
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Utilisation de la microfluidique et de la microscopie à fluorescence pour étudier la dynamique d’assemblage des filaments et des faisceaux d’actine unique
Dans cet article
Résumé
Nous présentons des protocoles pour des tests microfluidiques de filaments d’actine simples, en combinaison avec la microscopie à fluorescence, qui permettent de surveiller avec précision les filaments d’actine individuels en temps réel tout en les exposant séquentiellement à différentes solutions protéiques.
Résumé
Afin de déchiffrer les mécanismes moléculaires complexes qui régulent l’assemblage et le désassemblage des filaments d’actine, il est un grand atout de surveiller les réactions individuelles en direct dans des conditions bien contrôlées. Pour ce faire, des expériences en direct à filament unique ont émergé au cours des 20 dernières années, principalement en utilisant la microscopie à fluorescence à réflexion interne totale (TIRF), et ont fourni une mine de résultats clés. En 2011, afin d’élargir davantage les possibilités de ces expériences et d’éviter des artefacts problématiques récurrents, nous avons introduit une microfluidique simple dans ces essais. Cette étude détaille notre protocole de base, où les filaments d’actine individuels sont ancrés par une extrémité à la surface de la couverture passivée, s’alignent sur l’écoulement et peuvent être exposés successivement à différentes solutions protéiques. Nous présentons également les protocoles pour des applications spécifiques et expliquons comment des forces mécaniques contrôlées peuvent être appliquées, grâce à la traînée visqueuse de la solution fluide. Nous soulignons les mises en garde techniques de ces expériences et présentons brièvement les développements possibles basés sur cette technique. Ces protocoles et explications, ainsi que la disponibilité actuelle d’équipements microfluidiques faciles à utiliser, devraient permettre aux non-spécialistes de mettre en œuvre ce test dans leurs laboratoires.
Introduction
L’assemblage et le démontage des filaments d’actine et des réseaux de filaments d’actine sont contrôlés par plusieurs réactions biochimiques et dépendent du contexte mécanique. Afin de mieux comprendre ces mécanismes complexes, il est inestimable de pouvoir observer des réactions individuelles sur des filaments individuels (en nombre suffisant). Au cours des dernières décennies, l’observation des filaments d’actine dynamiques en temps réel, principalement à l’aide de la microscopie à fluorescence par réflexion interne totale (TIRF), est devenue une technique clé et a fourni une liste impressionnante de résultats qui n’auraient pas pu être obtenus avec des tests biochimiques en solution en vrac1.
Pour ce faire, il faut maintenir des filaments d’actine marqués par fluorescence près de la surface du couvercle du microscope tout en les exposant à des solutions de protéines de liaison à l’actine (ABP), qui peuvent également être marquées par fluorescence. Cela permet de surveiller les événements qui se produisent sur des filaments individuels dans des conditions biochimiques bien contrôlées et de quantifier ainsi les taux de réaction. Cependant, un certain nombre de limitations spécifiques devraient être prises en compte. Maintenir artificiellement les filaments près de la surface, souvent grâce à plusieurs points d’ancrage ou en utilisant un agent d’encombrement tel que la méthylcellulose, peut modifier leur comportement (par exemple, provoquer des pauses dans leur polymérisation et leur dépolymérisation2). Le suivi du contour de chaque filament peut être difficile, en particulier si de nouveaux filaments ou fragments de filament s’accumulent dans le champ de vision au fil du temps. Les réactions ont lieu dans un volume fini où la concentration de monomères d’actine et d’ABP peut varier au fil du temps, ce qui peut rendre difficile la détermination de constantes de vitesse précises. Enfin, le renouvellement ou la modification de la solution d’ABP est difficile à réaliser en moins de 30 s et conduira souvent à une teneur en protéines inhomogène dans l’échantillon.
Il y a un peu plus de 10 ans, inspirés par ce qui a déjà été fait pour étudier les brins individuels d’acide désoxyribonucléique (ADN)3, nous avons introduit une nouvelle technique basée sur la microfluidique pour observer et manipuler des filaments d’actine individuels4. Il permet de contourner les limitations susmentionnées des techniques classiques à filament unique. Dans ces essais microfluidiques, les filaments d’actine sont cultivés à partir de graines de spectrine-actine adsorbées sur le couvercle. Les filaments sont ainsi ancrés par une extrémité uniquement au fond de la chambre microfluidique et fluctuent au-dessus de la surface sans coller. Les filaments s’alignent sur le flux des solutions entrantes, ce qui facilite la surveillance de leur longueur de contour et les maintient dans une région peu profonde au-dessus de la glissière de couverture où le TIRF peut être utilisé. Différentes solutions sont simultanément acheminées dans la chambre sans mélange, et les filaments peuvent y être exposés séquentiellement et rapidement.
Ici, nous proposons une série de protocoles de base pour mettre en place des tests microfluidiques à filament d’actine unique en laboratoire. Les couvercles et les chambres microfluidiques peuvent être préparés à l’avance (en une demi-journée), et l’expérience elle-même, où plusieurs conditions biochimiques peuvent être testées, est réalisée en moins d’une journée.
Protocole
1. Préparation de la chambre microfluidique
- Sélectionnez un moule maître SU-8 avec plusieurs motifs de chambre. Les chambres typiques sont en forme de croix avec trois entrées et une sortie, de 20 μm de haut et 800 μm de large (Figure 1). Ces moules maîtres peuvent être achetés auprès d’entreprises externes ou fabriqués dans des laboratoires universitaires (par exemple, Gicquel, Y. et al.5).
- Placez du ruban adhésif autour du bord du moule.
- Placez ~50 cm de long, 19 mm de large, ruban de bureau transparent standard (voir Table des matériaux) sur un banc, côté collant vers le haut. Placez le moule verticalement à une extrémité et le long de la ligne médiane du ruban.
- Roulez le moule à l’autre extrémité du ruban adhésif pour créer une bordure de 1 cm autour du moule. Rabattez le ruban adhésif au fond du moule.
- Préparer une solution de polydiméthylsiloxane (PDMS).
- Dans un plat de pesage jetable, versez directement 25 à 30 g de base PDMS (Table des matériaux). Ajouter 10 % poids/poids d’agent de durcissement PDMS (Table des matériaux) avec une pipette Pasteur en plastique jetable.
- Mélanger manuellement et soigneusement avec un bâton en plastique. Assurez-vous que l’agent de durcissement est bien incorporé dans la base PDMS, même si l’agitation crée beaucoup de bulles.
- Dégazez la solution PDMS dans un dessiccateur sous vide (Table des matériaux) pendant au moins 5 min à température ambiante (RT). Les bulles se dilatent, remontent à la surface et éclatent lorsque le vide est brisé.
- Versez la solution PDMS sur le moule SU-8. Utilisez un bâton en plastique pour gratter et transférer autant de mélange que possible.
- Dégazez PDMS une deuxième fois (5 min dans le dessiccateur sous vide). Assurez-vous de vous débarrasser de la plupart des bulles (quelques petites bulles sur la surface supérieure sont bien).
- Placer le moule dans un four à 70 °C pendant au moins 5 h pour que le PDMS réticule et se solidifie.
- Retirez les chambres PDMS solides du moule.
ATTENTION: Les plaquettes de silicium pour les moules SU-8 sont extrêmement fragiles, il faut donc faire très attention lors de la séparation du PDMS des plaquettes. Travaillez sur une surface dure et plane et gardez la plaquette plate sur la surface.- Avec une lame de rasoir, faites une coupe circulaire dans le PDMS, à environ 1 cm du bord du moule. Tous les motifs doivent mesurer au moins 0,5 cm à l’intérieur de la coupe. Décollez doucement le bloc PDMS central avec des remorqueurs doux.
ATTENTION : Lors du décollement, maintenez le moule SU-8 à plat sur la paillasse pour éviter de le casser. - Placez pdMS sur une feuille d’aluminium propre, la surface moulée faisant face à la feuille d’aluminium, pour protéger sa surface de la poussière et rendre les motifs plus visibles.
- Avec une lame de rasoir, faites une coupe circulaire dans le PDMS, à environ 1 cm du bord du moule. Tous les motifs doivent mesurer au moins 0,5 cm à l’intérieur de la coupe. Décollez doucement le bloc PDMS central avec des remorqueurs doux.
- Choisissez et coupez une chambre avec une lame de rasoir à au moins 0,5 cm du motif. Le bloc PDMS résultant mesure environ 0,5 cm de haut, 1,5 cm de large et 3 cm de long. Percer trois entrées et une sortie avec un poinçon de biopsie de 0,75 mm I.D. (Table des matériaux).
- Nettoyez la chambre PDMS avec de l’éthanol ultrapur (Table des matériaux) et séchez-la à l’air à l’aide d’une sarbacane de sécurité (Table des matériaux). Placez le PDMS avec le motif tourné vers le haut dans une boîte de Petri propre et fermez le plat avec son couvercle.
2. Nettoyage des couvercles en verre
REMARQUE: Ici, une procédure standard de nettoyage du couvercle, basée sur une série d’étapes de sonication, est détaillée. D’autres procédures de nettoyage des couvercles en verre ont été décrites dans de nombreuses autres publications qui peuvent obtenirdes résultats satisfaisants similaires 6,7,8,9.
- Placez 10 à 20 couvercles (40 mm de long) sur un support en polytétrafluoroéthylène (PTFE) (table des matériaux). Sonicer les couvercles dans 0,5 L de solution de nettoyage du verre à 2% (Table des matériaux) dans un bécher en verre de 1 L (35 °C, 30 min).
- Jetez la solution de nettoyage du verre et rincez abondamment les couvercles avec du dH2O dans au moins trois bains successifs de 0,5 L.
- Préparer 0,5 L de 2 M KOH dans un bécher en verre de 1 L. Sonicate les couvercles en KOH (RT, 30 min). Jetez koh et rincez les couvercles avec du dH2O dans au moins trois bains de 0,5 L.
ATTENTION : Utilisez l’équipement de protection de sécurité de laboratoire approprié (gants, lunettes et blouse de laboratoire). - Transférer et soniquer les couvercles dans 0,5 L d’éthanol ultrapur (RT, 30 min). Les couvercles peuvent être conservés dans de l’éthanol jusqu’à 2 semaines. Fermez le bécher avec un film thermoplastique (Table des matériaux) pour éviter l’évaporation. Avant utilisation, séchez le couvercle avec un flux d’air.
3. Assemblage de la chambre PDMS
- Préchauffer la plaque chauffante à 100 °C. Placez jusqu’à trois chambres PDMS nettoyées et des couvercles en verre dans une boîte de Petri propre. Placez la boîte de Petri ouverte dans un nettoyant ultraviolet (UV) profond (λ = 185 nm, voir Tableau des matériaux) et exposez-la à la lumière UV pendant 3 à 5 minutes.
REMARQUE: Alternativement, les chambres PDMS et les couvercles peuvent être exposés à l’air ou au plasma d’oxygène pendant 30 s. - Positionnez doucement la chambre PDMS sur le couvercle. Assurez-vous que les deux surfaces mises en contact ont été directement exposées aux UV. Le PDMS adhère automatiquement au verre et la chambre devient clairement visible.
- Pour éliminer tout air emprisonné à l’interface PDMS-coverslip, appuyez très doucement sur la surface avec un doigt. Pour un collage plus serré, appuyez plus fortement sur les coins et les côtés. Assurez-vous que le plafond de la chambre n’entre pas en contact avec la surface vitrée.
- Placez la chambre avec le fond en verre face à la plaque chauffante à 100 °C pendant 5 min. Après cette étape, les liaisons verre-PDMS deviennent permanentes et les chambres ne peuvent être utilisées qu’une seule fois. Utilisez la chambre immédiatement ou conservez-la dans une boîte de Petri propre jusqu’à une semaine.
4. [FACULTATIF] Passivation directe et fonctionnalisation
REMARQUE: Selon l’application, les chambres peuvent être passivées et fonctionnalisées soit une fois connectées au dispositif de contrôle microfluidique (voir tableau des matériaux), soit en injectant manuellement des solutions directement dans la chambre avec une pipette avant sa connexion au dispositif microfluidique. Ce dernier offre l’avantage de consommer moins de réactif et d’éviter une contamination potentielle en faisant circuler la solution à travers le tube en polyéther éther cétone (PEEK) du dispositif microfluidique. Dans toutes les étapes suivantes, les solutions sont injectées en collant directement la pointe de la pipette dans la sortie. Afin d’éviter de créer des bulles à l’intérieur de la chambre, assurez-vous d’avoir une petite gouttelette qui dépasse de l’extrémité de la pipette lorsque vous branchez la pointe dans la sortie de la chambre PDMS. De même, retirez l’embout de la pipette avant que tout le volume n’ait été injecté.
- Injecter 20 μL de PLL-PEG (1 mg/mL dans une solution saline tamponnée au phosphate (PBS)). Incuber pendant au moins 1 h (ou pendant la nuit) à RT. Pour éviter l’évaporation, placez la chambre PDMS dans une boîte humide (par exemple, une boîte à embouts vide avec de l’eau dans le compartiment inférieur et la chambre PDMS sur la plate-forme de maintien des pointes).
- Injecter 20 μL de graines de 100 pM de spectrine-actine (dans le tampon F, voir tableau 1 et tableau 2). N’attendez pas plus de 1 min. Ajustez la concentration et le moment des graines pour ajuster la densité de surface des graines, suffisamment élevée pour les statistiques volumineuses et suffisamment faible pour que les filaments ne se chevauchent pas.
REMARQUE: Alternativement, si les graines de spectrine-actine ne sont pas disponibles, utilisez des segments de filament courts fonctionnalisés à la biotine qui seront immobilisés sur un couvercle recouvert de streptavidine 9,10. - [FACULTATIF] Injecter 20 μL d’albumine sérique bovine (BSA) à 5 % dans un tampon F. Départ à RT pendant 10 min.
- [FACULTATIF] Injecter 20 μL de 1 mg/mL β-caséine dans un tampon F. Départ à RT pendant 10 min.
REMARQUE: Suivez les étapes 4.3 et / ou 4.4 pour passer la chambre. Le choix de la passivation dépend des protéines utilisées et ne fonctionne pas aussi bien sur tous les ABP. Lors de l’utilisation de l’actine seule, PLL-PEG ou BSA est suffisant.
5. Connectez un dispositif microfluidique
REMARQUE : Utilisez un système microfluidique basé sur la pression avec jusqu’à quatre canaux pour contrôler les écoulements dans la chambre microfluidique (Figure 1A, voir Tableau des matériaux). Pour éviter la formation de bulles dans le tube microfluidique et perturber la stabilité de l’écoulement, dégazez toutes les solutions. Placer 5 mL de dH20 et 10 mL de stock tampon F dans un dessiccateur à vide relié à une pompe à vide (vide ultime <250 mbar) et dégazer pendant au moins 1 h à RT.
- Rincer les entrées + tubes de sortie avec dH2O (500 μL, 300 mbar).
- Remplissez tous les tubes de réservoir de 2 mL (voir tableau des matériaux) avec 300 μL de tampon F. Réglez la pression à 300 mbar et laissez cinq à huit gouttes se perdre. Répétez l’opération pour chaque canal et réglez la pression sur 0.
- Connectez la prise et rincez abondamment la chambre.
- Réglez la pression pour le tube de réservoir 4 (sortie) à 50 mbar. Une fois qu’une gouttelette sort de l’extrémité du tube, connectez le tube à la sortie de la chambre PDMS. Le liquide remplit la chambre et sort de toutes les entrées.
- [FACULTATIF] Si la chambre a été directement passivée (section 4), réglez la pression à 100 mbar pour rincer la chambre avec 50-100 μL de tampon F (3-5 min). Enlevez l’excès de liquide aux entrées avec un mouchoir de nettoyage.
- Réglez la pression à 20 mbar.
- Connectez les entrées.
- Réglez la pression pour le tube du réservoir de 1 à 50 mbar. Pour éviter d’introduire des bulles d’air, assurez-vous qu’une gouttelette sort du tube et de l’entrée PDMS.
- Connectez le tuyau à l’entrée 1 (les deux gouttelettes fusionnant lors de la connexion). Réglez la pression à 30 mbar.
- Répétez les étapes 5.4.1 à 5.4.2 pour connecter les entrées 2 et 3.
- Réglez la pression de toutes les entrées à 20 mbar et la pression de sortie à 0 mbar. Assurez-vous que les débits dans les entrées sont à peu près égaux (voir la section Dépannage).

Figure 1 : Injection de solutions à travers une chambre microfluidique. (A) Configuration microfluidique standard pour les expériences sur les filaments d’actine simples. Les solutions protéiques, placées dans les réservoirs 1 à 3, sont poussées dans la chambre en ajustant la pression dans la phase gazeuse. Les débits générés sont mesurés par des débitmètres. À l’intérieur des chambres microfluidiques, les solutions ne se mélangent pas et occupent de l’espace en fonction des pressions relatives appliquées (ici, pression égale sur toutes les entrées). Dimensions typiques: les tubes de réservoir contiennent jusqu’à 2 mL de solution. Les tubes en PEEK (0,25 mm de diamètre intérieur) relient les réservoirs aux débitmètres (après 10 cm de tubes) puis à la chambre PDMS (après 70 cm supplémentaires). Des raccords de tubes en silicium et en acier inoxydable sont utilisés pour connecter le tube EN PEEK aux entrées PDMS. Le canal microfluidique principal mesure de 20 à 60 μm de haut, environ 1 mm de large et 1 cm de long. (B,C) Profils d’écoulement à l’intérieur de la chambre microfluidique. (B) Le fluide génère un profil parabolique sur toute la hauteur de la chambre : v(z) = 6z(h-z)R/h3w, où h et w sont la hauteur et la largeur de la chambre, et R est le débit total. En bas: Filament d’actine unique polymérisé à partir de graines de spectrine-actine ancrées en surface. (C) Lorsque la largeur de la chambre est considérablement supérieure à sa hauteur, le débit est presque uniforme à travers la chambre, sauf sur les surfaces PDMS, où il passe à zéro. Veuillez cliquer ici pour voir une version agrandie de cette figure.
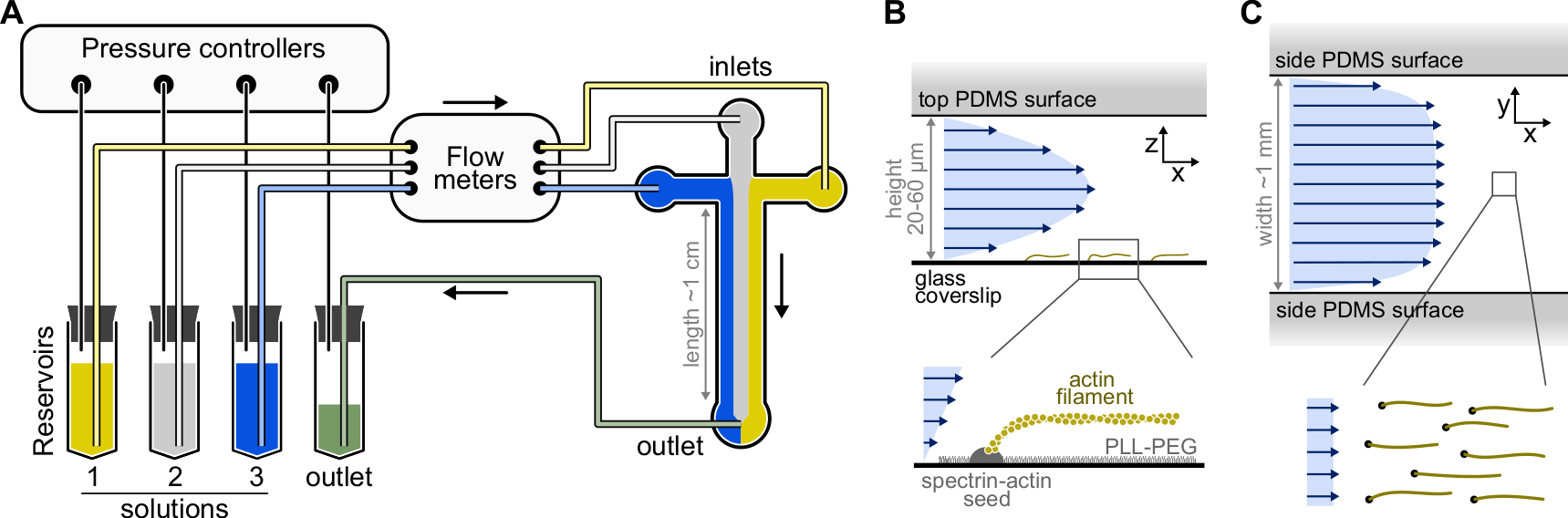{kind=link}
6. Configuration de la configuration avec des débits standard
REMARQUE: Le système de pression contrôlé par ordinateur permet un réglage facile et précis des pressions de toutes les entrées / sorties connectées à la chambre PDMS, donc le contrôle des débits entrants et sortants. Les configurations prédéfinies peuvent être enregistrées et activées/désactivées en un seul clic de souris. Vous trouverez ci-dessous les configurations recommandées (sauf indication contraire, la pression de sortie est réglée sur 0 mbar). Voir le tableau 3 pour connaître les débits attendus pour ces configurations prédéfinies. Les pressions indiquées ici doivent être ajustées en fonction de la géométrie de la chambre et de la configuration du système.
-
Modification : utilisez ce paramètre prédéfini lorsque vous changez un ou plusieurs réservoirs. Il crée un léger écoulement vers l’arrière dans le tube d’intérêt pour éviter l’introduction de bulles.
- Réglez toutes les pressions d’entrée sur 12 mbar et la pression de sortie sur 5 mbar (Figure 2B).
-
High Flow 'All': Utilisez ce préréglage pour injecter rapidement trois solutions en parallèle. Ils atteindront la chambre en 4 min.
- Réglez toutes les pressions d’entrée à 150 mbar.
-
High Flow 'x' : Utilisez ce préréglage pour injecter rapidement une solution. Il atteindra la chambre en 3 min (Figure 3A-C).
- Réglez la pression d’entrée 'x' à 150 mbar (~15 μL/min). La pression dans les autres entrées est ajustée à environ 100 mbar, de sorte que le débit résultant dans ces entrées est d’environ 500 nL / min.
-
Mid Flow 'All' : Utilisez ce préréglage pour mettre le système en pause.
- Réglez toutes les entrées sur 20 mbar (Figure 2A).
-
Mid Flow 'x' : utilisez ce préréglage pour permettre à la solution 'x' de remplir la majeure partie de la largeur du canal principal (voir Figure 2C,D), tout en limitant les autres solutions d’entrée aux côtés du canal. Les filaments d’actine dans la chambre seront ainsi exposés à la condition biochimique imposée par la solution 'x' uniquement.
- Réglez la pression d’entrée 'x' à 12 mbar. Réglez la pression dans les autres entrées et ajustez-la à ~9 mbar, de sorte que leurs débits respectifs soient ~150 nL/min.

Figure 2 : La pression appliquée à chaque réservoir contrôle la répartition/distribution spatiale des solutions à l’intérieur de la chambre microfluidique. (A) Avec une pression égale appliquée aux réservoirs, chaque solution occupe un tiers de la chambre. (B) Lors du changement d’un tube de réservoir (ici réservoir 3), la pression effective tombe à zéro, créant un écoulement vers l’arrière. (C,D) L’augmentation de la pression relative sur l’un des réservoirs permet l’exposition de la surface du verre à une seule solution. Le champ de vision au milieu de la chambre peut être exposé séquentiellement aux solutions 1 et 2 en alternant entre la configuration Mid Flow 1 (C) et Mid Flow 2 (D). Veuillez cliquer ici pour voir une version agrandie de cette figure.
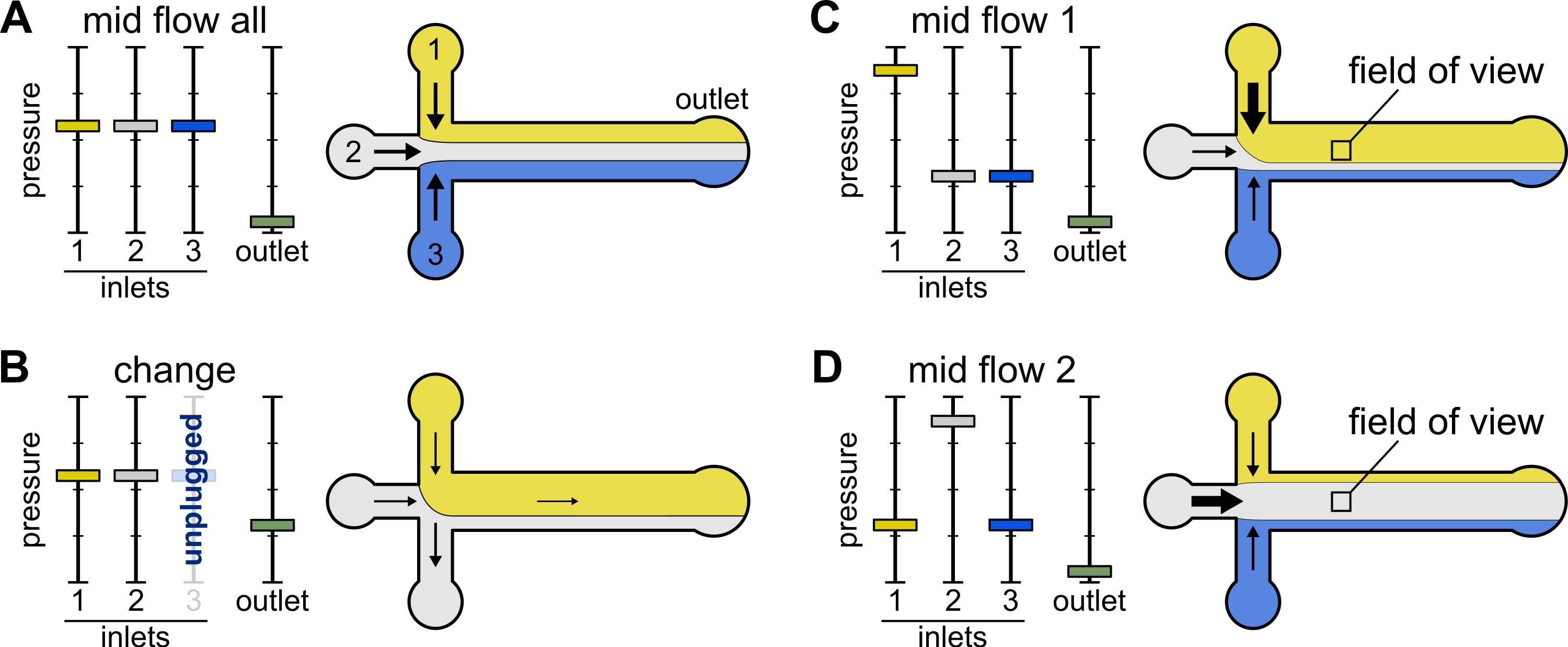{kind=link}
7. Changer la solution 'x'
REMARQUE: Comme le montre la figure 3A-C, il est important de garder à l’esprit que les solutions prennent quelques minutes pour s’écouler d’un tube de réservoir vers le canal principal de la chambre. Ce temps « mort » minimal est imposé par le volume de liquide contenu dans le tube et le profil d’écoulement à l’intérieur du tube (figure 3A-C).
- Préparer 200-300 μL de solution dans un nouveau tube de réservoir. Réglez la pression sur Modifier le réglage (voir rubrique 6).
- Dévissez le tube de réservoir de l’entrée 'x'. La solution dans le tube s’écoulera lentement vers l’arrière, de la chambre à la pointe du tube libre. Le débit mesuré devient négatif (Figure 2B).
- Une fois qu’une petite gouttelette s’est formée à l’extrémité du tube, vissez le nouveau tube avec la solution fraîche. Une fois que le tube est correctement serré au système de pression, le débit de l’entrée revient à positif.
- Réglez le réglage de pression sur High Flow 'x'.
- Selon la configuration microfluidique et la géométrie de la chambre, attendez 3 à 5 minutes pour que la solution remplisse complètement le tube et atteigne la chambre.
- [FACULTATIF] Suivez ce processus en mesurant l’augmentation de la fluorescence au fil du temps (Figure 3C).

Figure 3 : Arrivée retardée des solutions des réservoirs à la chambre PDMS et changement rapide des conditions biochimiques. (A-C) Arrivée retardée des solutions des réservoirs à la chambre PDMS. (A) Selon la géométrie de la chambre, la longueur du tube et la pression appliquée à l’entrée ou aux entrées, le remplacement d’une solution par une autre n’est pas instantané. Après avoir remplacé le tube du réservoir par un tube contenant une solution fluorescente (0 min), la solution remplit progressivement le tube (0,4 min) et la chambre PDMS (1-2 min). Un timing indicatif est donné pour une pression appliquée de 150 mbar, un tube PEEK de 80 cm et une chambre PDMS de 1600 μm de large et de 20 μm de haut. (B) Le profil d’écoulement parabolique à l’intérieur du tube en PEEK génère un gradient efficace de fluorescence le long du profil radial du tube et à l’intérieur de la chambre (voir également la figure 1B). (C) L’arrivée retardée des solutions peut être quantifiée en mesurant le signal d’épifluorescence de fond dans la chambre en fonction du temps. Conditions expérimentales: 0,5 μM 10% de G-actine marquée Alexa-568 est injectée avec 150 mbar à travers un débitmètre et un tube PEEK de 80 cm. (D,E) Changement rapide des conditions biochimiques. (D) Schéma des solutions entrantes dans deux conditions de débit moyen. (E) Augmentation de la fluorescence de fond comme lecture de la concentration d’actine. Le temps t = 0 est défini lorsque le début de la fluorescence augmente. Solution 1: 0,5 μM 10% de G-actine marquée Alexa-488, solution 2: tampon F. (C,E) Chambre PDMS : 20 μm de haut et 1600 μm de large. L’intensité de l’épifluorescence, ~2 μm au-dessus de la surface, a été quantifiée en faisant la moyenne du signal sur tout le champ de vision, normalisée à 0 en l’absence de fluorophore et à 1 à l’intensité maximale. Veuillez cliquer ici pour voir une version agrandie de cette figure.
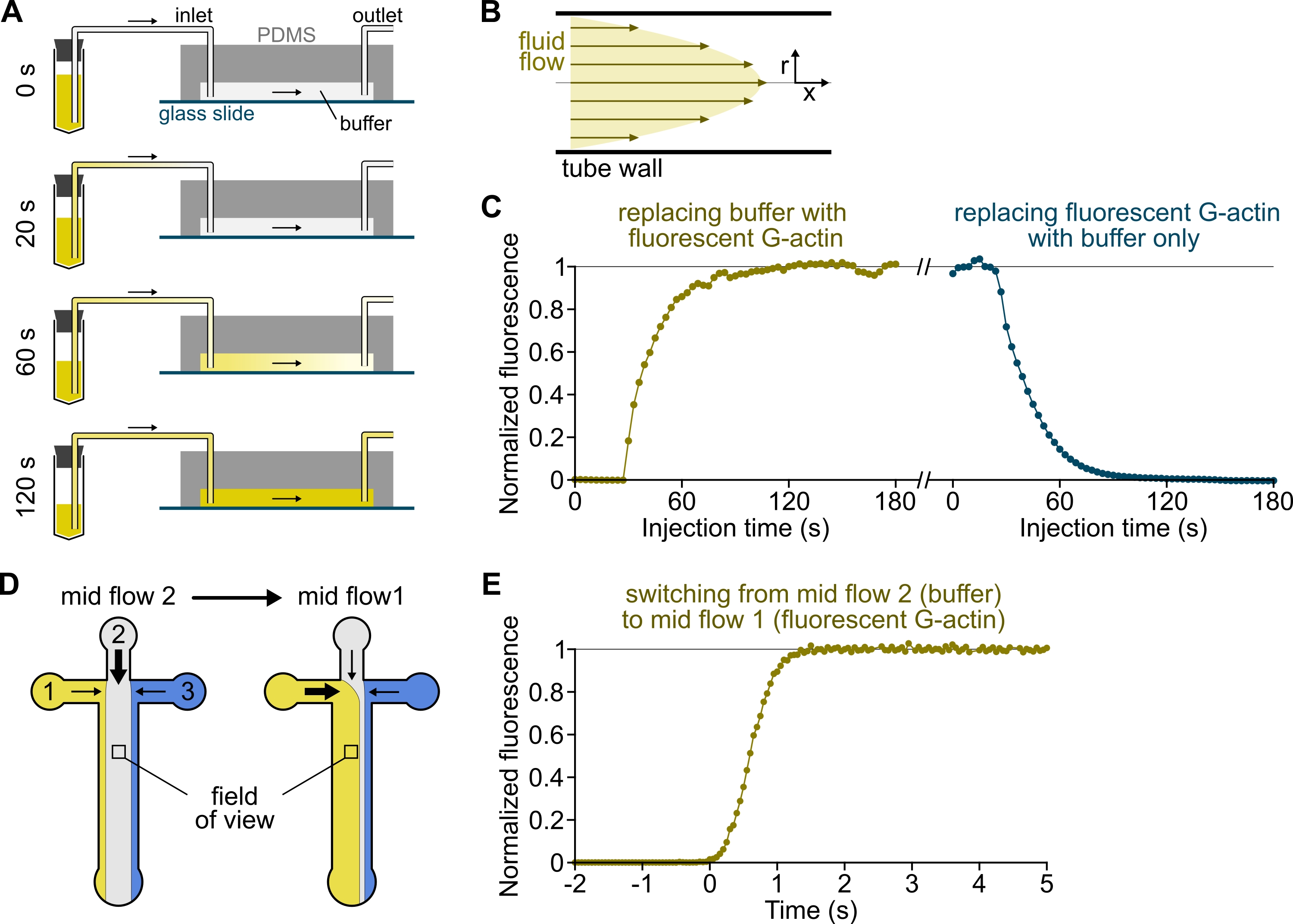{kind=link}
8. Expérience de base à filament unique : dépolymérisation de l’extrémité barbelée de l’adénosine diphosphate (ADP)-actine
REMARQUE : Cette section suppose une chambre non fonctionnalisée (section 5 seulement). Si la chambre a été directement fonctionnalisée (section 4), commencez à l’étape 8.4.
- Fonctionnalisation de surface avec des graines de filament d’actine:
- Changer la solution 3 à 200 μL de graines de spectrine-actine11 de 50 pM dans un tampon F (voir rubrique 7).
REMARQUE: Alternativement, si les graines de spectrine-actine ne sont pas disponibles, on peut utiliser des segments de filament courts fonctionnalisés par la biotine qui seront immobilisés sur un couvercle recouvert de streptavidine (voir 9,10 pour plus de détails). - Injecter pendant 2 min avec High Flow 3.
REMARQUE: Ajustez la concentration et le temps en fonction de la densité finale des graines.
- Changer la solution 3 à 200 μL de graines de spectrine-actine11 de 50 pM dans un tampon F (voir rubrique 7).
- Passivation de surface:
- Changer le tube 3 avec 300 μL de 5% de BSA dans le tampon F.
- Injecter pendant 5 min à High Flow 3, puis 5 min à Mid Flow 3. Au cours de cette deuxième étape, réduisez la pression dans les canaux 1 et 2 à 7-8 mbar pour obtenir un contre-courant ~ -100 nL / min, de sorte que toutes les surfaces de la chambre soient passivées BSA.
REMARQUE: Étant donné que la solution de BSA est plus visqueuse, les pressions doivent être ajustées en conséquence.
- Changer le tube 3 en tampon F et rincer le canal (5 min, High Flow 3).
- Préparer les solutions suivantes de 200 à 300 μL, toutes les protéines étant diluées dans un tampon F :
Entrée 1, solution de polymérisation : 1 μM 10 % de G-actine marquée Alexa-488, 1 μM de profiline (Tableau 1).
Entrée 2, solution vieillissante : 0,15 μM 10 % Alexa-488 étiqueté G-actine.
Entrée 3, solution de dépolymérisation : tampon F uniquement.
REMARQUE: La profiline est utilisée ici pour prévenir la nucléation spontanée et pour maintenir une concentration constante de G-actine. - Changer les tubes 1 à 3 (section 7). Injectez à l’aide du préréglage High Flow All pendant 3-4 min. Les trois solutions ont maintenant rempli le tube en PEEK et atteint la chambre (Figure 3A). La surface vitrée peut être exposée à n’importe quelle solution d’entrée sans temps mort (<1 s, Figure 3D,E).
- Allumez le microscope. Réglez les réglages: laser d’excitation de 150 mW 488 nm à une puissance de 10% à 20%, temps d’exposition de la caméra de 100 à 200 ms, profondeur de pénétration TIRF de 200 à 300 nm, objectif 60x. Ces paramètres sont utilisés tout au long du manuscrit.
- Polymérisation des filaments (Figure 4A) :
- Réglez le réglage de pression sur Mid Flow 1 pendant environ 10 min.
- [FACULTATIF] Polymérisation record (1 cadre/20 s, TIRF). Les filaments doivent polymériser à environ 10 sous-unités/seconde (sub/s)1,12.
- Vieillissement du filament: Réglez le réglage de pression sur Mid Flow 2 pendant 15 min. À la concentration critique, 0,15 μM de G-actine, la longueur du filament restera constante et les filaments se transformeront en ADP-F-actine 4 à 99 %>.
- Dépolymérisation (Figure 4A) :
- Démarrer l’acquisition à 1 image/5 s, en mode épifluorescence. Comme il y a un fond de fluorescence très faible dans les canaux 2 et 3, il n’est pas nécessaire d’utiliser le TIRF.
- Après une à deux images, passez à Mid Flow 3. Les filaments doivent dépolymériser à environ 10 sub/s (référence12).
- Pour réinitialiser l’expérience, cassez tous les filaments marqués par fluorescence en les exposant continuellement au laser à une puissance maximale pendant environ 2 minutes. Pour tester différentes conditions, changez les solutions 1, 2 ou 3 et injectez-les (High Flow, 3-4 min). Répétez les étapes 8.7 à 8.9.
9. Autres expériences sur un seul filament
- Tester les interactions des ABP avec la F-actine
REMARQUE: La microfluidique a été utilisée avec succès pour quantifier l’activité de plusieurs ABP à liaison latérale, tels que la cofiline, la tropomyosine et Arp2/3. Suivant le protocole de l’article 8 :- Remplacez le canal 3 par le PBA fluorescent qui vous intéresse dans le tampon F. Injecter (High Flow 3, 3 min).
- Polymérisation du filament: Réglez le réglage de pression sur Mid Flow 1 pendant 10 min.
- Liaison ABP : Commencez l’acquisition avec TIRF. Ajustez la fréquence d’images en fonction de la concentration ABP. Après 1-2 images, passez à Mid Flow 3.
REMARQUE: Selon l’ABP, il peut également être possible de passer rapidement (par exemple, pendant moins de 5 s) à Mid Flow 2 pour réduire davantage la fluorescence d’arrière-plan lors de la prise d’une image. - Déliaison ABP : Tout en poursuivant l’acquisition, passez à Mid Flow 2.
- Polymérisation avec formin à l’extrémité barbelée libre
REMARQUE: Il a été démontré que les formines affectent la polymérisation de l’extrémité barbelée du filament. La microfluidique est particulièrement adaptée pour mesurer les taux de liaison et de déliaison de la formine et leur impact sur l’allongement des filaments.- Préparez les solutions suivantes :
Canal 1 : 10 nM formin dans le tampon F (Tableau 1).
Canal 2: 1 μM 10% Alexa-488 étiqueté G-actine, 4 μM profiline.
Canal 3: F-buffer. - Changez les tubes 1, 2 et 3 (section 7). Injectez à l’aide du préréglage High Flow All pendant 3-4 min.
- Initier la polymérisation du filament : Réglez le réglage de pression sur Mid Flow 2 pendant 2 min.
- Liaison formin à l’extrémité barbelée du filament : Réglez les réglages de pression sur Mid Flow 1 pendant 30 s.
- Polymérisation médiée par Formin : Réglez le réglage de pression sur Mid Flow 2. Avec la formine mDia1 à leur extrémité barbelée, les filaments devraient polymériser à environ 50 sub/s 13,14,15.
- Préparez les solutions suivantes :
- Polymérisation/dépolymérisation à partir de formine ancrée en surface
REMARQUE: Il a été démontré que les taux de polymérisation et de dépolymérisation des extrémités barbelées décorées de formin dépendent de la tension appliquée au filament. En microfluidique, le frottement de l’écoulement du fluide le long du côté du filament génère une tension proportionnelle à la longueur du filament et au débit14,16.- Utilisez la méthode décrite ci-dessus pour remplacer les étapes 8.1, 8.2 et 8.3 pour la passivation de surface par :
- Changer le tube 3 à 1 μg/mL anticorps anti-His dans le tampon F. Injecter pendant 2 min avec High Flow 3.
- Changez le tube 3 avec 5% de BSA dans le tampon F. Injecter pendant 5 min à High Flow 3, puis 5 min à Mid Flow 3. Au cours de cette deuxième étape, réduisez la pression dans les canaux 1 et 2 à 7-8 mbar pour obtenir un contre-courant ~ -100 nL / min afin que toutes les surfaces de la chambre soient passivées BSA.
- Changer le tube 3 à 100 nM His-tagged formin dans F-buffer. Injecter pendant 5 min avec High Flow 3. Changez le tube 3 avec tampon F. Injecter pendant 5 min avec High Flow 3 pour s’assurer qu’il ne reste pas de formins dans le tube.
- Préparer et injecter les solutions suivantes (200-300 μL chacune, dans un tampon F) :
Canal 1: 1 μM 10% Alexa-488 étiqueté G-actine.
Canal 2 : 1 μM de G-actine non marquée, 4 μM de profiline.
Canal 3 : tampon F uniquement. - Nucléation du filament : Exposez les formines ancrées en surface à la G-actine (réglage Mid Flow 1).
- Polymérisation du filament : Exposez la chambre à la profiline-actine à l’aide de Mid Flow 2.
- Début de l’acquisition : 1 image/2 s, épifluorescence. Avec formin mDia1, les filaments doivent polymériser à 50-80 sub/s, en fonction de la longueur du filament et du débit14.
- Dépolymérisation du filament : Début de l’acquisition (1 trame/4 s, épifluorescence). Après 1-2 images, exposez les filaments à F-buffer, Mid Flow 3. Avec formin mDia1, les filaments doivent se dépolymeriser à 5-15 sub/s, en fonction de la longueur du filament et du débit14.
- Utilisez la méthode décrite ci-dessus pour remplacer les étapes 8.1, 8.2 et 8.3 pour la passivation de surface par :
- Filaments d’actine avec segments non marqués
REMARQUE: Le marquage fluorescent à l’actine crée plusieurs artefacts, tels que des pauses pendant la dépolymérisation17 et une liaison altérée à la tropomyosine18. Une solution de contournement pour ces artefacts consiste à utiliser la microfluidique pour assembler des filaments affichant des segments non marqués.- Préparer et injecter les solutions suivantes (200-300 μL dans le tampon F) :
Canal 1 : 1 μM de G-actine non marquée, 1 μM de profiline.
Canal 2: 0,3 μM 10% Alexa-488 étiqueté G-actine. - Exposez séquentiellement la surface au canal 2 (5 min), au canal 1 (10 min) et au canal 2 (15 min) pour générer des segments non marqués ADP-actine avec des segments marqués par fluorescence à chaque extrémité.
- Préparer et injecter les solutions suivantes (200-300 μL dans le tampon F) :
- Filaments barbelés à ancrage d’extrémité avec gelsolin
REMARQUE: Avec les graines de spectrine-actine, les filaments polymérisent à leur extrémité barbelée libre tandis que l’extrémité pointue est stabilisée par la graine de spectrine-actine. Une alternative consiste à ancrer les filaments avec un capper d’extrémité barbelé tel que la gelsoline.- Préparer une solution de F-actine de 4 μM 10% Alexa-488 étiqueté g-actine dans 20 μL de tampon F. Laisser l’actine se nucléer spontanément et polymériser à TA pendant au moins 30 min sur le banc. Enveloppez le tube dans du papier d’aluminium pour le protéger de la lumière.
- Pendant ce temps, préparez la chambre microfluidique et passivatez la surface avec un mélange de 5% de BSA et de 1% de biotine-BSA (voir étape 8.2).
- Rincer le canal 3 avec tampon F (2 min à high flow 3). Injecter 10 μg/mL de néutravidine dans un tampon F (4 min à haut débit 3).
- Changez les tubes pour :
Canal 1 : 10 nM de biotine-gelsoline (tableau 1).
Canal 2: F-buffer.
Canal 3 : F-actine prépolymérisée de 0,4 μM. - Injectez toutes les solutions ensemble à l’aide du réglage High Flow All pendant 3 min.
- Exposer toute la chambre à la gelsoline (Mid Flow 1, 30 s).
- Fixez les filaments à la surface (Low Flow 3: Canal 3 à 3 mbar, Canaux 1 et 2 à ~ 2 mbar, pendant environ 2 min).
- [FACULTATIF] Si la densité du filament est trop faible, répétez les étapes 9.5.6 et 9.5.7.
- Dépolymérisation à extrémité pointue : Début de l’acquisition (1 trame/30 s, épifluorescence). Après 1-2 images, exposez les filaments à la mémoire tampon uniquement, Mid Flow 2. Les filaments doivent dépolymériser à environ 0,2 sub/s.
10. Formation et désassemblage de faisceaux de filaments induits par la fascine par ADF/cofiline
REMARQUE: Pour former des faisceaux de filaments d’actine, assurez-vous d’avoir une densité de graines de filament suffisamment élevée à la surface de la chambre. Lorsqu’ils sont exposés à la protéine de la fascine, les filaments voisins qui fluctuent latéralement seront réticulés dynamiquement par les protéines de la fascine. Comme la fascine se délie rapidement du côté du filament19, la fascine doit être constamment présente dans la solution principale afin de maintenir le regroupement du filament.
- Suivez les étapes 8.1 à 8.3.
- Préparez les solutions suivantes (200-300 μL dans le tampon F) :
Canal 1, solution de polymérisation : 1 μM 10% de G-actine marquée Alexa-488, profiline 1 μM.
Canal 2, Solution groupée : 200 nM de fascine (Tableau 1), 0,15 μM 10 % Alexa-488 étiqueté G-actine.
Canal 3, Solution de démontage : 200 nM ADF/cofiline (Tableau 1), 100 nM de fascine, 0,15 μM 10 % Alexa-488 étiqueté G-actine. - Changer les tubes 1 à 3 (section 7). Injectez à l’aide du préréglage High Flow All , pendant 3-4 min.
- Polymérisation du filament: Réglez le réglage de pression sur Mid Flow 1 pendant environ 10 min. La polymérisation peut être imagée avec le TIRF.
- Regroupement de filaments (Figure 4C) : Début de l’acquisition d’images (1 image/5 s, épifluorescence). Après 1-2 images, réglez le réglage de pression sur Mid Flow 2 et observez le regroupement du filament.
- Fragmentation du faisceau : Démarrer l’acquisition d’images (1 image/5 s, épifluorescence). Après 1-2 images, réglez le réglage de pression sur Mid Flow 3 et observez le désassemblage induit par la cofiline des filaments simples et des faisceaux.
11. Procédure de nettoyage des dispositifs microfluidiques
REMARQUE: Pour éviter toute contamination d’une expérience à l’autre, il est essentiel de nettoyer en profondeur et de sécher complètement tous les tubes et débitmètres après chaque expérience.
- Débranchez tous les tubes de la chambre PDMS et jetez la chambre.
- Pour nettoyer les tubes et les débitmètres en PEEK, collez les extrémités du tube dans un tube en plastique vide de 15 mL et injectez les solutions suivantes à la pression maximale jusqu’à ce que le réservoir soit presque vide:
400 μL de tampon F.
400 μL de 0,5 M NaOH.
400 μL d’eau pure.
200 μL d’isopropanol. - Remplacer par un réservoir vide et souffler de l’air jusqu’à ce que les tubes soient complètement secs (~ 2-4 min, pression maximale).
12. Analyse d’images
REMARQUE: Alors que ce manuscrit se concentre sur la méthode d’assemblage, de manipulation et de visualisation de filaments d’actine simples en microfluidique, une brève méthode d’analyse des films acquis est fournie ici. L’analyse est effectuée sur des images 16 bits, à l’aide d’ImageJ, conformément à la section 8.
- Le traitement de l’image est minime :
- Importez la pile d’images de polymérisation ou de dépolymérisation.
- [FACULTATIF] Homogénéiser l’intensité de l’image avec la fonction Soustraire l’arrière-plan (paramètres par défaut (c’est-à-dire 'Rayon de la boule roulante' = 50 pixels)). Ceci est particulièrement utile si la fluorescence de fond change au cours d’un film ou si l’éclairage de fluorescence n’est pas homogène sur le champ de vision.
- Ajustez la luminosité et le contraste (arrière-plan proche de zéro, filaments proches du maximum).
- Créer un kymographe de filament:
- Sélectionnez un filament qui ne se met pas en pause, ne se casse pas et ne se détache pas. Ne sélectionnez pas en fonction du comportement autrement. Tracez une ligne de 1 à 2 pixels au-dessus (outil Ligne droite ). Enregistrez le numéro de filament (Ajouter dans ROI Manager).
- Appliquez la fonction Reslice (Nombre de tranches : 5 pixels). Calculer l’intensité maximale (fonction Zprojection).
- Mesurer le taux de polymérisation/dépolymérisation :
- Sur le kymographe, tracez une ligne le long de l’extrémité barbelée du filament (outil Ligne droite , Figure 4A). Mesurez la largeur et la hauteur de la ligne (fonction Mesurer).
- Répétez les étapes 12.2-12.3 sur plusieurs filaments. Calculer les taux de polymérisation/dépolymérisation (Figure 4A) :
 , où v est le taux (en sous/s), w la largeur de ligne (pixels), pix la taille de pixel (nm), h la hauteur de ligne (images) et dt le temps entre les images (en seconde). Ici, 2,7 nm correspond à la contribution effective d’une sous-unité d’actine à la longueur du filament.
, où v est le taux (en sous/s), w la largeur de ligne (pixels), pix la taille de pixel (nm), h la hauteur de ligne (images) et dt le temps entre les images (en seconde). Ici, 2,7 nm correspond à la contribution effective d’une sous-unité d’actine à la longueur du filament.
Résultats
Pour toutes les expériences décrites ci-dessus, les filaments d’actine marqués par fluorescence doivent être clairement visibles, avec un bon contraste, indiquant une faible fluorescence de fond de la surface (Figure 4, voir le fichier supplémentaire 1 pour le dépannage des problèmes courants). Les filaments d’actine ne doivent pas non plus coller à la surface : lorsque le débit dominant est faible, les fluctuations latérales des filaments d’actine doivent ê...
Discussion
Par rapport aux méthodes standard à filament unique où les filaments d’actine sont ancrés à la surface par plusieurs points le long de leur longueur ou maintenus à proximité de celle-ci par un agent d’encombrement tel que la méthylcellulose, la microfluidique offre un certain nombre d’avantages. Comme les interactions avec la surface sont minimes, les pauses artificielles que ces interactions peuvent induire pendant l’allongement et la dépolymérisation sont évitées. Les filaments sont alignés par l?...
Déclarations de divulgation
Les auteurs ne déclarent aucun conflit d’intérêts.
Remerciements
Nous sommes reconnaissants à B. Ladoux et R.-M. Laboratoire Mège pour l’utilisation de leur équipement de nettoyage UV, et J. Heuvingh et 0. du Roure pour la formation initiale que nous avons reçue sur la préparation de moules sur plaquettes de silicium et fournissant des conseils sur la microfluidique. Nous reconnaissons le financement de la subvention StG-679116 du Conseil européen de la recherche (à A.J.) et des subventions de l’Agence nationale de la recherche Muscactin et Conformin (à G.R.-L.).
matériels
| Name | Company | Catalog Number | Comments |
| β-Casein | Merck | C6905 | Used at 8 mg/mL |
| Biopsy punch (with plunger) | Ted Pella | 15115-2 | ID 0.75 mm, OD 1.07 mm |
| Biotin-BSA | Merck | A8549 | Used at 1 mg/mL |
| BSA | Merck | A8022 | Used at 50 mg/mL |
| Coverslip Mini-Rack Teflon holder | Invitrogen | C14784 | for 8 coverslips |
| Coverslips 22x40mm Thickness #1.5 | Menzel Gläser | 631-1370 | |
| DABCO | Merck | D27802 | component in f-buffer |
| DTT | Euromedex | EU0006-D | component in f-buffer |
| Ester NHS Alexa Fluor 488 | Invitrogen | A20000 | Fluorophore for actin labeling on Lys328. |
| EZ-Link Sulfo-NHS-Biotin | Thermo Scientific | 21338 | To biotinylate actin on Lys328 |
| Hellmanex III | Hellma | 9-307-011-4-507 | Glass cleaning detergent |
| ImageJ | NIH | N/A | open source software |
| Laboport | KNF | 811kn.18 | vacuum pump (ultimate vacuum: 240 mbar) |
| Magic invisible tape | Scotch | 7100024666 | standard transparent office tape |
| Micrewtube | Simport | T341-6T | 2 mL microfluidic reservoir tubes |
| Microfluidic device Part 1: Flow Unit S | Fluigent | FLU-S-D-PCKB | Flowmeter |
| Microfluidic device Part 2: Fluiwell-4C-2 mL | Fluigent | 14002001PCK | Reservoir holder |
| Microfluidic device Part 3: MFCS-EZ | Fluigent | EZ-11000001 EZ-00345001 | Pressure controller |
| Model 42 - UVO-Cleaner | Jelight Inc. | 42-220 | Ultraviolet cleaner |
| N6-(6-Aminohexyl)-ATP-ATTO-488 | Jena Bioscience | NU-805-488 | ATP-ATTO used to label actin |
| neutravidin | Thermo Scientific | 31000 | |
| PLL-PEG | SuSoS | PLL(20)-g[3.5]- PEG(2) | Use at 1 mg/mL in PBS. |
| Polydimethylsiloxane (PDMS) Sylgard 184 Silicon Elastomer | Dow Corning | 1673921 | Contains PDMS base and curing agent |
| Polyetheretherketone (PEEK) tubing | Merck | Z226661 | “Blue” : I.D. = 0.25 mm |
| Safety blow gun | Coilhose Pneumatics | 700-S | filtered air |
| Silicon tubing | VWR | 228-0701P | connect PEEK to coupler |
| Stainless steel catheter coupler | Prime Bioscience | SC22/15 | Inserted into PDMS inlets and outlet to connect to PEEK tubing |
| Thermoplastic film | Sigma Aldrich | PM996 | Standard "parafilm" |
| Ultrapure ethanol | VWR | 64-17-5 | |
| Ultrasonic cleaning bath | VWR | USC200TH | To accomodate 1 L beakers |
| Vacuum dessicator | SP Bel-Art | F42022-0000 | to degas the PDMS or solutions |
Références
- Wioland, H., Jégou, A., Romet-Lemonne, G. Celebrating 20 years of live single-actin-filament studies with five golden rules. Proceedings of the National Academy of Sciences of the United States of America. 119 (3), 2109506119 (2022).
- Kuhn, J. R., Pollard, T. D. Real-time measurements of actin filament polymerization by total internal reflection fluorescence microscopy. Biophysical Journal. 88 (2), 1387-1402 (2005).
- Brewer, L. R., Bianco, P. R. Laminar flow cells for single-molecule studies of DNA-protein interactions. Nature Methods. 5 (6), 517-525 (2008).
- Jégou, A., et al. Individual actin filaments in a microfluidic flow reveal the mechanism of ATP hydrolysis and give insight into the properties of profilin. PLoS Biology. 9 (9), 1001161 (2011).
- Gicquel, Y., et al. Microfluidic chips for in situ crystal x-ray diffraction and in situ dynamic light scattering for serial crystallography. Journal of Visualized Experiments: JoVE. (134), e57133 (2018).
- Chandradoss, S. D., et al. Surface passivation for single-molecule protein studies. Journal of Visualized Experiments: JoVE. (86), e50549 (2014).
- Schaedel, L., et al. Microtubules self-repair in response to mechanical stress. Nature Materials. 14 (11), 1156-1163 (2015).
- Zimmermann, D., Morganthaler, A. N., Kovar, D. R., Suarez, C. In vitro biochemical characterization of cytokinesis actin-binding proteins. Methods in Molecular Biology. 1369, 151-179 (2016).
- Funk, J., et al. Profilin and formin constitute a pacemaker system for robust actin filament growth. eLife. 8, 50963 (2019).
- Pandit, N. G., et al. Force and phosphate release from Arp2/3 complex promote dissociation of actin filament branches. Proceedings of the National Academy of Sciences of the United States of America. 117 (24), 13519-13528 (2020).
- Wioland, H., et al. ADF/Cofilin accelerates actin dynamics by severing filaments and promoting their depolymerization at both ends. Current Biology: CB. 27 (13), 1956-1967 (2017).
- Pollard, T. D., Mooseker, M. S. Direct measurement of actin polymerization rate constants by electron microscopy of actin filaments nucleated by isolated microvillus cores. The Journal of Cell Biology. 88 (3), 654-659 (1981).
- Kovar, D. R., Harris, E. S., Mahaffy, R., Higgs, H. N., Pollard, T. D. Control of the assembly of ATP- and ADP-actin by formins and profilin. Cell. 124 (2), 423-435 (2006).
- Jégou, A., Carlier, M. -. F., Romet-Lemonne, G. Formin mDia1 senses and generates mechanical forces on actin filaments. Nature Communications. 4, 1883 (2013).
- Breitsprecher, D., et al. Rocket launcher mechanism of collaborative actin assembly defined by single-molecule imaging. Science. 336 (6085), 1164-1168 (2012).
- Courtemanche, N., Lee, J. Y., Pollard, T. D., Greene, E. C. Tension modulates actin filament polymerization mediated by formin and profilin. Proceedings of the National Academy of Sciences of the United States of America. 110 (24), 9752-9757 (2013).
- Niedermayer, T., et al. Intermittent depolymerization of actin filaments is caused by photo-induced dimerization of actin protomers. Proceedings of the National Academy of Sciences. 109 (27), 10769-10774 (2012).
- Gateva, G., et al. Tropomyosin isoforms specify functionally distinct actin filament populations in vitro. Current Biology: CB. 27 (5), 705-713 (2017).
- Aratyn, Y. S., Schaus, T. E., Taylor, E. W., Borisy, G. G. Intrinsic dynamic behavior of fascin in filopodia. Molecular Biology of the Cell. 18 (10), 3928-3940 (2007).
- Pollard, T. D. Rate constants for the reactions of ATP- and ADP-actin with the ends of actin filaments. The Journal of Cell Biology. 103, 2747-2754 (1986).
- Wioland, H., Jegou, A., Romet-Lemonne, G. Torsional stress generated by ADF/cofilin on cross-linked actin filaments boosts their severing. Proceedings of the National Academy of Sciences of the United States of America. 116 (7), 2595-2602 (2019).
- Colombo, J., et al. A functional family of fluorescent nucleotide analogues to investigate actin dynamics and energetics. Nature Communications. 12 (1), 548 (2021).
- Spudich, J. A., Watt, S. The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. The Journal of Biological Chemistry. 246 (15), 4866-4871 (1971).
- Romet-Lemonne, G., Guichard, B., Jégou, A. Using microfluidics single filament assay to study formin control of actin assembly. Methods in Molecular Biology. 1805, 75-92 (2018).
- Gieselmann, R., Kwiatkowski, D. J., Janmey, P. A., Witke, W. Distinct biochemical characteristics of the two human profilin isoforms. European Journal of Biochemistry. 229 (3), 621-628 (1995).
- Lin, D. C., Lin, S. Actin polymerization induced by a motility-related high-affinity cytochalasin binding complex from human erythrocyte membrane. Proceedings of the National Academy of Sciences of the United States of America. 76 (5), 2345-2349 (1979).
- Casella, J. F., Maack, D. J., Lin, S. Purification and initial characterization of a protein from skeletal muscle that caps the barbed ends of actin filaments. The Journal of Biological Chemistry. 261 (23), 10915-10921 (1986).
- Kremneva, E., et al. Cofilin-2 controls actin filament length in muscle sarcomeres. Developmental Cell. 31 (2), 215-226 (2014).
- Le Clainche, C., Carlier, M. -. F. Actin-based motility assay. Current Protocols in Cell Biology. , 1-20 (2004).
- Vignjevic, D., et al. Formation of filopodia-like bundles in vitro from a dendritic network. The Journal of Cell Biology. 160 (6), 951-962 (2003).
- Duellberg, C., Cade, N. I., Holmes, D., Surrey, T. The size of the EB cap determines instantaneous microtubule stability. eLife. 5, 13470 (2016).
- Duellberg, C., Cade, N. I., Surrey, T. Microtubule aging probed by microfluidics-assisted tubulin washout. Molecular Biology of the Cell. 27 (22), 3563-3573 (2016).
- Suzuki, E. L., et al. Geometrical constraints greatly hinder formin mDia1 activity. Nano Letters. 20 (1), 22-32 (2020).
- Wioland, H., Suzuki, E., Cao, L., Romet-Lemonne, G., Jegou, A. The advantages of microfluidics to study actin biochemistry and biomechanics. Journal of Muscle Research and Cell Motility. 41 (1), 175-188 (2020).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.