diferenciar, sofrer apoptose, e até mesmo entrar e sair da quiescência. Muitos desses processos são controlados pela complexa interação entre programas genéticos intrínsecos do NSC com fatores extrínsecos do NSC, locais e sistêmicos. No organismo modelo genético, Drosophila melanogaster, NSCs, conhecidos como neuroblastos (NBs), mudam de quiescência para proliferação durante a transição embrionária para larval. Durante esse tempo, as larvas emergem de suas cascas de ovos e começam a engatinhar, buscando nutrientes dietéticos. Em resposta à alimentação animal, o corpo gordo, um órgão endócrino com capacidade de armazenamento lipídico, produz um sinal, que é liberado sistematicamente no hemoglifo circulante. Em resposta ao sinal derivado do corpo gordo (FBDS), peptídeos semelhantes à insulina drosophila (Dilps) são produzidos e liberados de neurônios neurosecretores cerebrais e glia, levando à ativação a jusante da sinalização de crescimento PI3-quinase em NBs e seu nicho glial e traqueial. Embora este seja o modelo atual de como os NBs mudam da quiescência para a proliferação, a natureza da sugestão extrínseca da FBDS permanece indescritível. Para entender melhor como as pistas sistêmicas extrínsecas da NB regulam a saída da quiescência, um método foi desenvolvido para cultivar cérebros larvais precoces in vitro antes da alimentação animal. Com este método, fatores exógenos podem ser fornecidos aos meios de comunicação cultural e à saída do NB da quiescência avaliada. Descobrimos que insulina exógena é suficiente para reativar NBs de quiescência em explantas cerebrais inteiras. Como este método é adequado para telas de grande escala, pretendemos identificar pistas extrínsecas adicionais que regulam as decisões de quiescência versus proliferação da NB. Como os genes e caminhos que regulam as decisões de proliferação do NSC são evolutivamente conservados, os resultados deste ensaio poderiam fornecer insights sobre a melhoria das terapias regenerativas na clínica.
Introdução
As células-tronco são de grande interesse devido ao seu potencial de uso na medicina regenerativa 1,2. Muitos animais, especialmente aqueles que são de longa duração, mantêm células-tronco dentro de seus tecidos adultos. Essas células-tronco residentes funcionam para manter a homeostase tecidual e são utilizadas para reparo após lesões físicas ou doença 3,4. A maioria das células-tronco em animais adultos são quiescentes, um estado relativamente adormecido caracterizado pela parada do ciclo celular e pela inativação da sinalização de crescimento5. Em resposta a sinais extrínsecos, as células-tronco saem da quiescência, entram no ciclo celular e começam a gerar progêneria filha específica ao seu tipo de tecido. Por exemplo, a fim de montar uma resposta imune eficaz, as células que apresentam antígenos induzem células T ingênuas quiescentes a entrar no ciclo celular e expandir clonalmente6. Em resposta à lesão muscular esquelética, células-tronco de satélite muscular entram no ciclo celular e geram myoblasts da filha para substituir myofibrils danificados 5,7. Embora esteja claro que as células-tronco quiescentes respondem a sinais extrínsecos, em muitos casos, a natureza da sugestão extrínseca permanece incerta, bem como o mecanismo de ativação de células-tronco induzidas por sinais. Obter uma melhor compreensão de como as células-tronco quiescentes respondem a sinais extrínsecos e entram no ciclo celular ajudará no desenvolvimento de melhores terapias com células-tronco na clínica e aumentarão o conhecimento científico.
Há décadas, organismos modelo têm sido usados para descobrir os genes e caminhos de sinalização celular que regulam a proliferação de células-tronco durante o desenvolvimento e na idade adulta. Em Drosophila, as células-tronco neurais (NSCs), conhecidas como neuroblastos (NBs), se dividem ao longo do desenvolvimento para gerar todos os neurônios e glia que finalmente se integram, formando o circuito neural necessário para a função cerebral 8,9. Como outras células-tronco, os NBs se dividem assimetricamente para se auto-renovar e, em alguns casos, simetricamente para expandir o pool de células-tronco. Os NBs são especificados durante a embriogênese e a maioria entra na quiescência no final, coincidentemente com o declínio dos estoques de nutrientes maternos (Figura 1). Depois que a embriogênese estiver completa, as larvas eclodem e comecem a se alimentar. Em resposta à alimentação animal, os NBs reativam a partir da quiescência e retomam as divisões celulares 10,11,12,13,14,15,16. Porque o CNS de Drosophila é relativamente simples e porque os NBs entram e saem da quiescência em horários definidos, usar drosophila para investigar a regulação da quiescência, entrada e saída, prova o ideal.

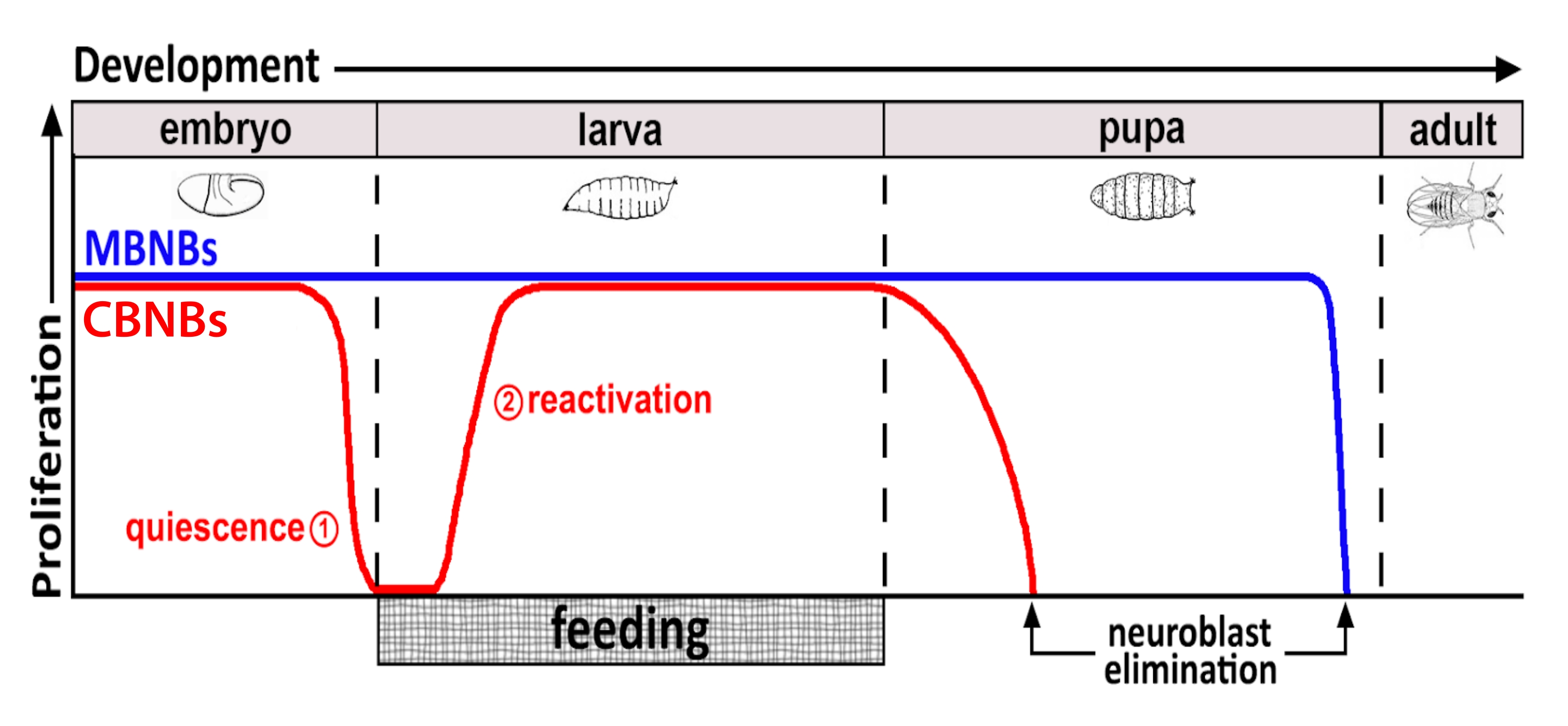
Figura 1: Proliferação relativa de NBs CB (neuroblastos cerebrais centrais, vermelho) e MB NBs (neuroblastos do corpo de cogumelos, azul) ao longo do tempo de desenvolvimento. No final da embriogênese, a maioria dos NBs (linha vermelha) cessam a proliferação e entram na quiescência. A quiescência continua até que larvas recém-eclodidas consumam sua primeira refeição completa. Os pontos de foco desta metodologia são denotados em círculos vermelhos (1, quiescência e 2, reativação). MB NBs (azul) são um subconjunto de NBs cerebrais centrais que se dividem continuamente durante o desenvolvimento (4 por hemisfério cerebral). Clique aqui para ver uma versão maior desta figura.
{kind=link}
Em resposta à alimentação animal, as vias de sinalização de crescimento PI3-quinase e TOR tornam-se ativas em NBs e em seu nicho glial e traqueal 10,11,15,16. Quando os nutrientes dietéticos são retirados ou quando os níveis de PI3-quinase são reduzidos, os NBs não reativam e o crescimento da glia e traqueia também são reduzidosem 10,11,15,16. O modelo atual afirma que a reativação da NB está acoplada ao crescimento larval pelo corpo de gordura, que libera um sinal sistêmico em resposta à alimentação animal 12,17,18. Este sinal, que permanece indescritível, provavelmente promove a expressão e liberação de peptídeo semelhante à insulina Drosophila (Dilps) no cérebro, o que leva à ativação a jusante de PI3-kinase em NBs e seu nicho glial e traqueal. Para entender melhor a natureza das deixas sistêmicas, desenvolvemos um método para reativar NBs quiescentes em explantas cerebrais cultivadas. Com este método, a reativação de NBs pode ser avaliada na ausência de pistas sistêmicas animais inteiras. Fatores exógenos podem ser reabastecidos aos meios de cultura e à reativação de NB conforme a incorporação do analógico de timmidina, EdU. Usando este método, determinamos que a insulina exógena é suficiente para reativar NBs quiescentes em explantas cerebrais. O trabalho futuro será direcionado para identificar fatores adicionais que, quando adicionados de volta, regulam positiva ou negativamente a quiescência de NB em explantas cerebrais.
Protocolo
1. Coleta de larvas de Drosophila
NOTA: Prepare a placa de levedura, a pasta de uva e o condomínio Fly antes de iniciar:
- Pasta de levedura: Em um recipiente pequeno, misture 5 g de levedura seca ativa com 10 mL de água para formar uma pasta que tenha a consistência de manteiga de amendoim. Cubra a pasta de levedura com plástico e use um elástico para fixá-lo firmemente ao recipiente.
NOTA: A pasta de levedura fresca se expandirá em seu recipiente e sairá da tampa, a menos que firmemente presa. A pasta de levedura durará vários dias em temperatura ambiente (RT). - Pratos de uva: Siga a receita para fazer pratos de uva (Tabela 1). Se usar placas armazenadas a 4°C, certifique-se de pré-aquecer as placas antes de usar, colocando-as no RT por 1h.
- Misture água (750 mL) e ágar (18,75 g) em um frasco de 4 L, redemoinho e autoclave por 20 min (ciclo líquido).
- Misture o suco de uva (250 mL) e a sacarose (25 g) em um frasco de 1 L com uma grande barra de mexida em uma placa aquecida (fogo baixo). Quando a sacarose for dissolvida, desligue o fogo, espere até que o frasco possa ser tocado antes de adicionar Tegosept (10%, 4 mL) e ácido propiônico (5 mL). Mantenha a barra de agitação.
- Quando a autoclavação estiver completa, deixe esfriar até que o frasco possa ser tocado (~60 °C), em seguida, misture na mistura de suco de uva.
- Combine todas as soluções em um frasco e deixe mexer na placa.
- Pipeta a solução em tampas de placas de Petri de pequeno porte (35 mm). Pipeta aproximadamente 9 mL por tampa ou até que uma cúpula convexa seja obtida.
- OPCIONAL: Coloque as tampas para se livrar de qualquer bolha.
- Quando as placas se solidificarem, empilhe as placas de uva em uma caixa com uma tampa hermética e coloque a caixa a 4 °C. As placas podem ser armazenadas por até 1 mês.
- Voe condomínio: Soque ~20 furos em uma garrafa de polipropileno de fundo quadrado de 6 onças usando uma agulha de 18 G.
- Transfira moscas adultas (~100 OregonR ou qualquer genótipo) para um condomínio de moscas e tampe o condomínio com uma placa de ágar de uva coberta com um pouco de pasta de levedura. Coloque o dab em direção ao centro da placa e afixe a placa no condomínio com fita de laboratório.
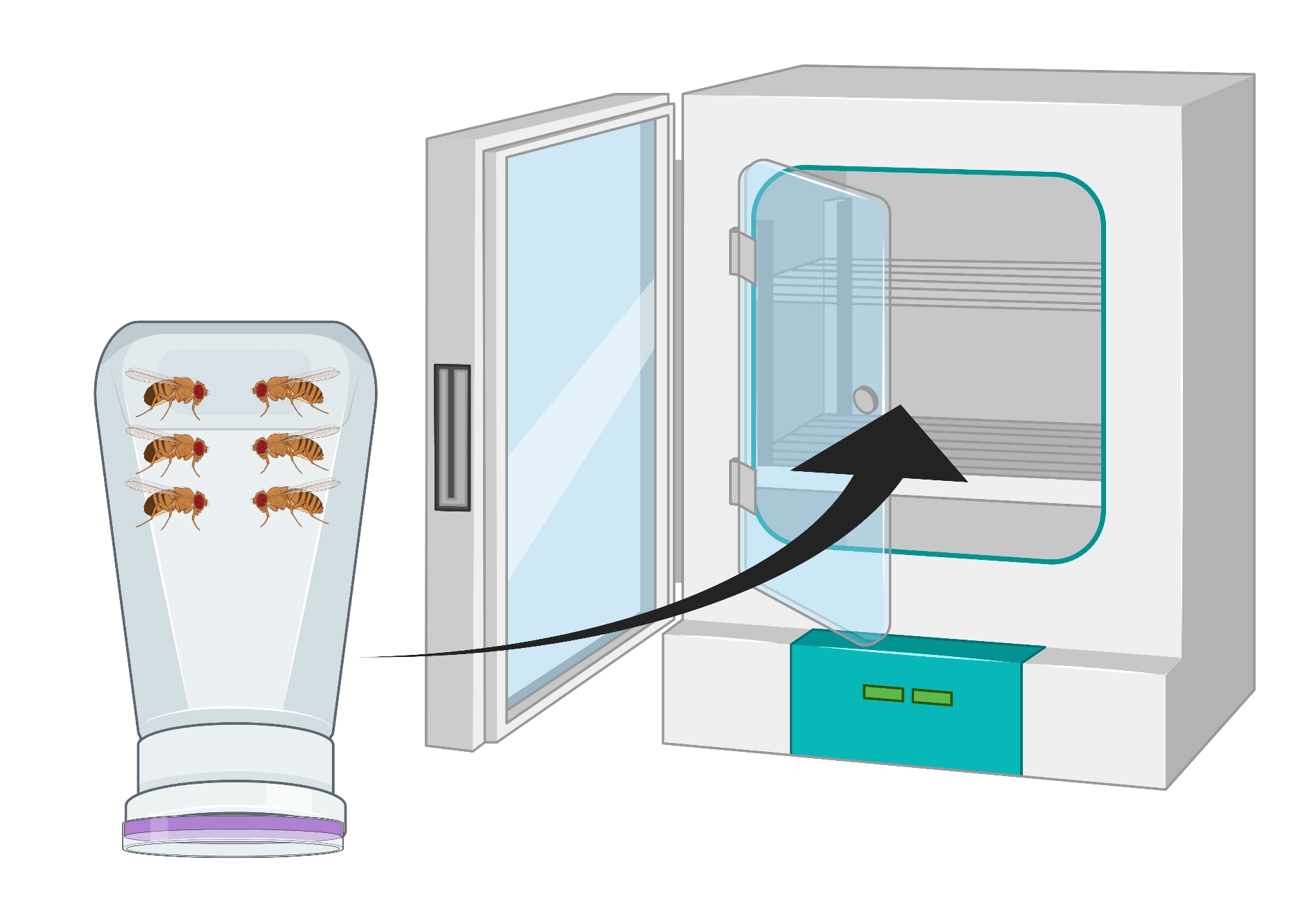
- Inverta o recipiente para que a placa de ágar de uva esteja na parte inferior e coloque-a em uma incubadora de 25 °C por 24 h (Figura 2).

Figura 2: Representação visual de garrafa de mosca invertida (condomínio) com adultos Drosophila masculino e feminino. A garrafa plástica tem pequenas perfurações, geradas com uma agulha de 18 G, para troca de oxigênio. A boca da garrafa é selada com uma tampa de suco de uva de ágar e é invertida e armazenada em uma incubadora de 25 °C. Clique aqui para ver uma versão maior desta figura.
{kind=link}
- Depois de 24 horas, troque a placa de ágar de uva e substitua-a por uma nova placa coberta com pasta de levedura. Troque rapidamente as duas placas enquanto toca continuamente o condomínio levemente no banco para que as moscas adultas não escapem.
- Examine a placa por olho e avalie o número de embriões na placa. Os embriões de drosophila são oblongos e brancos com dois apêndices parecidos com cordas.
- Se houver muito poucos embriões na placa (menos de 100), descarte a placa (raspe o ágar no lixo de mosca e guarde a tampa plástica para reutilização). Em muitos casos, as fêmeas adultas não colocarão muitos embriões na primeira noite em um novo condomínio. Se for esse o caso, dê ao adulto moscas mais 24 horas para se aclimatar.
- Se houver um grande número de embriões na placa (pelo menos 100), guarde-a e remova cuidadosamente a pasta de levedura usando uma espátula de fundo plana.
- Uma vez que a pasta de levedura seja removida, use uma palheira metálica para remover manualmente todas as larvas da placa de uva sob um microscópio dissecando. Ao olhar para a placa sob um microscópio dissecando, devem ser observadas larvas rastejantes, bem como embriões.
- Remova todas as larvas escovando a picareta metálica em direção ao lado de uma larva. Larvas são pegajosas e vão grudar na picareta. Uma vez que uma larva está na picareta, larvas adicionais podem ser facilmente recolhidas usando a larva na ferramenta para anexar mais.
NOTA: Larvas gostam de grudar uma na outra. Neste ponto, não importa se as larvas são danificadas. Essas larvas serão descartadas. - Depois de colher e remover todas as larvas, coloque a placa de volta na incubadora de 25 °C. Certifique-se de colocar a placa em um recipiente maior que possa ser selado. Coloque toalhas de papel molhadas no fundo do recipiente maior para manter a umidade.
- Depois de 30-60 min, leve a placa de volta ao microscópio dissecando e agora, pegue cuidadosamente ~20-25 larvas da mesma placa de ágar de uva para garantir que as larvas colhidas sejam recém-eclodidas dentro de uma janela de tempo de 30-60 minutos.
- Submerse a ponta da ferramenta com as larvas recém-eclodidas 20-25 em uma placa de Petri (60 mm) preenchida com 1-2 mL de Salina Tampão fosfato de 1x (PBS) por 2 min.
- Depois de 2 minutos, coloque o prato em um ângulo para juntar o líquido na parte inferior. Usando um pequeno pincel, escove as larvas do líquido até o fundo da placa de Petri.
- Colete todas as larvas no pincel e transfira as larvas para uma nova placa de Petri (60 mm) contendo 1-2 mL de 70% de etanol. Repita os passos 1.15 para coletar larvas com um pincel e transferi-las para uma nova placa de Petri com 1-2 mL de 1x PBS.
2. Mídia cultural e preparação de ferramentas
- Pulverize o banco e a área de trabalho com 70% de etanol e deixe secar.
- Pulverize as ferramentas de dissecção, fórceps e dois pratos de relógio de vidro, com 70% de etanol e deixe-os secar no banco.
- Faça a mídia de Schneider complementada (SSM, Tabela 2) e coloque-a no gelo.
- Pipeta 1 mL de SSM em cada um dos pratos de relógio de vidro.
- Usando uma micropipette com ponta estéril, transfira as larvas recém-eclodidas da placa de PBS para o SSM no primeiro prato de relógio de vidro. Usando uma micropipette com ponta estéril, transfira as larvas recém-eclodidas para o SSM no segundo prato de relógio de vidro.
3. Dissecções e culturas cerebrais
- Uma vez que as larvas estejam no segundo prato de relógio de vidro com SSM, disseque os cérebros das larvas usando fórceps e um microscópio dissecando. Ajuste a ampliação conforme necessário.
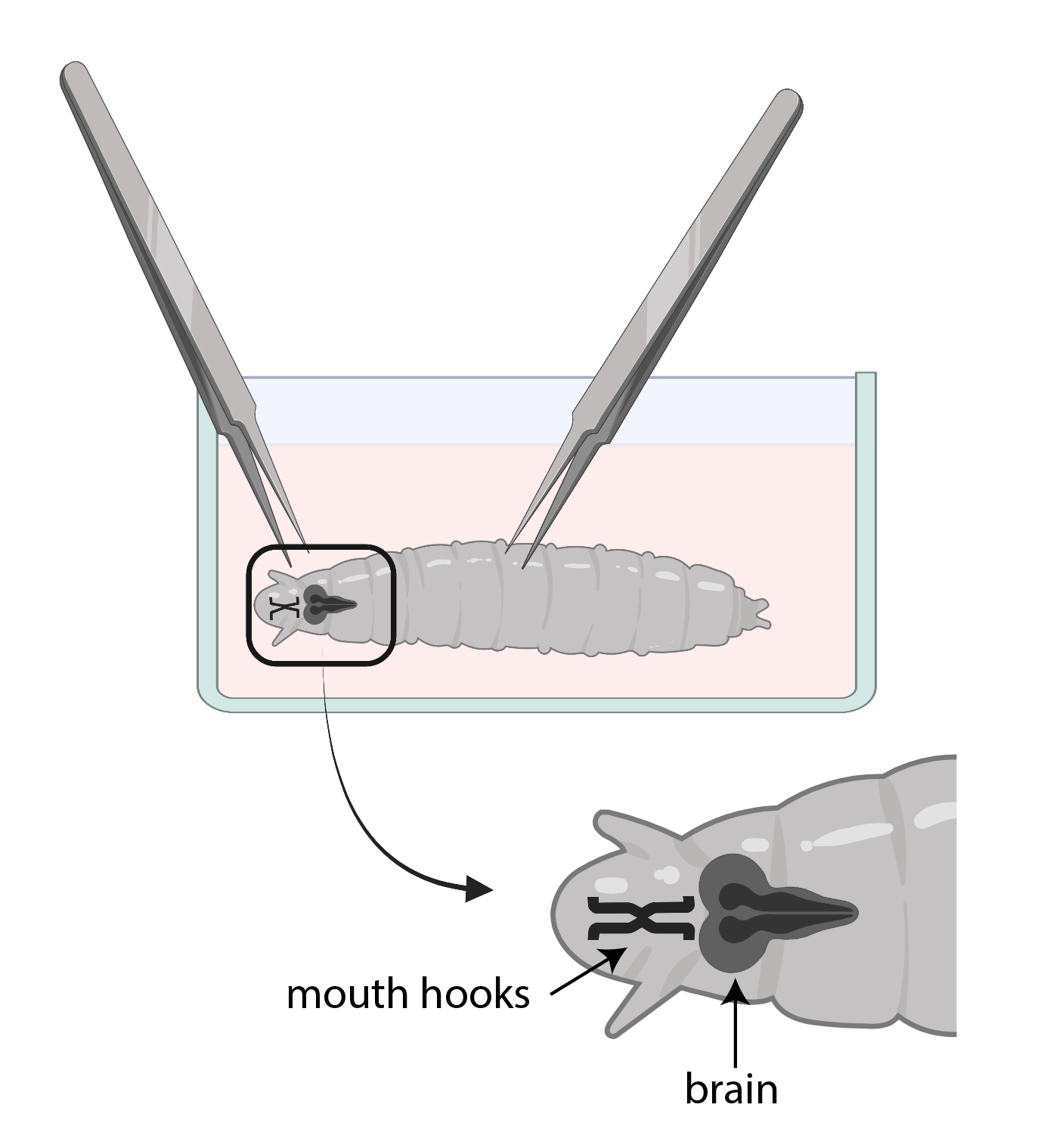
- Use um fórceps para pegar os ganchos da boca e com o outro, pegue suavemente o corpo no meio do caminho e puxe na direção oposta (Figura 3) para dividir a larva em dois pedaços.
NOTA: O cérebro estará localizado logo atrás dos ganchos da boca. Note que pode haver outros tecidos ao redor do cérebro. Tenha muito cuidado ao remover esses tecidos, pois pode resultar em danificar o cérebro

Figura 3: Larvas de drosophila em um prato de relógio de vidro com SSM. Os fórceps estão devidamente posicionados para dissecção. A localização do cérebro larval (cinza escuro) é posterior aos ganchos da boca (preto), e ambos são mostrados dentro da larva. Clique aqui para ver uma versão maior desta figura.
{kind=link}
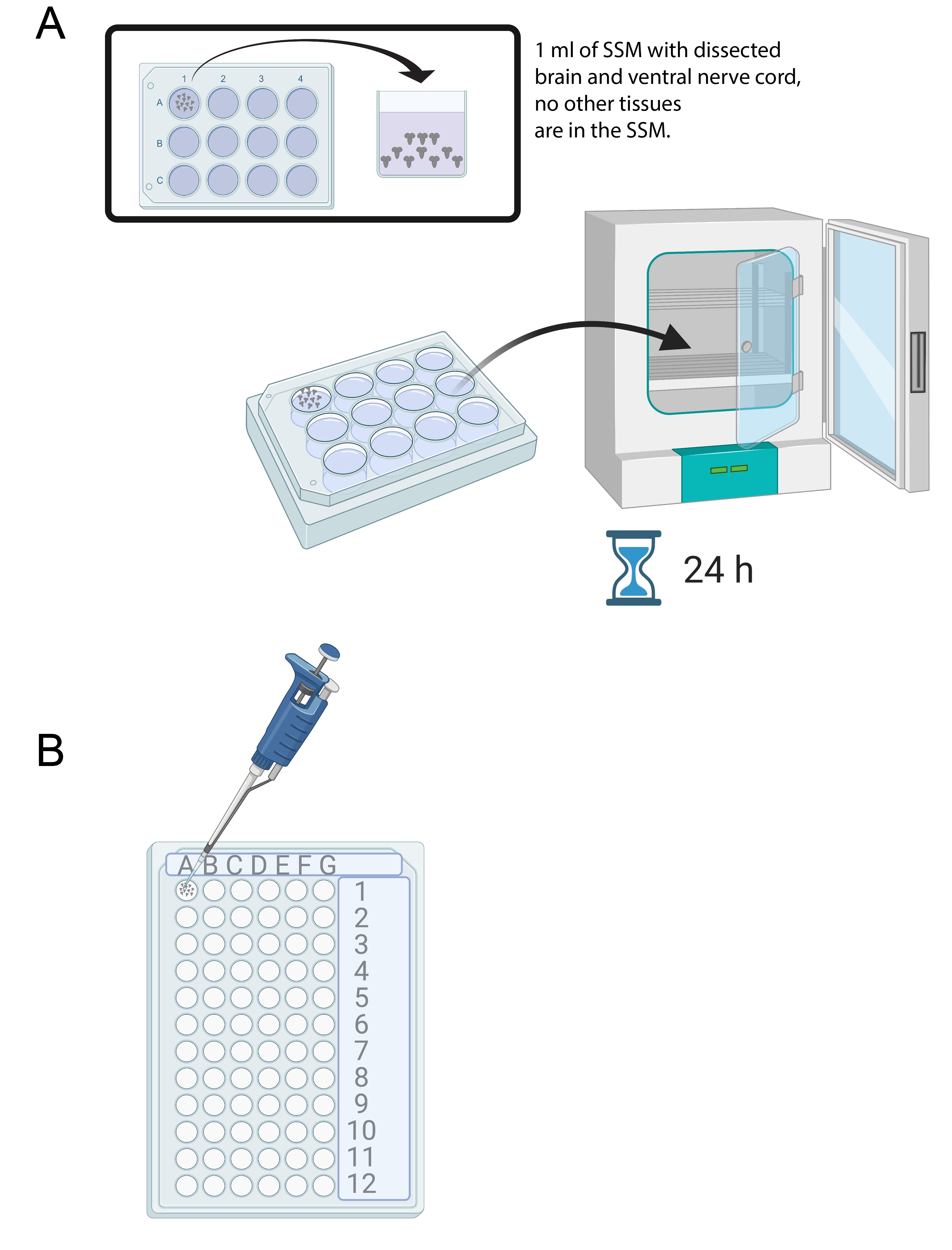
- Uma vez que 15-20 cérebros foram dissecados, adicione 1 mL de SSM em um poço de uma bandeja de cultura de 12 poços estéreis. Transfira os cérebros recém-dissecados para o SSM usando uma micropipette e uma ponta estéril (Figura 4A).
- Coloque os cérebros na mídia SSM na bandeja de cultura de 12 poços em uma incubadora a 25 °C por 24 h (Figura 4A).

Figura 4: Cultura cerebral e imunostaining. (A) Cérebros inteiros em um prato de cultura de 12 poços contendo 1 mL de SSM. O prato de cultura é então colocado em uma incubadora de 25 °C por 24 h. (B) mini bandeja de 72 poços que contém explantas cerebrais durante a imunossuagem. Cérebros são lavados e soluções transferidas usando uma micropipette P20 definida para 10 μL. Clique aqui para ver uma versão maior desta figura.
{kind=link}
4. Ensaio de proliferação, fixação cerebral e coloração de anticorpos
- No dia seguinte, faça 1 mL de solução SSM EdU. Pipeta 10 μL de um estoque de 10 mM de 5-Ethynyl-2′-desoxyuridina (EdU) com 990 μL de SSM (concentração final de EdU é igual a 0,1 mM) em um tubo e mistura de microcentrifuge estéril. Após a conclusão da incubação de 24 h, pipete os 1 mL de SSM EdU em um poço da bandeja de cultura estéril de 12 poços.
- Transfira os cérebros usando uma micropipette com uma ponta estéril do poço que contém SSM apenas para o novo poço que contém a solução SSM EdU. Incubar por 1h a 25 °C.
- Em seguida, transfira os cérebros rotulados pela EdU para outro poço na mesma bandeja de cultura contendo 1 mL de fixação (4% paraformaldeído, ver Tabela 3 para receita) por 20 minutos.
ATENÇÃO: Paraformaldeído é um risco biológico e deve ser descartado adequadamente. - Após a fixação, transfira rapidamente os cérebros para os poços de uma mini bandeja de 72 poços usando uma micropipette. Cada poço pode conter 10 cérebros e 10-15 μL de líquido (Figura 4B). Uma vez que os cérebros são transferidos para a mini bandeja (não mais de 10 cérebros por poço), remova a correção e enxágue os cérebros 3 vezes em 10 μL de 1x PBT (tampão fosfato, pH 7.4 contendo 0,1% Triton-X 100).
NOTA: Enxaguar significa pipetar 10 μL de 1x PBT no cérebro, remover e repetir 3 vezes. - Em seguida, lave os cérebros 3 vezes por 10 minutos cada, novamente em 10 μL 1x PBT. Certifique-se de que os cérebros estão cobertos com algum líquido o tempo todo.
- Depois que as lavagens estiverem completas, pipeta 10 μL de solução de bloqueio (1x PBT com soro de cabra 10% normal) no cérebro. Cubra a bandeja e sele-a usando uma tira de parafilm ao redor da borda.
- Uma vez selado, coloque a mini bandeja em uma caixa selada com toalhas molhadas para fornecer um ambiente úmido para evitar a evaporação. Coloque a caixa contendo a bandeja a 4 °C durante a noite.
- No dia seguinte, faça uma solução primária de anticorpos.
NOTA: Neste protocolo, o anti-rabisco de coelho foi usado para rotular membranas celulares e anti-deadpan de ratos para rotular neuroblastos, embora qualquer número de outros anticorpos primários pudesse ser usado.- Para fazer a solução primária de anticorpos, primeiro, fazer diluições de anticorpos primários na solução de bloqueio. Por exemplo, o anti-rabisco de coelho é usado em uma concentração final de 1:1000. Portanto, primeiro, diluir o anticorpo anti-rabisco de coelho às 1:100 (1 μL de anticorpo mais 99 μL de solução de bloqueio). Rat-deadpan é usado em uma concentração final de 1:100. Portanto, primeiro, diluir o anticorpo rat-deadpan às 1:10 (1 μL de anticorpo mais 9 μL de solução de bloqueio).
NOTA: Essas diluições podem ser armazenadas a longo prazo a 4 °C se a azida de sódio (0,05%) também for adicionada para inibir o crescimento bacteriano. - Em seguida, conte o número de poços que contêm cérebros. O número de poços determina o volume de solução de anticorpos primários a ser fazer. Por exemplo, se houver 2 poços de cérebros, prepare 20 μL de solução de anticorpos primários (para 10 poços, 100 μL, etc.). Para fazer uma solução de anticorpos primários de 20 μL, adicione 2 μL de cada diluição de anticorpos primários e 16 μL da solução de bloqueio.
NOTA: A concentração final de cada um dos anticorpos primários é de 1:1000 e 1:100, respectivamente. Em suma, faça a primeira diluição dos anticorpos primários em uma concentração para que a segunda diluição seja sempre 1:10 para chegar às respectivas concentrações finais. Neste caso, 1:1000 para coelhinho anti-rabisco e 1:100 para anti-morto.
- Para fazer a solução primária de anticorpos, primeiro, fazer diluições de anticorpos primários na solução de bloqueio. Por exemplo, o anti-rabisco de coelho é usado em uma concentração final de 1:1000. Portanto, primeiro, diluir o anticorpo anti-rabisco de coelho às 1:100 (1 μL de anticorpo mais 99 μL de solução de bloqueio). Rat-deadpan é usado em uma concentração final de 1:100. Portanto, primeiro, diluir o anticorpo rat-deadpan às 1:10 (1 μL de anticorpo mais 9 μL de solução de bloqueio).
- Remova a solução de bloqueio com o conjunto de micropipette a 10 μL e pipeta 10 μL de solução de anticorpos primários em cada poço.
- Cubra e sele a bandeja usando parafilm e coloque-a de volta na caixa selada com toalhas molhadas. Incubar durante a noite a 4 °C.
NOTA: O tremor não é necessário e é fortemente desencorajado. Anticorpos penetrarão nos cérebros sem tremer ou misturar. - No dia seguinte, remova a solução de anticorpos primários usando uma micropipette e enxágue os cérebros 3 vezes com 10 μL de 1x PBT. Em seguida, lave os cérebros 4 vezes com 10 μL de 1x PBT por 10 minutos cada. Durante as lavagens de 10 minutos, prepare a solução secundária de anticorpos.
- Para fazer a solução de anticorpos secundários, escolha anticorpos secundários que reconheçam os anticorpos primários. Neste protocolo, foram utilizados o anti-coelho de cabra Alexa Fluor 488 e a anti-rato de cabra Alexa 555.
- Pipeta 1 μL de cada um dos anticorpos secundários em um tubo de microcentrífuga com 298 μL da solução de bloqueio para fazer a concentração final 1:300 para cada anticorpo secundário.
- Após a última lavagem de 10 minutos, remova o PBT 1x e a pipeta 10 μL da solução de anticorpos secundários em cada poço. Sele a bandeja usando parafilm e coloque-a de volta na caixa com toalhas úmidas. Incubar durante a noite a 4 °C.
NOTA: Não se preocupe em remover todos os últimos μL nos poços entre enxaguas, lavagens ou ao adicionar soluções primárias e secundárias de anticorpos. Os cérebros sempre permanecerão submersos em alguns μL de líquido, o que é muito bom. - No dia seguinte, remova a solução de anticorpos secundários usando uma micropipette e enxágue os cérebros 3 vezes com 10 μL cada um de 1x PBT. Em seguida, lave os cérebros 4 vezes com 10 μL cada um de 1x PBT por 10 minutos cada.
- Durante as lavagens de 10 minutos, prepare a mistura de reação EdU para detectar a incorporação da EdU. Prepare o mix de reação EdU de acordo com as diretrizes dos fabricantes.
- Após a lavagem final, remova o PBT 1x e a pipeta 10 μL da reação EdU mistura em cada poço com cérebros. Sele a placa de microwell com parafilme e cubra com papel alumínio. Deixe a placa no banco por 30 minutos.
- Depois de 30 minutos, enxágue os cérebros 3 vezes com 10 μL cada um de 1x PBT e lave os cérebros 3 vezes com 10 μL cada um de 1x PBT por 5 min cada.
- Após a última lavagem, remova o PBT 1x e a pipeta 10 μL de uma solução de mídia de montagem baseada em glicerol. Sele a placa e coloque a 4 °C durante a noite.
5. Montagem e imagem dos cérebros
- No dia seguinte, preparem slides de microscópio (25 mm x 75 mm x 1 mm): Cola (por exemplo, supercola) um vidro de cobertura quadrada de 22 mm x 22 mm x 1 mm para cada extremidade do deslizamento do microscópio para criar uma 'ponte' sobre a qual as tampas maiores de 22 mm x 50 mm x 1 mm delip serão colocadas para fazer um espaço entre o slide e as tampas maiores (Figura 5A). Este espaço permitirá que o cérebro apenas movimento suficiente para ser corretamente orientado, evitando que ele seja esmagado.

Figura 5: Esquema mostrando tipos de deslizamento de microscópio, orientação e células no cérebro larval. (A) Representação visual do microscópio desliza sobre o qual um cérebro larval é montado e está pronto para ser imageado. (B) Uma diretriz também é mostrada para uso para orientação tecidual. (C) Slide de microscópio pronto para imagem em um microscópio confocal. (D) Desenho animado mostrando alguns dos tipos de células no cérebro larval. Clique aqui para ver uma versão maior desta figura.
{kind=link}
- Depois de colar os óculos de cobertura de 22 mm x 22 mm x 1 mm no slide do microscópio, pipeta 9,3 μL da solução de mídia de montagem baseada em glicerol contendo um cérebro de um poço da placa de microwell e colocá-lo no centro do slide (Figura 5A).
NOTA: Os cérebros larvais podem aderir à ponta da pipeta, por isso tenha cuidado. Para evitar a adesão, comece aspirando o antifato e, em seguida, aspirando o cérebro único no final do volume de 9,3 μL. - Uma vez que o cérebro esteja no slide, coloque delicadamente a tampa de 22 mm x 50 mm x 1 mm em cima. Posicione o cérebro como visto na Figura 5B. Mova suavemente a tampa para orientar o cérebro. A amostra está então pronta para a imagem.
- Use um microscópio confocal equipado com alta ampliação e um alto objetivo de abertura numérica (Figura 5C) para adquirir as melhores imagens. Por exemplo: 60x ou 63x, lente de imersão a óleo 1.4 NA.
- Imagem do cérebro com a superfície dorsal mais próxima da mancha (e objetivo). Adquira as pilhas Z através de todo o hemisfério cerebral começando na superfície ventral (mais distante do objetivo) em intervalos de 1 μm ou tamanho de passo Z.
NOTA: Os lasers utilizados dependem dos anticorpos secundários. Neste protocolo, as linhas laser utilizadas foram de 488 nm para detectar manchas de rabisco, 555 nm para detectar Deadpan e 633 nm para detectar EdU.
- Imagem do cérebro com a superfície dorsal mais próxima da mancha (e objetivo). Adquira as pilhas Z através de todo o hemisfério cerebral começando na superfície ventral (mais distante do objetivo) em intervalos de 1 μm ou tamanho de passo Z.
6. Análise de dados
- Use o software de código aberto fiji para analisar hemisférios cerebrais e usar o plugin de contador de células Fiji para contar as células.
Resultados
Cérebros selvagens recém-eclodidos do OregonR foram dissecados e cultivados por 24 h na mídia de Schneider (SSM) com insulina. Os tecidos foram fixados e manchados de acordo com o protocolo. Foram utilizados anticorpos primários gerados contra Deadpan (Dpn) para detectar NBs e Rabiscos para rotular membranas celulares. O analógico timmidina 5-Ethynyl-2′-desoxyuridina (Edu) foi adicionado para detectar a entrada da fase S e a reativação de NB. Encontramos NBs edu positivos de grande porte e DPN positivo após 24 ...
Discussão
O método descrito aqui para cultivar explanações cerebrais pode ser realizado na maioria dos ambientes de laboratório. As ferramentas necessárias, bem como o procedimento e a coleta de dados, são simples e simples. Com este método, pode-se testar uma variedade de hipóteses, incluindo aquelas relacionadas às cascatas de sinalização celular e fatores extrínsecos que regulam a reativação e proliferação do RN. Aqui, usando animais oregonr de tipo selvagem, descobrimos que a insulina exógena era suficiente pa...
Divulgações
Os autores não têm interesses concorrentes.
Agradecimentos
Reconhecemos o programa LSAMP Bridges to Doctorate para financiamento (CNK) e o NIH/NIGMS (R01-GM120421 e R35-GM141886). Somos gratos ao Dr. Conor Sipe pela Figura 1. Agradecemos também a todos os membros do laboratório Siegrist por seu apoio contínuo e mentoria. Agradecemos especialmente a Chhavi Sood e Gary Teeters por sua leitura cuidadosa do manuscrito e por fornecer comentários.
Materiais
| Name | Company | Catalog Number | Comments |
| 10 µL Pipette tips | Denville Sci | P2102 | |
| 1000 µL Pipette tips | Denville Sci | P2103-N | |
| 1000 µL Pipettor | Gilson | P1000 | |
| 16% paraformaldehyde (10 x 10 mL) | Electron Microscopy Sciences | 2912.60.0000 | Used for Fixation of Larval Brains |
| 20 µL Pipette | Gilson | P20 | |
| 200 µL Pipette tips | Denville Sci | 1158U56 | |
| 24-well multiwell culture plates | Fisher Scientific | 50-197-4477 | |
| 35 mm Petri dishes | Fisher Scientific | 08-757-100A | Grape Plate Ingredients |
| 4 °C refrigerator | Fisher Scientific | Provides an ideal temperature for >24 h incubations in antibody solution | |
| 63x Objective | Lecia | ||
| Active dry yeast | Most supermarkets | ||
| Agarose | Fisher Scientific | 214010 | Grape Plate Ingredients |
| Click-iT EdU Cell Proliferation Kit for Imaging, Alexa Fluor 647 dye | Thermo Fisher Scientific | C10340 | to label proliferating cells |
| Confocal Microscope | Leica | SP8 | |
| Coverslips 22 mm x 22 mm x 1 mm , 10 pack of 4 oz | Fisher Scientific | 12-544-10 | Two Coverslips are super glued to the ends of the microscope slide. This creates a space that allows for the brains to float in antifade while being imaged. |
| Coverslips, 22 mm x 50 mm x 1 mm | Fisher Scientific | 12-545E | The coverslip is placed on two square coverslips on the microscope slide ensuring that the brain in the antifade does not move while imaging. |
| Dissecting microscope | Zeiss | Stemi 2000 | |
| Ethanol 200 proof (100%), Decon Labs, 1 gallon bottle | Fisher Scientific | 2701 | Used to wash off the larvae before the 24 hr hold in culture medium |
| Fetal Bovine Serum (10%) | Sigma | F4135-100ML | Supplement for cell culture media. |
| Fine forceps for dissection | Fine Science Tools | 11295-20 | Forcepts used in disections. They work best when sharpened. |
| Fly Bottles for Crossing | Genessee Scientific | 32-130 | This bottle is used as a container that lets the flies lay eggs on the grape plate. |
| Glass Dissection Dish (3 well) | These are no longer available | ||
| Glutathione | Sigma | G6013 | Provides oxidative protection during cell culture. |
| Goat Serum | Sigma | G9023- 10ML | Blocking Agent |
| Grape Plates | Made in house | Made in house | Grape juice/agarose plates for collecting freshly hatched eggs |
| Image J | Imagej.net/fiji/downloads | Free Download: https://fiji.sc | Imaging platform that is used to count cells and Edu reactivation |
| Incubator | Thermo Fisher Scientific | Ensures that the temperature, humidity, and light exposure is exactly the same throughout experiment. | |
| Insulin | Sigma | I0516 | Independant variable of the experiment |
| Laminar flow hood | For aliquoting culture media | ||
| L-Glutamine | Sigma | G7513 | Provides support during cell culture |
| Nunc 72-well Microwell Mini Trays | Fisher Scientific | 12-565-154 | Immunostaining steps are performed in this tray |
| Parafilm | Fisher Scientific | S37440 | Film used to seal plates in order to prevent evaporation |
| Pen-Strep | Sigma | P4458-100ml | Antibiodics used to prevent bacterial contamination of cells during culture. |
| Phosphate Buffer, pH7.4 | Made in house | Made in house | Solvent used to wash the brains after fixing and staining steps |
| Pick | Fine Science Tools | 10140-01 | Used to pick larvae off of the grape plate |
| Propionic acid | Fisher Scientific | A-258 | Grape Plate Ingredients |
| Rabbit 405 | Abcam | ab175653 | Antibodies used for immunostaining |
| Rat 555 | Abcam | ab150166 | Antibodies used for immunostaining |
| Rb Scribble | A Gift from Chris Doe | Antibodies used for immunostaining | |
| Rt Deadpan | Abcam | ab195173 | Antibodies used for immunostaining |
| Schneiders Culture Medium | Life Tech | 21720024 | Contains nutrients that help the cells grow and proliferate |
| SlowFade Diamond Antifade (5 x 2 mL) | Life Tech | S36963 | Reagent that provides protection against fading fluorophores |
| Sterile Water | Autoclave Milli-Q water made in house | Needed for Solutions | |
| Sucrose | Fisher | S2-12 | Grape Plate Ingredients |
| Superfrost Microscope Slides | Fisher Scientific | 12-544-7 | |
| Superglue | Most supermarkets | ||
| Tegosept | Genesee Scientific | 20-259 | Grape Plate Ingredients |
| Triton-X 100 | Sigma | T9284-100ML | PBT |
| Welch's 100% grape grape juice | Most supermarkets | Grape Plate Ingredients |
Referências
- Suman, S., Domingues, A., Ratajczak, J., Ratajczak, M. Z. Potential clinical applications of stem cells in regenerative medicine. Advances in Experimental Medicine and Biology. 1201, 1-22 (2019).
- Tabar, V., Studer, L. Pluripotent stem cells in regenerative medicine: challenges and recent progress. Nature Reviews Genetics. 15, 82-92 (2014).
- Daley, G. Q. Stem cells and the evolving notion of cellular identity. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 370, 20140376 (2015).
- Rodrigues, M., Kosaric, N., Bonham, C. A., Gurtner, G. C. Wound healing: A cellular perspective. Physiological Reviews. 99, 665-706 (2019).
- van Velthoven, C. T. J., Rando, T. A. Stem cell quiescence: Dynamism, restraint, and cellular idling. Cell Stem Cell. 24, 213-225 (2019).
- Chapman, N. M., Boothby, M. R., Chi, H. Metabolic coordination of T cell quiescence and activation. Nature Reviews Immunology. 20, 55-70 (2020).
- Wosczyna, M. N., Rando, T. A. A muscle stem cell support group: Coordinated cellular responses in muscle regeneration. Developmental Cell. 46, 135-143 (2018).
- Homem, C. C., Knoblich, J. A. Drosophila neuroblasts: a model for stem cell biology. Development. 139, 4297-4310 (2012).
- Kang, K. H., Reichert, H. Control of neural stem cell self-renewal and differentiation in Drosophila. Cell and Tissue Research. 359, 33-45 (2015).
- Chell, J. M., Brand, A. H. Nutrition-responsive glia control exit of neural stem cells from quiescence. Cell. 143, 1161-1173 (2010).
- Sousa-Nunes, R., Yee, L. L., Gould, A. P. Fat cells reactivate quiescent neuroblasts via TOR and glial insulin relays in Drosophila. Nature. 471, 508-512 (2011).
- Britton, J. S., Edgar, B. A. Environmental control of the cell cycle in Drosophila: nutrition activates mitotic and endoreplicative cells by distinct mechanisms. Development. 125, 2149-2158 (1998).
- Lin, S., et al. Extremes of lineage plasticity in the Drosophila brain. Current biology : CB. 23, 1908-1913 (2013).
- Sipe, C. W., Siegrist, S. E. Eyeless uncouples mushroom body neuroblast proliferation from dietary amino acids in Drosophila. Elife. 6, 26343 (2017).
- Speder, P., Brand, A. H. Systemic and local cues drive neural stem cell niche remodelling during neurogenesis in Drosophila. Elife. 7, 30413 (2018).
- Yuan, X., Sipe, C. W., Suzawa, M., Bland, M. L., Siegrist, S. E. Dilp-2-mediated PI3-kinase activation coordinates reactivation of quiescent neuroblasts with growth of their glial stem cell niche. PLoS Biology. 18, 3000721 (2020).
- Colombani, J., et al. A nutrient sensor mechanism controls Drosophila growth. Cell. 114, 739-749 (2003).
- Geminard, C., Rulifson, E. J., Leopold, P. Remote control of insulin secretion by fat cells in Drosophila. Cell Metabolism. 10, 199-207 (2009).
- Siller, K. H., Serr, M., Steward, R., Hays, T. S., Doe, C. Q. Live imaging of Drosophila brain neuroblasts reveals a role for Lis1/dynactin in spindle assembly and mitotic checkpoint control. Molecular Biology of the Cell. 16, 5127-5140 (2005).
- Prithviraj, R., Trunova, S., Giniger, E. Ex vivo culturing of whole, developing Drosophila brains. Journal of Visualized Experiments: JoVE. (65), e4270 (2012).
- Bostock, M. P., et al. An immobilization technique for long-term time-lapse imaging of explanted drosophila tissues. Frontiers in Cell and Developmental Biology. 8, 590094 (2020).
- Datta, S. Activation of neuroblast proliferation in explant culture of the Drosophila larval CNS. Brain Research. 818, 77-83 (1999).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoExplore Mais Artigos
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados