
このコンテンツを視聴するには、JoVE 購読が必要です。 サインイン又は無料トライアルを申し込む。
Method Article
ショウ ジョウバエ 脳外植における神経幹細胞の再活性化
要約
培養ショウ ジョウバエ 脳外植体において静止期神経幹細胞を再活性化する方法が確立されている。この方法を用いて、神経幹細胞の静止、入口および退出の調節において、全身性シグナルの役割を組織内因性シグナルから切り離すことができる。
要約
神経幹細胞(NSC)は、増殖、分化、アポトーシスを受け、さらには静止に出入りする能力を有する。これらのプロセスの多くは、NSC内因性遺伝プログラムとNSC外因性因子(局所的および全身的)との間の複雑な相互作用によって制御される。遺伝子モデル生物である ショウジョウバエのメラノガスターでは、神経芽細胞(NB)として知られるNSCsは、胚から幼虫への移行中に静止から増殖に切り替わります。この間、幼虫は卵の殻から出てきて這い回り始め、食事の栄養素を探します。動物の摂食に応答して、脂質貯蔵能力を有する内分泌器官である脂肪体はシグナルを生成し、循環血リンパに全身的に放出される。脂肪体由来シグナル(FBDS)に応答して、 ショウジョウバエ インスリン様ペプチド(Dilps)が産生され、脳神経分泌ニューロンおよびグリアから放出され、NBおよびそれらのグリアおよび気管ニッチにおけるPI3キナーゼ成長シグナル伝達の下流活性化をもたらす。これは、NBが静止から増殖にどのように切り替わるかについての現在のモデルであるが、FBDS外因性手がかりの性質は依然として解明されていない。NB外因性全身の手がかりが静止からの出口をどのように調節するかをよりよく理解するために、動物給餌前に初期の幼虫脳をインビトロで培養する方法が開発された。この方法を用いると、外因性因子を培養培地に供給し、NBがアッセイされて静止状態から脱退することができる。我々は、外因性インスリンが全脳外植体の静止からNBを再活性化するのに十分であることを見出した。この方法は大規模なスクリーンに適しているため、NBの静止と拡散の決定を調節する追加の外因性手がかりを特定することを目指しています。NSCの増殖決定を調節する遺伝子および経路は進化的に保存されているため、このアッセイの結果は、診療所における再生療法の改善に関する洞察を提供する可能性がある。
概要
幹細胞は、再生医療での使用の可能性のために大きな関心事です1,2。多くの動物、特に長命の動物は、成体組織内で幹細胞を維持しています。これらの常在幹細胞は、組織の恒常性を維持するために機能し、身体的傷害または疾患後の修復に利用される3,4。成体動物におけるほとんどの幹細胞は静止状態であり、細胞周期停止および成長シグナル伝達の不活性化を特徴とする比較的休眠状態である5。外因性の手がかりに応答して、幹細胞は静止状態から抜け出し、細胞周期に入り、組織型に特異的な娘の子孫を生成し始める。例えば、効果的な免疫応答をマウントするために、抗原提示細胞は静止期のナイーブT細胞を誘導して細胞周期に入り、クローン的に拡張する6。骨格筋損傷に応答して、筋衛星幹細胞は細胞周期に入り、損傷した筋原線維を置き換えるために娘筋芽細胞を生成する5,7。静止幹細胞が外因性シグナルに応答することは明らかであるが、多くの場合、外因性手がかりの性質および手がかり誘導幹細胞活性化のメカニズムは不明のままである。静止幹細胞が外因性の手がかりにどのように反応し、細胞周期に入るかについてのより良い理解を得ることは、診療所におけるより良い幹細胞療法の開発に役立ち、科学的知識を増加させるでしょう。
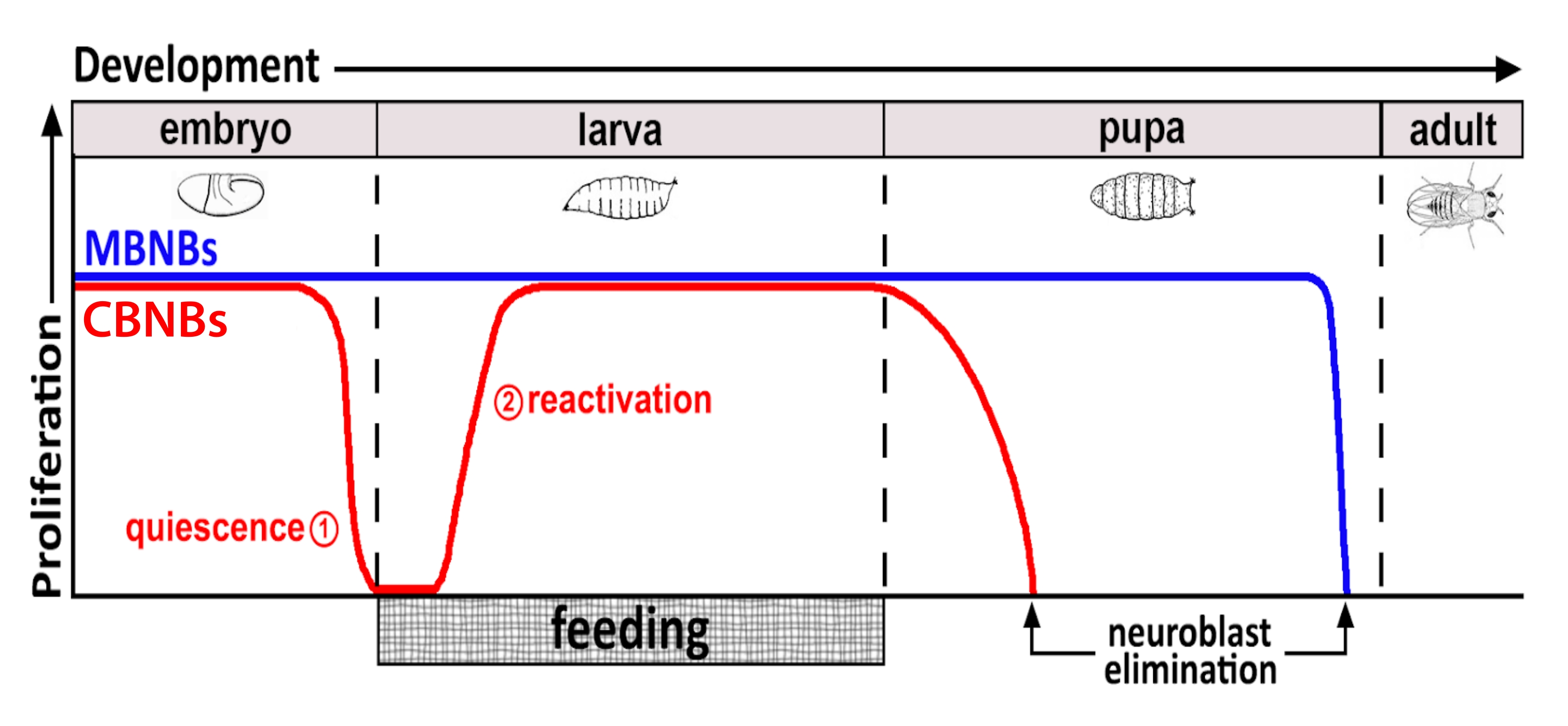
何十年もの間、モデル生物は、発生中および成人期の幹細胞増殖を調節する遺伝子および細胞シグナル伝達経路を明らかにするために使用されてきた。ショウジョウバエでは、神経芽細胞(NB)として知られる神経幹細胞(NSC)が発達中分裂し、最終的に統合するすべてのニューロンとグリアが生成され、脳機能に必要な神経回路を形成します8,9。他の幹細胞と同様に、NBは自己再生するために非対称的に分裂し、場合によっては対称的に分裂して幹細胞プールを拡張する。NBは胚発生時に特定され、そのほとんどは母親の栄養貯蔵量の減少と同時に、終わりに向かって静止状態に入る(図1)。胚発生が完了すると、幼虫は孵化し、摂食を開始する。動物摂食に応答して、NBは静止から再活性化し、細胞分裂を再開する10、11、12、13、14、15、16。ショウジョウバエのCNSは比較的単純であり、NBは定義された時間に静止に出入りするため、ショウジョウバエを使用して静止、出入りの調節を調査することは理想的であることが証明される。

図1:発生時間にわたるCB NB(中枢脳神経芽細胞、赤)およびMB NB(キノコ体神経芽細胞、青)の相対増殖。 胚発生の終わりに、ほとんどのNB(赤い線)は増殖を停止し、静止状態に入る。孵化したばかりの幼虫が最初の完全な食事を消費するまで、静止は続きます。この方法論の焦点の時間は、赤い円(1、静止および2、再活性化)で示されます。MB NB(青)は、発達中枢脳NBのサブセットであり、発達中(脳半球あたり4つ)に継続的に分裂する。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
動物摂食に応答して、PI3キナーゼおよびTOR成長シグナル伝達経路は、NBsおよびそれらのグリアおよび気管ニッチにおいて活性になる10、11、15、16。食事性栄養素が取り出されるか、またはPI3キナーゼのレベルが低下すると、NBは再活性化に失敗し、グリアおよび気管の成長も減少する10,11,15,16。現在のモデルは、NB再活性化が脂肪体による幼虫の成長に結合され、動物摂食に応答して全身シグナルを放出すると仮定している12、17、18。このシグナルは、とらえどころのないままであり、脳内のショウジョウバエインスリン様ペプチド(Dilps)の発現および放出を促進する可能性が高く、NBおよびそれらのグリアおよび気管ニッチにおけるPI3キナーゼの下流活性化をもたらす。全身の手がかりの性質をよりよく理解するために、我々は培養された脳外植体において静止性NBを再活性化する方法を開発した。この方法を使用すると、NBsの再活性化を、動物全体の全身的手がかりの非存在下でアッセイすることができる。外因性因子は、培養培地に再供給され得、そしてチミジン類似体EdUの組み込みに基づいてアッセイされたNB再活性化である。この方法を用いて、我々は、外因性インスリンが脳外植体における静止性NBを再活性化するのに十分であると決定した。今後の研究は、脳外植体におけるNB静止を正または負に調節する追加因子を特定することを目的としている。
プロトコル
ショウ ジョウバエ 幼虫コレクション
注:開始する前に、イーストプレート、グレープペースト、フライコンドミニアムを準備してください。
- 酵母ペースト:小さな容器に、5gの活性乾燥酵母を10mLの水と混合して、ピーナッツバターの粘稠度を有するペーストを形成する。酵母ペーストをプラスチックラップで覆い、ゴムバンドを使用して容器にしっかりと取り付けます。
注:新鮮な酵母ペーストは容器内で膨張し、しっかりと取り付けられていない限り蓋から飛び出します。酵母ペーストは室温(RT)で数日間持続する。 - グレーププレート:グレーププレートを作るためのレシピに従ってください(表1)。4°Cで保存したプレートを使用する場合は、RTで1時間置いて使用前にプレートを温めておくようにしてください。
- 水(750 mL)と寒天(18.75 g)を4 Lフラスコに混ぜ、旋回させ、オートクレーブで20分間(液体サイクル)混合します。
- ブドウジュース(250mL)とスクロース(25g)を、加熱したプレート(弱火)上の大きな攪拌子を備えた1Lフラスコで混合する。スクロースが溶解したら、火を止め、フラスコに触れることができるまで待ってから、テゴセプト(10%、4mL)およびプロピオン酸(5mL)を加える。攪拌棒をつけたままにします。
- オートクレーブ処理が完了したら、フラスコに触れることができるまで冷まし(〜60°C)、ブドウジュースミックスに混ぜる。
- すべての溶液を1つのフラスコにまとめ、プレート上でかき混ぜる。
- 溶液を小型のペトリ皿(35mm)の蓋にピペットで留める。ピペットは蓋あたりおよそ9mL、または凸状のドームが得られるまで。
- オプション:泡を取り除くために蓋に火をつけます。
- プレートが固まったら、ブドウプレートを気密蓋付きの箱に積み重ね、箱を4°Cに置きます。 プレートは最大1ヶ月間保管できます。
- フライコンドミニアム:18G針を使用して、6オンスの正方形の底ポリプロピレン ショウジョウバエ ボトルに約20個の穴を開けます。
- 成虫のハエ(〜100オレゴンRまたは任意の遺伝子型)をハエのコンドミニアムに移し、酵母ペーストのダブをトッピングしたブドウ寒天プレートでコンドミニアムをキャップします。プレートの中央に向かってダブを置き、ラボテープでプレートをコンドミニアムに貼り付けます。
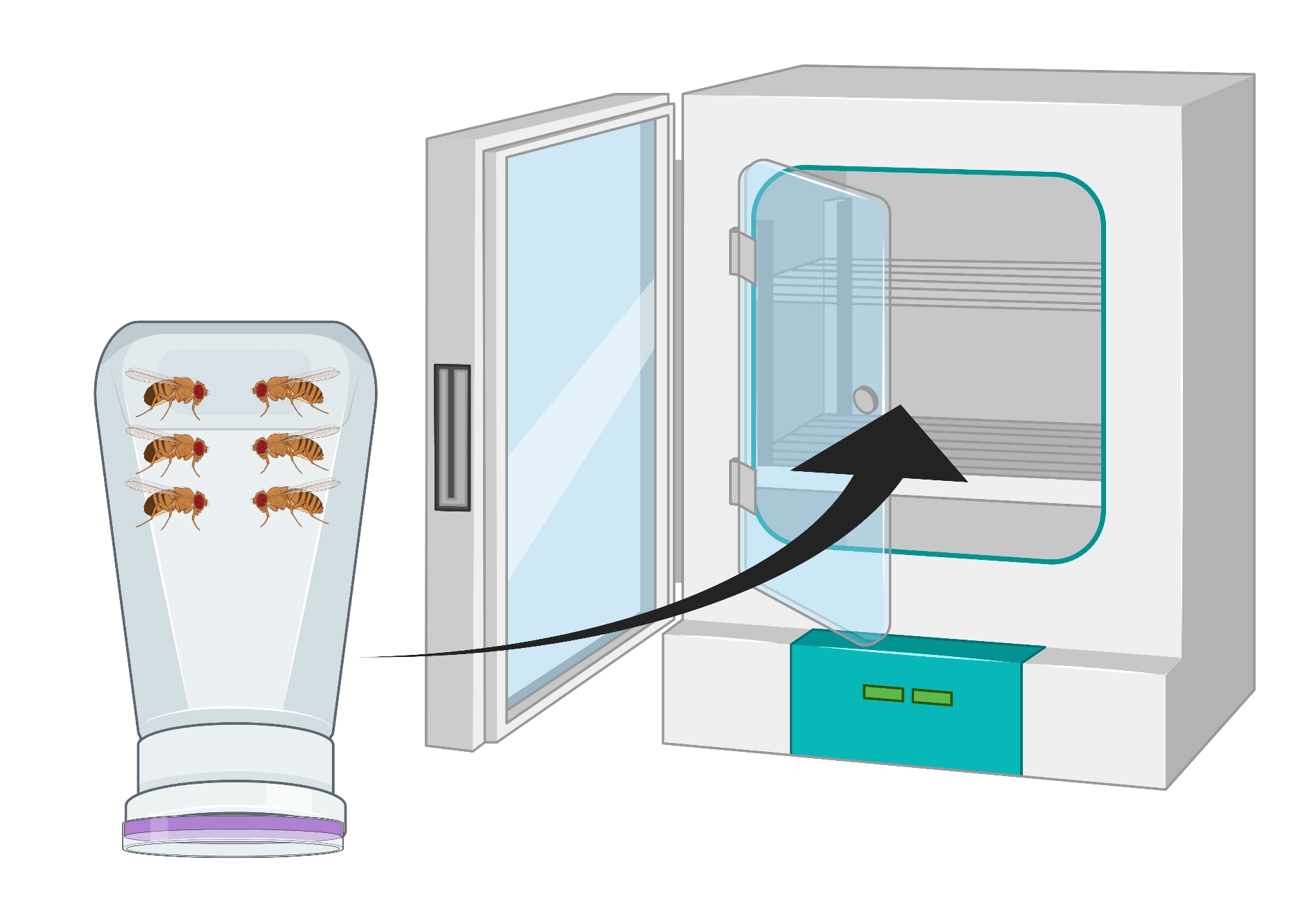
- ブドウ寒天プレートが底になるように容器をひっくり返し、25°Cのインキュベーターに24時間置きます(図2)。

図2:ショウ ジョウバエ の成虫の男性と女性の反転フライボトル(コンドミニアム)の視覚的表現。 ペットボトルには、酸素交換のために18G針で生成された小さな穿刺があります。瓶の口を寒天ブドウ果汁キャップで密封し、反転させて25°Cのインキュベーターに保管する。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
- 24時間後、ブドウ寒天プレートを交換し、酵母ペーストをトッピングした新しいプレートと交換します。大人のハエが逃げないように、ベンチでコンドミニアムを軽く軽く叩き続けながら、2つのプレートをすばやく切り替えます。
- プレートを目で見て、プレート上の胚の数を評価します。 ショウジョウバエ の胚は長方形で白色で、2つのひも状の付属器があります。
- プレート上に胚が非常に少ない(100未満)場合は、プレートを捨てます(寒天をフライゴミ箱にこすり落とし、再利用のためにプラスチック製の蓋を保存します)。多くの場合、成人女性は新しいコンドミニアムの最初の夜に多くの胚を産むことはありません。この場合、大人のハエに順応するためにさらに24時間を与えてください。
- プレート上に多数の胚(少なくとも100個)がある場合は、それを保管し、平底のヘラを使用して酵母ペーストを慎重に取り除きます。
- 酵母ペーストが除去されたら、金属ピックを使用して、解剖顕微鏡下でブドウ板からすべての幼虫を手動で除去する。解剖顕微鏡下でプレートを見ると、胚と同様に這う幼虫を観察する必要があります。
- 1つの幼虫の側面に向かって金属ピックをブラッシングすることによって、すべての幼虫を除去する。幼虫は粘着性があり、ピックに固執します。1匹の幼虫がピックについたら、ツールの幼虫を使用してさらに付着させることで、追加の幼虫を簡単に拾うことができます。
注:幼虫はお互いにくっつくのが好きです。この時点で、幼虫が損傷しても問題ありません。これらの幼虫は捨てられます。 - すべての幼虫を摘み取って取り除いた後、プレートを25°Cのインキュベーターに戻します。プレートは、密閉できる大きな容器に入れてください。濡れたペーパータオルを大きな容器の底に置き、湿気を保ちます。
- 30〜60分後、プレートを解剖顕微鏡に戻し、次に、同じブドウ寒天プレートから約20〜25匹の幼虫を慎重に選択して、摘み取った幼虫が30〜60分の時間枠内で新たに孵化するようにする。
- 孵化したばかりの幼虫20〜25匹のツールの先端を、1〜2mLの1xリン酸緩衝生理食塩水(PBS)で満たされたペトリ皿(60mm)に2分間浸漬する。
- 2分後、皿を斜めに傾けて、底に液体をプールします。小さなペイントブラシを使用して、幼虫を液体からペトリ皿の底までブラシで塗ります。
- 絵筆上のすべての幼虫を収集し、幼虫を1〜2mLの70%エタノールを含む新しいペトリ皿(60mm)に移す。ステップ1.15を繰り返して絵筆で幼虫を収集し、1〜2mLの1x PBSを含む新しいペトリ皿に移す。
2. 培地・器具作製
- ベンチと作業エリアに70%エタノールをスプレーし、乾燥させます。
- 解剖用具、鉗子、および2つのガラス製時計皿に70%エタノールを噴霧し、ベンチで乾燥させる。
- 補足したシュナイダーの培地(SSM、 表2)を作り、氷の上に置きます。
- SSM1mLをそれぞれのガラス時計皿にピペットで入れる。
- 滅菌チップを有するマイクロピペットを用いて、孵化したばかりの幼虫をPBSのプレートから第1のガラス時計皿中のSSMに移す。滅菌チップを備えたマイクロピペットを使用して、孵化したばかりの幼虫を2番目のガラス時計皿のSSMに移す。
3. 解剖と脳文化
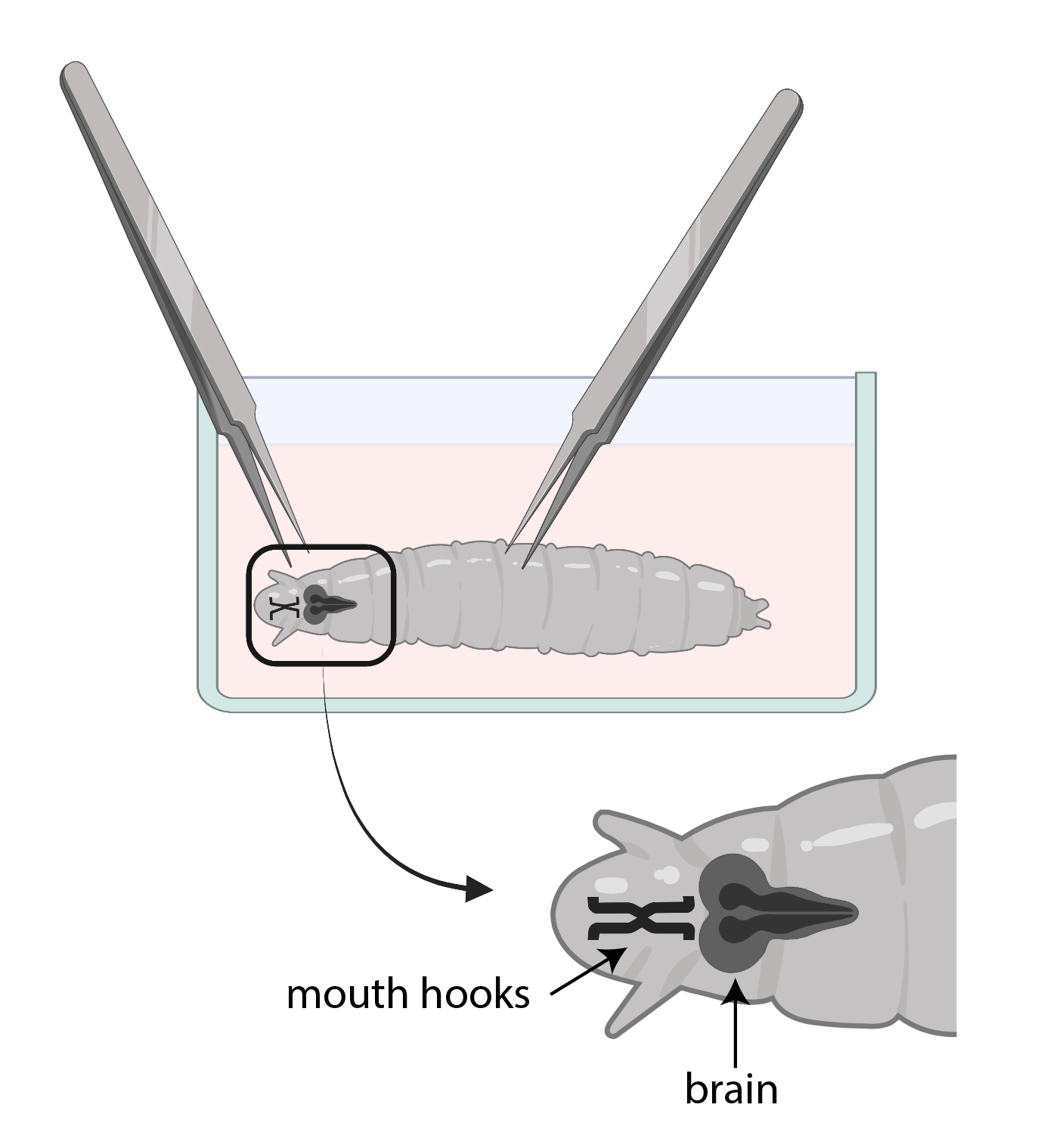
- 幼虫がSSMで2番目のガラス時計皿に入ったら、鉗子と解剖顕微鏡を使用して幼虫から脳を解剖します。必要に応じて倍率を調整します。
- 片方の鉗子を使って口のフックをつかみ、もう片方の鉗子を使って体を半分下にそっとつかみ、反対方向に引っ張って幼虫を2つに分けます。
注:脳は口のフックのすぐ後ろにあります。脳を取り囲む他の組織があるかもしれないことに注意してください。これらの組織を除去するときは、脳を損傷する可能性があるため、非常に注意してください

図3:ショウ ジョウバエ の幼虫をSSMでガラスの時計皿に入れた。 鉗子は解剖のために適切に配置されています。幼虫の脳の位置(濃い灰色)は口のフック(黒)の後部にあり、両方とも幼虫の内部に示されている。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
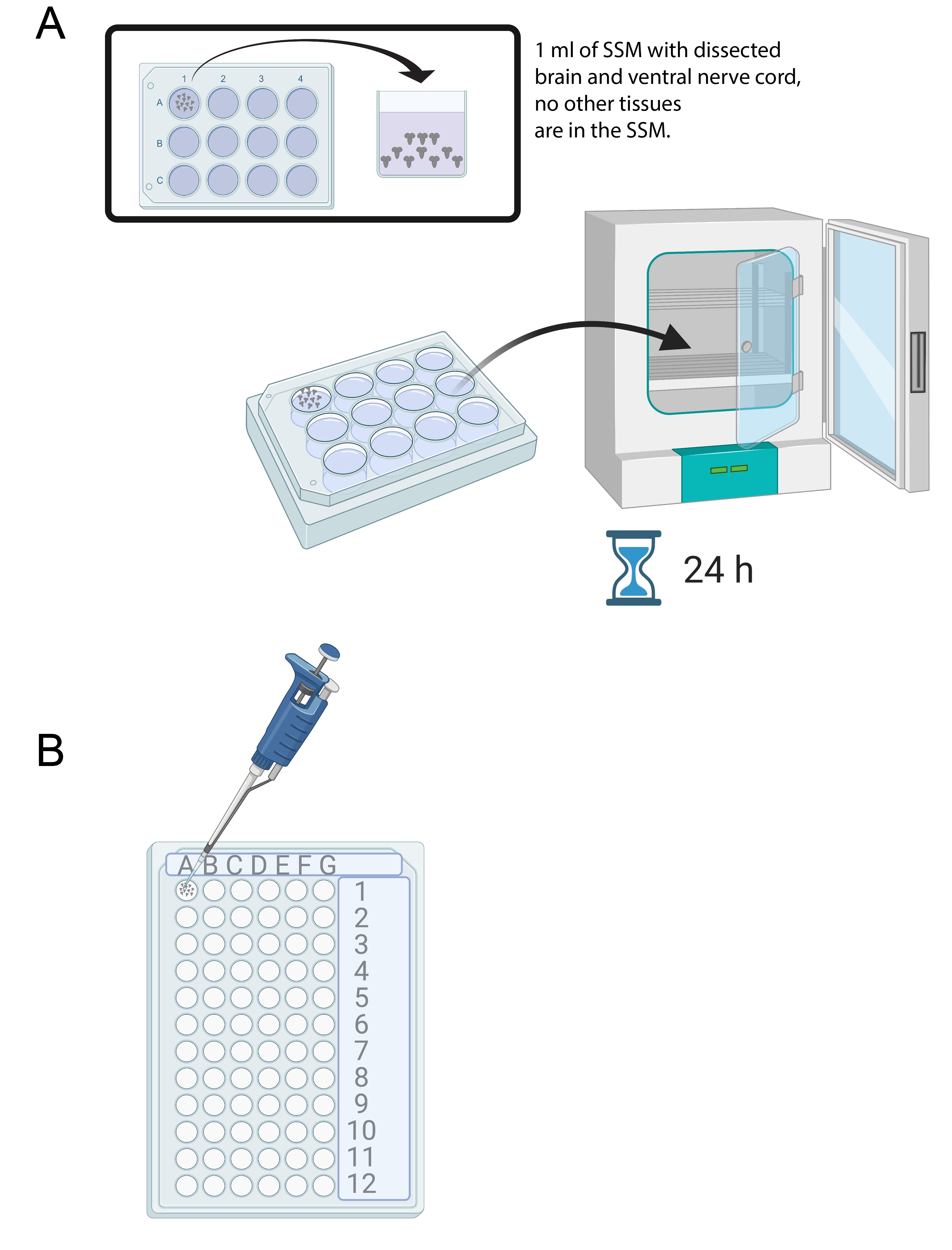
- 15〜20個の脳を解剖したら、滅菌12ウェル培養トレイの1ウェルに1mLのSSMを加える。新しく解剖した脳をマイクロピペットおよび滅菌チップを用いてSSMに移す(図4A)。
- 12ウェル培養トレイ内のSSM培地中の脳を25°Cで24時間インキュベーターに入れる(図4A)。

図4:脳培養および免疫染色。 (A)1mLのSSMを含む12ウェル培養皿中の全脳。次いで、培養皿を25°Cのインキュベーターに入れ、免疫染色中に脳外植体を保持する(B)72ウェルミニトレイに入れる。脳を洗浄し、溶液を10μLに設定したP20マイクロピペットを用いて移す。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
4. 増殖アッセイ、脳固定、抗体染色
- 翌日、1mLのEdU SSM溶液を作る。10 μL の 5-エチニル-2'-デオキシウリジン (EdU) ストック 10 μL と 990 μL の SSM (最終 EdU 濃度は 0.1 mM に等しい) を滅菌微量遠心チューブに入れ、混合します。24時間インキュベーションが完了した後、1mLのEdU SSMを滅菌12ウェル培養トレイの1ウェルにピペットで留める。
- SSMのみを含むウェルからEdU SSM溶液を含む新しいウェルに滅菌チップを備えたマイクロピペットを使用して脳を移す。25°Cで1時間インキュベートする。
- 次に、EdU標識脳を、1mLの固定液(4%パラホルムアルデヒド、レシピについては 表3 を参照)を含む同じ培養トレイ内の別のウェルに20分間移す。
警告: パラホルムアルデヒドは生物学的に危険を及ぼすものであり、適切に廃棄する必要があります。 - 固定後、マイクロピペットを使用して脳を72ウェルミニトレイのウェルに素早く移します。各ウェルは、10個の脳と10~15μLの液体を保持することができます(図4B)。脳をミニトレイ(ウェルあたり10脳以下)に移したら、固定を取り除き、10μLの1x PBT(リン酸緩衝液、0.1%Triton-X 100を含むpH 7.4)で脳を3回すすいでください。
注:すすぎとは、10 μLの1x PBTを脳にピペッティングし、除去し、3回繰り返すことを意味します。 - 次に、脳をそれぞれ10分間3回洗浄し、再び10μLの1x PBTで洗浄する。脳が常に液体で覆われていることを確認してください。
- 洗浄が完了したら、10 μLのブロッキング溶液(10%正常ヤギ血清を含む1x PBT)を脳にピペットで打ちます。トレイを覆い、端の周りにパラフィルムのストリップを使用して密封します。
- 密封したら、濡れたタオルの入った密閉された箱にミニトレイを入れて、蒸発を防ぐために湿った環境を提供します。トレイの入った箱を4°Cで一晩置く。
- 翌日、一次抗体溶液を作る。
注:このプロトコルでは、細胞膜を標識するためにウサギの抗落書きを使用し、神経芽細胞を標識するためにラットの抗デッドパンを使用したが、他の任意の数の一次抗体を使用することができた。- 一次抗体溶液を作製するには、まず、一次抗体の希釈物をブロッキング溶液中で作る。例えば、ウサギの落書き防止は、1:1000の最終濃度で使用される。したがって、まず、ウサギ落書き防止抗体を1:100(抗体1 μLに99 μLのブロッキング溶液を加えた)で希釈します。ラットデッドパンは、1:100の最終濃度で使用される。したがって、まず、ラットデッドパン抗体を1:10で希釈します(抗体1 μLに9 μLのブロッキング溶液を加えた)。
注:これらの希釈液は、アジ化ナトリウム(0.05%)も細菌の増殖を阻害するために添加されれば、4°Cで長期間保存することができます。 - 次に、脳を含む井戸の数を数えます。ウェルの数は、作る一次抗体溶液の量を決定する。例えば、脳のウェルが2つある場合は、20 μLの一次抗体溶液(10ウェル、100 μLなど)を調製します。20 μLの一次抗体溶液を作るには、各一次抗体希釈液を2 μL、ブロッキング溶液を16 μL加える。
注:それぞれの一次抗体の最終濃度は、それぞれ1:1000および1:100である。要するに、一次抗体の第1希釈を、第2希釈が常に1:10であるように、それぞれの最終濃度に到達する濃度で行う。この場合、ウサギの落書き防止は1:1000、ラットの抗デッドパンは1:100です。
- 一次抗体溶液を作製するには、まず、一次抗体の希釈物をブロッキング溶液中で作る。例えば、ウサギの落書き防止は、1:1000の最終濃度で使用される。したがって、まず、ウサギ落書き防止抗体を1:100(抗体1 μLに99 μLのブロッキング溶液を加えた)で希釈します。ラットデッドパンは、1:100の最終濃度で使用される。したがって、まず、ラットデッドパン抗体を1:10で希釈します(抗体1 μLに9 μLのブロッキング溶液を加えた)。
- 10 μLに設定したマイクロピペットと10 μLの一次抗体溶液のピペットでブロッキング溶液を各ウェルに取り出します。
- パラフィルムを使用してトレイを覆い、密封し、濡れたタオルで密閉された箱に戻します。4°Cで一晩インキュベートする。
メモ: 振る必要はありませんので、強くお勧めします。抗体は、揺れたり混ざったりすることなく脳に浸透します。 - 翌日、マイクロピペットを用いて一次抗体溶液を除去し、10μLの1x PBTで脳を3回すすいだ。次に、10μLの1x PBTでそれぞれ10分間、脳を4回洗浄する。10分間の洗浄の間に、二次抗体溶液を調製する。
- 二次抗体溶液を作製するには、一次抗体を認識する二次抗体を選択する。このプロトコルでは、ヤギ抗ウサギAlexa Fluor 488およびヤギ抗ラットAlexa 555が使用された。
- 各二次抗体のピペット1 μLを298 μLのブロッキング溶液と共に微量遠心管に入れ、各二次抗体について最終濃度1:300にした。
- 最後の10分間の洗浄後、1x PBTおよび10 μLの二次抗体溶液のピペットを各ウェルに除去する。パラフィルムを使用してトレイを密封し、湿ったタオルで箱に戻します。4°Cで一晩インキュベートする。
注:すすぎ、洗浄、または一次抗体溶液と二次抗体溶液を加えるときに、ウェル内の最後のμLをすべて除去することを心配しないでください。脳は常に数μLの液体に沈んだままですが、これは問題ありません。 - 翌日、マイクロピペットを用いて二次抗体溶液を除去し、1x PBTの各10μLで脳を3回すすいだ。次に、1x PBTの各10μLでそれぞれ10分間、脳を4回洗浄する。
- 10分間の洗浄の間に、EdU反応混合物を調製し、EdUの取り込みを検出した。EdU反応ミックスは、メーカーのガイドラインに従って調製します。
- 最終洗浄後、1x PBTおよび10 μLのEdU反応ミックスのピペットを脳と共に各ウェルに除去する。マイクロウェルプレートをパラフィルムでシールし、アルミホイルで覆います。プレートをベンチに30分間放置します。
- 30分後、1x PBTのそれぞれ10 μLで脳を3回すすぎ、1x PBTのそれぞれ10 μLでそれぞれ5分間脳を3回洗い流します。
- 最後の洗浄後、1x PBTおよびピペット10 μLのグリセロールベースのマウント培地溶液を除去します。プレートを密封し、4°Cで一晩置く。
5. 脳の取り付けとイメージング
- 翌日、顕微鏡スライド(25 mm x 75 mm x 1 mm)を準備します:顕微鏡スライドの両端に22 mm x 22 mm x 1 mmの正方形のカバーガラスを1枚接着して「ブリッジ」を作成し、その上にスライドと大きなカバースリップの間にスペースを作ります(図5A)。この空間は、脳が押しつぶされるのを防ぎながら、脳が正しく向くのに十分な動きを可能にします。

図5:幼虫脳内の顕微鏡スライド、向き、および細胞型を示す模式図。 (A)幼虫脳が装着され、画像化の準備が整った顕微鏡スライドの視覚的表現。(B)組織の配向に使用するガイドラインも示されている。(C)共焦点顕微鏡でイメージング可能な顕微鏡スライド。(D)幼虫の脳内の細胞型のいくつかを示す漫画。この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
- 22 mm x 22 mm x 1 mmのカバーグラスを顕微鏡スライドに接着した後、マイクロウェルプレートのウェルから1つの脳を含むグリセロールベースのマウント培地溶液9.3 μLをピペットし、スライドの中央に置きます(図5A)。
注:幼虫の脳はピペットチップに付着することがありますので、注意してください。癒着を避けるために、まず退色防止剤を吸引し、次に9.3μLの容量の終わりに向かって単一の脳を吸引することから始めます。 - 脳がスライドの上に着いたら、22 mm x 50 mm x 1 mmのカバースリップをそっと上に置きます。 図5Bに示すように脳を配置します。カバースリップをそっと動かして、脳の向きを変えます。これで、サンプルをイメージングする準備が整います。
- 高倍率と高開口数対物レンズを搭載した共焦点顕微鏡(図5C)を使用して、最適な画像を取得します。例:60xまたは63x、1.4 NA油浸レンズ。
- カバースリップ(および目的)に最も近い背側表面を持つ脳をイメージします。腹面(対物レンズから最も遠い)から始まる脳半球全体を通るZスタックを、1μm間隔またはZステップサイズで取得します。
注:使用されるレーザーは、二次抗体に依存する。このプロトコルでは、使用されたレーザーラインは、落書き染色を検出するために488nm、デッドパンを検出するために555nm、およびEdUを検出するために633nmであった。
- カバースリップ(および目的)に最も近い背側表面を持つ脳をイメージします。腹面(対物レンズから最も遠い)から始まる脳半球全体を通るZスタックを、1μm間隔またはZステップサイズで取得します。
6. データ解析
- フィジーのオープンソースソフトウェアを使用して脳半球を分析し、フィジーセルカウンタープラグインを使用してセルをカウントします。
結果
孵化したばかりのオレゴンR野生型脳を解剖し、シュナイダー培地(SSM)にインスリンを補給して24時間培養した。組織は、プロトコールに従って固定および染色された。NBsを検出するためにデッドパン(Dpn)に対して生成された一次抗体および細胞膜を標識するために落書きが用いられた。チミジン類似体5-エチニル-2'-デオキシウリジン(Edu)を添加し、S相の侵入およびNB再活性化を検出した。培養?...
ディスカッション
脳外植体を培養するためにここで記載した方法は、ほとんどの実験室環境で実施することができる。必要なツール、手順、データ収集はシンプルで簡単です。この方法では、細胞シグナル伝達カスケードおよびNBの再活性化および増殖を調節する外因性因子に関連するものを含む、様々な仮説を検定することができる。ここで、野生型オレゴンR動物を用いて、我々は、外因性インスリンが、?...
開示事項
著者は競合する利害関係を持っていません。
謝辞
我々は、LSAMP Bridges to Doctorate Program for Funding(CNK)並びにNIH/NIGMS(R01-GM120421及びR35-GM141886)を認める。図1のコナー・サイプ博士に感謝しています。また、Siegrist のラボメンバーの皆さんの継続的なサポートとメンターシップにも感謝します。特に、Chhavi SoodとGary Teetersが原稿を注意深く読み、コメントを提供してくれたことに感謝します。
資料
| Name | Company | Catalog Number | Comments |
| 10 µL Pipette tips | Denville Sci | P2102 | |
| 1000 µL Pipette tips | Denville Sci | P2103-N | |
| 1000 µL Pipettor | Gilson | P1000 | |
| 16% paraformaldehyde (10 x 10 mL) | Electron Microscopy Sciences | 2912.60.0000 | Used for Fixation of Larval Brains |
| 20 µL Pipette | Gilson | P20 | |
| 200 µL Pipette tips | Denville Sci | 1158U56 | |
| 24-well multiwell culture plates | Fisher Scientific | 50-197-4477 | |
| 35 mm Petri dishes | Fisher Scientific | 08-757-100A | Grape Plate Ingredients |
| 4 °C refrigerator | Fisher Scientific | Provides an ideal temperature for >24 h incubations in antibody solution | |
| 63x Objective | Lecia | ||
| Active dry yeast | Most supermarkets | ||
| Agarose | Fisher Scientific | 214010 | Grape Plate Ingredients |
| Click-iT EdU Cell Proliferation Kit for Imaging, Alexa Fluor 647 dye | Thermo Fisher Scientific | C10340 | to label proliferating cells |
| Confocal Microscope | Leica | SP8 | |
| Coverslips 22 mm x 22 mm x 1 mm , 10 pack of 4 oz | Fisher Scientific | 12-544-10 | Two Coverslips are super glued to the ends of the microscope slide. This creates a space that allows for the brains to float in antifade while being imaged. |
| Coverslips, 22 mm x 50 mm x 1 mm | Fisher Scientific | 12-545E | The coverslip is placed on two square coverslips on the microscope slide ensuring that the brain in the antifade does not move while imaging. |
| Dissecting microscope | Zeiss | Stemi 2000 | |
| Ethanol 200 proof (100%), Decon Labs, 1 gallon bottle | Fisher Scientific | 2701 | Used to wash off the larvae before the 24 hr hold in culture medium |
| Fetal Bovine Serum (10%) | Sigma | F4135-100ML | Supplement for cell culture media. |
| Fine forceps for dissection | Fine Science Tools | 11295-20 | Forcepts used in disections. They work best when sharpened. |
| Fly Bottles for Crossing | Genessee Scientific | 32-130 | This bottle is used as a container that lets the flies lay eggs on the grape plate. |
| Glass Dissection Dish (3 well) | These are no longer available | ||
| Glutathione | Sigma | G6013 | Provides oxidative protection during cell culture. |
| Goat Serum | Sigma | G9023- 10ML | Blocking Agent |
| Grape Plates | Made in house | Made in house | Grape juice/agarose plates for collecting freshly hatched eggs |
| Image J | Imagej.net/fiji/downloads | Free Download: https://fiji.sc | Imaging platform that is used to count cells and Edu reactivation |
| Incubator | Thermo Fisher Scientific | Ensures that the temperature, humidity, and light exposure is exactly the same throughout experiment. | |
| Insulin | Sigma | I0516 | Independant variable of the experiment |
| Laminar flow hood | For aliquoting culture media | ||
| L-Glutamine | Sigma | G7513 | Provides support during cell culture |
| Nunc 72-well Microwell Mini Trays | Fisher Scientific | 12-565-154 | Immunostaining steps are performed in this tray |
| Parafilm | Fisher Scientific | S37440 | Film used to seal plates in order to prevent evaporation |
| Pen-Strep | Sigma | P4458-100ml | Antibiodics used to prevent bacterial contamination of cells during culture. |
| Phosphate Buffer, pH7.4 | Made in house | Made in house | Solvent used to wash the brains after fixing and staining steps |
| Pick | Fine Science Tools | 10140-01 | Used to pick larvae off of the grape plate |
| Propionic acid | Fisher Scientific | A-258 | Grape Plate Ingredients |
| Rabbit 405 | Abcam | ab175653 | Antibodies used for immunostaining |
| Rat 555 | Abcam | ab150166 | Antibodies used for immunostaining |
| Rb Scribble | A Gift from Chris Doe | Antibodies used for immunostaining | |
| Rt Deadpan | Abcam | ab195173 | Antibodies used for immunostaining |
| Schneiders Culture Medium | Life Tech | 21720024 | Contains nutrients that help the cells grow and proliferate |
| SlowFade Diamond Antifade (5 x 2 mL) | Life Tech | S36963 | Reagent that provides protection against fading fluorophores |
| Sterile Water | Autoclave Milli-Q water made in house | Needed for Solutions | |
| Sucrose | Fisher | S2-12 | Grape Plate Ingredients |
| Superfrost Microscope Slides | Fisher Scientific | 12-544-7 | |
| Superglue | Most supermarkets | ||
| Tegosept | Genesee Scientific | 20-259 | Grape Plate Ingredients |
| Triton-X 100 | Sigma | T9284-100ML | PBT |
| Welch's 100% grape grape juice | Most supermarkets | Grape Plate Ingredients |
参考文献
- Suman, S., Domingues, A., Ratajczak, J., Ratajczak, M. Z. Potential clinical applications of stem cells in regenerative medicine. Advances in Experimental Medicine and Biology. 1201, 1-22 (2019).
- Tabar, V., Studer, L. Pluripotent stem cells in regenerative medicine: challenges and recent progress. Nature Reviews Genetics. 15, 82-92 (2014).
- Daley, G. Q. Stem cells and the evolving notion of cellular identity. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 370, 20140376 (2015).
- Rodrigues, M., Kosaric, N., Bonham, C. A., Gurtner, G. C. Wound healing: A cellular perspective. Physiological Reviews. 99, 665-706 (2019).
- van Velthoven, C. T. J., Rando, T. A. Stem cell quiescence: Dynamism, restraint, and cellular idling. Cell Stem Cell. 24, 213-225 (2019).
- Chapman, N. M., Boothby, M. R., Chi, H. Metabolic coordination of T cell quiescence and activation. Nature Reviews Immunology. 20, 55-70 (2020).
- Wosczyna, M. N., Rando, T. A. A muscle stem cell support group: Coordinated cellular responses in muscle regeneration. Developmental Cell. 46, 135-143 (2018).
- Homem, C. C., Knoblich, J. A. Drosophila neuroblasts: a model for stem cell biology. Development. 139, 4297-4310 (2012).
- Kang, K. H., Reichert, H. Control of neural stem cell self-renewal and differentiation in Drosophila. Cell and Tissue Research. 359, 33-45 (2015).
- Chell, J. M., Brand, A. H. Nutrition-responsive glia control exit of neural stem cells from quiescence. Cell. 143, 1161-1173 (2010).
- Sousa-Nunes, R., Yee, L. L., Gould, A. P. Fat cells reactivate quiescent neuroblasts via TOR and glial insulin relays in Drosophila. Nature. 471, 508-512 (2011).
- Britton, J. S., Edgar, B. A. Environmental control of the cell cycle in Drosophila: nutrition activates mitotic and endoreplicative cells by distinct mechanisms. Development. 125, 2149-2158 (1998).
- Lin, S., et al. Extremes of lineage plasticity in the Drosophila brain. Current biology : CB. 23, 1908-1913 (2013).
- Sipe, C. W., Siegrist, S. E. Eyeless uncouples mushroom body neuroblast proliferation from dietary amino acids in Drosophila. Elife. 6, 26343 (2017).
- Speder, P., Brand, A. H. Systemic and local cues drive neural stem cell niche remodelling during neurogenesis in Drosophila. Elife. 7, 30413 (2018).
- Yuan, X., Sipe, C. W., Suzawa, M., Bland, M. L., Siegrist, S. E. Dilp-2-mediated PI3-kinase activation coordinates reactivation of quiescent neuroblasts with growth of their glial stem cell niche. PLoS Biology. 18, 3000721 (2020).
- Colombani, J., et al. A nutrient sensor mechanism controls Drosophila growth. Cell. 114, 739-749 (2003).
- Geminard, C., Rulifson, E. J., Leopold, P. Remote control of insulin secretion by fat cells in Drosophila. Cell Metabolism. 10, 199-207 (2009).
- Siller, K. H., Serr, M., Steward, R., Hays, T. S., Doe, C. Q. Live imaging of Drosophila brain neuroblasts reveals a role for Lis1/dynactin in spindle assembly and mitotic checkpoint control. Molecular Biology of the Cell. 16, 5127-5140 (2005).
- Prithviraj, R., Trunova, S., Giniger, E. Ex vivo culturing of whole, developing Drosophila brains. Journal of Visualized Experiments: JoVE. (65), e4270 (2012).
- Bostock, M. P., et al. An immobilization technique for long-term time-lapse imaging of explanted drosophila tissues. Frontiers in Cell and Developmental Biology. 8, 590094 (2020).
- Datta, S. Activation of neuroblast proliferation in explant culture of the Drosophila larval CNS. Brain Research. 818, 77-83 (1999).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved