
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Utilizzo della microscopia a microfluidica e fluorescenza per studiare la dinamica di assemblaggio di singoli filamenti e fasci di actina
In questo articolo
Riepilogo
Presentiamo protocolli per semplici saggi microfluidici di filamenti di actina, in combinazione con la microscopia a fluorescenza, che consentono di monitorare con precisione i singoli filamenti di actina in tempo reale esponendoli sequenzialmente a diverse soluzioni proteiche.
Abstract
Al fine di decifrare i complessi meccanismi molecolari che regolano l'assemblaggio e lo smontaggio dei filamenti di actina, è una grande risorsa monitorare le singole reazioni dal vivo in condizioni ben controllate. Per fare ciò, negli ultimi 20 anni sono emersi esperimenti dal vivo a singolo filamento, per lo più utilizzando la microscopia a fluorescenza a riflessione interna totale (TIRF) e hanno fornito una miniera di risultati chiave. Nel 2011, al fine di espandere ulteriormente le possibilità di questi esperimenti ed evitare artefatti problematici ricorrenti, abbiamo introdotto semplici microfluidiche in questi test. Questo studio descrive in dettaglio il nostro protocollo di base, in cui i singoli filamenti di actina sono ancorati da un'estremità alla superficie passivata del coverslip, si allineano con il flusso e possono essere successivamente esposti a diverse soluzioni proteiche. Presentiamo anche i protocolli per applicazioni specifiche e spieghiamo come possono essere applicate forze meccaniche controllate, grazie alla resistenza viscosa della soluzione scorrevole. Evidenziamo gli avvertimenti tecnici di questi esperimenti e presentiamo brevemente i possibili sviluppi basati su questa tecnica. Questi protocolli e spiegazioni, insieme alla disponibilità odierna di apparecchiature di microfluidica facili da usare, dovrebbero consentire ai non specialisti di implementare questo test nei loro laboratori.
Introduzione
L'assemblaggio e lo smontaggio di filamenti di actina e reti di filamenti di actina sono controllati da diverse reazioni biochimiche e dipendono dal contesto meccanico. Al fine di ottenere informazioni su questi complessi meccanismi, è inestimabile essere in grado di osservare le singole reazioni sui singoli filamenti (in numero sufficientemente grande). Negli ultimi decenni, l'osservazione di filamenti dinamici di actina in tempo reale, per lo più utilizzando la microscopia a fluorescenza a riflessione interna totale (TIRF), è emersa come una tecnica chiave e ha fornito un elenco impressionante di risultati che non avrebbero potuto essere ottenuti con i saggi biochimici di soluzione di massa1.
Per raggiungere questo obiettivo, è necessario mantenere filamenti di actina marcati fluorescentemente vicino alla superficie del coverslip del microscopio mentre li si espone a soluzioni di proteine leganti l'actina (ABP), che possono anche essere etichettate fluorescentemente. Ciò fornisce un mezzo per monitorare gli eventi che si verificano su singoli filamenti in condizioni biochimiche ben controllate e quindi quantificare le velocità di reazione. Tuttavia, dovrebbero essere prese in considerazione una serie di limitazioni specifiche. Mantenere artificialmente i filamenti vicini alla superficie, spesso grazie a più punti di ancoraggio o utilizzando un agente di affollamento come la metilcellulosa, può alterare il loro comportamento (ad esempio, causando pause nella loro polimerizzazione e depolimerizzazione2). Tracciare il contorno di ciascun filamento può essere difficile, in particolare se nuovi filamenti o frammenti di filamento si accumulano nel campo visivo nel tempo. Le reazioni avvengono in un volume finito in cui la concentrazione di monomeri di actina e ABP può variare nel tempo, rendendo potenzialmente difficile ricavare costanti di velocità accurate. Infine, rinnovare o modificare la soluzione di ABP è difficile da ottenere in meno di 30 s e spesso porterà a un contenuto proteico disomogeneo nel campione.
Poco più di 10 anni fa, ispirati da quanto già fatto per studiare i singoli filamenti di Acido Desossiribonucleico (DNA)3, abbiamo introdotto una nuova tecnica basata sulla microfluidica per osservare e manipolare i singoli filamenti di actina4. Permette di aggirare le suddette limitazioni delle tecniche classiche a singolo filamento. In questi saggi di microfluidica, i filamenti di actina vengono coltivati da semi di spettrina-actina adsorbiti sul coperchio. I filamenti sono quindi ancorati da un'estremità solo al fondo della camera microfluidica e fluttuano sopra la superficie senza attaccarsi. I filamenti si allineano con il flusso delle soluzioni in entrata, facilitando così il monitoraggio della loro lunghezza del contorno e mantenendoli in una regione poco profonda sopra il coperchio in cui è possibile utilizzare TIRF. Diverse soluzioni vengono simultaneamente fatte fluire nella camera senza miscelazione e i filamenti possono essere esposti a loro in sequenza e rapidamente.
Qui, proponiamo una serie di protocolli di base per impostare saggi di microfluidica a filamento singolo di actina in laboratorio. Coverslips e camere microfluidiche possono essere preparati in anticipo (in mezza giornata) e l'esperimento stesso, in cui è possibile testare diverse condizioni biochimiche, viene eseguito in meno di un giorno.
Protocollo
1. Preparazione della camera microfluidica
- Selezionate uno stampo master SU-8 con diversi modelli di camera. Le camere tipiche sono a forma di croce con tre ingressi e un'uscita, 20 μm di altezza e 800 μm di larghezza (Figura 1). Tali stampi master possono essere acquistati da aziende esterne o realizzati in laboratori accademici (ad esempio, Gicquel, Y. et al.5).
- Posizionare il nastro attorno al bordo dello stampo.
- Metti ~ 50 cm di lunghezza, 19 mm di larghezza, nastro da ufficio trasparente standard (vedi Tabella dei materiali) su una panca, lato appiccicoso verso l'alto. Posizionare lo stampo verticalmente a un'estremità e lungo la linea mediana del nastro.
- Arrotolare lo stampo all'altra estremità del nastro per creare un bordo di 1 cm attorno allo stampo. Piegare il nastro sul fondo dello stampo.
- Preparare la soluzione di polidimetilsilossano (PDMS).
- In una pesata monouso, versare direttamente 25-30 g di base PDMS (Table of Materials). Aggiungere il 10% di peso/peso dell'agente polimerizzante PDMS (Tabella dei materiali) con una pipetta Pasteur in plastica monouso.
- Mescolare manualmente e accuratamente con un bastoncino di plastica. Assicurarsi che l'agente polimerizzante sia ben incorporato nella base PDMS, anche se l'agitazione crea molte bolle.
- Degasare la soluzione PDMS in un essiccatore sottovuoto (Table of Materials) per almeno 5 min a temperatura ambiente (RT). Le bolle si espanderanno, saliranno in superficie e scoppieranno quando il vuoto si romperà.
- Versare la soluzione PDMS sullo stampo SU-8. Utilizzare un bastoncino di plastica per raschiare e trasferire il più possibile la miscela.
- Degas PDMS per la seconda volta (5 min nell'essiccatore sottovuoto). Assicurati di sbarazzarti della maggior parte delle bolle (alcune piccole bolle sulla superficie superiore vanno bene).
- Posizionare lo stampo in forno a 70 °C per almeno 5 ore affinché il PDMS reticoli e solidifichi.
- Rimuovere le camere PDMS solide dallo stampo.
ATTENZIONE: i wafer di silicio per stampi SU-8 sono estremamente fragili, quindi è necessario prestare molta attenzione quando si separa il PDMS dai wafer. Lavora su una superficie dura e piana e mantieni il wafer piatto sulla superficie.- Con una lama di rasoio, effettuare un taglio circolare nel PDMS, a circa 1 cm di distanza dal bordo dello stampo. Tutti i modelli devono essere di almeno 0,5 cm all'interno del taglio. Staccare delicatamente il blocco PDMS centrale con delicati rimorchiatori.
ATTENZIONE: Quando si stacca, mantenere lo stampo SU-8 piatto sul piano di lavoro per evitare di romperlo. - Posizionare PDMS su un foglio di alluminio pulito, la superficie stampata rivolta verso il foglio di alluminio, per proteggere la sua superficie dalla polvere e per rendere i modelli più visibili.
- Con una lama di rasoio, effettuare un taglio circolare nel PDMS, a circa 1 cm di distanza dal bordo dello stampo. Tutti i modelli devono essere di almeno 0,5 cm all'interno del taglio. Staccare delicatamente il blocco PDMS centrale con delicati rimorchiatori.
- Scegli e taglia una camera con una lama di rasoio ad almeno 0,5 cm di distanza dal modello. Il blocco PDMS risultante è alto circa 0,5 cm, largo 1,5 cm e lungo 3 cm. Forare tre ingressi e un'uscita con un punzone per biopsia 0,75 mm ID (Tabella dei materiali).
- Pulire la camera PDMS con etanolo ultrapuro (Table of Materials) e asciugare all'aria utilizzando una pistola di sicurezza (Table of Materials). Posizionare il PDMS con il motivo rivolto verso l'alto in una capsula di Petri pulita e chiudere il piatto con il coperchio.
2. Pulizia del coperchio del vetro
NOTA: Qui, una procedura standard di pulizia del coverslip, basata su una serie di passaggi di sonicazione, è dettagliata. Altre procedure di pulizia del coperchio del vetro sono state descritte in molte altre pubblicazioni che possono ottenere risultati soddisfacenti simili 6,7,8,9.
- Posizionare 10-20 coverslips (40 mm di lunghezza) su un supporto in politetrafluoroetilene (PTFE) (Tabella dei materiali). Sonicare i coperchi in 0,5 L di soluzione detergente per vetro al 2% (Tabella dei materiali) in un becher di vetro da 1 L (35 °C, 30 min).
- Smaltire la soluzione detergente per vetri e risciacquare abbondantemente i coperchi con dH2O in almeno tre bagni successivi da 0,5 L.
- Preparare 0,5 L di 2 M KOH in un becher di vetro da 1 L. Sonicare i coverslips in KOH (RT, 30 min). Smaltire KOH e risciacquare i coverslip con dH2O in almeno tre bagni da 0,5 L.
ATTENZIONE: utilizzare dispositivi di protezione di sicurezza di laboratorio appropriati (guanti, occhiali e camice da laboratorio). - Trasferire e sonicare i coverslip in 0,5 L di etanolo ultrapuro (RT, 30 min). Coverslips può essere mantenuto in etanolo per un massimo di 2 settimane. Chiudere il becher con film termoplastico (Table of Materials) per evitare l'evaporazione. Prima dell'uso, asciugare il coperchio con flusso d'aria.
3. Assemblaggio della camera PDMS
- Preriscaldare la piastra calda a 100 °C. Posizionare fino a tre camere PDMS pulite e coperture in vetro in una capsula di Petri pulita. Posizionare la capsula di Petri aperta in un detergente ultravioletto (UV) profondo (λ = 185 nm, vedere Tabella dei materiali) ed esporla alla luce UV per 3-5 minuti.
NOTA: In alternativa, le camere PDMS e i coverslip possono essere esposti all'aria o al plasma di ossigeno per 30 s. - Posizionare delicatamente la camera PDMS sopra il coperchio. Assicurarsi che le due superfici messe a contatto siano state esposte direttamente ai raggi UV. Il PDMS si attacca automaticamente al vetro e la camera diventa chiaramente visibile.
- Per rimuovere l'aria intrappolata nell'interfaccia PDMS-coverslip, premere delicatamente la superficie con un dito. Per un incollaggio più stretto, premere più forte su angoli e lati. Assicurarsi che il soffitto della camera non entri in contatto con la superficie del vetro.
- Posizionare la camera con il fondo di vetro rivolto verso la piastra calda a 100 °C per 5 minuti. Dopo questo passaggio, i legami vetro-PDMS diventano permanenti e le camere possono essere utilizzate solo una volta. Utilizzare immediatamente la camera o conservarla in una capsula di Petri pulita per un massimo di una settimana.
4. [OPZIONALE] Passivazione diretta e funzionalizzazione
NOTA: A seconda dell'applicazione, le camere possono essere passivate e funzionalizzate una volta collegate al dispositivo di controllo microfluidico (vedere Tabella dei materiali) o iniettando manualmente soluzioni direttamente nella camera con una pipetta prima del suo collegamento al dispositivo microfluidico. Quest'ultimo offre il vantaggio di consumare meno reagente ed evitare potenziali contaminazioni facendo scorrere la soluzione attraverso il tubo in polietere etere chetone (PEEK) del dispositivo microfluidico. In tutti i passaggi seguenti, le soluzioni vengono iniettate attaccando direttamente la punta della pipetta nell'uscita. Per evitare di creare bolle all'interno della camera, assicurarsi di avere una piccola goccia che sporge dalla punta della pipetta quando si collega la punta all'uscita della camera PDMS. Allo stesso modo, rimuovere la punta della pipetta prima che l'intero volume sia stato iniettato.
- Iniettare 20 μL di PLL-PEG (1 mg/mL in soluzione salina tamponata con fosfato (PBS)). Incubare per un minimo di 1 ora (o durante la notte) a RT. Per evitare l'evaporazione, posizionare la camera PDMS in una scatola umida (ad esempio, una scatola di punta vuota con acqua nel compartimento inferiore e la camera PDMS sulla piattaforma di tenuta della punta).
- Iniettare 20 μL di 100 pM di semi di spettrina-actina (in F-buffer, vedere Tabella 1 e Tabella 2). Attendere non più di 1 minuto. Regola la concentrazione e i tempi del seme per regolare la densità della superficie del seme, abbastanza alta per statistiche di grandi dimensioni e abbastanza bassa da non sovrapporre i filamenti.
NOTA: In alternativa, se i semi di spettrina-actina non sono disponibili, utilizzare segmenti di filamento corto funzionalizzati alla biotina che saranno immobilizzati su una coverliprivestita di streptavidina 9,10. - [FACOLTATIVO] Iniettare 20 μL di albumina sierica bovina al 5% (BSA) in F-buffer. Lasciare a RT per 10 min.
- [FACOLTATIVO] Iniettare 20 μL di 1 mg/mL β-caseina in F-buffer. Lasciare a RT per 10 min.
NOTA: seguire i passaggi 4.3 e/o 4.4 per passivare ulteriormente la camera. La scelta della passivazione dipende dalle proteine utilizzate e non funziona ugualmente bene su tutti gli ABP. Quando si utilizza l'actina da sola, PLL-PEG o BSA è sufficiente.
5. Collegare il dispositivo microfluidico
NOTA: Utilizzare un sistema microfluidico basato sulla pressione con un massimo di quattro canali per controllare i flussi nella camera microfluidica (Figura 1A, vedere Tabella dei materiali). Per evitare la formazione di bolle nel tubo microfluidico e perturbare la stabilità del flusso, degasare tutte le soluzioni. Posizionare 5 mL di dH20 e 10 mL di F-buffer stock in un essiccatore per vuoto collegato a una pompa per vuoto (vuoto finale <250 mbar) e degassare per almeno 1 ora a RT.
- Risciacquare ingressi + tubi di uscita con dH2O (500 μL, 300 mbar).
- Riempire tutti i tubi del serbatoio da 2 mL (vedere Tabella dei materiali) con 300 μL di F-buffer. Impostare la pressione a 300 mbar e lasciare che da cinque a otto gocce vadano sprecate. Ripetere l'operazione per ogni canale e impostare la pressione su 0.
- Collegare la presa e risciacquare ampiamente la camera.
- Impostare la pressione per il tubo del serbatoio 4 (uscita) a 50 mbar. Una volta che una goccia esce dall'estremità del tubo, collegare il tubo all'uscita della camera PDMS. Il liquido si riempie nella camera ed esce da tutte le prese.
- [FACOLTATIVO] Se la camera è stata passivata direttamente (sezione 4), impostare la pressione su 100 mbar per risciacquare la camera con 50-100 μL di F-buffer (3-5 min). Rimuovere il liquido in eccesso alle prese con un fazzoletto detergente.
- Impostare la pressione su 20 mbar.
- Collegare gli ingressi.
- Impostare la pressione per il tubo del serbatoio da 1 a 50 mbar. Per evitare l'introduzione di bolle d'aria, assicurarsi che una goccia esca dal tubo e dall'ingresso PDMS.
- Collegare il tubo all'ingresso 1 (le due goccioline si fondono durante il collegamento). Impostare la pressione su 30 mbar.
- Ripetere i passaggi 5.4.1-5.4.2 per collegare gli ingressi 2 e 3.
- Impostare la pressione di tutte le prese a 20 mbar e la pressione di uscita a 0 mbar. Assicurarsi che le portate nelle prese d'ingresso siano approssimativamente uguali (vedere la sezione Risoluzione dei problemi).

Figura 1: Iniezione di soluzioni attraverso una camera microfluidica. (A) Configurazione microfluidica standard per esperimenti di filamenti di actina singola. Le soluzioni proteiche, collocate nei serbatoi 1-3, vengono spinte nella camera regolando la pressione nella fase gassosa. Le portate generate sono misurate da misuratori di portata. All'interno delle camere microfluidiche, le soluzioni non si mescolano e occupano spazio a seconda delle pressioni relative applicate (qui, uguale pressione su tutte le prese). Dimensioni tipiche: i tubi del serbatoio contengono fino a 2 ml di soluzione. Il tubo in PEEK (diametro interno 0,25 mm) collega i serbatoi ai misuratori di portata (dopo 10 cm di tubo) e quindi alla camera PDMS (dopo altri 70 cm). Tubi in silicone e accoppiatori per tubi in acciaio inossidabile vengono utilizzati per collegare il tubo PEEK agli ingressi PDMS. Il canale microfluidico principale è alto 20-60 μm, largo circa 1 mm e lungo 1 cm. (B,C) Profili di flusso all'interno della camera microfluidica. (B) Il fluido genera un profilo parabolico attraverso l'altezza della camera: v(z) = 6z(h-z)R/h3w, dove h e w sono l'altezza e la larghezza della camera, e R è la portata totale. In basso: singolo filamento di actina polimerizzato da semi di spettrina-actina ancorati alla superficie. (C) Quando la larghezza della camera è considerevolmente maggiore della sua altezza, il flusso è quasi uniforme in tutta la camera, tranne che sulle superfici PDMS, dove va a zero. Fare clic qui per visualizzare una versione più grande di questa figura.
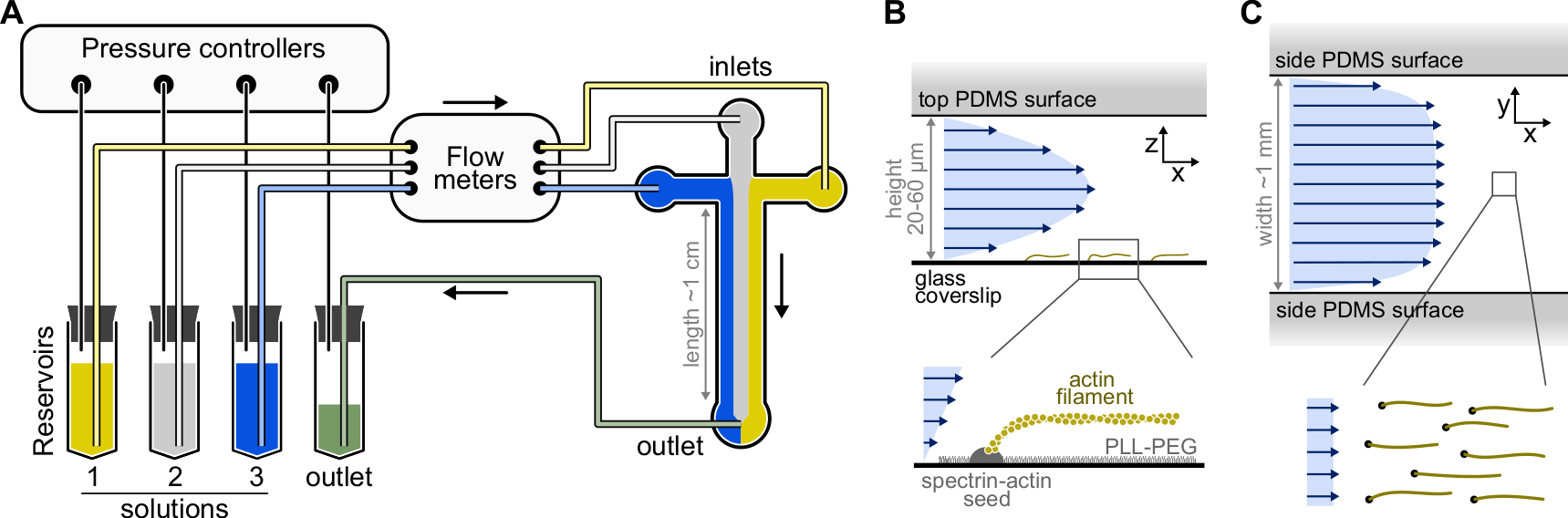{kind=link}
6. Configurazione del setup con portate standard
NOTA: Il sistema di pressione controllato da computer consente una regolazione facile e precisa delle pressioni di tutte le prese/uscite collegate alla camera PDMS, quindi il controllo delle portate in entrata e in uscita. Le configurazioni preimpostate possono essere salvate e attivate / disattivate con un solo clic del mouse. Di seguito sono riportate le configurazioni consigliate (se non diversamente specificato, la pressione di uscita è impostata su 0 mbar). Vedere la Tabella 3 per le portate previste per queste configurazioni preimpostate. Le pressioni qui indicate devono essere regolate in base alla geometria della camera e alla configurazione del sistema.
-
Modifica: utilizzare questo predefinito quando si cambiano uno o più serbatoi. Crea un lieve flusso all'indietro nel tubo di interesse per prevenire l'introduzione di bolle.
- Impostare tutte le pressioni di ingresso su 12 mbar e la pressione di uscita su 5 mbar (Figura 2B).
-
High Flow 'All': utilizzare questo preset per iniettare rapidamente tre soluzioni in parallelo. Raggiungeranno la camera entro 4 minuti.
- Impostare tutte le pressioni di ingresso su 150 mbar.
-
High Flow 'x': utilizzare questo preset per iniettare rapidamente una soluzione. Raggiungerà la camera entro 3 minuti (Figura 3A-C).
- Impostare la pressione di ingresso 'x' a 150 mbar (~15 μL/min). La pressione nelle altre prese viene regolata a circa 100 mbar, in modo tale che la portata risultante in queste prese sia di ~ 500 nL / min.
-
Mid Flow 'All': usa questo preset per mettere in pausa il sistema.
- Impostare tutte le prese su 20 mbar (Figura 2A).
-
Mid Flow 'x': utilizzare questo predefinito per consentire alla soluzione 'x' di riempire la maggior parte della larghezza del canale principale (vedere la Figura 2C,D), limitando al contempo le altre soluzioni di ingresso ai lati del canale. I filamenti di actina nella camera saranno quindi esposti alla condizione biochimica imposta dalla sola soluzione 'x'.
- Impostare la pressione di ingresso 'x' a 12 mbar. Impostare la pressione negli altri ingressi e regolare a ~ 9 mbar, in modo tale che le rispettive portate siano ~ 150 nL / min.

Figura 2: La pressione applicata a ciascun serbatoio controlla la distribuzione divisoria/spaziale delle soluzioni all'interno della camera microfluidica. (A) A parità di pressione applicata ai serbatoi, ogni soluzione occupa un terzo della camera. (B) Quando si cambia un tubo del serbatoio (qui serbatoio 3), la pressione effettiva scende a zero, creando un flusso all'indietro. (C,D) L'aumento della pressione relativa su uno dei serbatoi consente l'esposizione della superficie del vetro a una singola soluzione. Il campo visivo al centro della camera può essere esposto in sequenza alle soluzioni 1 e 2 alternando la configurazione Mid Flow 1 (C) e Mid Flow 2 (D). Fare clic qui per visualizzare una versione più grande di questa figura.
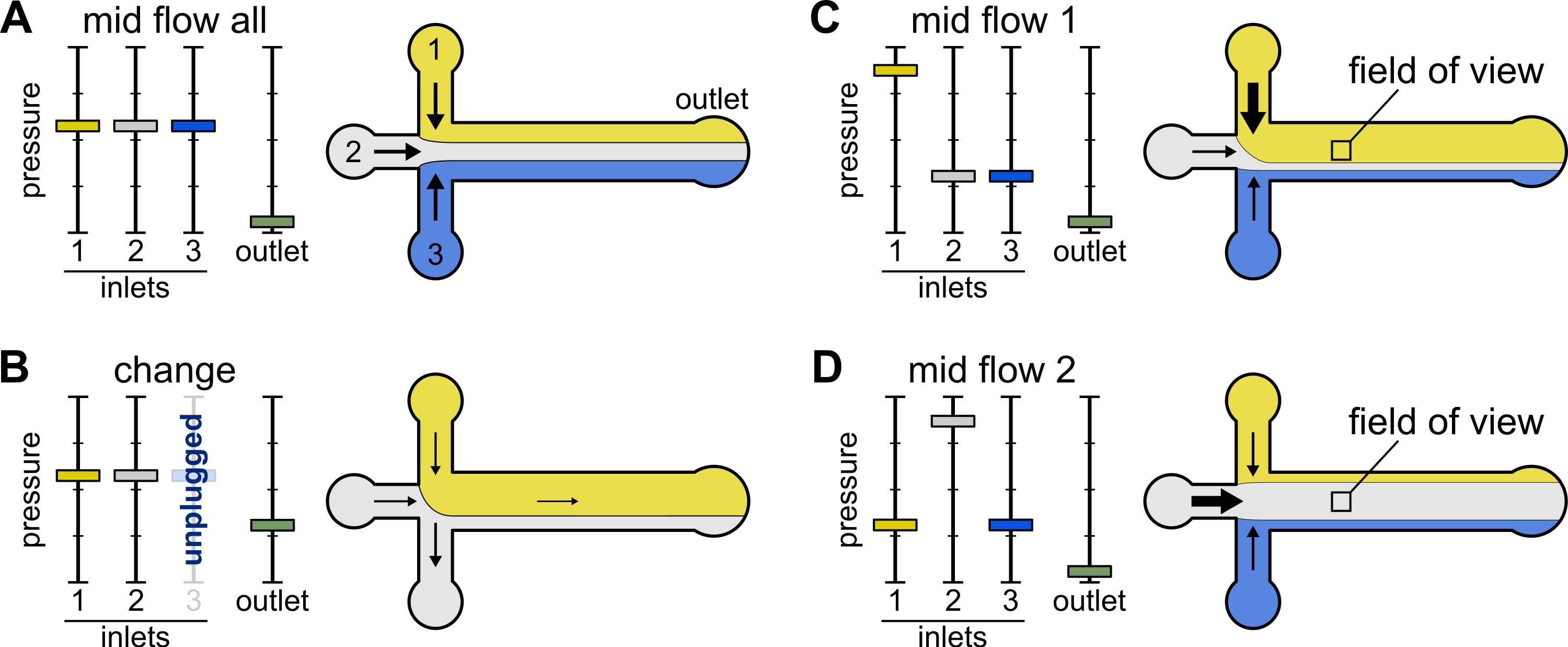{kind=link}
7. Modifica della soluzione 'x'
NOTA: come mostrato nella Figura 3A-C, è importante tenere presente che le soluzioni impiegano pochi minuti per fluire da un tubo del serbatoio al canale principale della camera. Questo tempo minimo di "morte" è imposto dal volume di liquido contenuto nel tubo e dal profilo di flusso all'interno del tubo (Figura 3A-C).
- Preparare 200-300 μL di soluzione in un nuovo tubo del serbatoio. Impostare la pressione su Modifica impostazione (vedere paragrafo 6).
- Svitare il tubo del serbatoio di ingresso 'x'. La soluzione nel tubo scorrerà lentamente all'indietro, dalla camera alla punta libera del tubo. La portata misurata diventa negativa (Figura 2B).
- Una volta che una piccola goccia si è formata sulla punta del tubo, avvitare il nuovo tubo con la soluzione fresca. Una volta che il tubo è correttamente stretto al sistema di pressione, la portata dell'ingresso ritorna positiva.
- Impostare l'impostazione della pressione su High Flow 'x'.
- A seconda della configurazione microfluidica e della geometria della camera, attendere 3-5 minuti affinché la soluzione riempia completamente il tubo e raggiunga la camera.
- [FACOLTATIVO] Seguire questo processo misurando l'aumento della fluorescenza nel tempo (Figura 3C).

Figura 3: Arrivo ritardato delle soluzioni dai serbatoi alla camera PDMS e rapido cambiamento delle condizioni biochimiche. (A-C) Arrivo ritardato delle soluzioni dai serbatoi alla camera PDMS. (A) A seconda della geometria della camera, della lunghezza del tubo e della pressione applicata all'ingresso o agli ingressi, la sostituzione di una soluzione con un'altra non è istantanea. Dopo aver sostituito il tubo del serbatoio con uno contenente una soluzione fluorescente (0 min), la soluzione riempie progressivamente il tubo (0,4 min) e la camera PDMS (1-2 min). La temporizzazione indicativa è data per una pressione applicata di 150 mbar, un tubo peek da 80 cm e una camera PDMS larga 1600 μm e alta 20 μm. (B) Il profilo di flusso parabolico all'interno del tubo peek genera un gradiente efficace di fluorescenza lungo il profilo radiale del tubo e all'interno della camera (vedere anche Figura 1B). (C) L'arrivo ritardato delle soluzioni può essere quantificato misurando il segnale di epifluorescenza di fondo nella camera in funzione del tempo. Condizioni sperimentali: 0,5 μM 10% G-actina marcata Alexa-568 viene iniettata con 150 mbar attraverso un flussometro e un tubo PEEK da 80 cm. (D,E) Rapido cambiamento delle condizioni biochimiche. (D) Modello delle soluzioni in entrata in due condizioni di flusso medio. (E) Aumento della fluorescenza di fondo come lettura della concentrazione di actina. Il tempo t = 0 è impostato all'inizio dell'aumento della fluorescenza. Soluzione 1: 0,5 μM 10% G-actina etichettata con Alexa-488, soluzione 2: F-buffer. (C,E) Camera PDMS: alta 20 μm e larga 1600 μm. L'intensità dell'epifluorescenza, ~2 μm sopra la superficie, è stata quantificata facendo la media del segnale sull'intero campo visivo, normalizzato a 0 in assenza di fluoroforo e 1 alla massima intensità. Fare clic qui per visualizzare una versione più grande di questa figura.
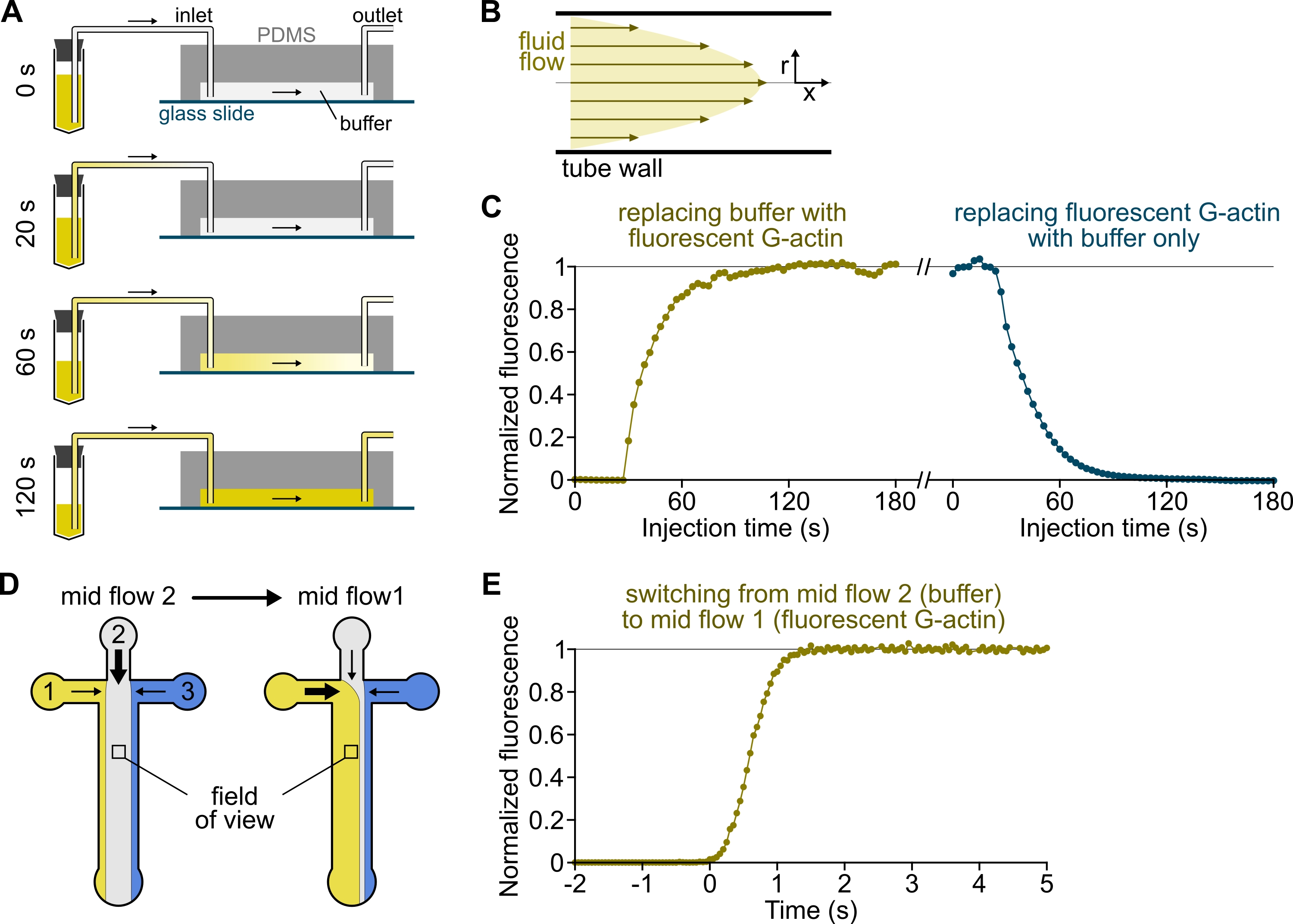{kind=link}
8. Esperimento di base a singolo filamento: depolimerizzazione dell'estremità dell'adenosina difosfato (ADP) -actina
NOTA: questa sezione presuppone una camera non funzionalizzata (solo sezione 5). Se la camera è stata funzionalizzata direttamente (sezione 4), iniziare dal punto 8.4.
- Funzionalizzazione superficiale con semi di filamento di actina:
- Modificare la soluzione da 3 a 200 μL di semi di 50 pM di spettrina-actina11 in F-buffer (vedere paragrafo 7).
NOTA: In alternativa, se i semi di spettrina-actina non sono disponibili, si possono utilizzare segmenti di filamento corto funzionalizzati alla biotina che saranno immobilizzati su una copertura rivestita di streptavidina (fare riferimento a 9,10 per i dettagli). - Iniettare per 2 minuti con High Flow 3.
NOTA: Regolare la concentrazione e il tempo in base alla densità finale del seme.
- Modificare la soluzione da 3 a 200 μL di semi di 50 pM di spettrina-actina11 in F-buffer (vedere paragrafo 7).
- Passivazione superficiale:
- Cambiare il tubo 3 con 300 μL del 5% di BSA in F-buffer.
- Iniettare per 5 minuti ad High Flow 3, seguito da 5 min a Mid Flow 3. Durante questa seconda fase, ridurre la pressione nei canali 1 e 2 a 7-8 mbar per ottenere un controcorrente ~ -100 nL / min, in modo che l'intera superficie della camera sia passivata BSA.
NOTA: Poiché la soluzione BSA è più viscosa, le pressioni devono essere regolate di conseguenza.
- Cambiare il tubo 3 in F-buffer e risciacquare il canale (5 min, High Flow 3).
- Preparare le seguenti soluzioni da 200-300 μL, tutte le proteine diluite in F-buffer:
Ingresso 1, soluzione di polimerizzazione: 1 μM 10% Alexa-488 marcato G-actina, 1 μM profilina (Tabella 1).
Ingresso 2, soluzione di invecchiamento: 0,15 μM 10% Alexa-488 etichettato G-actina.
Ingresso 3, soluzione di depolimerizzazione: solo F-buffer.
NOTA: Profilina è qui usato per prevenire la nucleazione spontanea e per mantenere una concentrazione costante di G-actina. - Cambiare i tubi da 1 a 3 (sezione 7). Iniettare utilizzando il preset High Flow All per 3-4 min. Le tre soluzioni hanno ora riempito il tubo in PEEK e raggiunto la camera (Figura 3A). La superficie del vetro può essere esposta a qualsiasi soluzione di ingresso senza tempo morto (<1 s, Figura 3D,E).
- Accendere il microscopio. Impostare le impostazioni: laser di eccitazione 150 mW 488 nm al 10% -20% di potenza, tempo di esposizione della fotocamera 100-200 ms, profondità di penetrazione TIRF 200-300 nm, obiettivo 60x. Queste impostazioni sono utilizzate in tutto il manoscritto.
- Polimerizzazione del filamento (Figura 4A):
- Impostare l'impostazione della pressione su Mid Flow 1 per ~ 10 min.
- [FACOLTATIVO] Polimerizzazione record (1 frame/20 s, TIRF). I filamenti dovrebbero polimerizzare a circa 10 subunità/secondo (sub/s)1,12.
- Invecchiamento del filamento: impostare l'impostazione della pressione su Mid Flow 2 per 15 minuti. Alla concentrazione critica, 0,15 μM G-actina, la lunghezza del filamento rimarrà costante e i filamenti si trasformeranno in >99% ADP-F-actina4.
- Depolimerizzazione (Figura 4A):
- Avviare l'acquisizione a 1 frame/5 s, in modalità epifluorescenza. Poiché nei canali 2 e 3 è presente uno sfondo a fluorescenza molto bassa, non è necessario utilizzare TIRF.
- Dopo uno o due fotogrammi, passare a Mid Flow 3. I filamenti dovrebbero depolimerizzare a circa 10 sub/s (riferimento12).
- Per ripristinare l'esperimento, rompere tutti i filamenti etichettati in modo fluorescente esponendoli continuamente al laser alla massima potenza per ~ 2 minuti. Per testare condizioni diverse, cambiare le soluzioni 1, 2 o 3 e iniettarle (High Flow, 3-4 min). Ripetere i passaggi da 8.7 a 8.9.
9. Altri esperimenti a filamento singolo
- Test delle interazioni degli ABP con F-actina
NOTA: La microfluidica è stata utilizzata con successo per quantificare l'attività di diversi ABP leganti lateralmente, come cofilina, tropomiosina e Arp2/3. Seguendo il protocollo di cui al paragrafo 8:- Cambia canale 3 con l'ABP fluorescente di interesse in F-buffer. Iniezione (High Flow 3, 3 min).
- Polimerizzazione del filamento: impostare l'impostazione della pressione su Mid Flow 1 per 10 min.
- ABP binding: Inizia l'acquisizione con TIRF. Regola la frequenza dei fotogrammi in base alla concentrazione ABP. Dopo 1-2 fotogrammi, passare a Mid Flow 3.
NOTA: a seconda dell'ABP, può anche essere possibile passare rapidamente (ad esempio, per meno di 5 s) a Mid Flow 2 per ridurre ulteriormente la fluorescenza di sfondo quando si scatta un'immagine. - ABP unbinding: mentre continua l'acquisizione, passa a Mid Flow 2.
- Polimerizzazione con formina all'estremità libera spinata
NOTA: Formins ha dimostrato di influenzare la polimerizzazione dell'estremità spinata del filamento. La microfluidica è particolarmente adatta per misurare i tassi di legame e slegamento della formina e il loro impatto sull'allungamento del filamento.- Preparare le soluzioni seguenti:
Canale 1: 10 nM formin in F-buffer (Tabella 1).
Canale 2: 1 μM 10% Alexa-488 marcato G-actina, 4 μM profilina.
Canale 3: F-buffer. - Cambiare i tubi 1, 2 e 3 (sezione 7). Iniettare utilizzando il preset High Flow All per 3-4 min.
- Avviare la polimerizzazione del filamento: impostare l'impostazione della pressione su Mid Flow 2 per 2 min.
- Legame Formin all'estremità spinata del filamento: impostare le impostazioni di pressione su Mid Flow 1 per 30 s.
- Polimerizzazione mediata da formin: impostare l'impostazione della pressione su Mid Flow 2. Con formin mDia1 alla loro estremità spinata, i filamenti dovrebbero polimerizzare a circa 50 sub / s 13,14,15.
- Preparare le soluzioni seguenti:
- Polimerizzazione/depolimerizzazione da formina ancorata in superficie
NOTA: È stato dimostrato che i tassi di polimerizzazione e depolimerizzazione delle estremità spinate decorate con formina dipendono dalla tensione applicata al filamento. In microfluidica, l'attrito del flusso del fluido lungo il lato del filamento genera una tensione proporzionale alla lunghezza del filamento e alla portata14,16.- Utilizzare il metodo descritto nella sezione 8 sopra descritta, sostituendo i passaggi 8.1, 8.2 e 8.3 per la passivazione superficiale con:
- Cambiare il tubo da 3 a 1 μg/mL anticorpo anti-His in F-buffer. Iniettare per 2 minuti con High Flow 3.
- Cambio tubo 3 con BSA al 5% in F-buffer. Iniettare per 5 minuti ad High Flow 3, seguito da 5 min a Mid Flow 3. Durante questa seconda fase, ridurre la pressione nei canali 1 e 2 a 7-8 mbar per ottenere un controflusso ~ -100 nL / min in modo che l'intera superficie della camera sia passivata BSA.
- Cambia il tubo da 3 a 100 nM His-tagged formin in F-buffer. Iniettare per 5 minuti con High Flow 3. Cambiare il tubo 3 con F-buffer. Iniettare per 5 minuti con High Flow 3 per garantire che non rimangano formin nel tubo.
- Preparare e iniettare le seguenti soluzioni (200-300 μL ciascuna, in F-buffer):
Canale 1: 1 μM 10% Alexa-488 etichettato G-actina.
Canale 2: 1 μM G-actina non etichettata, 4 μM profilina.
Canale 3: solo F-buffer. - Nucleazione del filamento: esporre le formine ancorate alla superficie alla G-actina (impostazione Mid Flow 1).
- Polimerizzazione del filamento: Esporre la camera alla profilina-actina utilizzando Mid Flow 2.
- Inizio acquisizione: 1 frame/2 s, epifluorescenza. Con formin mDia1, i filamenti dovrebbero polimerizzare a 50-80 sub/s, a seconda della lunghezza del filamento e della portata14.
- Depolimerizzazione del filamento: Avvio dell'acquisizione (1 frame/4 s, epifluorescenza). Dopo 1-2 fotogrammi, esporre i filamenti all'F-buffer, Mid Flow 3. Con formin mDia1, i filamenti dovrebbero depolimerizzare a 5-15 sub/s, a seconda della lunghezza del filamento e della portata14.
- Utilizzare il metodo descritto nella sezione 8 sopra descritta, sostituendo i passaggi 8.1, 8.2 e 8.3 per la passivazione superficiale con:
- Filamenti di actina con segmenti non etichettati
NOTA: l'etichettatura fluorescente dell'actina crea diversi artefatti, come le pause durante la depolimerizzazione17 e il legame alterato della tropomiosina18. Una soluzione alternativa per questi artefatti consiste nell'utilizzare la microfluidica per assemblare filamenti che mostrano segmenti senza etichetta.- Preparare e iniettare le seguenti soluzioni (200-300 μL in F-buffer):
Canale 1: 1 μM G-actina non etichettata, 1 μM profilina.
Canale 2: 0,3 μM 10% Alexa-488 etichettato G-actina. - Esporre in sequenza la superficie al canale 2 (5 min), al canale 1 (10 min) e al canale 2 (15 min) per generare segmenti ADP-actina non etichettati con segmenti marcati in modo fluorescente a ciascuna estremità.
- Preparare e iniettare le seguenti soluzioni (200-300 μL in F-buffer):
- Filamenti spinati ancorati all'estremità con gelsolina
NOTA: Con i semi di spettrina-actina, i filamenti polimerizzano alla loro estremità spinata libera mentre l'estremità appuntita è stabilizzata dal seme di spettrina-actina. Un'alternativa è quella di ancorare i filamenti con un tappo terminale spinato come la gelsolina.- Preparare una soluzione di F-actina di 4 μM 10% Alexa-488 marcata G-actina in 20 μL di F-buffer. Lasciare che l'actina nucleanti spontaneamente e polimerizzi a RT per almeno 30 minuti sul banco. Avvolgere il tubo in un foglio di alluminio per proteggerlo dalla luce.
- Nel frattempo, preparare la camera microfluidica e passivare la superficie con una miscela di 5% BSA e 1% biotina-BSA (vedi passo 8.2).
- Risciacquare il canale 3 con F-buffer (2 min ad Alta Portata 3). Iniettare 10 μg/mL neutravidina in F-buffer (4 min ad alta portata 3).
- Cambia i tubi in:
Canale 1: 10 nM biotina-gelsolina (Tabella 1).
Canale 2: F-buffer.
Canale 3: 0,4 μM di F-actina prepolimerizzata. - Iniettare tutte le soluzioni insieme utilizzando l'impostazione High Flow All per 3 min.
- Esporre l'intera camera alla gelsolina (Mid Flow 1, 30 s).
- Attaccare i filamenti alla superficie (Low Flow 3: Canale 3 a 3 mbar, Canali 1 e 2 a ~ 2 mbar, per circa 2 min).
- [FACOLTATIVO] Se la densità del filamento è troppo bassa, ripetere i passaggi 9.5.6 e 9.5.7.
- Depolimerizzazione dell'estremità appuntita: acquisizione iniziale (1 frame/30 s, epifluorescenza). Dopo 1-2 fotogrammi, esporre i filamenti solo al buffer, Mid Flow 2. I filamenti dovrebbero depolimerizzare a circa 0,2 sub/s.
10. Formazione e smontaggio di fasci di filamenti indotti da fascin da ADF / cofilin
NOTA: Per formare fasci di filamenti di actina, assicurarsi di avere una densità di semi di filamento sufficientemente elevata sulla superficie della camera. Se esposti alla proteina fascina, i filamenti vicini che fluttuano lateralmente saranno dinamicamente reticolati da proteine fascine. Poiché il fascio si slega rapidamente dal lato del filamento19, il fascio deve essere costantemente presente nella soluzione principale che scorre per mantenere l'impacchettamento del filamento.
- Seguire i passaggi 8.1-8.3.
- Preparare le seguenti soluzioni (200-300 μL in F-buffer):
Canale 1, soluzione di polimerizzazione: 1 μM 10% Alexa-488 marcato G-actina, 1 μM profilina.
Canale 2, Soluzione bundling: fascina 200 nM (Tabella 1), 0,15 μM 10% Alexa-488 marcata G-actina.
Canale 3, Soluzione di smontaggio: 200 nM ADF/cofilin (Tabella 1), 100 nM fascina, 0,15 μM 10% Alexa-488 etichettato G-actina. - Cambiare i tubi da 1 a 3 (sezione 7). Iniettare utilizzando il preset High Flow All , per 3-4 min.
- Polimerizzazione del filamento: impostare l'impostazione della pressione su Mid Flow 1 per ~ 10 min. La polimerizzazione può essere ripresa con TIRF.
- Raggruppamento di filamenti (Figura 4C): avviare l'acquisizione di immagini (1 fotogramma/5 s, epifluorescenza). Dopo 1-2 fotogrammi, impostare l'impostazione della pressione su Mid Flow 2 e osservare il raggruppamento dei filamenti.
- Frammentazione del bundle: inizia l'acquisizione dell'immagine (1 frame/5 s, epifluorescenza). Dopo 1-2 fotogrammi, impostare l'impostazione della pressione su Mid Flow 3 e osservare lo smontaggio indotto dalla cofilina sia dei singoli filamenti che dei fasci.
11. Procedura di pulizia del dispositivo microfluidico
NOTA: per evitare qualsiasi contaminazione da un esperimento all'altro, è fondamentale pulire e asciugare completamente tutti i tubi e i misuratori di portata dopo ogni esperimento.
- Scollegare tutti i tubi dalla camera PDMS ed eliminare la camera.
- Per pulire i tubi in PEEK e i misuratori di portata, inserire il tubo in un tubo di plastica vuoto da 15 ml e iniettare le seguenti soluzioni alla massima pressione fino a quando il serbatoio non è quasi vuoto:
400 μL di F-buffer.
400 μL di 0,5 M NaOH.
400 μL di acqua pura.
200 μL di isopropanolo. - Sostituire con un serbatoio vuoto e soffiare aria fino a quando i tubi sono completamente asciutti (~ 2-4 min, pressione massima).
12. Analisi delle immagini
NOTA: Mentre questo manoscritto si concentra sul metodo per assemblare, manipolare e visualizzare singoli filamenti di actina in microfluidica, un breve metodo per analizzare i film acquisiti è fornito qui. L'analisi viene eseguita su immagini a 16 bit, utilizzando ImageJ, seguendo la sezione 8.
- Il trattamento dell'immagine è minimo:
- Importare lo stack di immagini di polimerizzazione o depolimerizzazione.
- [FACOLTATIVO] Omogeneizza l'intensità dell'immagine con la funzione Sottrai sfondo (impostazioni predefinite (ad esempio "Raggio della palla rotante" = 50 pixel)). Ciò è particolarmente utile se la fluorescenza di fondo cambia nel corso di un filmato o se l'illuminazione a fluorescenza non è omogenea sul campo visivo.
- Regola luminosità e contrasto (sfondo vicino allo zero, filamenti vicini al massimo).
- Crea il kymograph a filamento:
- Selezionare un filamento che non si fermi, non si rompa o non si stacchi. Non selezionare in base al comportamento in altro modo. Disegna una linea 1-2 pixel sopra (strumento Linea retta). Salva il numero del filamento (Aggiungi ROI Manager).
- Applicare la funzione Reslice (Conteggio sezioni: 5 pixel). Calcolare l'intensità massima (funzione Zprojection).
- Misurare il tasso di polimerizzazione/depolimerizzazione:
- Sul kymograph, disegnare una linea lungo l'estremità spinata del filamento (strumento Linea retta , Figura 4A). Misurare la larghezza e l'altezza della linea (funzione Misura).
- Ripetere i passaggi 12.2-12.3 su più filamenti. Calcolare i tassi di polimerizzazione/depolimerizzazione (Figura 4A):
 , dove v è la velocità (in sub/s), w la larghezza della linea (pixel), pix la dimensione dei pixel (nm), h l'altezza della linea (fotogrammi) e dt il tempo tra i fotogrammi (in secondo). Qui, 2,7 nm corrispondono al contributo effettivo di una subunità di actina alla lunghezza del filamento.
, dove v è la velocità (in sub/s), w la larghezza della linea (pixel), pix la dimensione dei pixel (nm), h l'altezza della linea (fotogrammi) e dt il tempo tra i fotogrammi (in secondo). Qui, 2,7 nm corrispondono al contributo effettivo di una subunità di actina alla lunghezza del filamento.
Risultati
Per tutti gli esperimenti sopra descritti, i filamenti di actina marcati fluorescentemente dovrebbero essere chiaramente visibili, con un buon contrasto, indicativo di una bassa fluorescenza di fondo dalla superficie (Figura 4, vedere il file supplementare 1 per la risoluzione dei problemi comuni). Anche i filamenti di actina non dovrebbero attaccarsi alla superficie: quando la portata dominante è bassa, le fluttuazioni laterali dei filamenti di actina dovrebbero essere per...
Discussione
Rispetto ai metodi standard a filamento singolo in cui i filamenti di actina sono ancorati alla superficie da più punti lungo la loro lunghezza o mantenuti vicino ad essa da un agente di affollamento come la metilcellulosa, la microfluidica offre una serie di vantaggi. Poiché le interazioni con la superficie sono minime, vengono evitate le pause artificiali che queste interazioni possono indurre sia durante l'allungamento che durante la depolimerizzazione. I filamenti sono allineati dal flusso, paralleli tra loro, faci...
Divulgazioni
Gli autori non dichiarano conflitti di interesse.
Riconoscimenti
Siamo grati a B. Ladoux e R.-M. Laboratorio Mège per l'uso delle loro apparecchiature di pulizia UV, e J. Heuvingh e 0. du Roure per la formazione iniziale che abbiamo ricevuto sulla preparazione di stampi su wafer di silicio e fornendo suggerimenti sulla microfluidica. Riconosciamo i finanziamenti della sovvenzione del Consiglio europeo della ricerca StG-679116 (ad A.J.) e delle sovvenzioni agence Nationale de la Recherche Muscactin e Conformin (a G.R.-L.).
Materiali
| Name | Company | Catalog Number | Comments |
| β-Casein | Merck | C6905 | Used at 8 mg/mL |
| Biopsy punch (with plunger) | Ted Pella | 15115-2 | ID 0.75 mm, OD 1.07 mm |
| Biotin-BSA | Merck | A8549 | Used at 1 mg/mL |
| BSA | Merck | A8022 | Used at 50 mg/mL |
| Coverslip Mini-Rack Teflon holder | Invitrogen | C14784 | for 8 coverslips |
| Coverslips 22x40mm Thickness #1.5 | Menzel Gläser | 631-1370 | |
| DABCO | Merck | D27802 | component in f-buffer |
| DTT | Euromedex | EU0006-D | component in f-buffer |
| Ester NHS Alexa Fluor 488 | Invitrogen | A20000 | Fluorophore for actin labeling on Lys328. |
| EZ-Link Sulfo-NHS-Biotin | Thermo Scientific | 21338 | To biotinylate actin on Lys328 |
| Hellmanex III | Hellma | 9-307-011-4-507 | Glass cleaning detergent |
| ImageJ | NIH | N/A | open source software |
| Laboport | KNF | 811kn.18 | vacuum pump (ultimate vacuum: 240 mbar) |
| Magic invisible tape | Scotch | 7100024666 | standard transparent office tape |
| Micrewtube | Simport | T341-6T | 2 mL microfluidic reservoir tubes |
| Microfluidic device Part 1: Flow Unit S | Fluigent | FLU-S-D-PCKB | Flowmeter |
| Microfluidic device Part 2: Fluiwell-4C-2 mL | Fluigent | 14002001PCK | Reservoir holder |
| Microfluidic device Part 3: MFCS-EZ | Fluigent | EZ-11000001 EZ-00345001 | Pressure controller |
| Model 42 - UVO-Cleaner | Jelight Inc. | 42-220 | Ultraviolet cleaner |
| N6-(6-Aminohexyl)-ATP-ATTO-488 | Jena Bioscience | NU-805-488 | ATP-ATTO used to label actin |
| neutravidin | Thermo Scientific | 31000 | |
| PLL-PEG | SuSoS | PLL(20)-g[3.5]- PEG(2) | Use at 1 mg/mL in PBS. |
| Polydimethylsiloxane (PDMS) Sylgard 184 Silicon Elastomer | Dow Corning | 1673921 | Contains PDMS base and curing agent |
| Polyetheretherketone (PEEK) tubing | Merck | Z226661 | “Blue” : I.D. = 0.25 mm |
| Safety blow gun | Coilhose Pneumatics | 700-S | filtered air |
| Silicon tubing | VWR | 228-0701P | connect PEEK to coupler |
| Stainless steel catheter coupler | Prime Bioscience | SC22/15 | Inserted into PDMS inlets and outlet to connect to PEEK tubing |
| Thermoplastic film | Sigma Aldrich | PM996 | Standard "parafilm" |
| Ultrapure ethanol | VWR | 64-17-5 | |
| Ultrasonic cleaning bath | VWR | USC200TH | To accomodate 1 L beakers |
| Vacuum dessicator | SP Bel-Art | F42022-0000 | to degas the PDMS or solutions |
Riferimenti
- Wioland, H., Jégou, A., Romet-Lemonne, G. Celebrating 20 years of live single-actin-filament studies with five golden rules. Proceedings of the National Academy of Sciences of the United States of America. 119 (3), 2109506119 (2022).
- Kuhn, J. R., Pollard, T. D. Real-time measurements of actin filament polymerization by total internal reflection fluorescence microscopy. Biophysical Journal. 88 (2), 1387-1402 (2005).
- Brewer, L. R., Bianco, P. R. Laminar flow cells for single-molecule studies of DNA-protein interactions. Nature Methods. 5 (6), 517-525 (2008).
- Jégou, A., et al. Individual actin filaments in a microfluidic flow reveal the mechanism of ATP hydrolysis and give insight into the properties of profilin. PLoS Biology. 9 (9), 1001161 (2011).
- Gicquel, Y., et al. Microfluidic chips for in situ crystal x-ray diffraction and in situ dynamic light scattering for serial crystallography. Journal of Visualized Experiments: JoVE. (134), e57133 (2018).
- Chandradoss, S. D., et al. Surface passivation for single-molecule protein studies. Journal of Visualized Experiments: JoVE. (86), e50549 (2014).
- Schaedel, L., et al. Microtubules self-repair in response to mechanical stress. Nature Materials. 14 (11), 1156-1163 (2015).
- Zimmermann, D., Morganthaler, A. N., Kovar, D. R., Suarez, C. In vitro biochemical characterization of cytokinesis actin-binding proteins. Methods in Molecular Biology. 1369, 151-179 (2016).
- Funk, J., et al. Profilin and formin constitute a pacemaker system for robust actin filament growth. eLife. 8, 50963 (2019).
- Pandit, N. G., et al. Force and phosphate release from Arp2/3 complex promote dissociation of actin filament branches. Proceedings of the National Academy of Sciences of the United States of America. 117 (24), 13519-13528 (2020).
- Wioland, H., et al. ADF/Cofilin accelerates actin dynamics by severing filaments and promoting their depolymerization at both ends. Current Biology: CB. 27 (13), 1956-1967 (2017).
- Pollard, T. D., Mooseker, M. S. Direct measurement of actin polymerization rate constants by electron microscopy of actin filaments nucleated by isolated microvillus cores. The Journal of Cell Biology. 88 (3), 654-659 (1981).
- Kovar, D. R., Harris, E. S., Mahaffy, R., Higgs, H. N., Pollard, T. D. Control of the assembly of ATP- and ADP-actin by formins and profilin. Cell. 124 (2), 423-435 (2006).
- Jégou, A., Carlier, M. -. F., Romet-Lemonne, G. Formin mDia1 senses and generates mechanical forces on actin filaments. Nature Communications. 4, 1883 (2013).
- Breitsprecher, D., et al. Rocket launcher mechanism of collaborative actin assembly defined by single-molecule imaging. Science. 336 (6085), 1164-1168 (2012).
- Courtemanche, N., Lee, J. Y., Pollard, T. D., Greene, E. C. Tension modulates actin filament polymerization mediated by formin and profilin. Proceedings of the National Academy of Sciences of the United States of America. 110 (24), 9752-9757 (2013).
- Niedermayer, T., et al. Intermittent depolymerization of actin filaments is caused by photo-induced dimerization of actin protomers. Proceedings of the National Academy of Sciences. 109 (27), 10769-10774 (2012).
- Gateva, G., et al. Tropomyosin isoforms specify functionally distinct actin filament populations in vitro. Current Biology: CB. 27 (5), 705-713 (2017).
- Aratyn, Y. S., Schaus, T. E., Taylor, E. W., Borisy, G. G. Intrinsic dynamic behavior of fascin in filopodia. Molecular Biology of the Cell. 18 (10), 3928-3940 (2007).
- Pollard, T. D. Rate constants for the reactions of ATP- and ADP-actin with the ends of actin filaments. The Journal of Cell Biology. 103, 2747-2754 (1986).
- Wioland, H., Jegou, A., Romet-Lemonne, G. Torsional stress generated by ADF/cofilin on cross-linked actin filaments boosts their severing. Proceedings of the National Academy of Sciences of the United States of America. 116 (7), 2595-2602 (2019).
- Colombo, J., et al. A functional family of fluorescent nucleotide analogues to investigate actin dynamics and energetics. Nature Communications. 12 (1), 548 (2021).
- Spudich, J. A., Watt, S. The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. The Journal of Biological Chemistry. 246 (15), 4866-4871 (1971).
- Romet-Lemonne, G., Guichard, B., Jégou, A. Using microfluidics single filament assay to study formin control of actin assembly. Methods in Molecular Biology. 1805, 75-92 (2018).
- Gieselmann, R., Kwiatkowski, D. J., Janmey, P. A., Witke, W. Distinct biochemical characteristics of the two human profilin isoforms. European Journal of Biochemistry. 229 (3), 621-628 (1995).
- Lin, D. C., Lin, S. Actin polymerization induced by a motility-related high-affinity cytochalasin binding complex from human erythrocyte membrane. Proceedings of the National Academy of Sciences of the United States of America. 76 (5), 2345-2349 (1979).
- Casella, J. F., Maack, D. J., Lin, S. Purification and initial characterization of a protein from skeletal muscle that caps the barbed ends of actin filaments. The Journal of Biological Chemistry. 261 (23), 10915-10921 (1986).
- Kremneva, E., et al. Cofilin-2 controls actin filament length in muscle sarcomeres. Developmental Cell. 31 (2), 215-226 (2014).
- Le Clainche, C., Carlier, M. -. F. Actin-based motility assay. Current Protocols in Cell Biology. , 1-20 (2004).
- Vignjevic, D., et al. Formation of filopodia-like bundles in vitro from a dendritic network. The Journal of Cell Biology. 160 (6), 951-962 (2003).
- Duellberg, C., Cade, N. I., Holmes, D., Surrey, T. The size of the EB cap determines instantaneous microtubule stability. eLife. 5, 13470 (2016).
- Duellberg, C., Cade, N. I., Surrey, T. Microtubule aging probed by microfluidics-assisted tubulin washout. Molecular Biology of the Cell. 27 (22), 3563-3573 (2016).
- Suzuki, E. L., et al. Geometrical constraints greatly hinder formin mDia1 activity. Nano Letters. 20 (1), 22-32 (2020).
- Wioland, H., Suzuki, E., Cao, L., Romet-Lemonne, G., Jegou, A. The advantages of microfluidics to study actin biochemistry and biomechanics. Journal of Muscle Research and Cell Motility. 41 (1), 175-188 (2020).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati