
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Uso de microfluídica y microscopía de fluorescencia para estudiar la dinámica de ensamblaje de filamentos y haces de actina única
En este artículo
Resumen
Presentamos protocolos para ensayos microfluídicos de filamentos de actina simples, en combinación con microscopía de fluorescencia, que permiten monitorear con precisión filamentos de actina individuales en tiempo real mientras se exponen secuencialmente a diferentes soluciones de proteínas.
Resumen
Con el fin de descifrar los complejos mecanismos moleculares que regulan el ensamblaje y desmontaje de los filamentos de actina, es un gran activo monitorear las reacciones individuales en vivo en condiciones bien controladas. Para hacerlo, han surgido experimentos en vivo de un solo filamento en los últimos 20 años, principalmente utilizando microscopía de fluorescencia de reflexión interna total (TIRF), y han proporcionado un tesoro de resultados clave. En 2011, con el fin de ampliar aún más las posibilidades de estos experimentos y evitar artefactos problemáticos recurrentes, introdujimos microfluídica simple en estos ensayos. Este estudio detalla nuestro protocolo básico, donde los filamentos de actina individuales se anclan por un extremo a la superficie pasivada de la cubierta, se alinean con el flujo y pueden exponerse sucesivamente a diferentes soluciones proteicas. También presentamos los protocolos para aplicaciones específicas y explicamos cómo se pueden aplicar las fuerzas mecánicas controladas, gracias al arrastre viscoso de la solución que fluye. Destacamos las advertencias técnicas de estos experimentos y presentamos brevemente los posibles desarrollos basados en esta técnica. Estos protocolos y explicaciones, junto con la disponibilidad actual de equipos de microfluídica fáciles de usar, deberían permitir a los no especialistas implementar este ensayo en sus laboratorios.
Introducción
El montaje y desmontaje de filamentos de actina y redes de filamentos de actina están controlados por varias reacciones bioquímicas y dependen del contexto mecánico. Para obtener información sobre estos complejos mecanismos, es invaluable poder observar reacciones individuales en filamentos individuales (en cantidades suficientemente grandes). En las últimas décadas, la observación de filamentos dinámicos de actina en tiempo real, principalmente utilizando microscopía de fluorescencia de reflexión interna total (TIRF), se ha convertido en una técnica clave y ha proporcionado una impresionante lista de resultados que no se podrían haber obtenido con ensayos bioquímicos de solución a granel1.
Para lograr esto, es necesario mantener los filamentos de actina marcados fluorescentemente cerca de la superficie de la cubierta del microscopio mientras se exponen a soluciones de proteínas de unión a actina (ABP), que también pueden ser marcadas fluorescentemente. Hacerlo proporciona un medio para monitorear los eventos que tienen lugar en filamentos individuales en condiciones bioquímicas bien controladas y, por lo tanto, cuantificar las velocidades de reacción. Sin embargo, deben considerarse una serie de limitaciones específicas. El mantenimiento artificial de los filamentos cerca de la superficie, a menudo gracias a múltiples puntos de anclaje o mediante el uso de un agente de apiñamiento como la metilcelulosa, puede alterar su comportamiento (por ejemplo, causando pausas en su polimerización y despolimerización2). El seguimiento del contorno de cada filamento puede ser un desafío, especialmente si se acumulan nuevos filamentos o fragmentos de filamento en el campo de visión con el tiempo. Las reacciones tienen lugar en un volumen finito donde la concentración de monómeros de actina y ABP puede variar con el tiempo, lo que potencialmente dificulta la obtención de constantes de velocidad precisas. Finalmente, renovar o cambiar la solución de ABP es difícil de lograr en menos de 30 s y a menudo conducirá a un contenido de proteína no homogéneo en la muestra.
Hace poco más de 10 años, inspirados por lo que ya se hizo para estudiar las hebras individuales de ácido desoxirribonucleico (ADN)3, introdujimos una nueva técnica basada en microfluídica para observar y manipular filamentos de actina individuales4. Permite eludir las limitaciones antes mencionadas de las técnicas clásicas de filamento único. En estos ensayos de microfluídica, los filamentos de actina se cultivan a partir de semillas de espectrina-actina adsorbidas en el cobertor. Por lo tanto, los filamentos se anclan por un extremo solo a la parte inferior de la cámara microfluídica y fluctúan por encima de la superficie sin pegarse. Los filamentos se alinean con el flujo de las soluciones entrantes, lo que facilita el monitoreo de la longitud de su contorno y los mantiene en una región poco profunda por encima del espolón donde se puede usar TIRF. Diferentes soluciones fluyen simultáneamente en la cámara sin mezclarse, y los filamentos se pueden exponer a ellas secuencial y rápidamente.
Aquí, proponemos una serie de protocolos básicos para establecer ensayos de microfluídica de filamento de actina única en el laboratorio. Las cámaras de coverslips y microfluidos se pueden preparar con anticipación (en medio día), y el experimento en sí, donde se pueden probar varias condiciones bioquímicas, se realiza en menos de un día.
Protocolo
1. Preparación de la cámara microfluídica
- Seleccione un molde maestro SU-8 con varios patrones de cámara. Las cámaras típicas tienen forma de cruz con tres entradas y una salida, 20 μm de alto y 800 μm de ancho (Figura 1). Dichos moldes maestros pueden comprarse a empresas externas o fabricarse en laboratorios académicos (por ejemplo, Gicquel, Y. et al.5).
- Coloque cinta adhesiva alrededor del borde del molde.
- Coloque ~ 50 cm de largo, 19 mm de ancho, cinta de oficina transparente estándar (consulte la Tabla de materiales) en un banco, con el lado pegajoso hacia arriba. Coloque el molde verticalmente en un extremo y a lo largo de la línea media de la cinta.
- Enrolle el molde hasta el otro extremo de la cinta para crear un borde de 1 cm alrededor del molde. Doble la cinta sobre la parte inferior del molde.
- Preparar la solución de polidimetilsiloxano (PDMS).
- En un plato de pesaje desechable, vierta directamente 25-30 g de base PDMS (Tabla de materiales). Añadir un 10% de agente de curado PDMS peso/peso (Tabla de Materiales) con una pipeta Pasteur de plástico desechable.
- Mezclar manualmente y a fondo con un palo de plástico. Asegúrese de que el agente de curado esté bien incorporado en la base de PDMS, incluso si la agitación crea muchas burbujas.
- Desgasificar la solución PDMS en un desecador al vacío (Tabla de Materiales) durante al menos 5 min a temperatura ambiente (RT). Las burbujas se expandirán, subirán a la superficie y estallarán cuando se rompa el vacío.
- Vierta la solución PDMS sobre el molde SU-8. Use un palo de plástico para raspar y transferir la mayor cantidad posible de la mezcla.
- Desgasificar PDMS por segunda vez (5 min en el desecador al vacío). Asegúrese de deshacerse de la mayoría de las burbujas (algunas burbujas pequeñas en la superficie superior están bien).
- Coloque el molde en un horno a 70 °C durante al menos 5 h para que el PDMS se reticule y solidifique.
- Retire las cámaras PDMS sólidas del molde.
PRECAUCIÓN: Las obleas de silicio para moldes SU-8 son extremadamente frágiles, por lo que se debe tener mucho cuidado al separar el PDMS de las obleas. Trabaje en una superficie dura y plana y mantenga la oblea plana en la superficie.- Con una cuchilla de afeitar, haga un corte circular en el PDMS, a aproximadamente 1 cm del borde del molde. Todos los patrones deben tener al menos 0,5 cm dentro del corte. Despegue suavemente el bloque central de PDMS con suaves tirones.
PRECAUCIÓN: Al despegar, mantenga el molde SU-8 plano en la mesa de trabajo para evitar que se rompa. - Coloque PDMS en papel de aluminio limpio, la superficie moldeada frente al papel de aluminio, para proteger su superficie del polvo y hacer que los patrones sean más visibles.
- Con una cuchilla de afeitar, haga un corte circular en el PDMS, a aproximadamente 1 cm del borde del molde. Todos los patrones deben tener al menos 0,5 cm dentro del corte. Despegue suavemente el bloque central de PDMS con suaves tirones.
- Elija y corte una cámara con una cuchilla de afeitar a al menos 0,5 cm de distancia del patrón. El bloque PDMS resultante mide alrededor de 0,5 cm de alto, 1,5 cm de ancho y 3 cm de largo. Perforar tres entradas y una salida con una punción de biopsia de 0,75 mm I.D. (Tabla de Materiales).
- Limpie la cámara PDMS con etanol ultrapuro (Tabla de Materiales) y seque al aire con una cerbatana de seguridad (Tabla de Materiales). Coloque el PDMS con el patrón hacia arriba en una placa de Petri limpia y cierre el plato con su tapa.
2. Limpieza de la cubierta de vidrio
NOTA: Aquí, se detalla un procedimiento estándar de limpieza de la cubierta, basado en una serie de pasos de sonicación. Otros procedimientos de limpieza de la cubierta de vidrio se han descrito en muchas otras publicaciones que pueden lograr resultados satisfactorios similares 6,7,8,9.
- Coloque 10-20 fundas (40 mm de largo) en un soporte de politetrafluoroetileno (PTFE) (Tabla de materiales). Sonicar las cubiertas en 0,5 L de solución de limpieza de vidrio al 2% (Tabla de Materiales) en un vaso de precipitados de vidrio de 1 L (35 °C, 30 min).
- Deseche la solución de limpieza de vidrio y enjuague las cubiertas extensamente con dH2O en al menos tres baños sucesivos de 0,5 L.
- Preparar 0,5 L de 2 M KOH en un vaso de precipitados de vidrio de 1 L. Sonicar los coverslips en KOH (RT, 30 min). Deseche el KOH y enjuague los cubrehojas con dH2O en al menos tres baños de 0,5 L.
PRECAUCIÓN: Use el equipo de protección de seguridad de laboratorio apropiado (guantes, anteojos y bata de laboratorio). - Transfiera y sonice los coverslips en 0,5 L de etanol ultrapuro (RT, 30 min). Los coverslips se pueden mantener en etanol hasta por 2 semanas. Cierre el vaso de precipitados con película termoplástica (Tabla de Materiales) para evitar la evaporación. Antes de usar, seque la cubierta con flujo de aire.
3. Montaje de la cámara PDMS
- Precaliente la placa caliente a 100 °C. Coloque hasta tres cámaras PDMS limpias y cubiertas de vidrio en una placa de Petri limpia. Coloque la placa de Petri abierta en un limpiador ultravioleta (UV) profundo (λ = 185 nm, consulte la Tabla de materiales) y expóngala a la luz UV durante 3-5 min.
NOTA: Alternativamente, las cámaras PDMS y los cubrehojas pueden exponerse al aire o al plasma de oxígeno durante 30 s. - Coloque suavemente la cámara PDMS sobre el deslizamiento de la cubierta. Asegúrese de que las dos superficies puestas en contacto estén expuestas directamente a los rayos UV. El PDMS se adhiere automáticamente al vidrio y la cámara se vuelve claramente visible.
- Para eliminar cualquier aire atrapado en la interfaz PDMS-coverslip, presione muy suavemente la superficie con un dedo. Para una unión más estrecha, presione con más fuerza sobre las esquinas y los lados. Asegúrese de que el techo de la cámara no entre en contacto con la superficie de vidrio.
- Coloque la cámara con el fondo de vidrio mirando hacia la placa caliente a 100 °C durante 5 min. Después de este paso, los enlaces vidrio-PDMS se vuelven permanentes, y las cámaras solo se pueden usar una vez. Use la cámara inmediatamente o guárdela en una placa de Petri limpia hasta por una semana.
4. [OPCIONAL] Pasivación directa y funcionalización
NOTA: Dependiendo de la aplicación, las cámaras se pueden pasivar y funcionalizar una vez conectadas al dispositivo de control microfluídico (consulte la Tabla de materiales) o inyectando manualmente soluciones directamente en la cámara con una pipeta antes de su conexión al dispositivo microfluídico. Este último ofrece la ventaja de consumir menos reactivo y evitar la contaminación potencial al hacer fluir la solución a través del tubo de poliéter éter cetona (PEEK) del dispositivo microfluídico. En todos los pasos siguientes, las soluciones se inyectan pegando directamente la punta de la pipeta en la salida. Para evitar la creación de burbujas dentro de la cámara, asegúrese de tener una pequeña gota que sobresalga de la punta de la pipeta cuando conecte la punta a la salida de la cámara PDMS. Del mismo modo, retire la punta de la pipeta antes de que se haya inyectado todo el volumen.
- Inyecte 20 μL de PLL-PEG (1 mg/ml en solución salina tamponada con fosfato (PBS)). Incubar durante un mínimo de 1 h (o durante la noche) en RT. Para evitar la evaporación, coloque la cámara PDMS en una caja húmeda (por ejemplo, una caja de punta vacía con agua en el compartimento inferior y la cámara PDMS en la plataforma de sujeción de punta).
- Inyectar 20 μL de semillas de espectrina-actina de 100 pM (en F-buffer, ver Tabla 1 y Tabla 2). Espere no más de 1 minuto. Ajuste la concentración y el tiempo de la semilla para ajustar la densidad de la superficie de la semilla, lo suficientemente alta para estadísticas grandes y lo suficientemente baja como para que los filamentos no se superpongan.
NOTA: Alternativamente, si las semillas de espectrina-actina no están disponibles, use segmentos de filamento corto funcionalizados con biotina que se inmovilizarán en un cobertor recubierto de estreptavidina 9,10. - [OPCIONAL] Inyecte 20 μL de albúmina sérica bovina (BSA) al 5% en F-buffer. Dejar en RT durante 10 min.
- [OPCIONAL] Inyecte 20 μL de 1 mg/ml de β-caseína en F-buffer. Dejar en RT durante 10 min.
NOTA: Siga los pasos 4.3 y/o 4.4 para pasivar aún más la cámara. La elección de la pasivación depende de las proteínas utilizadas y no funciona igual de bien en todos los ABP. Cuando se usa actina sola, PLL-PEG o BSA es suficiente.
5. Conecte el dispositivo microfluídico
NOTA: Utilice un sistema microfluídico basado en presión con hasta cuatro canales para controlar los flujos en la cámara microfluídica (Figura 1A, consulte la Tabla de materiales). Para evitar la formación de burbujas en el tubo microfluídico y perturbar la estabilidad del flujo, desgasifique todas las soluciones. Coloque 5 ml de dH20 y 10 ml de material tampón F en un desecador de vacío conectado a una bomba de vacío (vacío final <250 mbar) y desgasifique durante al menos 1 h en RT.
- Enjuague las entradas + tubos de salida con dH2O (500 μL, 300 mbar).
- Llene todos los tubos de depósito de 2 ml (consulte la Tabla de materiales) con 300 μL de tampón F. Ajuste la presión a 300 mbar y deje que se desperdicien de cinco a ocho gotas. Repita para cada canal y ajuste la presión a 0.
- Conecte la toma de corriente y enjuague la cámara extensamente.
- Ajuste la presión para el tubo del depósito 4 (salida) a 50 mbar. Una vez que una gota salga del extremo del tubo, conecte el tubo a la salida de la cámara PDMS. El líquido se llena en la cámara y sale de todas las entradas.
- [OPCIONAL] Si la cámara ha sido pasivada directamente (sección 4), ajuste la presión a 100 mbar para enjuagar la cámara con 50-100 μL de F-buffer (3-5 min). Retire el exceso de líquido en las entradas con un pañuelo de limpieza.
- Ajuste la presión a 20 mbar.
- Conecte las entradas.
- Ajuste la presión para el tubo del depósito de 1 a 50 mbar. Para evitar la introducción de burbujas de aire, asegúrese de que una gota salga del tubo y de la entrada pdmS.
- Conecte el tubo a la entrada 1 (las dos gotas se fusionan al conectarse). Ajuste la presión a 30 mbar.
- Repita los pasos 5.4.1-5.4.2 para conectar las entradas 2 y 3.
- Ajuste la presión de todas las entradas a 20 mbar y la presión de salida a 0 mbar. Asegúrese de que los caudales de las entradas sean aproximadamente iguales (consulte la sección Solución de problemas).

Figura 1: Inyección de soluciones a través de una cámara microfluídica. (A) Configuración microfluídica estándar para experimentos de filamentos de actina individuales. Las soluciones proteicas, colocadas en reservorios 1-3, se empujan hacia la cámara ajustando la presión en la fase gaseosa. Los caudales generados se miden mediante caudalímetros. Dentro de las cámaras microfluídicas, las soluciones no se mezclan y ocupan espacio dependiendo de las presiones relativas aplicadas (aquí, presión igual en todas las entradas). Dimensiones típicas: los tubos del depósito contienen hasta 2 ml de solución. Los tubos PEEK (0,25 mm de diámetro interior) conectan los depósitos a los caudalímetros (después de 10 cm de tubo) y luego a la cámara PDMS (después de otros 70 cm). Los tubos de silicio y los acopladores de tubos de acero inoxidable se utilizan para conectar los tubos PEEK a las entradas pdmS. El canal microfluídico principal tiene una altura de 20-60 μm, alrededor de 1 mm de ancho y 1 cm de largo. (B,C) Perfiles de flujo dentro de la cámara microfluídica. (B) El fluido genera un perfil parabólico a través de la altura de la cámara: v(z) = 6z(h-z)R/h3w, donde h y w son la altura y anchura de la cámara, y R es el caudal total. Abajo: Filamento de actina simple polimerizado a partir de semillas de espectrina-actina ancladas a la superficie. (C) Cuando el ancho de la cámara es considerablemente mayor que su altura, el flujo es casi uniforme a través de la cámara, excepto en las superficies PDMS, donde llega a cero. Haga clic aquí para ver una versión más grande de esta figura.
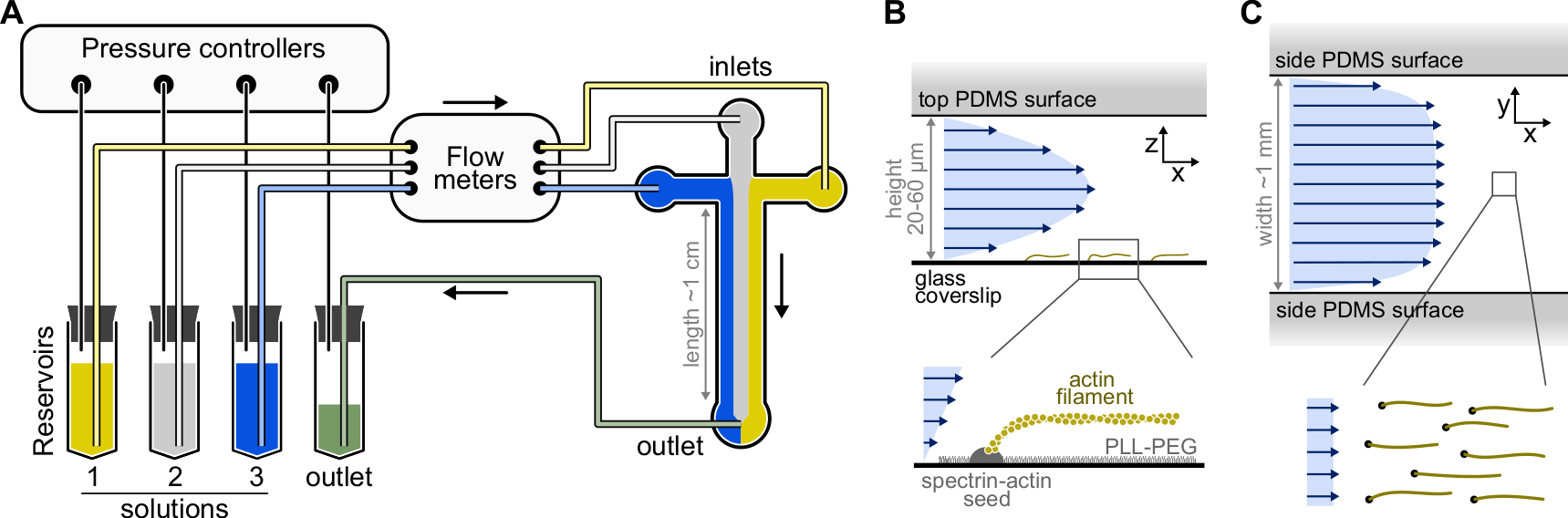{kind=link}
6. Configuración de la configuración con caudales estándar
NOTA: El sistema de presión controlado por ordenador permite un ajuste fácil y preciso de las presiones de todas las entradas/salidas conectadas a la cámara PDMS, por lo tanto, el control de los caudales de entrada y salida. Las configuraciones preestablecidas se pueden guardar y activar /desactivar con un solo clic del mouse. A continuación se muestran las configuraciones recomendadas (a menos que se indique lo contrario, la presión de salida se establece en 0 mbar). Consulte la Tabla 3 para conocer los caudales esperados para estas configuraciones preestablecidas. Las presiones indicadas aquí deben ajustarse en función de la geometría de la cámara y la configuración del sistema.
-
Cambio: Utilice este ajuste preestablecido al cambiar uno o más depósitos. Crea un flujo descendente leve en el tubo de interés para evitar la introducción de burbujas.
- Ajuste todas las presiones de entrada a 12 mbar y la presión de salida a 5 mbar (Figura 2B).
-
'All' de alto flujo: Utilice este ajuste preestablecido para inyectar rápidamente tres soluciones en paralelo. Llegarán a la cámara en 4 minutos.
- Ajuste todas las presiones de entrada a 150 mbar.
-
Alto flujo 'x': Utilice este ajuste preestablecido para inyectar rápidamente una solución. Llegará a la cámara en 3 minutos (Figura 3A-C).
- Ajuste la presión de entrada 'x' a 150 mbar (~15 μL/min). La presión en las otras entradas se ajusta a alrededor de 100 mbar, de modo que el caudal resultante en estas entradas es de ~ 500 nL / min.
-
Mid Flow 'All': Utilice este ajuste preestablecido para pausar el sistema.
- Establezca todas las entradas en 20 mbar (Figura 2A).
-
Flujo medio 'x': Utilice este ajuste preestablecido para permitir que la solución 'x' rellene la mayor parte del ancho del canal principal (consulte la Figura 2C,D), al tiempo que restringe las otras soluciones de entrada a los lados del canal. Por lo tanto, los filamentos de actina en la cámara estarán expuestos a la condición bioquímica impuesta por la solución 'x' solamente.
- Ajuste la presión de entrada 'x' a 12 mbar. Ajuste la presión en las otras entradas y ajuste a ~ 9 mbar, de modo que sus respectivos caudales sean ~ 150 nL / min.

Figura 2: La presión aplicada a cada reservorio controla la partición/distribución espacial de las soluciones dentro de la cámara microfluídica. (A) Con la misma presión aplicada a los reservorios, cada solución ocupa un tercio de la cámara. (B) Al cambiar un tubo de depósito (aquí depósito 3), la presión efectiva cae a cero, creando un flujo hacia atrás. (C,D) El aumento de la presión relativa en uno de los depósitos permite la exposición de la superficie de vidrio a una sola solución. El campo de visión en el centro de la cámara se puede exponer secuencialmente a las soluciones 1 y 2 alternando entre la configuración Mid Flow 1 (C) y Mid Flow 2 (D). Haga clic aquí para ver una versión más grande de esta figura.
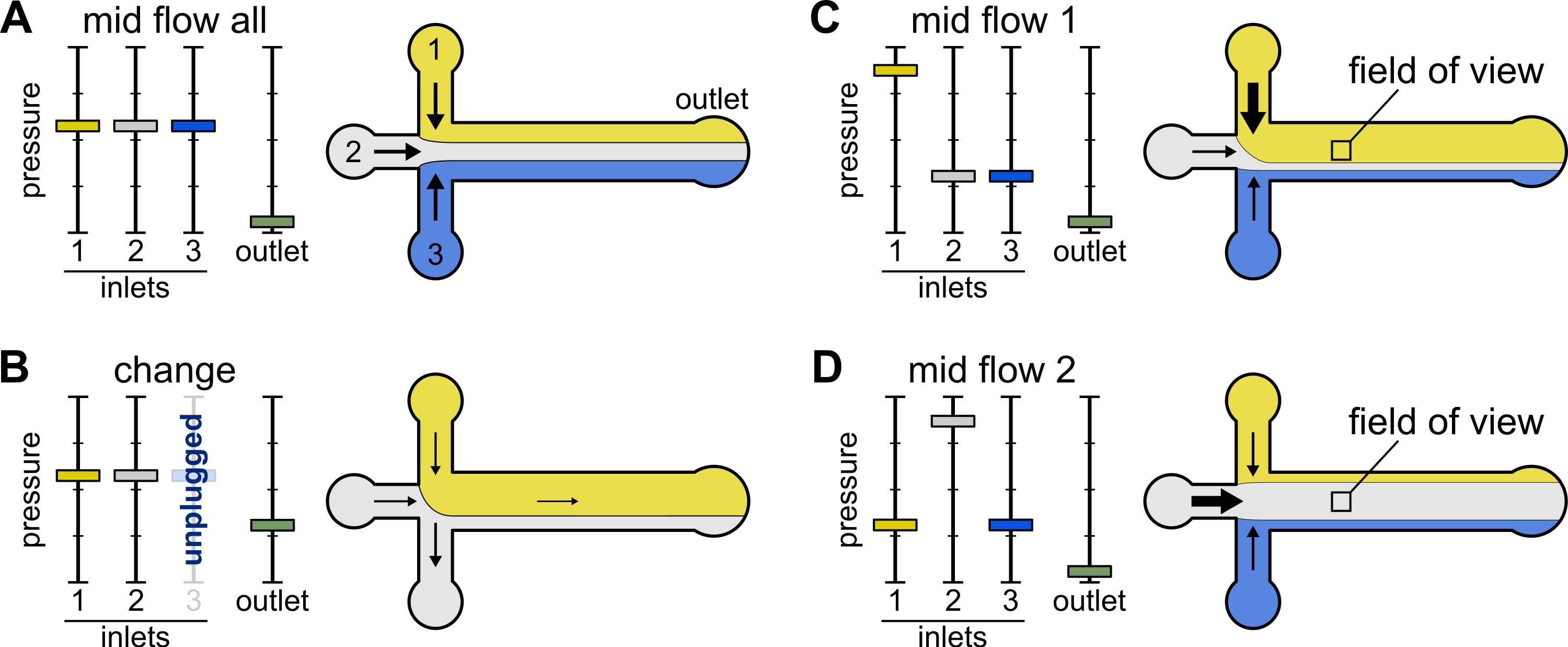{kind=link}
7. Cambiar la solución 'x'
NOTA: Como se muestra en la Figura 3A-C, es importante tener en cuenta que las soluciones tardan minutos en fluir desde un tubo de depósito hasta el canal principal de la cámara. Este tiempo mínimo de "muerto" se impone por el volumen de líquido contenido en la tubería y el perfil de flujo dentro de la tubería (Figura 3A-C).
- Preparar 200-300 μL de solución en un nuevo tubo de depósito. Establezca la presión en Cambiar configuración (ver sección 6).
- Desenrosque el tubo del depósito de entrada 'x'. La solución en el tubo fluirá lentamente hacia atrás, desde la cámara hasta la punta libre del tubo. El caudal medido se vuelve negativo (Figura 2B).
- Una vez que se haya formado una pequeña gota en la punta del tubo, atornille el tubo nuevo con la solución fresca. Una vez que el tubo se aprieta correctamente al sistema de presión, el caudal de la entrada vuelve a ser positivo.
- Establezca el ajuste de presión en Alto flujo 'x'.
- Dependiendo de la configuración microfluídica y la geometría de la cámara, espere de 3 a 5 minutos para que la solución llene completamente el tubo y llegue a la cámara.
- [OPCIONAL] Siga este proceso midiendo el aumento de la fluorescencia a lo largo del tiempo (Figura 3C).

Figura 3: Retraso en la llegada de soluciones de los reservorios a la cámara PDMS y cambio rápido de las condiciones bioquímicas. (A-C) Retraso en la llegada de soluciones de los embalses a la cámara PDMS. (A) Dependiendo de la geometría de la cámara, la longitud del tubo y la presión aplicada en la(s) entrada(s), el reemplazo de una solución por otra no es instantánea. Después de cambiar el tubo del depósito a uno que contenga una solución fluorescente (0 min), la solución llena progresivamente el tubo (0,4 min) y la cámara PDMS (1-2 min). Se da un tiempo indicativo para una presión aplicada de 150 mbar, un tubo PEEK de 80 cm y una cámara PDMS de 1600 μm de ancho y 20 μm de alto. (B) El perfil de flujo parabólico dentro de la tubería PEEK genera un gradiente efectivo de fluorescencia a lo largo del perfil radial de la tubería y dentro de la cámara (ver también figura 1B). (C) La llegada tardía de soluciones se puede cuantificar midiendo la señal de epifluorescencia de fondo en la cámara en función del tiempo. Condiciones experimentales: 0.5 μM 10% de G-actina marcada con Alexa-568 se inyecta con 150 mbar a través de un medidor de flujo y un tubo PEEK de 80 cm. (D,E) Cambio rápido de las condiciones bioquímicas. (D) Patrón de soluciones entrantes en dos condiciones de flujo medio. (E) Aumento de la fluorescencia de fondo como lectura de la concentración de actina. El tiempo t = 0 se establece como el inicio del aumento de la fluorescencia. Solución 1: 0.5 μM 10% Alexa-488-etiquetado G-actina, solución 2: F-buffer. (C,E) Cámara PDMS: 20 μm de alto y 1600 μm de ancho. La intensidad de la epifluorescencia, ~2 μm sobre la superficie, se cuantificó promediando la señal sobre el campo de visión completo, normalizada a 0 en ausencia de fluoróforo y 1 a máxima intensidad. Haga clic aquí para ver una versión más grande de esta figura.
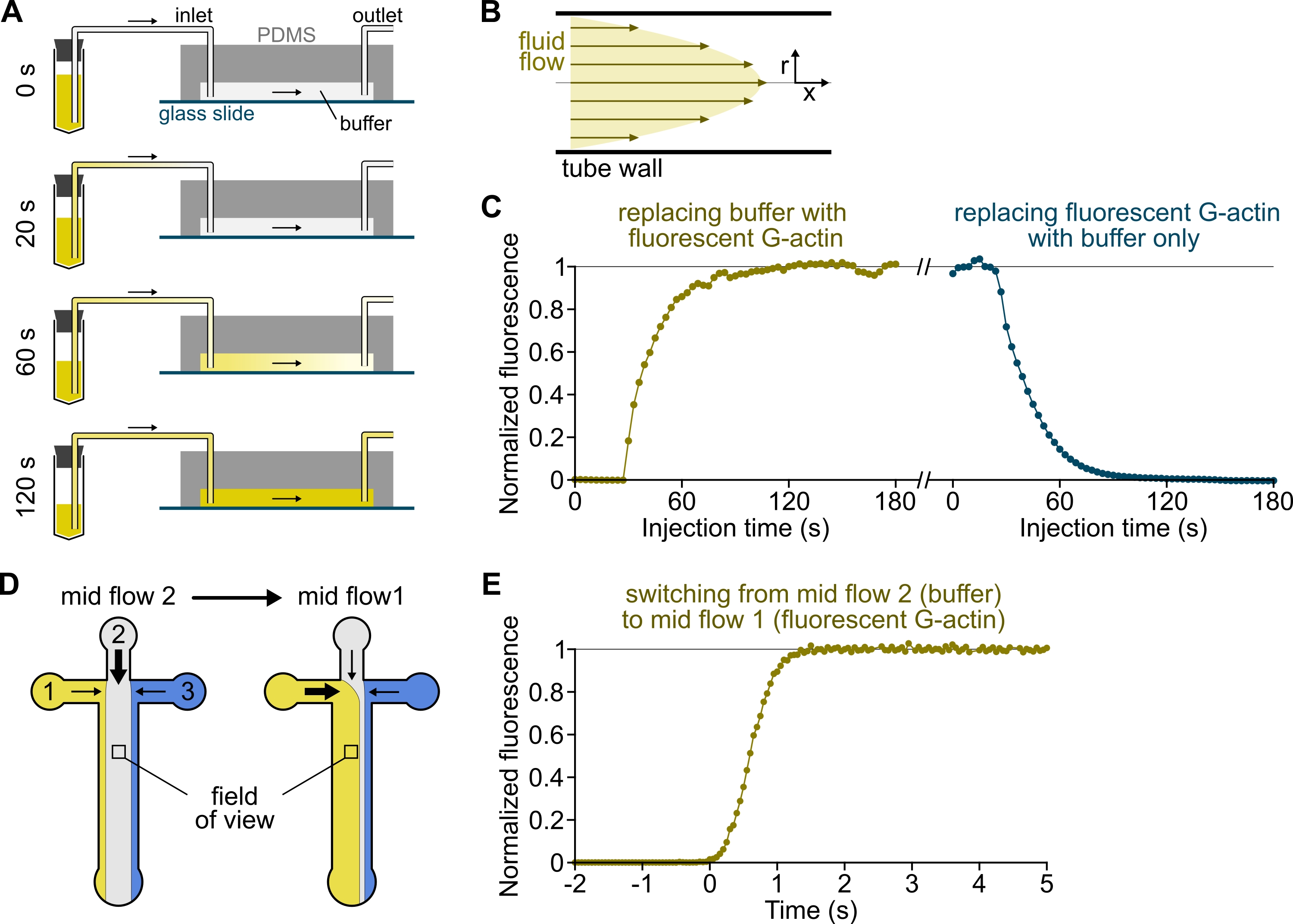{kind=link}
8. Experimento básico de filamento único: despolimerización del extremo de púas de adenosina difosfato (ADP)-actina
NOTA: Esta sección asume una cámara no funcionalizada (solo la sección 5). Si la cámara se ha funcionalizado directamente (sección 4), comience en el paso 8.4.
- Funcionalización superficial con semillas de filamento de actina:
- Cambie la solución 3 a 200 μL de semillas de espectrina-actina de 50 pM11 en F-buffer (ver sección 7).
NOTA: Alternativamente, si las semillas de espectrina-actina no están disponibles, se pueden usar segmentos de filamento corto funcionalizados con biotina que se inmovilizarán en un capa recubierto de estreptavidina (consulte 9,10 para obtener más detalles). - Inyectar durante 2 min con High Flow 3.
NOTA: Ajuste la concentración y el tiempo dependiendo de la densidad final de la semilla.
- Cambie la solución 3 a 200 μL de semillas de espectrina-actina de 50 pM11 en F-buffer (ver sección 7).
- Pasivación superficial:
- Tubo de cambio 3 con 300 μL de BSA al 5% en F-buffer.
- Inyecte durante 5 min en High Flow 3, seguido de 5 min en Mid Flow 3. Durante este segundo paso, reduzca la presión en los canales 1 y 2 a 7-8 mbar para obtener un contraflujo ~ -100 nL / min, de modo que todas las superficies de la cámara estén pasivadas por BSA.
NOTA: Dado que la solución BSA es más viscosa, las presiones deben ajustarse en consecuencia.
- Cambie el tubo 3 a F-buffer y enjuague el canal (5 min, High Flow 3).
- Preparar las siguientes soluciones de 200-300 μL, todas las proteínas diluidas en F-buffer:
Entrada 1, solución de polimerización: 1 μM 10% Alexa-488 marcado G-actina, 1 μM de profilina (Tabla 1).
Entrada 2, solución de envejecimiento: 0.15 μM 10% Alexa-488 etiquetado G-actina.
Entrada 3, solución de despolimerización: solo F-buffer.
NOTA: La profilina se usa aquí para prevenir la nucleación espontánea y para mantener una concentración constante de G-actina. - Cambie los tubos 1 a 3 (sección 7). Inyecte usando el ajuste preestablecido High Flow All durante 3-4 min. Las tres soluciones han llenado el tubo PEEK y han llegado a la cámara (Figura 3A). La superficie de vidrio se puede exponer a cualquier solución de entrada sin tiempo muerto (<1 s, Figura 3D, E).
- Encienda el microscopio. Ajuste la configuración: láser de excitación de 150 mW 488 nm a una potencia del 10%-20%, tiempo de exposición de la cámara de 100-200 ms, profundidad de penetración TIRF de 200-300 nm, objetivo 60x. Estos ajustes se utilizan en todo el manuscrito.
- Polimerización de filamentos (Figura 4A):
- Ajuste el ajuste de presión a Mid Flow 1 durante ~10 min.
- [OPCIONAL] Polimerización de registro (1 frame/20 s, TIRF). Los filamentos deben polimerizarse a unas 10 subunidades/segundo (sub/s)1,12.
- Envejecimiento del filamento: Ajuste el ajuste de presión a Mid Flow 2 durante 15 min. A la concentración crítica, 0.15 μM G-actina, la longitud del filamento permanecerá constante y los filamentos se convertirán en >99% ADP-F-actina4.
- Despolimerización (Figura 4A):
- Comience la adquisición a 1 fotograma/5 s, en modo de epifluorescencia. Como hay un fondo de fluorescencia muy bajo en los canales 2 y 3, no es necesario usar TIRF.
- Después de uno o dos fotogramas, cambie a Mid Flow 3. Los filamentos deben despolimerizarse a unos 10 sub/s (referencia12).
- Para restablecer el experimento, rompa todos los filamentos marcados fluorescentemente exponiéndolos continuamente al láser a la máxima potencia durante ~ 2 min. Para probar diferentes condiciones, cambie las soluciones 1, 2 o 3 e inyértelas (alto flujo, 3-4 min). Repita los pasos 8.7-8.9.
9. Otros experimentos de filamento único
- Probar las interacciones de los ABP con F-actina
NOTA: La microfluídica se ha utilizado con éxito para cuantificar la actividad de varios ABP de unión lateral, como la cofilina, la tropomiosina y Arp2/3. Siguiendo el protocolo de la sección 8:- Cambie el canal 3 al ABP fluorescente de interés en F-buffer. Inyección (Alto flujo 3, 3 min).
- Polimerización de filamentos: Ajuste el ajuste de presión a Mid Flow 1 durante 10 min.
- Enlace ABP: Iniciar la adquisición con TIRF. Ajuste la velocidad de fotogramas en función de la concentración de ABP. Después de 1-2 fotogramas, cambie a Mid Flow 3.
NOTA: Dependiendo del ABP, también puede ser posible cambiar rápidamente (por ejemplo, por menos de 5 s) a Mid Flow 2 para reducir aún más la fluorescencia de fondo al tomar una imagen. - Desvinculación de ABP: Mientras continúa la adquisición, cambie a Mid Flow 2.
- Polimerización con formina en el extremo libre de púas
NOTA: Se ha demostrado que las forminas afectan la polimerización final de púas de filamento. La microfluídica está particularmente adaptada para medir las tasas de unión y desvinculación de formina y su impacto en el alargamiento del filamento.- Prepare las siguientes soluciones:
Canal 1: 10 nM formin en F-buffer (Tabla 1).
Canal 2: 1 μM 10% Alexa-488 etiquetado G-actina, 4 μM profilina.
Canal 3: F-buffer. - Cambie los tubos 1, 2 y 3 (sección 7). Inyecte usando el ajuste preestablecido High Flow All durante 3-4 min.
- Iniciar la polimerización del filamento: Ajuste el ajuste de presión a Mid Flow 2 durante 2 min.
- Unión de Formin al extremo de púas del filamento: Ajuste los ajustes de presión a Mid Flow 1 durante 30 s.
- Polimerización mediada por formina: Ajuste el ajuste de presión a Mid Flow 2. Con formin mDia1 en su extremo de púas, los filamentos deben polimerizarse a alrededor de 50 sub/s 13,14,15.
- Prepare las siguientes soluciones:
- Polimerización/despolimerización a partir de formina anclada en superficie
NOTA: Se ha demostrado que las tasas de polimerización y despolimerización de los extremos de púas decorados con formina dependen de la tensión aplicada al filamento. En microfluídica, la fricción del flujo de fluido a lo largo del lado del filamento genera una tensión proporcional a la longitud del filamento y al caudal14,16.- Utilice el método de la sección 8 descrito anteriormente, reemplazando los pasos 8.1, 8.2 y 8.3 para la pasivación superficial con:
- Cambie el tubo 3 a 1 μg/ml de anticuerpos anti-His en F-buffer. Inyectar durante 2 min con High Flow 3.
- Tubo de cambio 3 con BSA al 5% en F-buffer. Inyecte durante 5 min en High Flow 3, seguido de 5 min en Mid Flow 3. Durante este segundo paso, reduzca la presión en los canales 1 y 2 a 7-8 mbar para obtener un flujo de contador ~ -100 nL / min para que todas las superficies de la cámara sean pasivadas por BSA.
- Cambie el tubo 3 a 100 nM His-tagged formin en F-buffer. Inyecte durante 5 min con High Flow 3. Cambie el tubo 3 con F-buffer. Inyecte durante 5 minutos con High Flow 3 para asegurarse de que no queden formins en el tubo.
- Preparar e inyectar las siguientes soluciones (200-300 μL cada una, en F-buffer):
Canal 1: 1 μM 10% Alexa-488 etiquetado G-actina.
Canal 2: 1 μM de G-actina sin etiquetar, 4 μM de profilina.
Canal 3: F-buffer solamente. - Nucleación de filamentos: Exponga las forminas ancladas en la superficie a la G-actina (ajuste de flujo medio 1).
- Polimerización de filamentos: Exponga la cámara a la profilina-actina usando Mid Flow 2.
- Inicio adquisición: 1 frame/2 s, epifluorescencia. Con formin mDia1, los filamentos deben polimerizarse a 50-80 sub/s, dependiendo de la longitud del filamento y el caudal14.
- Despolimerización de filamentos: Iniciar adquisición (1 frame/4 s, epifluorescencia). Después de 1-2 fotogramas, exponga los filamentos al F-buffer, Mid Flow 3. Con formin mDia1, los filamentos deben despolimerizarse a 5-15 sub/s, dependiendo de la longitud del filamento y el caudal14.
- Utilice el método de la sección 8 descrito anteriormente, reemplazando los pasos 8.1, 8.2 y 8.3 para la pasivación superficial con:
- Filamentos de actina con segmentos sin etiquetar
NOTA: El etiquetado fluorescente de actina crea varios artefactos, como pausas durante la despolimerización17 y alteración de la unión a la tropomiosina18. Una solución alternativa para estos artefactos es usar microfluídica para ensamblar filamentos que muestran segmentos sin etiquetar.- Preparar e inyectar las siguientes soluciones (200-300 μL en F-buffer):
Canal 1: 1 μM de G-actina sin etiquetar, 1 μM de profilina.
Canal 2: 0.3 μM 10% Alexa-488 etiquetado G-actina. - Exponga secuencialmente la superficie al canal 2 (5 min), canal 1 (10 min) y canal 2 (15 min) para generar segmentos sin etiquetar ADP-actina con segmentos marcados fluorescentemente en cada extremo.
- Preparar e inyectar las siguientes soluciones (200-300 μL en F-buffer):
- Filamentos de púas con gelsolin
NOTA: Con las semillas de espectrina-actina, los filamentos polimerizan en su extremo libre de púas, mientras que el extremo puntiagudo es estabilizado por la semilla de espectrina-actina. Una alternativa es anclar filamentos con una tapa de extremo de púas como la gelsolina.- Prepare una solución de F-actina de 4 μM 10% Alexa-488 etiquetada G-actina en 20 μL de F-buffer. Deje que la actina nuclese espontáneamente y polimerice en RT durante al menos 30 minutos en el banco. Envuelva el tubo en papel de aluminio para protegerlo de la luz.
- Mientras tanto, prepare la cámara microfluídica y pasiva la superficie con una mezcla de 5% de BSA y 1% de biotina-BSA (ver paso 8.2).
- Enjuague el canal 3 con F-buffer (2 min en High Flow 3). Inyecte 10 μg/ml de neutravidina en F-buffer (4 min a alto flujo 3).
- Cambie los tubos a:
Canal 1: 10 nM biotina-gelsolina (Tabla 1).
Canal 2: F-buffer.
Canal 3: F-actina prepolimerizada de 0,4 μM. - Inyecte todas las soluciones juntas utilizando el ajuste High Flow All durante 3 min.
- Exponga toda la cámara a la gelsolina (flujo medio 1, 30 s).
- Conecte filamentos a la superficie (bajo flujo 3: canal 3 a 3 mbar, canales 1 y 2 a ~ 2 mbar, durante aproximadamente 2 min).
- [OPCIONAL] Si la densidad del filamento es demasiado baja, repita los pasos 9.5.6 y 9.5.7.
- Despolimerización del extremo puntiagudo: Inicio de adquisición (1 fotograma/30 s, epifluorescencia). Después de 1-2 fotogramas, exponga los filamentos solo al búfer, Mid Flow 2. Los filamentos deben despolimerizarse a alrededor de 0,2 sub/s.
10. Formación y desmontaje de haces de filamentos inducidos por fasina por ADF/cofilina
NOTA: Para formar haces de filamentos de actina, asegúrese de tener una densidad de semillas de filamentos suficientemente alta en la superficie de la cámara. Cuando se exponen a la proteína fascin, los filamentos vecinos que fluctúan lateralmente estarán dinámicamente reticulados por las proteínas fascin. Como la fascin se desata rápidamente del lado del filamento19, la fascin tiene que estar constantemente presente en la solución principal que fluye para mantener la agrupación del filamento.
- Siga los pasos 8.1-8.3.
- Prepare las siguientes soluciones (200-300 μL en F-buffer):
Canal 1, solución de polimerización: 1 μM 10% Alexa-488 marcado G-actina, 1 μM profilin.
Canal 2, Solución de empaquetado: fascin 200 nM (Tabla 1), 0.15 μM 10% Alexa-488 etiquetado G-actina.
Canal 3, Solución de desmontaje: 200 nM ADF/cofilina (Tabla 1), fascin 100 nM, 0,15 μM 10% Alexa-488 etiquetado G-actina. - Cambie los tubos 1 a 3 (sección 7). Inyecte usando el ajuste preestablecido High Flow All , durante 3-4 min.
- Polimerización de filamentos: Ajuste el ajuste de presión a Mid Flow 1 durante ~ 10 min. La polimerización se puede visualizar con TIRF.
- Agrupación de filamentos (Figura 4C): Iniciar la adquisición de imágenes (1 fotograma/5 s, epifluorescencia). Después de 1-2 cuadros, ajuste el ajuste de presión a Mid Flow 2 y observe el empaquetado de filamentos.
- Fragmentación del haz: Iniciar la adquisición de imágenes (1 fotograma/5 s, epifluorescencia). Después de 1-2 fotogramas, ajuste el ajuste de presión a Mid Flow 3 y observe el desmontaje inducido por cofilina tanto de filamentos individuales como de haces.
11. Procedimiento de limpieza de dispositivos microfluídicos
NOTA: Para evitar cualquier contaminación de un experimento a otro, es fundamental limpiar y secar completamente todos los tubos y medidores de flujo después de cada experimento.
- Desconecte todos los tubos de la cámara PDMS y deseche la cámara.
- Para limpiar los tubos PEEK y los medidores de flujo, tape los extremos de los tubos en un tubo de plástico vacío de 15 ml e inyecte las siguientes soluciones a la presión máxima hasta que el depósito esté casi vacío:
400 μL de F-buffer.
400 μL de 0,5 M NaOH.
400 μL de agua pura.
200 μL de isopropanol. - Reemplace con un depósito vacío y sople aire hasta que los tubos estén completamente secos (~ 2-4 min, presión máxima).
12. Análisis de imágenes
NOTA: Si bien este manuscrito se centra en el método para ensamblar, manipular y visualizar filamentos de actina individuales en microfluídica, aquí se proporciona un breve método para analizar las películas adquiridas. El análisis se realiza en imágenes de 16 bits, utilizando ImageJ, siguiendo la sección 8.
- El tratamiento de la imagen es mínimo:
- Importe la pila de imágenes de polimerización o despolimerización.
- [OPCIONAL] Homogeneice la intensidad de la imagen con la función Restar fondo (configuración predeterminada (es decir, 'Radio de bola rodante' = 50 píxeles)). Esto es particularmente útil si la fluorescencia de fondo cambia durante el transcurso de una película o si la iluminación de fluorescencia no es homogénea en el campo de visión.
- Ajuste el brillo y el contraste (fondo cerca de cero, filamentos cerca del máximo).
- Crear kymograph de filamento:
- Seleccione un filamento que no se detenga, rompa o desprenda. No seleccione en función del comportamiento de lo contrario. Dibuje una línea 1-2 píxeles por encima (herramienta Línea recta ). Guarde el número de filamento (Agregar en ROI Manager).
- Aplique la función Reslice (recuento de sectores: 5 píxeles). Calcular la intensidad máxima (función Zprojection).
- Mida la tasa de polimerización/despolimerización:
- En el kymograph, dibuje una línea a lo largo del extremo de púas del filamento (herramienta Línea recta , Figura 4A). Mida el ancho y el alto de la línea (función Medir).
- Repita los pasos 12.2-12.3 sobre varios filamentos. Calcular las tasas de polimerización/despolimerización (Figura 4A):
 , donde v es la velocidad (en sub/s), w el ancho de línea (píxeles), pix el tamaño de píxel (nm), h la altura de línea (fotogramas) y dt el tiempo entre fotogramas (en segundo). Aquí, 2,7 nm corresponde a la contribución efectiva de una subunidad de actina a la longitud del filamento.
, donde v es la velocidad (en sub/s), w el ancho de línea (píxeles), pix el tamaño de píxel (nm), h la altura de línea (fotogramas) y dt el tiempo entre fotogramas (en segundo). Aquí, 2,7 nm corresponde a la contribución efectiva de una subunidad de actina a la longitud del filamento.
Resultados
Para todos los experimentos descritos anteriormente, los filamentos de actina marcados fluorescentemente deben ser claramente visibles, con buen contraste, indicativos de baja fluorescencia de fondo desde la superficie (Figura 4, consulte el Archivo complementario 1 para la solución de problemas comunes). Los filamentos de actina tampoco deben adherirse a la superficie: cuando el caudal dominante es bajo, las fluctuaciones laterales de los filamentos de actina deben ser per...
Discusión
En comparación con los métodos estándar de filamento único en los que los filamentos de actina están anclados a la superficie por múltiples puntos a lo largo de su longitud o mantenidos cerca de ella por un agente de apiñamiento como la metilcelulosa, la microfluídica ofrece una serie de ventajas. Como las interacciones con la superficie son mínimas, se evitan las pausas artificiales que estas interacciones pueden inducir durante el alargamiento y la despolimerización. Los filamentos están alineados por el flu...
Divulgaciones
Los autores declaran que no hay conflictos de intereses.
Agradecimientos
Agradecemos a B. Ladoux y R.-M. Mège lab para el uso de sus equipos de limpieza UV, y J. Heuvingh y 0. du Roure para la capacitación inicial que recibimos sobre la preparación de moldes en obleas de silicio y la provisión de consejos sobre microfluídica. Reconocemos la financiación de la subvención StG-679116 del Consejo Europeo de Investigación (a A.J.) y las becas de la Agence Nationale de la Recherche Muscactin y Conformin (a G.R.-L.).
Materiales
| Name | Company | Catalog Number | Comments |
| β-Casein | Merck | C6905 | Used at 8 mg/mL |
| Biopsy punch (with plunger) | Ted Pella | 15115-2 | ID 0.75 mm, OD 1.07 mm |
| Biotin-BSA | Merck | A8549 | Used at 1 mg/mL |
| BSA | Merck | A8022 | Used at 50 mg/mL |
| Coverslip Mini-Rack Teflon holder | Invitrogen | C14784 | for 8 coverslips |
| Coverslips 22x40mm Thickness #1.5 | Menzel Gläser | 631-1370 | |
| DABCO | Merck | D27802 | component in f-buffer |
| DTT | Euromedex | EU0006-D | component in f-buffer |
| Ester NHS Alexa Fluor 488 | Invitrogen | A20000 | Fluorophore for actin labeling on Lys328. |
| EZ-Link Sulfo-NHS-Biotin | Thermo Scientific | 21338 | To biotinylate actin on Lys328 |
| Hellmanex III | Hellma | 9-307-011-4-507 | Glass cleaning detergent |
| ImageJ | NIH | N/A | open source software |
| Laboport | KNF | 811kn.18 | vacuum pump (ultimate vacuum: 240 mbar) |
| Magic invisible tape | Scotch | 7100024666 | standard transparent office tape |
| Micrewtube | Simport | T341-6T | 2 mL microfluidic reservoir tubes |
| Microfluidic device Part 1: Flow Unit S | Fluigent | FLU-S-D-PCKB | Flowmeter |
| Microfluidic device Part 2: Fluiwell-4C-2 mL | Fluigent | 14002001PCK | Reservoir holder |
| Microfluidic device Part 3: MFCS-EZ | Fluigent | EZ-11000001 EZ-00345001 | Pressure controller |
| Model 42 - UVO-Cleaner | Jelight Inc. | 42-220 | Ultraviolet cleaner |
| N6-(6-Aminohexyl)-ATP-ATTO-488 | Jena Bioscience | NU-805-488 | ATP-ATTO used to label actin |
| neutravidin | Thermo Scientific | 31000 | |
| PLL-PEG | SuSoS | PLL(20)-g[3.5]- PEG(2) | Use at 1 mg/mL in PBS. |
| Polydimethylsiloxane (PDMS) Sylgard 184 Silicon Elastomer | Dow Corning | 1673921 | Contains PDMS base and curing agent |
| Polyetheretherketone (PEEK) tubing | Merck | Z226661 | “Blue” : I.D. = 0.25 mm |
| Safety blow gun | Coilhose Pneumatics | 700-S | filtered air |
| Silicon tubing | VWR | 228-0701P | connect PEEK to coupler |
| Stainless steel catheter coupler | Prime Bioscience | SC22/15 | Inserted into PDMS inlets and outlet to connect to PEEK tubing |
| Thermoplastic film | Sigma Aldrich | PM996 | Standard "parafilm" |
| Ultrapure ethanol | VWR | 64-17-5 | |
| Ultrasonic cleaning bath | VWR | USC200TH | To accomodate 1 L beakers |
| Vacuum dessicator | SP Bel-Art | F42022-0000 | to degas the PDMS or solutions |
Referencias
- Wioland, H., Jégou, A., Romet-Lemonne, G. Celebrating 20 years of live single-actin-filament studies with five golden rules. Proceedings of the National Academy of Sciences of the United States of America. 119 (3), 2109506119 (2022).
- Kuhn, J. R., Pollard, T. D. Real-time measurements of actin filament polymerization by total internal reflection fluorescence microscopy. Biophysical Journal. 88 (2), 1387-1402 (2005).
- Brewer, L. R., Bianco, P. R. Laminar flow cells for single-molecule studies of DNA-protein interactions. Nature Methods. 5 (6), 517-525 (2008).
- Jégou, A., et al. Individual actin filaments in a microfluidic flow reveal the mechanism of ATP hydrolysis and give insight into the properties of profilin. PLoS Biology. 9 (9), 1001161 (2011).
- Gicquel, Y., et al. Microfluidic chips for in situ crystal x-ray diffraction and in situ dynamic light scattering for serial crystallography. Journal of Visualized Experiments: JoVE. (134), e57133 (2018).
- Chandradoss, S. D., et al. Surface passivation for single-molecule protein studies. Journal of Visualized Experiments: JoVE. (86), e50549 (2014).
- Schaedel, L., et al. Microtubules self-repair in response to mechanical stress. Nature Materials. 14 (11), 1156-1163 (2015).
- Zimmermann, D., Morganthaler, A. N., Kovar, D. R., Suarez, C. In vitro biochemical characterization of cytokinesis actin-binding proteins. Methods in Molecular Biology. 1369, 151-179 (2016).
- Funk, J., et al. Profilin and formin constitute a pacemaker system for robust actin filament growth. eLife. 8, 50963 (2019).
- Pandit, N. G., et al. Force and phosphate release from Arp2/3 complex promote dissociation of actin filament branches. Proceedings of the National Academy of Sciences of the United States of America. 117 (24), 13519-13528 (2020).
- Wioland, H., et al. ADF/Cofilin accelerates actin dynamics by severing filaments and promoting their depolymerization at both ends. Current Biology: CB. 27 (13), 1956-1967 (2017).
- Pollard, T. D., Mooseker, M. S. Direct measurement of actin polymerization rate constants by electron microscopy of actin filaments nucleated by isolated microvillus cores. The Journal of Cell Biology. 88 (3), 654-659 (1981).
- Kovar, D. R., Harris, E. S., Mahaffy, R., Higgs, H. N., Pollard, T. D. Control of the assembly of ATP- and ADP-actin by formins and profilin. Cell. 124 (2), 423-435 (2006).
- Jégou, A., Carlier, M. -. F., Romet-Lemonne, G. Formin mDia1 senses and generates mechanical forces on actin filaments. Nature Communications. 4, 1883 (2013).
- Breitsprecher, D., et al. Rocket launcher mechanism of collaborative actin assembly defined by single-molecule imaging. Science. 336 (6085), 1164-1168 (2012).
- Courtemanche, N., Lee, J. Y., Pollard, T. D., Greene, E. C. Tension modulates actin filament polymerization mediated by formin and profilin. Proceedings of the National Academy of Sciences of the United States of America. 110 (24), 9752-9757 (2013).
- Niedermayer, T., et al. Intermittent depolymerization of actin filaments is caused by photo-induced dimerization of actin protomers. Proceedings of the National Academy of Sciences. 109 (27), 10769-10774 (2012).
- Gateva, G., et al. Tropomyosin isoforms specify functionally distinct actin filament populations in vitro. Current Biology: CB. 27 (5), 705-713 (2017).
- Aratyn, Y. S., Schaus, T. E., Taylor, E. W., Borisy, G. G. Intrinsic dynamic behavior of fascin in filopodia. Molecular Biology of the Cell. 18 (10), 3928-3940 (2007).
- Pollard, T. D. Rate constants for the reactions of ATP- and ADP-actin with the ends of actin filaments. The Journal of Cell Biology. 103, 2747-2754 (1986).
- Wioland, H., Jegou, A., Romet-Lemonne, G. Torsional stress generated by ADF/cofilin on cross-linked actin filaments boosts their severing. Proceedings of the National Academy of Sciences of the United States of America. 116 (7), 2595-2602 (2019).
- Colombo, J., et al. A functional family of fluorescent nucleotide analogues to investigate actin dynamics and energetics. Nature Communications. 12 (1), 548 (2021).
- Spudich, J. A., Watt, S. The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. The Journal of Biological Chemistry. 246 (15), 4866-4871 (1971).
- Romet-Lemonne, G., Guichard, B., Jégou, A. Using microfluidics single filament assay to study formin control of actin assembly. Methods in Molecular Biology. 1805, 75-92 (2018).
- Gieselmann, R., Kwiatkowski, D. J., Janmey, P. A., Witke, W. Distinct biochemical characteristics of the two human profilin isoforms. European Journal of Biochemistry. 229 (3), 621-628 (1995).
- Lin, D. C., Lin, S. Actin polymerization induced by a motility-related high-affinity cytochalasin binding complex from human erythrocyte membrane. Proceedings of the National Academy of Sciences of the United States of America. 76 (5), 2345-2349 (1979).
- Casella, J. F., Maack, D. J., Lin, S. Purification and initial characterization of a protein from skeletal muscle that caps the barbed ends of actin filaments. The Journal of Biological Chemistry. 261 (23), 10915-10921 (1986).
- Kremneva, E., et al. Cofilin-2 controls actin filament length in muscle sarcomeres. Developmental Cell. 31 (2), 215-226 (2014).
- Le Clainche, C., Carlier, M. -. F. Actin-based motility assay. Current Protocols in Cell Biology. , 1-20 (2004).
- Vignjevic, D., et al. Formation of filopodia-like bundles in vitro from a dendritic network. The Journal of Cell Biology. 160 (6), 951-962 (2003).
- Duellberg, C., Cade, N. I., Holmes, D., Surrey, T. The size of the EB cap determines instantaneous microtubule stability. eLife. 5, 13470 (2016).
- Duellberg, C., Cade, N. I., Surrey, T. Microtubule aging probed by microfluidics-assisted tubulin washout. Molecular Biology of the Cell. 27 (22), 3563-3573 (2016).
- Suzuki, E. L., et al. Geometrical constraints greatly hinder formin mDia1 activity. Nano Letters. 20 (1), 22-32 (2020).
- Wioland, H., Suzuki, E., Cao, L., Romet-Lemonne, G., Jegou, A. The advantages of microfluidics to study actin biochemistry and biomechanics. Journal of Muscle Research and Cell Motility. 41 (1), 175-188 (2020).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados