
Для просмотра этого контента требуется подписка на Jove Войдите в систему или начните бесплатную пробную версию.
Method Article
Использование микрофлюидики и флуоресцентной микроскопии для изучения динамики сборки одноактиновых нитей и пучков
В этой статье
Резюме
Мы представляем протоколы для простых микрофлюидных анализов актиновой нити в сочетании с флуоресцентной микроскопией, которые позволяют точно контролировать отдельные актиновые нити в режиме реального времени, последовательно подвергая их воздействию различных белковых растворов.
Аннотация
Чтобы расшифровать сложные молекулярные механизмы, которые регулируют сборку и разборку актиновых нитей, является большим преимуществом для мониторинга отдельных реакций, живущих в хорошо контролируемых условиях. Для этого за последние 20 лет появились живые эксперименты с одной нитью, в основном с использованием микроскопии полной флуоресценции внутреннего отражения (TIRF), и дали множество ключевых результатов. В 2011 году, чтобы еще больше расширить возможности этих экспериментов и избежать повторяющихся проблемных артефактов, мы ввели в эти анализы простую микрофлюидику. В этом исследовании подробно описывается наш основной протокол, где отдельные актиновые нити закреплены одним концом на пассивированной поверхности покрова, выравниваются с потоком и могут последовательно подвергаться воздействию различных белковых растворов. Мы также представляем протоколы для конкретных применений и объясняем, как можно применять контролируемые механические силы благодаря вязкому сопротивлению текучего раствора. Мы выделяем технические предостережения этих экспериментов и кратко представляем возможные разработки, основанные на этой технике. Эти протоколы и объяснения, наряду с сегодняшним наличием простого в использовании оборудования для микрофлюидики, должны позволить неспециалистам реализовать этот анализ в своих лабораториях.
Введение
Сборка и разборка актиновых нитей и сетей актиновых нитей контролируется несколькими биохимическими реакциями и зависит от механического контекста. Чтобы получить представление об этих сложных механизмах, бесценно иметь возможность наблюдать индивидуальные реакции на отдельных нитях (в достаточно большом количестве). За последние десятилетия наблюдение динамических актиновых нитей в режиме реального времени, главным образом с использованием флуоресцентной микроскопии полного внутреннего отражения (TIRF), стало ключевым методом и обеспечило впечатляющий список результатов, которые не могли быть получены с помощью биохимических анализов объемного раствора1.
Для достижения этого необходимо поддерживать флуоресцентно меченые актиновые нити близко к поверхности крышки микроскопа, подвергая их воздействию растворов актин-связывающих белков (ABP), которые также могут быть флуоресцентно помечены. Это обеспечивает средства для мониторинга событий, происходящих на отдельных нитях в хорошо контролируемых биохимических условиях, и, таким образом, для количественной оценки скорости реакции. Однако следует рассмотреть ряд конкретных ограничений. Искусственное поддержание нитей близко к поверхности, часто благодаря множественным точкам крепления или с помощью скученного агента, такого как метилцеллюлоза, может изменить их поведение (например, вызывая паузы в их полимеризации и деполимеризации2). Отслеживание контура каждой нити накала может быть сложной задачей, особенно если новые нити или фрагменты нити накапливаются в поле зрения с течением времени. Реакции происходят в конечном объеме, где концентрация мономеров актина и ABP может изменяться с течением времени, что потенциально затрудняет получение точных констант скорости. Наконец, возобновление или изменение раствора ABP трудно достичь менее чем за 30 с и часто приводит к неоднородному содержанию белка в образце.
Чуть более 10 лет назад, вдохновленные тем, что уже было сделано для изучения отдельных нитей дезоксирибонуклеиновой кислоты (ДНК)3, мы представили новую технику, основанную на микрофлюидике, для наблюдения и манипулирования отдельными актиновыми нитями4. Это позволяет обойти вышеупомянутые ограничения классических однонитевых техник. В этих анализах микрофлюидики актиновые нити выращиваются из спектрин-актиновых семян, адсорбированных на покровном листе. Таким образом, нити закреплены одним концом только на дне микрофлюидной камеры и колеблются над поверхностью, не прилипая. Нити накаливания выравниваются с потоком поступающих растворов, тем самым облегчая контроль длины их контура и поддерживая их в неглубокой области над крышкой, где можно использовать TIRF. Различные растворы одновременно поступают в камеру без перемешивания, и нити могут подвергаться их последовательному и быстрому воздействию.
Здесь мы предлагаем ряд основных протоколов для создания одноактиновых микрофлюидных анализов в лаборатории. Чехлы и микрофлюидные камеры могут быть подготовлены заранее (за полдня), а сам эксперимент, где можно проверить несколько биохимических состояний, проводится менее чем за сутки.
протокол
1. Подготовка микрофлюидной камеры
- Выберите мастер-форму СУ-8 с несколькими камерными узорами. Типичные камеры имеют крестообразную форму с тремя входами и одним выходом, высотой 20 мкм и шириной 800 мкм (рисунок 1). Такие мастер-формы могут быть приобретены у внешних компаний или изготовлены в академических лабораториях (например, Gicquel, Y. et al.5).
- Поместите ленту по краю формы.
- Нанесите на скамейку ~ 50 см длиной, шириной 19 мм, стандартную прозрачную офисную ленту (см. Таблицу материалов), липкой стороной вверх. Поместите форму вертикально на одном конце и вдоль средней линии ленты.
- Сверните форму на другой конец ленты, чтобы создать границу 1 см вокруг формы. Сложите ленту поверх нижней части формы.
- Готовят раствор полидиметилсилоксана (PDMS).
- В одноразовую весовую посуду непосредственно влить 25-30 г основы PDMS (Таблица материалов). Добавьте 10% веса/веса отверждающего агента PDMS (Таблица материалов) с одноразовой пластиковой пипеткой Pasteur.
- Перемешайте вручную и тщательно с помощью пластиковой палочки. Убедитесь, что отверждающий агент хорошо встроен в основу PDMS, даже если при перемешивании образуется много пузырьков.
- Дегазация раствора PDMS в вакуумном осушителе (Таблица материалов) в течение не менее 5 мин при комнатной температуре (RT). Пузырьки будут расширяться, подниматься на поверхность и лопаться, когда вакуум нарушается.
- Налейте раствор PDMS на форму СУ-8. Используйте пластиковую палочку, чтобы соскоблить и перенести как можно больше смеси.
- Degas PDMS во второй раз (5 мин в вакуумном осушителе). Обязательно избавьтесь от большинства пузырьков (несколько маленьких пузырьков на верхней поверхности прекрасны).
- Поместите форму в духовку при температуре 70 °C в течение не менее 5 ч, чтобы PDMS сетчаться и затвердеть.
- Извлеките твердые камеры PDMS из формы.
ВНИМАНИЕ: Кремниевые пластины для пресс-форм SU-8 чрезвычайно хрупкие, поэтому при отделении PDMS от пластин необходимо соблюдать большую осторожность. Работайте на твердой, плоской поверхности и держите пластину плоской на поверхности.- Лезвием бритвы сделайте круговой разрез в PDMS, примерно в 1 см от края формы. Все узоры должны быть не менее 0,5 см внутри разреза. Аккуратно отклейте центральный блок PDMS мягкими подергиваниями.
ВНИМАНИЕ: При отслаивании держите форму SU-8 плоской на столешнице, чтобы предотвратить ее разрушение. - Поместите PDMS на чистую алюминиевую фольгу, формованную поверхность, обращенную к алюминиевой фольге, чтобы защитить ее поверхность от пыли и сделать узоры более заметными.
- Лезвием бритвы сделайте круговой разрез в PDMS, примерно в 1 см от края формы. Все узоры должны быть не менее 0,5 см внутри разреза. Аккуратно отклейте центральный блок PDMS мягкими подергиваниями.
- Выберите и вырежьте камеру лезвием бритвы на расстоянии не менее 0,5 см от узора. Полученный блок PDMS имеет высоту около 0,5 см, ширину 1,5 см и длину 3 см. Пронзите три входа и одно выходное отверстие с помощью биопсийного перфоратора 0,75 мм I.D. (Таблица материалов).
- Очистите камеру PDMS сверхчистым этанолом (Таблица материалов) и высушите на воздухе с помощью предохранительного паяльного пистолета (Таблица материалов). Поместите PDMS с рисунком вверх в чистую чашку Петри и закройте тарелку крышкой.
2. Очистка стеклянных крышек
ПРИМЕЧАНИЕ: Здесь подробно описана стандартная процедура очистки крышки, основанная на серии этапов обработки ультразвуком. Другие процедуры очистки стеклянных крышек были описаны во многих других публикациях, которые могут достичь аналогичных удовлетворительных результатов 6,7,8,9.
- Поместите 10-20 чехлов (длиной 40 мм) на держатель политетрафторэтилена (PTFE) (Таблица материалов). Обработать крышки ультразвуком в 0,5 л 2% раствора для очистки стекла (Таблица материалов) в стеклянном стакане объемом 1 л (35 °C, 30 мин).
- Утилизируйте раствор для очистки стекла и тщательно промойте крышки с помощью dH2O по крайней мере в трех последовательных ваннах объемом 0,5 л.
- Приготовьте 0,5 л 2 М КОН в стеклянном стакане объемом 1 л. Обработка обшивки оболочкой в KOH (RT, 30 мин). Утилизируйте KOH и промойте крышки с dH2O по крайней мере в трех ваннах объемом 0,5 л.
ВНИМАНИЕ: Используйте соответствующее лабораторное оборудование для защиты безопасности (перчатки, очки и лабораторный халат). - Переложить и обработать обтекателями ультрачистым спиртом в 0,5 л сверхчистого этанола (RT, 30 мин). Покровные листы можно хранить в этаноле до 2 недель. Закройте стакан термопластичной пленкой (Таблица материалов), чтобы предотвратить испарение. Перед использованием высушите крышку потоком воздуха.
3. Сборка камеры PDMS
- Разогрейте конфорку до 100 °C. Поместите до трех очищенных камер PDMS и стеклянных крышек в чистую чашку Петри. Поместите открытую чашку Петри в глубокий ультрафиолетовый (УФ) очиститель (λ = 185 нм, см. Таблицу материалов) и подвергайте ее воздействию ультрафиолетового излучения в течение 3-5 мин.
ПРИМЕЧАНИЕ: Альтернативно, камеры и крышки PDMS могут подвергаться воздействию воздуха или кислородной плазмы в течение 30 с. - Осторожно расположите камеру PDMS над крышкой. Убедитесь, что две поверхности, введенные в контакт, подвергались непосредственному воздействию ультрафиолета. PDMS автоматически прилипает к стеклу, и камера становится хорошо видимой.
- Чтобы удалить воздух, захваченный на интерфейсе PDMS-coverslip, очень осторожно надавите пальцем на поверхность. Для более плотного склеивания прижмите сильнее к углам и сторонам. Следите за тем, чтобы потолок камеры не соприкасался со стеклянной поверхностью.
- Поместите камеру со стеклянным дном лицом к конфорке при 100 °C в течение 5 мин. После этого этапа связи стекло-PDMS становятся постоянными, и камеры можно использовать только один раз. Используйте камеру немедленно или храните ее в чистой чашке Петри до недели.
4. [НЕОБЯЗАТЕЛЬНО] Прямая пассивация и функционализация
ПРИМЕЧАНИЕ: В зависимости от области применения камеры могут быть пассивированы и функционализированы либо после подключения к микрофлюидному управляющему устройству (см. Таблицу материалов), либо путем ручного впрыскивания растворов непосредственно в камеру с помощью пипетки перед ее подключением к микрофлюидному устройству. Последний предлагает преимущество в потреблении меньшего количества реагента и предотвращении потенциального загрязнения путем протекания раствора через трубку полиэфирного эфира кетона (PEEK) микрофлюидного устройства. На всех следующих этапах растворы впрыскиваются путем непосредственного втыкания наконечника пипетки в розетку. Чтобы избежать создания пузырьков внутри камеры, убедитесь, что крошечная капля торчит из наконечника пипетки при подключении наконечника к выходному отверстию камеры PDMS. Аналогичным образом, снимите наконечник пипетки до того, как весь объем будет введен.
- Вводят 20 мкл ФАПЧ-ПЭГ (1 мг/мл в фосфатно-буферном физиологическом растворе (PBS)). Инкубировать в течение минимум 1 ч (или на ночь) в RT. Для предотвращения испарения поместите камеру PDMS во влажный ящик (например, пустой наконечник с водой в нижнем отсеке и камеру PDMS на платформе для удержания наконечника).
- Вводят 20 мкл семян спектрин-актина 100 пМ (в F-буфере, см. таблицу 1 и таблицу 2). Подождите не более 1 минуты. Отрегулируйте концентрацию семян и время для настройки поверхностной плотности семян, достаточно высокой для больших статистических данных и достаточно низкой, чтобы нити не перекрывались.
ПРИМЕЧАНИЕ: Альтернативно, если спектрин-актиновые семена недоступны, используйте биотин-функционализированные короткие сегменты нитей, которые будут иммобилизованы на покрытом стрептавидином покровном листе 9,10. - [НЕОБЯЗАТЕЛЬНО] Ввести 20 мкл 5% бычьего сывороточного альбумина (BSA) в F-буфер. Оставьте в RT на 10 минут.
- [НЕОБЯЗАТЕЛЬНО] Вводят 20 мкл 1 мг/мл β-казеина в F-буфер. Оставьте в RT на 10 минут.
ПРИМЕЧАНИЕ: Выполните шаги 4.3 и/или 4.4 для дальнейшего пассивации камеры. Выбор пассивации зависит от используемых белков и не работает одинаково хорошо на всех ABP. При использовании только актина достаточно PLL-PEG или BSA.
5. Подключите микрофлюидное устройство
ПРИМЕЧАНИЕ: Используйте микрофлюидную систему на основе давления с четырьмя каналами для управления потоками в микрофлюидной камере (рисунок 1A, см. Таблицу материалов). Чтобы избежать образования пузырьков в микрофлюидных трубках и нарушения стабильности потока, дегазация всех растворов. Поместите 5 мл dH20 и 10 мл F-буферного запаса в вакуумный осушитель, подключенный к вакуумному насосу (конечный вакуум <250 мбар) и дегазацию в течение не менее 1 ч на RT.
- Промывные воздухозаборники + выпускные трубки с dH2O (500 мкл, 300 мбар).
- Заполните все резервуарные трубы объемом 2 мл (см. Таблицу материалов) 300 мкл F-буфера. Установите давление до 300 мбар и пусть пять-восемь капель пойдут впустую. Повторите для каждого канала и установите давление равным 0.
- Подключите розетку и тщательно промойте камеру.
- Установите давление для резервуарной трубы 4 (выпускное отверстие) до 50 мбар. Как только капля выйдет из конца трубки, подключите трубку к выходному отверстию камеры PDMS. Жидкость заполняется в камере и выходит из всех входов.
- [НЕОБЯЗАТЕЛЬНО] Если камера была непосредственно пассивирована (секция 4), установите давление до 100 мбар для промывки камеры 50-100 мкл F-буфера (3-5 мин). Удалите лишнюю жидкость на входах с помощью очищающей ткани.
- Установите давление до 20 мбар.
- Подключите входные отверстия.
- Установите давление для резервуарной трубы от 1 до 50 мбар. Чтобы избежать попадания пузырьков воздуха, убедитесь, что капля выходит из трубки и входного отверстия PDMS.
- Подключите трубку к входу 1 (две капли сливаются при подключении). Установите давление на 30 мбар.
- Повторите шаги 5.4.1-5.4.2 для подключения входных отверстий 2 и 3.
- Установите давление всех входных отверстий на 20 мбар и давление на выходе на 0 мбар. Убедитесь, что расход во входных отверстиях примерно равен (см. раздел Устранение неполадок).

Рисунок 1: Введение растворов через микрофлюидную камеру. (A) Стандартная микрофлюидная установка для экспериментов с одноактиновыми нитями. Белковые растворы, помещенные в резервуары 1-3, выталкивают в камеру путем регулировки давления в газовой фазе. Генерируемые расходомеры измеряются расходомерами. Внутри микрофлюидных камер растворы не смешиваются и занимают пространство в зависимости от применяемых относительных давлений (здесь равное давление на все входы). Типичные размеры: резервуарные трубы содержат до 2 мл раствора. Трубки PEEK (внутренний диаметр 0,25 мм) соединяют резервуары с расходомерами (после 10 см НКТ), а затем с камерой PDMS (еще через 70 см). Силиконовые трубки и соединительные трубы из нержавеющей стали используются для подключения труб PEEK к входным отверстиям PDMS. Основной микрофлюидный канал имеет высоту 20-60 мкм, ширину около 1 мм и длину 1 см. (В,С) Профили потока внутри микрофлюидной камеры. (B) Жидкость создает параболический профиль по всей высоте камеры: v(z) = 6z(h-z)R/h3w, где h и w - высота и ширина камеры, а R - общий расход. Нижняя часть: Одиночная актиновая нить, полимеризованная из поверхностно-анкерных семян спектрин-актина. (C) Когда ширина камеры значительно превышает ее высоту, поток почти равномерен по всей камере, за исключением поверхностей PDMS, где он сводится к нулю. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
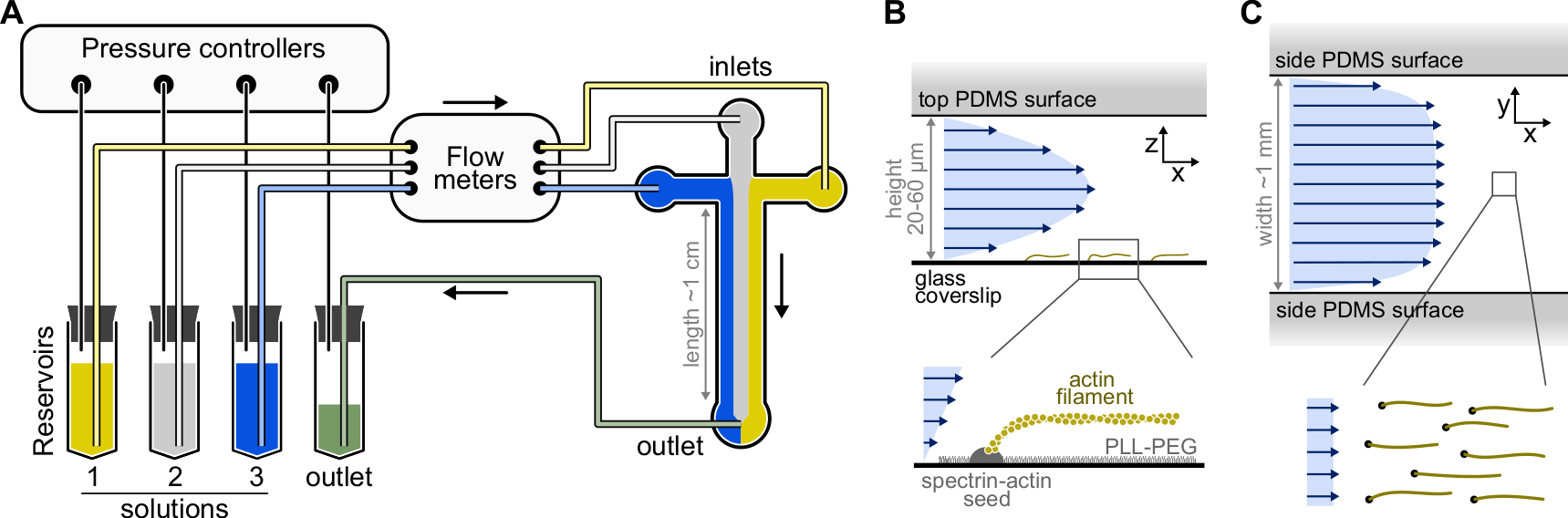{kind=link}
6. Настройка установки со стандартными скоростями потока
ПРИМЕЧАНИЕ: Система давления с компьютерным управлением позволяет легко и точно регулировать давление всех входов/выходов, подключенных к камере PDMS, что позволяет контролировать скорость входящего и исходящего потока. Предустановленные конфигурации можно сохранять и включать/выключать одним щелчком мыши. Ниже приведены рекомендуемые конфигурации (если не указано иное, давление на выходе установлено равным 0 мбар). Ожидаемые скорости потока для этих предустановленных конфигураций см. в таблице 3 . Указанное здесь давление должно регулироваться в зависимости от геометрии камеры и конфигурации системы.
-
Изменение: используйте этот пресет при изменении одного или нескольких резервуаров. Он создает мягкий обратный поток в интересующей трубке, чтобы предотвратить введение пузырьков.
- Установите все давления на входе в 12 мбар и давление на выходе на 5 мбар (рисунок 2B).
-
High Flow 'All': Используйте этот предустановленный набор для быстрого параллельного введения трех растворов. Они достигнут камеры в течение 4 минут.
- Установите все давления на входе в 150 мбар.
-
High Flow 'x': используйте этот предустановленный набор для быстрого введения раствора. Он достигнет камеры в течение 3 мин (рисунок 3A-C).
- Установите давление на входе 'x' на 150 мбар (~15 мкл/мин). Давление в других входных отверстиях регулируется примерно до 100 мбар, так что результирующая скорость потока в этих входах составляет ~ 500 нЛ / мин.
-
Mid Flow 'All': Используйте этот предустановку для приостановки работы системы.
- Установите для всех входных отверстий значение 20 мбар (рисунок 2A).
-
Mid Flow 'x': Используйте этот предустановку, чтобы позволить решению 'x' заполнять большую часть ширины основного канала (см. Рисунок 2C,D), ограничивая при этом другие входные решения сторонами канала. Таким образом, актиновые нити в камере будут подвергаться воздействию биохимического состояния, налагаемого только раствором «x».
- Установите давление на входе 'x' на 12 мбар. Установите давление в других входных отверстиях и отрегулируйте до ~9 мбар, чтобы их соответствующие скорости потока составляли ~ 150 нл/мин.

Рисунок 2: Давление, приложенное к каждому резервуару, контролирует разделение/пространственное распределение растворов внутри микрофлюидной камеры. (A) При равном давлении, приложенном к резервуарам, каждый раствор занимает одну треть камеры. (B) При замене резервуарной трубы (здесь резервуар 3) эффективное давление падает до нуля, создавая обратный поток. (С,Г) Увеличение относительного давления на одном из резервуаров позволяет подвергать поверхность стекла воздействию одного раствора. Поле зрения в середине камеры может быть последовательно подвергнуто воздействию решений 1 и 2 путем чередования конфигурации Mid Flow 1 (C) и Mid Flow 2 (D). Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
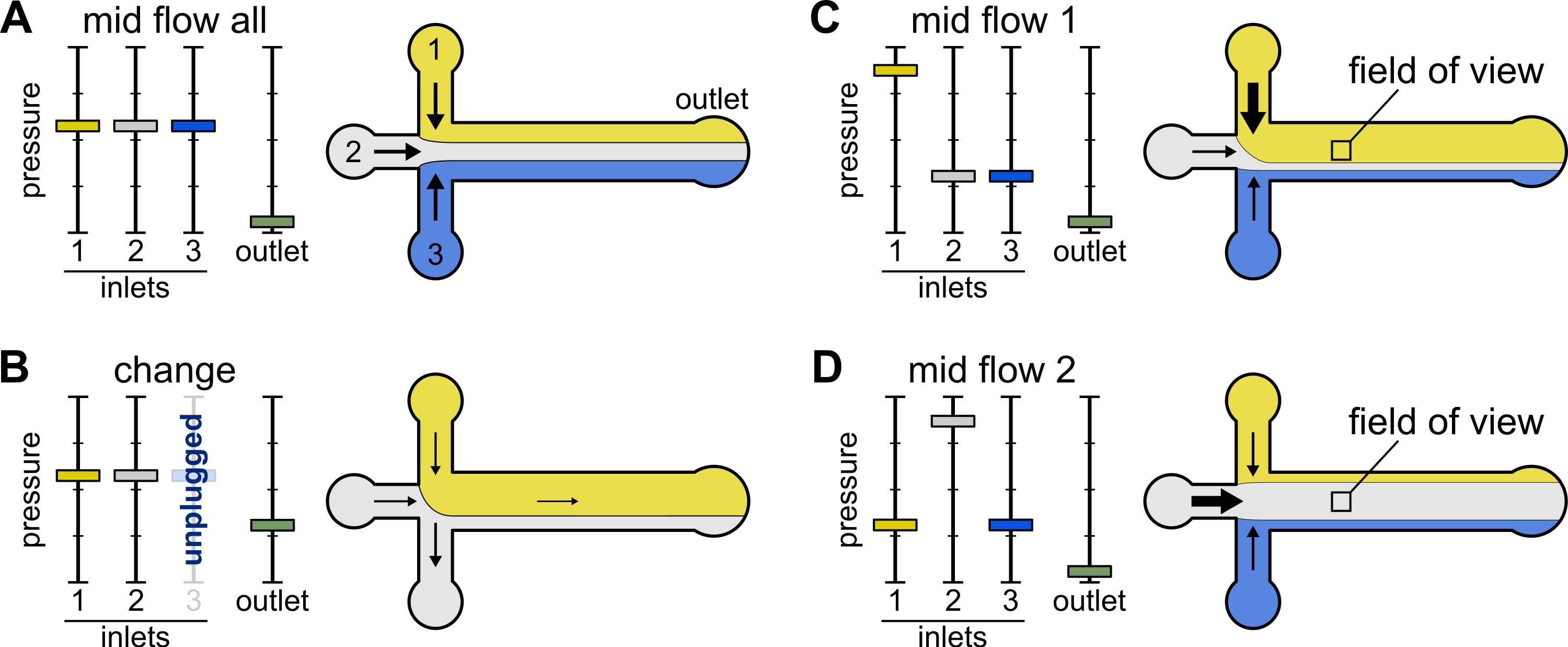{kind=link}
7. Изменение решения 'x'
ПРИМЕЧАНИЕ: Как показано на рисунке 3A-C, важно иметь в виду, что растворам требуются минуты, чтобы течь из резервуарной трубы в основной канал камеры. Это минимальное «мертвое» время обусловлено объемом жидкости, содержащейся в трубке, и профилем потока внутри трубки (рисунок 3A-C).
- Приготовьте 200-300 мкл раствора в новой резервуарной трубе. Установите значение Параметр « Изменить » (см. раздел 6).
- Открутите резервуарную трубу входного отверстия 'x'. Раствор в трубке будет медленно течь назад, от камеры к свободному наконечнику трубки. Измеренный расход становится отрицательным (рисунок 2B).
- После того, как крошечная капля образовалась на кончике трубки, вкрутите новую трубку со свежим раствором. Как только трубка правильно затянута к системе давления, скорость потока входного отверстия возвращается к положительной.
- Установите для параметра давления значение High Flow 'x'.
- В зависимости от микрофлюидной конфигурации и геометрии камеры подождите 3-5 мин, пока раствор полностью заполнит трубку и достигнет камеры.
- [НЕОБЯЗАТЕЛЬНО] Следуйте этому процессу, измеряя увеличение флуоресценции с течением времени (рисунок 3C).

Рисунок 3: Задержка поступления растворов из резервуаров в камеру PDMS и быстрое изменение биохимических условий. (А-С) Задержка поступления растворов из резервуаров в камеру PDMS. (A) В зависимости от геометрии камеры, длины трубы и приложенного давления на входе (входах) замена одного раствора другим не является мгновенной. После замены резервуарной трубки на ту, которая содержит флуоресцентный раствор (0 мин), раствор постепенно заполняют трубкой (0,4 мин) и камерой PDMS (1-2 мин). Ориентировочное время указано для приложенного давления 150 мбар, трубки PEEK 80 см и камеры PDMS шириной 1600 мкм и высотой 20 мкм. (B) Профиль параболического потока внутри трубки PEEK генерирует эффективный градиент флуоресценции вдоль радиального профиля трубки и внутри камеры (см. также рисунок 1B). (C) Задержка прибытия растворов может быть количественно определена путем измерения фонового сигнала эпифлуоресценции в камере в зависимости от времени. Экспериментальные условия: 0,5 мкМ 10% G-актина, меченного Alexa-568, вводят 150 мбар через расходомер и трубку PEEK 80 см. (D,E) Быстрое изменение биохимических условий. (D) Структура входящих решений в двух условиях mid flow. (E) Увеличение фоновой флуоресценции в качестве показаний концентрации актина. Время t = 0 устанавливается по мере наступления флуоресцентного увеличения. Раствор 1: 0,5 мкМ 10% Меченый Alexa-488 G-актин, раствор 2: F-буфер. (С,Э) Камера PDMS: высота 20 мкм и ширина 1600 мкм. Интенсивность эпифлуоресценции, ~2 мкм над поверхностью, количественно определяли путем усреднения сигнала по всему полю зрения, нормализованного до 0 при отсутствии флуорофора и 1 при максимальной интенсивности. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
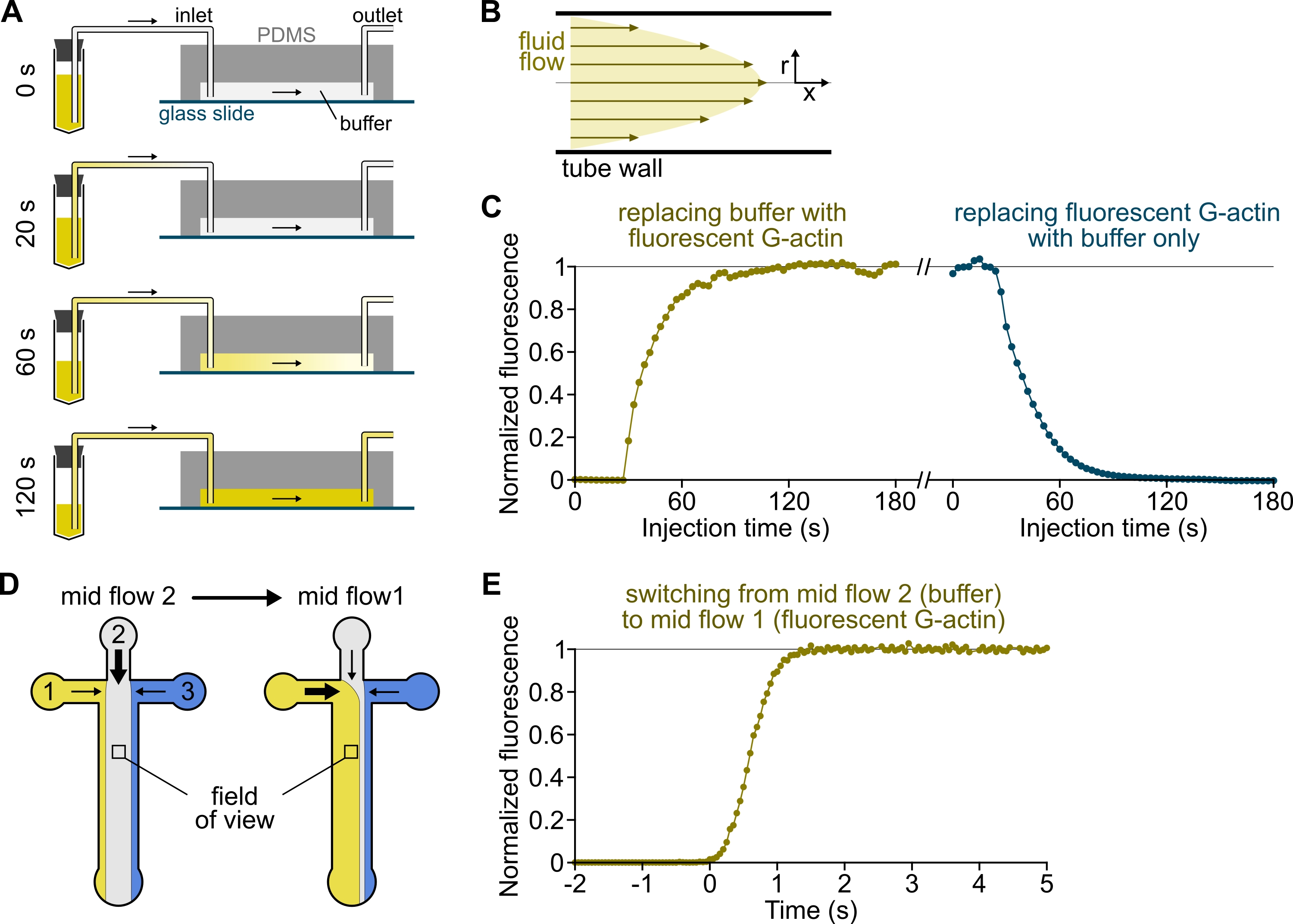{kind=link}
8. Основной эксперимент с одной нитью: Аденозиндифосфат (АДФ)-актин колючей концевой деполимеризации
ПРИМЕЧАНИЕ: В настоящем разделе предполагается нефункционализированная камера (только в разделе 5). Если камера была непосредственно функционализирована (раздел 4), начните с шага 8.4.
- Функционализация поверхности с помощью семян актиновой нити:
- Изменить раствор от 3 до 200 мкл 50 пМ спектрин-актиновых семян11 в F-буфере (см. раздел 7).
ПРИМЕЧАНИЕ: Альтернативно, если семена спектрин-актина недоступны, можно использовать биотин-функционализированные короткие сегменты нитей, которые будут иммобилизованы на покрытом стрептавидином покровном листе (подробнее см. 9,10). - Вводить в течение 2 мин с высоким расходом 3.
ПРИМЕЧАНИЕ: Отрегулируйте концентрацию и время в зависимости от конечной плотности семян.
- Изменить раствор от 3 до 200 мкл 50 пМ спектрин-актиновых семян11 в F-буфере (см. раздел 7).
- Пассивация поверхности:
- Замените трубку 3 с 300 мкл 5% BSA в F-буфере.
- Вводят в течение 5 мин при высоком потоке 3, затем 5 мин при среднем потоке 3. Во время этой второй стадии уменьшите давление в каналах 1 и 2 до 7-8 мбар, чтобы получить встречный поток ~-100 нЛ/мин, чтобы вся поверхность камеры была пассивирована BSA.
ПРИМЕЧАНИЕ: Поскольку раствор BSA является более вязким, давление необходимо соответствующим образом регулировать.
- Замените трубку 3 на F-буфер и промойте канал (5 мин, High Flow 3).
- Приготовьте следующие растворы по 200-300 мкл, все белки разбавляя в F-буфере:
Входное отверстие 1, раствор полимеризации: 1 мкМ 10% Alexa-488 с маркировкой G-актин, 1 мкМ профилина (таблица 1).
Вход 2, раствор для выдержки: 0,15 мкМ 10% Alexa-488 с маркировкой G-актин.
Вход 3, раствор для деполимеризации: только F-буфер.
ПРИМЕЧАНИЕ: Профилин используется здесь для предотвращения спонтанного зарождения и поддержания постоянной концентрации G-актина. - Замените трубки с 1 на 3 (раздел 7). Впрыскивание с помощью предустановленного high Flow All в течение 3-4 мин. Три решения заполнили трубку PEEK и достигли камеры (рисунок 3A). Поверхность стекла может подвергаться воздействию любого входного раствора без мертвого времени (<1 с, рисунок 3D, E).
- Включите микроскоп. Установите настройки: 150 мВт 488 нм лазер возбуждения при мощности 10%-20%, время экспозиции камеры 100-200 мс, глубина проникновения TIRF 200-300 нм, объектив 60x. Эти настройки используются во всей рукописи.
- Полимеризация нити (рисунок 4А):
- Установите для параметра давления значение Mid Flow 1 в течение ~10 мин.
- [НЕОБЯЗАТЕЛЬНО] Запись полимеризации (1 кадр/20 с, TIRF). Нити должны полимеризоваться со скоростью около 10 субъединиц в секунду (суб/с)1,12.
- Старение нити накала: Установите для установки давления значение Mid Flow 2 в течение 15 минут. При критической концентрации, 0,15 мкМ G-актина, длина нити накаливания останется постоянной, а нити накаливания превратятся в >99% АДФ-F-актин4.
- Деполимеризация (рисунок 4А):
- Начните съемку со скоростью 1 кадр/5 с, в режиме эпифлюоресценции. Поскольку в каналах 2 и 3 очень низкий фон флуоресценции, нет необходимости использовать TIRF.
- После одного-двух кадров переключитесь на Mid Flow 3. Нити накала должны деполимеризоваться со скоростью около 10 суб/с (ссылка12).
- Чтобы сбросить эксперимент, разорвите все флуоресцентно меченые нити, непрерывно подвергая их воздействию лазера с максимальной мощностью в течение ~ 2 мин. Для тестирования различных условий измените растворы 1, 2 или 3 и введите их (High Flow, 3-4 мин). Повторите шаги 8.7–8.9.
9. Другие эксперименты с одной нитью накаливания
- Тестирование взаимодействия ABP с F-актином
ПРИМЕЧАНИЕ: Микрофлюидика была успешно использована для количественной оценки активности нескольких побочных связывающих ABP, таких как кофилин, тропомиозин и Arp2/3. В соответствии с протоколом, содержащимся в разделе 8:- Измените канал 3 на флуоресцентный ABP, представляющий интерес в F-буфере. Впрыск (высокий расход 3, 3 мин).
- Полимеризация нити: Установите для установки давления значение Mid Flow 1 в течение 10 мин.
- Привязка ABP: Начните приобретение с TIRF. Отрегулируйте частоту кадров в зависимости от концентрации ABP. Через 1-2 кадра переключитесь на Mid Flow 3.
ПРИМЕЧАНИЕ: В зависимости от ABP также можно быстро (например, менее 5 с) переключиться на Mid Flow 2 для дальнейшего уменьшения фоновой флуоресценции при съемке изображения. - ABP unbinding: Продолжая приобретение, переключитесь на Mid Flow 2.
- Полимеризация формином на свободном колючем конце
ПРИМЕЧАНИЕ: Было показано, что формины влияют на полимеризацию колючего конца нити. Микрофлюидика особенно адаптирована для измерения скорости связывания и развязывания формина и их влияния на удлинение нити накала.- Подготовьте следующие решения:
Канал 1: 10 нМ формин в F-буфере (таблица 1).
Канал 2: 1 мкМ 10% Alexa-488, меченый G-актином, 4 мкМ профилина.
Канал 3: F-буфер. - Замените трубки 1, 2 и 3 (раздел 7). Впрыскивание с помощью предустановленного high Flow All в течение 3-4 мин.
- Начало полимеризации нити накаливания: Установите для установки давления значение Mid Flow 2 в течение 2 мин.
- Привязка Формана к колючему концу нити: Установите для настроек давления значение Mid Flow 1 в течение 30 с.
- Формин-опосредованная полимеризация: Установите для установки давления значение Mid Flow 2. С формином mDia1 на колючем конце нити должны полимеризоваться со скоростью около 50 суб/с 13,14,15.
- Подготовьте следующие решения:
- Полимеризация/деполимеризация из поверхностно-анкерового формина
ПРИМЕЧАНИЕ: Было показано, что скорость полимеризации и деполимеризации колючих концов, украшенных формином, зависит от напряжения, приложенного к нити накаливания. В микрофлюидике трение потока жидкости вдоль стороны нити накала создает напряжение, пропорциональное длине нити накала и скорости потока14,16.- Используйте метод, описанный в разделе 8, описанный выше, заменив этапы 8.1, 8.2 и 8.3 для пассивации поверхности следующим текстом:
- Изменение трубки от 3 до 1 мкг/мл анти-Его антитела в F-буфере. Вводить в течение 2 мин с высоким расходом 3.
- Замените трубку 3 с 5% BSA в F-буфере. Вводят в течение 5 мин при высоком потоке 3, затем 5 мин при среднем потоке 3. На этом втором этапе уменьшите давление в каналах 1 и 2 до 7-8 мбар, чтобы получить встречный поток ~-100 нЛ/мин, чтобы вся поверхность камеры была пассивирована BSA.
- Замените трубку от 3 до 100 нМ His-tagged formin в F-буфере. Вводить в течение 5 мин с высоким расходом 3. Замените трубку 3 на F-буфер. Впрыскивайте в течение 5 мин с High Flow 3 , чтобы убедиться, что в трубке не осталось форминов.
- Приготовить и ввести следующие растворы (по 200-300 мкл каждый, в F-буфере):
Канал 1: 1 мкМ 10% Alexa-488 с маркировкой G-актина.
Канал 2: 1 мкМ немаркированного G-актина, 4 мкМ профилина.
Канал 3: только F-буфер. - Нуклеация нити: Подвергает поверхностно-закрепленные формины воздействию G-актина (установка Mid Flow 1).
- Полимеризация нити: Подвергайте камеру воздействию профилина-актина с помощью Mid Flow 2.
- Начало съемки: 1 кадр/2 с, эпифлуоресценция. При применении formin mDia1 нити накаливания должны полимеризоваться со скоростью 50-80 суб/с, в зависимости от длины нити накала и расхода14.
- Деполимеризация нити: Начало съемки (1 кадр/4 с, эпифлуоресценция). Через 1-2 кадра подвергните нити F-буферу, Mid Flow 3. При применении formin mDia1 нити накала должны деполимеризоваться со скоростью 5-15 суб/с, в зависимости от длины нити накала и скорости потока14.
- Используйте метод, описанный в разделе 8, описанный выше, заменив этапы 8.1, 8.2 и 8.3 для пассивации поверхности следующим текстом:
- Актиновые нити с немаркированными сегментами
ПРИМЕЧАНИЕ: Актиновая флуоресцентная маркировка создает несколько артефактов, таких как паузы во время деполимеризации17 и измененное связывание тропомиозина18. Обходным путем для этих артефактов является использование микрофлюидики для сборки нитей, отображающих немаркированные сегменты.- Готовят и вводят следующие растворы (200-300 мкл в F-буфере):
Канал 1: 1 мкМ немаркированного G-актина, 1 мкМ профилина.
Канал 2: 0,3 мкМ 10% Alexa-488 с маркировкой G-актина. - Последовательно подвергайте поверхность воздействию канала 2 (5 мин), канала 1 (10 мин) и канала 2 (15 мин) для получения ADP-актиновых немаркированных сегментов с флуоресцентно мечеными сегментами на каждом конце.
- Готовят и вводят следующие растворы (200-300 мкл в F-буфере):
- Колючие торцевые нити с гельзолином
ПРИМЕЧАНИЕ: С семенами спектрин-актина нити полимеризуются на их свободном колючем конце, в то время как заостренный конец стабилизируется семя спектрин-актина. Альтернативой является закрепление нитей с колючим концом, таким как гельсолин.- Готовят раствор F-актина 4 мкМ 10% Alexa-488 с меченым G-актином в 20 мкл F-буфера. Дайте актину спонтанно зародиться и полимеризоваться при RT в течение не менее 30 минут на скамейке. Оберните трубку в алюминиевую фольгу, чтобы защитить ее от света.
- Тем временем подготовьте микрофлюидную камеру и пассивируйте поверхность смесью 5% BSA и 1% биотина-BSA (см. шаг 8.2).
- Промывочный канал 3 с F-буфером (2 мин при высоком потоке 3). Вводят 10 мкг/мл нейтравидина в F-буфер (4 мин при высоком потоке 3).
- Замените трубки на:
Канал 1: 10 нМ биотин-гельсолин (табл. 1).
Канал 2: F-буфер.
Канал 3: 0,4 мкМ преполимеризованный F-актин. - Впрыскивайте все растворы вместе, используя настройку High Flow All в течение 3 минут.
- Подвергните всю камеру воздействию гелсолина (Mid Flow 1, 30 с).
- Прикрепите нити на поверхность (Low Flow 3: Канал 3 при 3 мбар, Каналы 1 и 2 при ~2 мбар, в течение примерно 2 мин).
- [НЕОБЯЗАТЕЛЬНО] Если плотность нити накала слишком низкая, повторите шаги 9.5.6 и 9.5.7.
- Деполимеризация заостренного конца: Начальная съемка (1 кадр/30 с, эпифлуоресценция). После 1-2 кадров подвергайте нити только буферу, Mid Flow 2. Нити накала должны деполимеризоваться со скоростью около 0,2 суб/с.
10. Образование и разборка пучков нитей, индуцированных фасцином, методом АДФ/кофилина
ПРИМЕЧАНИЕ: Для образования пучков актиновых нитей убедитесь, что на поверхности камеры имеется достаточно высокая плотность семян нити. При воздействии белка фасцина соседние нити, которые колеблются латерально, будут динамически сшиты белками фасцина. Поскольку фасцин быстро развязывается со сторонынити 19, фасцин должен постоянно присутствовать в основном текучем растворе, чтобы поддерживать соединение нитей.
- Выполните шаги 8.1-8.3.
- Приготовьте следующие растворы (200-300 мкл в F-буфере):
Канал 1, раствор полимеризации: 1 мкМ 10% Алекса-488 меченый G-актин, 1 мкМ профилина.
Канал 2, Пакетное решение: 200 нМ фасцина (таблица 1), 0,15 мкМ 10% Alexa-488 с маркировкой G-актина.
Канал 3, Раствор для разборки: 200 нМ АДФ/кофилин (таблица 1), 100 нМ фасцина, 0,15 мкМ 10% Alexa-488 с маркировкой G-актина. - Замените трубки с 1 на 3 (раздел 7). Вводить с помощью предустановки High Flow All в течение 3-4 мин.
- Полимеризация нити: Установите для установки давления значение Mid Flow 1 в течение ~10 мин. Полимеризация может быть изображена с помощью TIRF.
- Комплектация нити накаливания (рисунок 4C): Начало получения изображения (1 кадр/5 с, эпифлуоресценция). После 1-2 кадров установите для установки давления значение Mid Flow 2 и наблюдайте за объединением нити накала.
- Фрагментация пучка: Начало получения изображения (1 кадр/5 с, эпифлуоресценция). После 1-2 кадров установите для установки давления значение Mid Flow 3 и наблюдайте за вызванной кофилином разборкой как одиночных нитей, так и пучков.
11. Процедура очистки микрофлюидного устройства
ПРИМЕЧАНИЕ: Чтобы избежать загрязнения от одного эксперимента к другому, крайне важно тщательно очистить и полностью высушить все трубки и расходомеры после каждого эксперимента.
- Отсоедините все трубки от камеры PDMS и выбросьте камеру.
- Для очистки труб PEEK и расходомеров заклейте концы труб в пустую пластиковую трубку объемом 15 мл и впрыскивайте следующие растворы под максимальным давлением до тех пор, пока резервуар не станет почти пустым:
400 мкл F-буфера.
400 мкл 0,5 М NaOH.
400 мкл чистой воды.
200 мкл изопропанола. - Замените пустым резервуаром и пейте воздухом до полного высыхания трубки (~2-4 мин, максимальное давление).
12. Анализ изображений
ПРИМЕЧАНИЕ: В то время как эта рукопись фокусируется на методе сборки, манипулирования и визуализации одиночных актиновых нитей в микрофлюидике, здесь представлен краткий метод анализа приобретенных фильмов. Анализ выполняется на 16-битных изображениях с помощью ImageJ, следуя разделу 8.
- Обработка изображений минимальна:
- Импорт стека изображений полимеризации или деполимеризации.
- [НЕОБЯЗАТЕЛЬНО] Гомогенизируйте интенсивность изображения с помощью функции Вычитания фона (настройки по умолчанию (например, 'Радиус катящегося шара' = 50 пикселей)). Это особенно полезно, если фоновая флуоресценция изменяется в течение фильма или если флуоресцентное освещение не однородно в поле зрения.
- Отрегулируйте яркость и контрастность (фон около нуля, нити накаливания около максимума).
- Создать нитевидный кимограф:
- Выберите нить накала, которая не приостанавливается, не разрывается и не отсоединяется. Не выбирайте на основе поведения в противном случае. Нарисуйте линию на 1–2 пикселя выше (инструмент «Прямая линия »). Сохраните номер нити накала (Добавить в менеджере ROI).
- Примените функцию Reslice (Количество фрагментов: 5 пикселей). Рассчитайте максимальную интенсивность (функция Zprojection).
- Измерьте скорость полимеризации/деполимеризации:
- На кимографе проведите линию вдоль колючего конца нити накаливания (инструмент «Прямая линия», рисунок 4А). Измерьте ширину и высоту линии (функция Измерение).
- Повторите шаги 12.2–12.3 на нескольких нитях. Рассчитайте скорости полимеризации/деполимеризации (рисунок 4А):
 , где v — скорость (в суб/с), w — ширина линии (пиксели), pix — размер пикселя (нм), h — высота линии (кадры) и dt — время между кадрами (во второй секунде). Здесь 2,7 нм соответствует эффективному вкладу актиновой субъединицы в длину нити накала.
, где v — скорость (в суб/с), w — ширина линии (пиксели), pix — размер пикселя (нм), h — высота линии (кадры) и dt — время между кадрами (во второй секунде). Здесь 2,7 нм соответствует эффективному вкладу актиновой субъединицы в длину нити накала.
Результаты
Для всех экспериментов, описанных выше, флуоресцентно меченые актиновые нити должны быть хорошо видны, с хорошим контрастом, указывающим на низкую фоновую флуоресценцию с поверхности (рисунок 4, см. Дополнительный файл 1 для устранения общих проблем). Актиновые ...
Обсуждение
По сравнению со стандартными методами с одной нитью, где актиновые нити закреплены на поверхности несколькими точками по их длине или поддерживаются близко к ней с помощью агента скученности, такого как метилцеллюлоза, микрофлюидика предлагает ряд преимуществ. Поскольку взаимодейств...
Раскрытие информации
Авторы заявляют об отсутствии конфликта интересов.
Благодарности
Мы благодарны Б. Ладу и Р.-М. Лаборатория Mège для использования своего УФ-очистительного оборудования, а также J. Heuvingh и 0. du Roure за первоначальное обучение, которое мы получили по подготовке пресс-форм на кремниевых пластинах и предоставлению советов по микрофлюидике. Мы признаем финансирование из гранта Европейского исследовательского совета StG-679116 (для A.J.) и Agence Nationale de la Recherche Grants Muscactin and Conformin (для G.R.-L.).
Материалы
| Name | Company | Catalog Number | Comments |
| β-Casein | Merck | C6905 | Used at 8 mg/mL |
| Biopsy punch (with plunger) | Ted Pella | 15115-2 | ID 0.75 mm, OD 1.07 mm |
| Biotin-BSA | Merck | A8549 | Used at 1 mg/mL |
| BSA | Merck | A8022 | Used at 50 mg/mL |
| Coverslip Mini-Rack Teflon holder | Invitrogen | C14784 | for 8 coverslips |
| Coverslips 22x40mm Thickness #1.5 | Menzel Gläser | 631-1370 | |
| DABCO | Merck | D27802 | component in f-buffer |
| DTT | Euromedex | EU0006-D | component in f-buffer |
| Ester NHS Alexa Fluor 488 | Invitrogen | A20000 | Fluorophore for actin labeling on Lys328. |
| EZ-Link Sulfo-NHS-Biotin | Thermo Scientific | 21338 | To biotinylate actin on Lys328 |
| Hellmanex III | Hellma | 9-307-011-4-507 | Glass cleaning detergent |
| ImageJ | NIH | N/A | open source software |
| Laboport | KNF | 811kn.18 | vacuum pump (ultimate vacuum: 240 mbar) |
| Magic invisible tape | Scotch | 7100024666 | standard transparent office tape |
| Micrewtube | Simport | T341-6T | 2 mL microfluidic reservoir tubes |
| Microfluidic device Part 1: Flow Unit S | Fluigent | FLU-S-D-PCKB | Flowmeter |
| Microfluidic device Part 2: Fluiwell-4C-2 mL | Fluigent | 14002001PCK | Reservoir holder |
| Microfluidic device Part 3: MFCS-EZ | Fluigent | EZ-11000001 EZ-00345001 | Pressure controller |
| Model 42 - UVO-Cleaner | Jelight Inc. | 42-220 | Ultraviolet cleaner |
| N6-(6-Aminohexyl)-ATP-ATTO-488 | Jena Bioscience | NU-805-488 | ATP-ATTO used to label actin |
| neutravidin | Thermo Scientific | 31000 | |
| PLL-PEG | SuSoS | PLL(20)-g[3.5]- PEG(2) | Use at 1 mg/mL in PBS. |
| Polydimethylsiloxane (PDMS) Sylgard 184 Silicon Elastomer | Dow Corning | 1673921 | Contains PDMS base and curing agent |
| Polyetheretherketone (PEEK) tubing | Merck | Z226661 | “Blue” : I.D. = 0.25 mm |
| Safety blow gun | Coilhose Pneumatics | 700-S | filtered air |
| Silicon tubing | VWR | 228-0701P | connect PEEK to coupler |
| Stainless steel catheter coupler | Prime Bioscience | SC22/15 | Inserted into PDMS inlets and outlet to connect to PEEK tubing |
| Thermoplastic film | Sigma Aldrich | PM996 | Standard "parafilm" |
| Ultrapure ethanol | VWR | 64-17-5 | |
| Ultrasonic cleaning bath | VWR | USC200TH | To accomodate 1 L beakers |
| Vacuum dessicator | SP Bel-Art | F42022-0000 | to degas the PDMS or solutions |
Ссылки
- Wioland, H., Jégou, A., Romet-Lemonne, G. Celebrating 20 years of live single-actin-filament studies with five golden rules. Proceedings of the National Academy of Sciences of the United States of America. 119 (3), 2109506119 (2022).
- Kuhn, J. R., Pollard, T. D. Real-time measurements of actin filament polymerization by total internal reflection fluorescence microscopy. Biophysical Journal. 88 (2), 1387-1402 (2005).
- Brewer, L. R., Bianco, P. R. Laminar flow cells for single-molecule studies of DNA-protein interactions. Nature Methods. 5 (6), 517-525 (2008).
- Jégou, A., et al. Individual actin filaments in a microfluidic flow reveal the mechanism of ATP hydrolysis and give insight into the properties of profilin. PLoS Biology. 9 (9), 1001161 (2011).
- Gicquel, Y., et al. Microfluidic chips for in situ crystal x-ray diffraction and in situ dynamic light scattering for serial crystallography. Journal of Visualized Experiments: JoVE. (134), e57133 (2018).
- Chandradoss, S. D., et al. Surface passivation for single-molecule protein studies. Journal of Visualized Experiments: JoVE. (86), e50549 (2014).
- Schaedel, L., et al. Microtubules self-repair in response to mechanical stress. Nature Materials. 14 (11), 1156-1163 (2015).
- Zimmermann, D., Morganthaler, A. N., Kovar, D. R., Suarez, C. In vitro biochemical characterization of cytokinesis actin-binding proteins. Methods in Molecular Biology. 1369, 151-179 (2016).
- Funk, J., et al. Profilin and formin constitute a pacemaker system for robust actin filament growth. eLife. 8, 50963 (2019).
- Pandit, N. G., et al. Force and phosphate release from Arp2/3 complex promote dissociation of actin filament branches. Proceedings of the National Academy of Sciences of the United States of America. 117 (24), 13519-13528 (2020).
- Wioland, H., et al. ADF/Cofilin accelerates actin dynamics by severing filaments and promoting their depolymerization at both ends. Current Biology: CB. 27 (13), 1956-1967 (2017).
- Pollard, T. D., Mooseker, M. S. Direct measurement of actin polymerization rate constants by electron microscopy of actin filaments nucleated by isolated microvillus cores. The Journal of Cell Biology. 88 (3), 654-659 (1981).
- Kovar, D. R., Harris, E. S., Mahaffy, R., Higgs, H. N., Pollard, T. D. Control of the assembly of ATP- and ADP-actin by formins and profilin. Cell. 124 (2), 423-435 (2006).
- Jégou, A., Carlier, M. -. F., Romet-Lemonne, G. Formin mDia1 senses and generates mechanical forces on actin filaments. Nature Communications. 4, 1883 (2013).
- Breitsprecher, D., et al. Rocket launcher mechanism of collaborative actin assembly defined by single-molecule imaging. Science. 336 (6085), 1164-1168 (2012).
- Courtemanche, N., Lee, J. Y., Pollard, T. D., Greene, E. C. Tension modulates actin filament polymerization mediated by formin and profilin. Proceedings of the National Academy of Sciences of the United States of America. 110 (24), 9752-9757 (2013).
- Niedermayer, T., et al. Intermittent depolymerization of actin filaments is caused by photo-induced dimerization of actin protomers. Proceedings of the National Academy of Sciences. 109 (27), 10769-10774 (2012).
- Gateva, G., et al. Tropomyosin isoforms specify functionally distinct actin filament populations in vitro. Current Biology: CB. 27 (5), 705-713 (2017).
- Aratyn, Y. S., Schaus, T. E., Taylor, E. W., Borisy, G. G. Intrinsic dynamic behavior of fascin in filopodia. Molecular Biology of the Cell. 18 (10), 3928-3940 (2007).
- Pollard, T. D. Rate constants for the reactions of ATP- and ADP-actin with the ends of actin filaments. The Journal of Cell Biology. 103, 2747-2754 (1986).
- Wioland, H., Jegou, A., Romet-Lemonne, G. Torsional stress generated by ADF/cofilin on cross-linked actin filaments boosts their severing. Proceedings of the National Academy of Sciences of the United States of America. 116 (7), 2595-2602 (2019).
- Colombo, J., et al. A functional family of fluorescent nucleotide analogues to investigate actin dynamics and energetics. Nature Communications. 12 (1), 548 (2021).
- Spudich, J. A., Watt, S. The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. The Journal of Biological Chemistry. 246 (15), 4866-4871 (1971).
- Romet-Lemonne, G., Guichard, B., Jégou, A. Using microfluidics single filament assay to study formin control of actin assembly. Methods in Molecular Biology. 1805, 75-92 (2018).
- Gieselmann, R., Kwiatkowski, D. J., Janmey, P. A., Witke, W. Distinct biochemical characteristics of the two human profilin isoforms. European Journal of Biochemistry. 229 (3), 621-628 (1995).
- Lin, D. C., Lin, S. Actin polymerization induced by a motility-related high-affinity cytochalasin binding complex from human erythrocyte membrane. Proceedings of the National Academy of Sciences of the United States of America. 76 (5), 2345-2349 (1979).
- Casella, J. F., Maack, D. J., Lin, S. Purification and initial characterization of a protein from skeletal muscle that caps the barbed ends of actin filaments. The Journal of Biological Chemistry. 261 (23), 10915-10921 (1986).
- Kremneva, E., et al. Cofilin-2 controls actin filament length in muscle sarcomeres. Developmental Cell. 31 (2), 215-226 (2014).
- Le Clainche, C., Carlier, M. -. F. Actin-based motility assay. Current Protocols in Cell Biology. , 1-20 (2004).
- Vignjevic, D., et al. Formation of filopodia-like bundles in vitro from a dendritic network. The Journal of Cell Biology. 160 (6), 951-962 (2003).
- Duellberg, C., Cade, N. I., Holmes, D., Surrey, T. The size of the EB cap determines instantaneous microtubule stability. eLife. 5, 13470 (2016).
- Duellberg, C., Cade, N. I., Surrey, T. Microtubule aging probed by microfluidics-assisted tubulin washout. Molecular Biology of the Cell. 27 (22), 3563-3573 (2016).
- Suzuki, E. L., et al. Geometrical constraints greatly hinder formin mDia1 activity. Nano Letters. 20 (1), 22-32 (2020).
- Wioland, H., Suzuki, E., Cao, L., Romet-Lemonne, G., Jegou, A. The advantages of microfluidics to study actin biochemistry and biomechanics. Journal of Muscle Research and Cell Motility. 41 (1), 175-188 (2020).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеСмотреть дополнительные статьи
This article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены