
Method Article
Captura de la conformación cromosómica a través de escalas de longitud
En este artículo
Resumen
Hi-C 3.0 es un protocolo Hi-C mejorado que combina reticulantes de formaldehído y glutarato de disuccinimidil con un cóctel de enzimas de restricción DpnII y DdeI para aumentar la relación señal-ruido y la resolución de la detección de interacción de cromatina.
Resumen
La captura de conformación cromosómica (3C) se utiliza para detectar interacciones tridimensionales de la cromatina. Por lo general, la reticulación química con formaldehído (FA) se utiliza para fijar las interacciones de la cromatina. Luego, la digestión de la cromatina con una enzima de restricción y la posterior religación de los extremos del fragmento convierte la proximidad tridimensional (3D) en productos de ligadura únicos. Finalmente, después de la reversión de los enlaces cruzados, la eliminación de proteínas y el aislamiento del ADN, el ADN se corta y se prepara para la secuenciación de alto rendimiento. La frecuencia de ligadura de proximidad de pares de loci es una medida de la frecuencia de su colocalización en el espacio tridimensional en una población celular.
Una biblioteca Hi-C secuenciada proporciona información de todo el genoma sobre las frecuencias de interacción entre todos los pares de loci. La resolución y precisión de Hi-C se basa en una reticulación eficiente que mantiene los contactos de la cromatina y la fragmentación frecuente y uniforme de la cromatina. Este documento describe un protocolo Hi-C in situ mejorado, Hi-C 3.0, que aumenta la eficiencia de la reticulación mediante la combinación de dos reticulantes (formaldehído [FA] y glutarato de disuccinimidil [DSG]), seguido de una digestión más fina utilizando dos enzimas de restricción (DpnII y DdeI). Hi-C 3.0 es un protocolo único para la cuantificación precisa de las características de plegamiento del genoma a escalas más pequeñas, como bucles y dominios de asociación topológica (TAD), así como características a escalas más grandes de todo el núcleo, como los compartimentos.
Introducción
La captura de conformación cromosómica se ha utilizado desde 20021. Fundamentalmente, cada variante de captura de conformación se basa en la fijación de las interacciones ADN-proteína y proteína-proteína para preservar la organización de la cromatina 3D. Esto es seguido por la fragmentación del ADN, generalmente por la digestión de restricción y, finalmente, la religación de los extremos de ADN cercanos para convertir loci espacialmente proximales en secuencias de ADN covalentes únicas. Los protocolos 3C iniciales utilizaron PCR para muestrear interacciones específicas "uno a uno". Los ensayos 4C posteriores permitieron la detección de interacciones "uno a todos"2, mientras que 5C detectó interacciones "muchos a muchos"3. La captura de la conformación cromosómica llegó a buen término después de implementar la secuenciación de alto rendimiento (NGS) de próxima generación, que permitió la detección de interacciones genómicas "todo a todos" utilizando Hi-C4 de todo el genoma y técnicas comparables como 3C-seq5, TCC6 y Micro-C 7,8 (ver también revisión de Denker y De Laat9).
En Hi-C, los nucleótidos biotinilados se utilizan para marcar voladizos 5' después de la digestión y antes de la ligadura (Figura 1). Esto permite la selección de fragmentos adecuadamente digeridos y religados utilizando perlas recubiertas de estreptavidina, diferenciándolo de GCC10. Una importante actualización del protocolo Hi-C fue implementada por Rao et al.11, quienes realizaron la digestión y religación en núcleos intactos (es decir, in situ) para reducir los productos de ligadura espuria. Además, la sustitución de la digestión HindIII con la digestión MboI (o DpnII) redujo el tamaño del fragmento y aumentó el potencial de resolución de Hi-C. Este aumento permitió la detección de estructuras de escala relativamente pequeña y una localización genómica más precisa de puntos de contacto, como bucles de ADN entre pequeños elementos cis, por ejemplo, bucles entre sitios unidos a CTCF generados por extrusión de bucle11,12. Sin embargo, este potencial tiene un costo. En primer lugar, un aumento de dos veces en la resolución requiere un aumento de cuatro veces (22) en las lecturas de secuenciación13. En segundo lugar, los pequeños tamaños de los fragmentos aumentan la posibilidad de confundir fragmentos vecinos no digeridos con fragmentos digeridos y religados14. Como se mencionó, en Hi-C, los fragmentos digeridos y religados difieren de los fragmentos no digeridos por la presencia de biotina en la unión de ligadura. Sin embargo, se requiere una eliminación adecuada de biotina de los extremos no ligados para asegurar que solo las uniones de ligadura se retiren14,15.
Con el costo decreciente de NGS, se hace factible estudiar el plegamiento cromosómico con mayor detalle. Para disminuir el tamaño de los fragmentos de ADN y, por lo tanto, aumentar la resolución, el protocolo Hi-C se puede adaptar para usar enzimas de restricción de corte con mayor frecuencia16 o para usar combinaciones de enzimas de restricción17,18,19. Alternativamente, la MNasa 7,8 en Micro-C y la DNasa en DNasa Hi-C20 pueden ajustarse para lograr una digestión óptima.
Una evaluación sistemática reciente de los fundamentos de los métodos 3C mostró que la detección de características de plegamiento cromosómico en cada escala de longitud mejoró enormemente con la reticulación secuencial con 1% FA seguida de 3 mM DSG17. Además, Hi-C con digestión HindIII fue la mejor opción para detectar características de plegado a gran escala, como compartimentos, y que Micro-C fue superior en la detección de características de plegamiento a pequeña escala, como bucles de ADN. Estos resultados condujeron al desarrollo de una única estrategia de alta resolución "Hi-C 3.0", que utiliza la combinación de reticulantes FA y DSG seguidos de doble digestión con endonucleasas DpnII y DdeI21. Hi-C 3.0 proporciona una estrategia eficaz para uso general porque detecta con precisión las características de plegado en todas las escalas de longitud17. La parte experimental del protocolo Hi-C 3.0 se detalla aquí y se muestran los resultados típicos que se pueden esperar después de la secuenciación.

Figura 1: Procedimiento Hi-C en seis pasos. Las celdas se fijan primero con FA y luego con DSG (1). Luego, la lisis precede a una doble digestión con DdeI y DpnII (2). La biotina se agrega por relleno saliente y los extremos romos proximales se ligan (3) antes de la purificación del ADN (4). La biotina se elimina de los extremos no ligados antes de la sonicación y la selección del tamaño (5). Finalmente, la extracción de biotina permite la ligadura del adaptador y la amplificación de la biblioteca por PCR (6). Abreviaturas: FA = formaldehído; DSG = glutarato de disuccinimidilo; B = Biotina. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Protocolo
1. Fijación por reticulación
- Fijación de formaldehído: a partir de células en monocapa
- Tener células sembradas en un medio apropiado para cosechar 5 × 106 células por placa de 150 mm.
NOTA: Los usuarios pueden elegir cualquier contenedor preferido que garantice el crecimiento óptimo de cualquier línea celular de mamíferos. Además, las células se pueden aislar del tejido. - Aspirar el medio con una pipeta Pasteur acoplada a una trampa de vacío de una placa de 150 mm, lavar 2x con ~10 ml HBSS.
- Inmediatamente antes de la reticulación, prepare una solución de reticulación de FA al 1% en un tubo de 50 ml combinando 22,5 ml de HBSS y 625 μL de FA al 37% hasta una concentración final del 1%. Mezclar suavemente meciéndose.
PRECAUCIÓN: Utilice campana extractora; El formaldehído es tóxico. - Para reticular las células, vierta 23.125 ml de la solución de FA al 1% en cada placa de 15 cm.
- Incubar a temperatura ambiente durante 10 minutos y mecer suavemente los platos a mano cada 2 minutos.
- Agregue 1,25 ml de glicina a 2,5 M (128 mM final) y agite suavemente la placa para apagar la reacción de reticulación.
- Incubar a temperatura ambiente durante 5 minutos y continuar la incubación en hielo durante al menos 15 minutos para detener la reticulación.
- Raspa las celdas de las placas con un raspador de celdas o un policía de goma.
- Transfiera la suspensión celular a un tubo cónico de 50 ml con una pipeta. Centrifugar a 1.000 × g durante 10 min a temperatura ambiente y desechar el sobrenadante por aspiración.
- Lave el pellet celular una vez con 10 ml de solución salina tamponada con fosfato (DPBS) de Dulbecco, usando una pipeta para resuspender. Luego, centrifugar a 1,000 × g durante 10 minutos a temperatura ambiente. Proceda inmediatamente a la reticulación DSG.
NOTA: Tenga cuidado al lavar el pellet de celda, ya que los gránulos celulares pueden estar sueltos y las células pueden perderse.
- Tener células sembradas en un medio apropiado para cosechar 5 × 106 células por placa de 150 mm.
- Fijación de formaldehído: a partir de células en suspensión
- Tener células sembradas en un medio apropiado para cosechar 5 × 106 células por vaso.
NOTA: Los usuarios pueden elegir cualquier contenedor preferido que garantice el crecimiento celular óptimo de cualquier línea celular de mamíferos. - Inmediatamente antes de la cosecha, contar las células y transferir 5 × 106 células a un tubo cónico de 50 ml.
- Granular suavemente las células centrifugando a 300 × g durante 10 min a temperatura ambiente.
- Prepare una solución de reticulación de FA al 1% agregando 1,25 ml de FA al 37% a 45 ml de HBSS y mezcle invirtiendo el tubo varias veces.
NOTA: Agregue todo el 1.25 ml de FA sin dividir la cantidad.
PRECAUCIÓN: El formaldehído es altamente tóxico. - Resuspender el pellet celular en los 46,25 ml de solución de reticulación de FA al 1% preparada en el paso anterior pipeteando hacia arriba y hacia abajo.
- Incubar a temperatura ambiente durante exactamente 10 minutos en rotador, balancín o mediante una inversión manual suave del tubo cada 1-2 minutos.
- Apagar la reacción de reticulación añadiendo 2,5 ml de glicina 2,5 M (128 mM final) y mezclar bien invirtiendo el tubo.
- Incubar durante 5 minutos a temperatura ambiente, y luego en hielo durante al menos 15 minutos para detener la reticulación por completo.
- Centrifugar a temperatura ambiente para granular las células reticuladas a 1.000 × g durante 10 min y desechar el sobrenadante por aspiración.
- Lave las celdas una vez con 10 ml de DPBS y luego centrifugue a 1,000 × g durante 10 minutos a temperatura ambiente. Deseche el sobrenadante completamente usando una pipeta e inmediatamente proceda a la reticulación DSG.
NOTA: Tenga cuidado al lavar el pellet de celda, ya que los gránulos celulares pueden estar sueltos y las células pueden perderse.
- Tener células sembradas en un medio apropiado para cosechar 5 × 106 células por vaso.
- Reticulación con disuccinimidil glutarato
- Resuspender las células granuladas en 9,9 ml de DPBS antes de añadir 100 μL de 300 mM DSG (3 mM final). Mezclar por inversión.
NOTA: DSG es sensible a la humedad. Es importante preparar un nuevo stock de 300 mM DSG en DMSO el día de la reticulación.
PRECAUCIÓN: DSG en DMSO es altamente tóxico. - Reticulación de las celdas a temperatura ambiente durante 40 minutos en un rotador.
- Añadir 1,925 ml de glicina 2,5 M (400 mM final), invertir para mezclar e incubar a temperatura ambiente durante 5 min.
- Centrifugar las células a 2.000 × g durante 15 min a temperatura ambiente.
NOTA: Tenga cuidado al retirar el sobrenadante de los gránulos de células sueltas. - Resuspender el pellet en 1 ml de albúmina sérica bovina (BSA)-DPBS al 0,05% y transferir a un tubo de 1,7 ml.
NOTA: La adición de BSA puede ayudar a reducir la aglutinación celular. - Centrifugar las células a 2.000 × g durante 15 min a 4 °C y extraer el sobrenadante por pipeta.
NOTA: Para evitar perder el pellet, retire rápida y completamente el sobrenadante. - Congele el pellet en nitrógeno líquido y guárdelo a -80 °C o continúe inmediatamente con el siguiente paso.
- Resuspender las células granuladas en 9,9 ml de DPBS antes de añadir 100 μL de 300 mM DSG (3 mM final). Mezclar por inversión.
2. Captura de la conformación cromosómica
- Lisis celular y digestión de la cromatina
- Resuspender (pipeta) las alícuotas celulares reticuladas (~5 × 106 células) en 1 ml de tampón de lisis helado (receta en la Tabla Suplementaria S1) que contiene 10 μL de cóctel inhibidor de proteasa y transferir a un homogeneizador de rebote para una incubación de 15 minutos en hielo.
NOTA: Agregue inhibidores de proteasa al tampón de lisis inmediatamente antes de usarlo. - Mueva lentamente el mortero A hacia arriba y hacia abajo 30 veces para homogeneizar las células en hielo e incubar en hielo durante 1 minuto para permitir que las células se enfríen, antes de otros 30 golpes.
- Transfiera el lisado a un tubo de microcentrífuga de 1,7 ml.
NOTA: Mantenga la suspensión en movimiento porque a veces las células se pegan en la punta de la pipeta. - Centrifugar la suspensión lisada a 2.500 × g durante 5 min a temperatura ambiente.
- Deseche el sobrenadante y mueva o haga un vórtice con el pellet húmedo para resuspenderlo. Retire la mayor cantidad posible de sobrenadante para obtener una sustancia similar al yogur con grumos mínimos.
- Resuspender el pellet en 500 μL de tampón de restricción 1x helado (a partir de 10x; ver receta en la Tabla Suplementaria S1) y centrifugar durante 5 min a 2.500 × g. Repita este paso para un segundo lavado.
NOTA: El pellet en 1x Tampón de restricción es más granular que el pellet anterior en tampón de lisis. - Resuspender las células en un volumen final de 360 μL de 1x tampón de restricción mediante pipeteo después de agregar ~ 340 μL al volumen de arrastre del pellet, que depende del tamaño de la celda.
- Reservar 18 μL de cada lisado para probar la integridad de la cromatina (IC). Conservar las muestras de IC a 4 °C.
- Añadir 38 μL de dodecil sulfato de sodio (SDS) al 1% a cada tubo Hi-C (volumen total de 380 μL) y mezclar cuidadosamente pipeteando sin introducir burbujas.
PRECAUCIÓN: SDS es tóxico. - Incubar las muestras a 65 °C sin agitar durante exactamente 10 minutos para abrir la cromatina.
- Coloque inmediatamente los tubos en hielo y prepare la mezcla de digestión con Triton X-100 para apagar la SDS como se describe en la Tabla 1.
- Agregue 107 μL de la mezcla de digestión al tubo Hi-C (487 μL en total) para digerir la cromatina durante la noche (~ 16 h) a 37 ° C en un termomezclador con agitación de intervalo (por ejemplo, 900 rpm, 30 s encendido, 4 minutos apagado).
NOTA: La adición de Tritón a una concentración final del 1% sirve para apagar la SDS.
- Resuspender (pipeta) las alícuotas celulares reticuladas (~5 × 106 células) en 1 ml de tampón de lisis helado (receta en la Tabla Suplementaria S1) que contiene 10 μL de cóctel inhibidor de proteasa y transferir a un homogeneizador de rebote para una incubación de 15 minutos en hielo.
- Finaliza la biotinilación del ADN
- Después de la digestión durante la noche, transferir las muestras a 65 °C durante 20 minutos para desactivar la actividad endonucleasa restante.
- Durante la incubación, prepare una mezcla maestra de relleno como se muestra en la Tabla 2.
- Después de la incubación, coloque las muestras inmediatamente en hielo.
- Reservar un control de digestión (DC) de 10 μL para cada muestra y almacenar a 4 °C.
- Retire la condensación de la tapa con una pipeta o girando. A cada muestra, añadir 58 μL de mezcla de relleno de biotina (volumen total de la muestra 535 μL) y pipetear suavemente sin formar burbujas.
- Incubar las muestras a 23 °C durante 4 h en un termomezclador (por ejemplo, 900 rpm, 30 s encendido, 4 min apagado).
- Ligadura de fragmentos de ADN proximal
- Prepare la mezcla de ligadura como se muestra en la Tabla 3 mientras se está incubando el relleno de biotina.
- Añadir 665 μL de la mezcla de ligadura a cada muestra (volumen total de la muestra 1.200 μL). Mezclar suavemente por pipeteo.
- Incubar las muestras a 16 °C durante 4 h en un termomezclador con agitación a intervalos (por ejemplo, 900 rpm, 30 s encendido, 4 min apagado). Almacene estas muestras que contienen cromatina unida covalentemente a 4 °C durante unos días.
- Reversión de la reticulación
- Lleve los volúmenes de las muestras de CI y CC a 50 μL con 1x Tris Low EDTA (TLE; consulte la receta en la Tabla suplementaria S1).
- Añadir 10 μL de 10 mg/ml de proteinasa K a las muestras de IC y DC.
- Incubar a 65 °C durante la noche con agitación a intervalos (p. ej., 900 rpm, 30 s encendido, 4 min apagado). Alternativamente, realice una inversión de 30 minutos de la reticulación para estos controles durante la purificación del ADN de las muestras Hi-C.
- A cada muestra de Hi-C, añadir 50 μL de 10 mg/ml de proteinasa K e incubar a 65 °C durante al menos 2 h con agitación a intervalos (p. ej., 900 rpm, 30 s encendido, 4 min apagado).
- Añadir otros 50 μL de 10 mg/ml de proteinasa K a cada tubo de Hi-C (volumen total de la muestra de 1.300 μL) y continuar incubando a 65 °C durante la noche. Conservar a 4 °C hasta la purificación del ADN.
NOTA: La división de las incubaciones de proteinasa K asegura la digestión total de proteínas.
- Purificación del ADN
- Deje que los tubos se enfríen desde 65 °C hasta temperatura ambiente.
- Transfiera cada muestra a un tubo cónico de 15 ml y agregue 2,6 ml (2 veces el volumen) de fenol: cloroformo: alcohol isoamílico a cada tubo.
PRECAUCIÓN: Fenol: cloroformo: alcohol isoamílico es un irritante muy tóxico y es potencialmente cancerígeno. - Vortex cada tubo durante 1 min y luego transfiera su contenido a un tubo de bloqueo de fase de 15 ml.
- Centrifugar las muestras durante 5 minutos a velocidad máxima (1.500-3.500 × g) en una centrífuga de sobremesa.
- Vierta con cuidado la fase acuosa en un tubo de ultracentrífuga de 35 ml y agregue agua ultrapura a un volumen final de 1.250 μL.
NOTA: Utilice tubos para adaptarse a la ultracentrífuga disponible o dividir en varios tubos de microcentrífuga. - Añadirun volumen de 1/10 (~125 μL) de acetato de sodio 3 M y mezclar bien por inversión.
- Agregue un volumen de 2.5x (~ 3.4 ml) de etanol 100% helado a cada muestra, equilibre los tubos para la ultracentrifugación agregando etanol 100% helado y mezcle bien por inversión.
- Incubar los tubos en hielo seco durante ~15 min (evitar la solidificación).
- Centrifugar los tubos a 18.000 × g durante 30 min a 4 °C.
NOTA: Para rotores en ángulo: marque los tubos donde estará el pellet. - Con una pipeta, retire y deseche completamente el sobrenadante del lado que no sea pellet.
NOTA: En este punto, el pellet debe hacerse visible y puede marcarse en el tubo, ya que puede no ser claramente visible después del secado en el siguiente paso. - Seque al aire las muestras durante aproximadamente 10 minutos o hasta que estén visiblemente secas.
- Solubilizar cada pellet en 450 μL de 1x TLE por pipeteo o remolino y transferir a 0,5 ml Unidad de filtro centrífugo (UFC) con un corte de peso molecular de 3 kDa.
- Centrifugar la UFC a velocidad máxima durante 10 minutos y desechar el caudal. Lave cada tubo de ultracentrífuga con 450 μL adicionales de 1x TLE y transfiéralo a su UFC para otro lavado.
NOTA: Lavar la UFC de esta manera limita la pérdida de ADN al tiempo que reduce la concentración de sal. - Centrifugar la UFC a velocidad máxima durante 10 minutos y desechar el caudal.
- Agregue 80 μL de 1x TLE a la columna y gire la columna en un nuevo tubo de recolección antes de centrifugar durante 2 min a velocidad máxima para obtener un volumen final de ~ 100 μL.
- Añadir 1 μL de RnaseA (1 mg/ml; dilución de 10 veces de 10 mg/ml de cepa) a cada muestra e incubar a 37 °C en un bloque de calor, en un baño maría o en un termomezclador durante al menos 30 min.
- Después del tratamiento con Rnase, retirar las muestras a 37 °C y conservar a 4 °C hasta la fase de control de calidad.
- Comprobación de la calidad de la cromatina, la digestión enzimática y la ligadura de muestras
- Enfríe las muestras de CI y CC a temperatura ambiente después de invertir los enlaces cruzados en el paso 2.4.5. Luego, transfiera a un tubo de bloqueo de fase de 2 ml.
NOTA: Asegúrese de que el contenido de bloqueo de fase se centrifuga a un pellet (velocidad máxima de 2 min). - Añadir 200 μL de fenol:cloroformo:alcohol isoamílico y mezclar las muestras mediante vórtice durante 1 min.
- Centrifugar los tubos durante 5 min a velocidad máxima.
- Transfiera la fase acuosa de cada muestra (~50 μL) a un nuevo tubo de microfuga de 1,7 ml.
- Añadir 1 μL de Rnasa A (a partir de 1 mg/ml) e incubar a 37 °C durante al menos 30 min.
- Cargue las muestras en un gel de agarosa al 0,8% como se recomienda en la Tabla 4.
NOTA: Los resultados esperados de un control de calidad se muestran en la Figura 2. - Cuantificar el ADN por densitometría del gel o usando Qubit o Nanodrop.
NOTA: La cuantificación precisa garantiza el volumen de entrada correcto en la siguiente parte del protocolo. Utilice varios estándares con una cantidad conocida para construir una curva estándar.
- Enfríe las muestras de CI y CC a temperatura ambiente después de invertir los enlaces cruzados en el paso 2.4.5. Luego, transfiera a un tubo de bloqueo de fase de 2 ml.
Tabla 1: Reactivos de digestión. Haga clic aquí para descargar esta tabla.
Tabla 2: Reactivos de relleno de biotina. *Tenga en cuenta que las enzimas cambiantes pueden requerir diferentes tampones y dNTP biotinilados. Haga clic aquí para descargar esta tabla.
Tabla 3: Reactivos de mezcla de ligadura. Abreviatura: BSA = albúmina sérica bovina. Haga clic aquí para descargar esta tabla.
Tabla 4: Parámetros de carga de gel para la evaluación de la calidad y la selección de tamaño. Haga clic aquí para descargar esta tabla.

Figura 2: Gel de agarosa que muestra los resultados típicos del control de calidad de la purificación posterior al ADN. (A) El control de IC debe indicar una banda de ADN de alto peso molecular. (B) Las muestras DC y Hi-C muestran un rango de tamaños de ADN. La muestra Hi-C, después de haber sido combinada en fragmentos más grandes, debe ser de mayor peso molecular que la DC. El rango de concentración de los marcadores permite generar una curva estándar. Tenga en cuenta que, en este ejemplo, el CI se cargó en un gel separado, pero se recomienda cargar y ejecutar todas las muestras y controles juntos. Abreviaturas: CI = integridad de la cromatina; DC = control de la digestión; Hi-C = ligadura de proximidad. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
3. Preparación de la biblioteca de secuenciación Hi-C
- Eliminación de biotina de extremos no ligados
- Preparar reacciones de eliminación de biotina como se muestra en la Tabla 5.
NOTA: Normalmente, 10 μg de ADN es suficiente, pero se pueden usar hasta 30 μg. - Distribuir 2 alícuotas de 65 μL de cada reacción de 130 μL en dos tubos de PCR.
- Transfiera a un termociclador o máquina de PCR e incube como se describe en la Tabla 5.
NOTA: Las muestras pueden almacenarse aquí a 4 °C (días a semanas), -20 °C (a largo plazo) o trasladarse inmediatamente a la sonicación.
- Preparar reacciones de eliminación de biotina como se muestra en la Tabla 5.
- Sonicación
- Agrupe los duplicados de la muestra del paso de eliminación de biotina (volumen total de muestra de 130 μL) en un tubo sonicador de 130 μL para la sonicación.
- Sonicar las muestras utilizando los parámetros dados en la Tabla 6 para lograr una distribución estrecha y estrecha por debajo de 500 pb.
NOTA: Se pueden usar diferentes tipos de sonicadores, pero para una distribución de fragmentos estrecha (100-500 pb), la configuración del sonicador puede requerir optimización.
- Selección de tamaño con cuentas magnéticas
- Pipet el ADN sonicado de los tubos sonicadores en un tubo de unión baja de 1,7 ml.
- Llevar cada muestra a un volumen total de 500 μL con 1x TLE. Trate de obtener el volumen lo más cerca posible de 500 μL, ya que la proporción de la mezcla de muestras a perlas magnéticas es esencial para la selección del tamaño.
- Agregue 400 μL de mezcla de perlas magnéticas a cada tubo para lograr una proporción de mezcla de perlas magnéticas con respecto al volumen de muestra de 0.8.
NOTA: En estas condiciones, las perlas capturan fragmentos de ADN >300 pb, que será la Fracción Superior. El sobrenadante contendrá fragmentos <300 pb, que serán la Fracción Inferior. - Mezclar los tubos por vórtice e incubar durante 10 minutos a temperatura ambiente en un rotador. Para esta y otras relaciones de selección de tamaño, asegúrese de que el volumen completo de la muestra se mezcle bien. Busque el "modo errático" que tienen algunos rotores, que funciona bien para volúmenes más pequeños.
- Incubar durante 5 min a temperatura ambiente en un separador magnético de partículas (MPS).
- Durante la incubación, agregue 500 μL de la mezcla de perlas magnéticas a un tubo fresco de 1,7 μL de baja unión para cada muestra.
NOTA: Estos tubos se utilizarán para la siguiente selección de tamaño de la fracción inferior mediante la generación de una mezcla de perlas magnéticas a una relación de muestra de 1.1: 1. - Deje los tubos en el MPS durante 5 minutos.
- Retire el sobrenadante de las perlas y vuelva a suspender con 150 μL de la mezcla de perlas magnéticas.
NOTA: Este paso evita la saturación de las perlas con ADN al aumentar el número de cuentas sin aumentar el volumen. - Transfiera el sobrenadante del paso 3.3.5 al tubo etiquetado preparado para seleccionar la fracción inferior (paso 3.3.8).
NOTA: 150 μL de mezcla de perlas magnéticas + 400 μL de mezcla de perlas magnéticas 0.8x (550 μL en total) dividido por 500 μL de muestra inicial = 1.1x mezcla de perlas magnéticas a relación muestral. - Mezclar los tubos de la fracción inferior mediante vórtice e incubar durante 10 minutos a temperatura ambiente en un rotador.
NOTA: Las perlas se unirán a fragmentos de ADN >100 pb, lo que resultará en una fracción final unida a cuentas de 100-300 pb. - Coloque los tubos de fracción inferior en el MPS durante 5 minutos (temperatura ambiente).
- Retire el sobrenadante y centrifugar los tubos brevemente para eliminar aún más el sobrenadante tanto como sea posible.
- Lave las perlas de ambas fracciones dos veces usando 200 μL de etanol al 70% y recupere las perlas durante 5 minutos en el MPS cada vez.
- Después de un giro rápido en una centrífuga, retire el etanol por completo y seque aún más las perlas en el MPS.
NOTA: Secar hasta que el alcohol se evapore por completo. El pellet debe verse como chocolate negro sin agrietarse (puede tomar ~ 10 minutos). - Resuspender ambas fracciones en 50 μL de tampón TLE 1x. Incubar a temperatura ambiente durante 10 minutos y golpear o mover los tubos cada dos minutos para estimular la mezcla y la elución.
- Separe las perlas del sobrenadante en el MPS durante 5 minutos para ambas fracciones.
- Mantenga el sobrenadante de cada muestra. Pipet el sobrenadante en un tubo de unión baja de 1,7 ml.
NOTA: Las muestras pueden conservarse a 4 °C durante unos días o a -20 °C durante largos plazos. - Ejecute un gel de agarosa al 2% como en la Tabla 4 para determinar la calidad y cantidad de la muestra. Consulte la Figura 3 para ver un ejemplo de dicho gel.
NOTA: Si por experiencia, la sonicación es altamente reproducible, uno puede omitir este gel y proceder a finalizar la reparación inmediatamente. Se recomienda que los usuarios conserven las fracciones superiores hasta después de la valoración PCR. Las cantidades subóptimas de ADN que requerirían mucha amplificación por PCR pueden ser rescatadas del material en la Fracción Superior. - Cuantifique la cantidad de ADN del gel directamente después de generar una curva estándar a partir de la entrada de escalera de ADN conocida o mediante el uso de un Qubit o Nanodrop.
- Reparación final
- Preparar la mezcla de reparación final como en la Tabla 7 (cantidad por reacción dada).
- Transfiera los 46 μL restantes de ADN eluyido de la fracción inferior a los tubos de PCR y agregue 24 μL de la mezcla de reparación final preparada. Incubar en una máquina de PCR, como se propone en la Tabla 7.
- Una vez completado el programa, mantener las muestras a 4 °C hasta que se desplieguen.
- Extracción de productos de ligadura biotinilada con perlas recubiertas de estreptavidina
- Determinar la cantidad de perlas recubiertas de estreptavidina para cada biblioteca a partir de la selección de tamaño cuantificada (paso 3.3.19).
NOTA: Estas perlas recubiertas de estreptavidina (solución de 10 mg/ml) pueden unir 20 μg de ADN bicatenario por mg de perlas (= 20 μg / 100 μL perlas). Use 2 μL por cada 1 μg de ADN Hi-C, pero no menos de 10 μL. - Mezcle las perlas recubiertas de estreptavidina y pipete el volumen de perlas necesarias para cada biblioteca (calculado en el paso anterior) en tubos individuales de 1,7 ml de baja unión.
- Resuspender las perlas en 400 μL de tampón de lavado Tween (TWB; ver receta en la Tabla suplementaria S1) e incubar durante ~3 min a temperatura ambiente en un rotador (ver instrucciones en el paso 3.3.4).
- Separe las perlas del sobrenadante en el MPS durante 1 minuto y retire el sobrenadante.
- Lave las perlas pipeteando otros 400 μL de TWB.
- Separe las perlas del sobrenadante en el MPS durante 1 minuto y retire el sobrenadante.
- Agregue 400 μL de 2x Binding Buffer (BB) (receta en la Tabla Suplementaria S1) a las perlas y vuelva a suspender. Además, añadir 330 μL de 1x TLE y la solución de reparación final (del paso 3.4.3).
- Incubar las muestras durante 15 minutos a temperatura ambiente mientras se mezclan en un rotador.
- Separe las perlas del sobrenadante en el MPS durante 1 minuto y retire el sobrenadante.
- Añadir 400 μL de 1x BB a las perlas y volver a suspender.
- Separe las perlas del sobrenadante en el MPS durante 1 minuto y retire el sobrenadante.
- Agregue 100 μL de 1x TLE para lavar las perlas.
- Separe las perlas del sobrenadante en el MPS durante 1 minuto y retire el sobrenadante.
- Finalmente, agregue 41 μL de 1x TLE para resuspender las perlas.
- Determinar la cantidad de perlas recubiertas de estreptavidina para cada biblioteca a partir de la selección de tamaño cuantificada (paso 3.3.19).
- Cola A
- Prepare la mezcla de cola A como en la Tabla 8.
- Pipet las reacciones en tubos de PCR e incubar como en la Tabla 8.
- Coloque los tubos de PCR en hielo inmediatamente después de retirarlos del termociclador y transfiera el contenido a tubos de unión baja de 1,7 ml.
- Separe las perlas del sobrenadante en el MPS durante 1 minuto y deseche el sobrenadante.
- Añadir 400 μL de 1x tampón de ligadura, diluido de 5x tampón ligasa de ADN T4 con agua ultrapura.
- Separe las perlas del sobrenadante en el MPS durante 1 minuto y luego deseche el sobrenadante.
- Añadir 1x tampón de ligadura a un volumen final de 40 μL.
- Oligos adaptadores de recocido
- Preparar oligoculatas adaptadoras de 100 μM (Tabla 9).
NOTA: Ordene 250 nmoles de oligos purificados por HPLC. - Analice los adaptadores en tubos de PCR como se describe en la Tabla 9.
- Utilice un termociclador de PCR para aumentar gradualmente la temperatura de 0,5 °C/s a 97,5 °C. Mantener a 97,5 °C durante 2,5 min.
- Utilice un termociclador de PCR para aumentar gradualmente la temperatura a 0,1 °C/s durante 775 ciclos (alcanzando los 20 °C). Mantener la temperatura a 4 °C hasta su uso posterior.
- Añadir 83 μL de 1x tampón de recocido (receta en la tabla suplementaria S1) para diluir los adaptadores a 15 μM. Conservar los adaptadores a -20 °C.
- Preparar oligoculatas adaptadoras de 100 μM (Tabla 9).
- Secuenciación del adaptador de ligadura
- Prepare la mezcla de ligadura del adaptador en un tubo de unión baja de 1,7 ml (Tabla 10).
- Ligar durante 2 h a temperatura ambiente.
- Separe las perlas del sobrenadante en el MPS durante 1 minuto y deseche el sobrenadante.
- Añadir 400 μL de TWB y pipetear las perlas hacia arriba y hacia abajo con cuidado antes de la incubación en el rotador durante 5 minutos a temperatura ambiente. Separe las perlas del sobrenadante en el MPS y repita este paso una vez más.
- Separe las perlas del sobrenadante en el MPS (~ 1 min), deseche el sobrenadante y agregue 200 μL de 1x BB.
- Separe las perlas del sobrenadante en el MPS (~ 1 min) y deseche el sobrenadante.
- Agregue 200 μL de 1x tampón pre-PCR (de 10x; receta en la Tabla Suplementaria S1) y transfiéralo a un nuevo tubo de unión baja de 1.7 ml.
- Separe las perlas del sobrenadante en el MPS (~ 1 min) y deseche el sobrenadante.
- Añadir 20 μL de 1x tampón pre-PCR y mezclar por pipeteo.
- Mantener los tubos sobre hielo mientras estén en uso o almacenar a 4 °C.
- Optimización del número de ciclo de PCR mediante titulación
- Configure 30 μL de reacciones de mezcla maestra por muestra como en la Tabla 11 (cantidad por reacción dada).
- Ejecute el menor número de ciclos y tome una alícuota de 5 μL. Ejecute 2-3 ciclos adicionales en la reacción restante antes de tomar la siguiente alícuota de 5 μL. Repita para recoger cuatro alícuotas.
- Utilice los parámetros de PCR de la Tabla 11 para cada alícuota.
- Agregue 5 μL de agua y 2 μL de colorante 6x a cada muestra de 5 μL. Ejecute con un gel TBE de agarosa al 2% (receta en la Tabla Suplementaria S1) con 25-150 ng de escalera de bajo peso molecular. Consulte la figura 4 para ver los resultados esperados.
NOTA: Un número óptimo de ciclos para la amplificación final de PCR de la biblioteca [paso 3.10] es el número más bajo de ciclos para obtener un producto visible en el gel menos un ciclo.
- Amplificación final de PCR de la biblioteca
- Configure reacciones de 12 x 30 μL para amplificar cada biblioteca final para la secuenciación como se muestra en la Tabla 11.
- Ciclar las reacciones de PCR de acuerdo con la Tabla 11 después de determinar el número de ciclos después de la titulación de PCR (paso 3.9.3).
- Una vez que se complete la PCR, agrupe las muestras replicadas en un tubo de microfuga de baja unión de 1,7 ml.
- Coloque los tubos en el MPS y transfiera el sobrenadante a un nuevo tubo de microfuga de baja unión de 1,7 ml.
- Resuspender las perlas recubiertas de estreptavidina sobrantes en 20 μL de 1x tampón pre-PCR.
NOTA: Esta plantilla se puede reutilizar cuando se almacena a 4 °C durante días o semanas o a largo plazo a -20 °C.
- Eliminación de imprimaciones con mezcla de perlas magnéticas
- Utilice 1x tampón TLE (receta en la Tabla suplementaria S1) para ajustar el volumen del paso 3.10.4 a exactamente 360 μL.
- A cada muestra, añadir 360 μL de la mezcla de perlas magnéticas y pipetear hacia arriba y hacia abajo para mezclar.
- En un rotador, mezclar las muestras durante 10 minutos a temperatura ambiente.
- Separe las perlas del sobrenadante en el MPS a temperatura ambiente (3-5 min).
- Lave las perlas dos veces usando 200 μL de etanol al 70% y recupere las perlas durante 5 minutos en el MPS cada vez.
- Centrifugar rápidamente en una centrífuga y pipetear completamente el etanol. Seque las perlas en el aire en el MPS para evaporar aún más el etanol.
NOTA: El pellet debe verse como chocolate negro sin agrietarse (puede tomar ~ 10 minutos). - Añadir 30 μL de agua ultrapura y resuspender para eluyer el ADN durante 10 min a temperatura ambiente. Mueva los tubos cada 2 minutos para ayudar a mezclar.
- Separe las perlas del sobrenadante en el MPS durante 5 minutos.
- Recoja el sobrenadante de cada muestra en un tubo fresco de 1,7 ml.
- Ejecute 1 μL de la biblioteca en un gel TBE de agarosa al 2% (receta en la Tabla Suplementaria S1) para obtener una distribución de tamaño de fragmento y cuantificar la biblioteca final (Figura 5).
NOTA: La digestión ClaI solo puede ocurrir para las uniones DpnII-DpnII y sirve como un control de ligadura positiva que debería resultar en una distribución de tamaño de fragmento más baja para la biblioteca final. Las bibliotecas finales pueden almacenarse durante unos días a 4 °C, a largo plazo a -20 °C, o diluirse inmediatamente y enviarse para su secuenciación.
Tabla 5: Reactivos de eliminación de biotina y temperaturas Haga clic aquí para descargar esta tabla.
Tabla 6: Parámetros para la sonicación. Haga clic aquí para descargar esta tabla.
Tabla 7: Reactivos de reparación final y temperaturas. Haga clic aquí para descargar esta tabla.
Tabla 8: Reactivos de relaves A y temperaturas. Haga clic aquí para descargar esta tabla.
Tabla 9: Cebadores de PCR y oligos de adaptadores pareados con recocido de reactivos para recocido. Abreviatura: 5PHOS = 5' fosfato. Los asteriscos indican bases de ADN fosforotioadas. # Combine un oligo indexado con el oligo universal para recocir en un adaptador indexado. Haga clic aquí para descargar esta tabla.
Tabla 10: Reactivos de ligadura adaptadora. Haga clic aquí para descargar esta tabla.
Tabla 11: Reactivos de PCR y parámetros de ciclo. Haga clic aquí para descargar esta tabla.

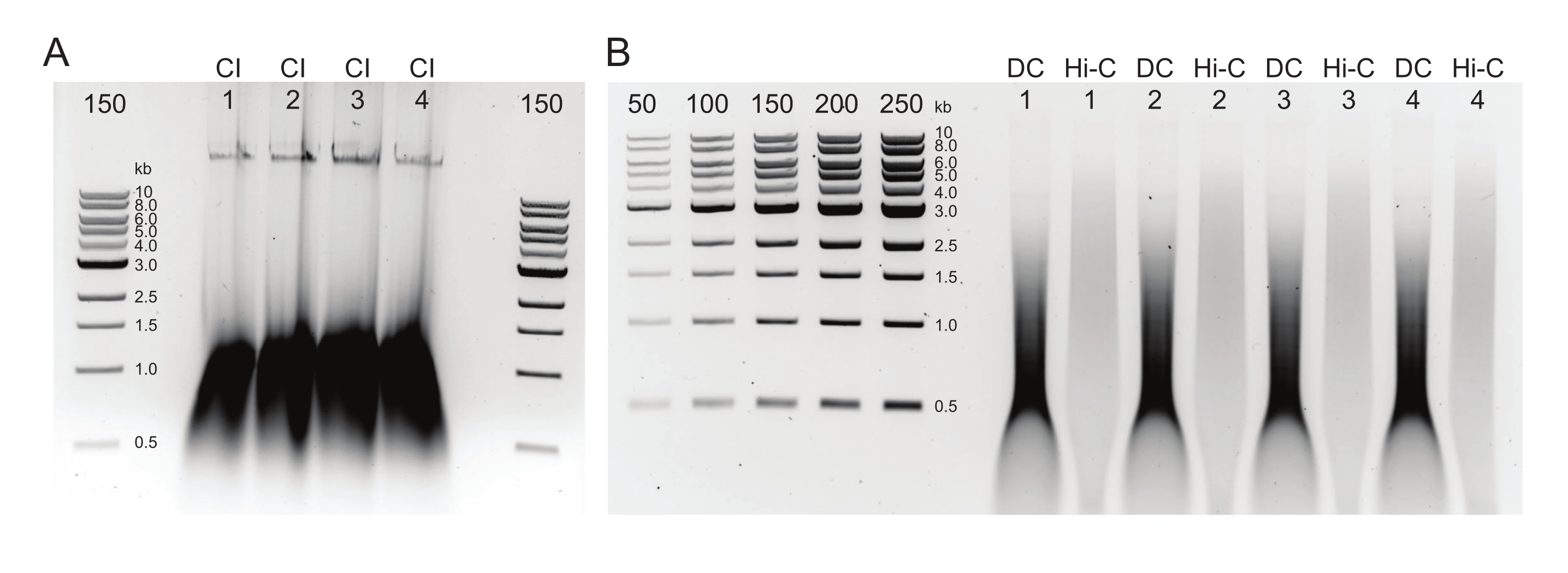
Figura 3: Gel de agarosa que muestra los resultados típicos de la selección posterior al tamaño. Se muestran las fracciones superior e inferior para cuatro muestras (numeradas del 1 al 4) de DpnII-DdeI Hi-C. El primer carril para cada muestra contiene la fracción superior, derivada de una mezcla de perlas magnéticas 0.8x, y los carriles segundo y tercero contienen una dilución de la fracción inferior derivada de una mezcla de perlas magnéticas 1.1x. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4: Gel de agarosa con resultados de titulación por PCR. A partir de 5 ciclos de PCR, se toman muestras después de cada 2 ciclos (5, 7, 9 y 11 ciclos) para cada una de las cuatro bibliotecas. Sobre la base de esta figura, se eligió 6 ciclos como el ciclo óptimo para cada muestra. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5: Productos finales de PCR. Después de la limpieza y la selección del tamaño, los productos de PCR (Hi-C) se cargaron junto a una fracción digerida por ClaI de la misma biblioteca (ClaI). Los fragmentos digeridos por ClaI indican la presencia de codiciadas ligaduras DpnII-DpnII. Tenga en cuenta que ClaI no digiere las uniones DpnII-DdeI y, por lo tanto, no todas las ligaduras contribuirán a una reducción de tamaño de esta restricción. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Resultados
Las figuras de este manuscrito fueron generadas a partir de un experimento separado y replicado del publicado previamente por Lafontaine et al.21. Después de obtener los datos de secuenciación de alto rendimiento, se utilizó Open Chromatin Collective (Open2C: https://github.com/open2c) para procesar los datos Hi-C. Una tubería similar se puede encontrar en el portal de datos del proyecto 4D Nucleome (https://data.4dnucleome.org/resources/data-analysis/hi_c-processing-pipeline). Brevemente, el destilador de tuberías Nextflow (https://github.com/open2c/distiller-nf) se implementó para (1) alinear las secuencias de moléculas Hi-C con el genoma de referencia, (2) analizar la alineación .sam y formar archivos con pares Hi-C, (3) filtrar duplicados de PCR y (4) pares agregados en matrices agrupadas de interacciones Hi-C. Estas matrices formateadas HDF5, llamadas coolers, pueden (1) verse en un servidor HiGlass (https://higlass.io/) y (2) analizarse utilizando un gran conjunto de herramientas computacionales de código abierto presentes en la colección "cooltools" mantenida por Open Chromatin Collective (https://github.com/open2c/cooltools) para extraer y cuantificar características de plegado como compartimentos, TAD y bucles.
Algunos indicadores de calidad de las bibliotecas Hi-C3.0 se pueden evaluar inmediatamente después de mapear pares de lectura a un genoma de referencia, utilizando algunas métricas / indicadores simples. Primero, típicamente ~ 50% de los pares de lectura secuenciados se pueden mapear de forma única para las células humanas. Debido a la naturaleza polimérica de los cromosomas, la mayoría de estas lecturas mapeadas (~ 60% -90%) representan interacciones dentro de un cromosoma (cis), con frecuencias de interacción que decaen rápidamente con el aumento de la distancia genómica (decaimiento dependiente de la distancia). La desintegración dependiente de la distancia se puede visualizar mejor en un "gráfico de escala", que muestra la probabilidad de contacto (por brazo cromosómico) en función de la distancia genómica. Encontramos que el uso de diferentes reticulantes y enzimas puede alterar la decaimiento dependiente de la distancia a distancias de largo y corto alcance17. La adición de reticulación DSG aumenta la detectabilidad de las interacciones a distancias cortas cuando se combina con enzimas como Mnase y combinaciones de DpnII-DdeI que producen fragmentos más pequeños (Figura 6A).
El decaimiento dependiente de la distancia también se puede observar directamente a partir de matrices de interacción 2D: las interacciones se vuelven más infrecuentes cuando se encuentran más lejos de la diagonal central (Figura 6B). Además, las características de plegamiento genómico, como compartimentos, TAD y bucles, se pueden identificar a partir de matrices Hi-C y gráficos de escala como desviaciones de la desintegración general dependiente de la distancia promedio de todo el genoma. Es importante destacar que la reticulación con DSG además de FA disminuye las ligaduras aleatorias, que no están restringidas debido a la naturaleza polimérica de los cromosomas y, por lo tanto, es más probable que ocurran entre cromosomas (en trans) (Figura 6C). La reducción de la ligadura aleatoria conduce a un aumento de las relaciones señal-ruido, especialmente para las interacciones intracromosómicas intercromosómicas y de muy largo alcance (>10-50 Mb).

Figura 6: Resultados representativos de bibliotecas Hi-C mapeadas y filtradas. (A) Gráficos de escala con probabilidad de contacto y su derivado para varias enzimas, ordenados por longitud de fragmento (arriba) y reticulación con FA o FA + DSG (abajo). La digestión con MNAse (microC) o DpnII-DdeI (Hi-C 3.0) aumenta significativamente los contactos de corto alcance (arriba) al igual que la adición de DSG a FA (abajo). (B) Las columnas muestran mapas de calor Hi-C de la digestión de DpnII después de la reticulación FA y la digestión de DpnII o DdeI después de la reticulación FA+DSG. Las flechas blancas muestran una fuerza creciente de los "puntos" después de la reticulación DSG y la digestión DdeI, lo que implica una mejor detección de los bucles de ADN. Las filas muestran diferentes partes del cromosoma 3 a resolución creciente, alineándose con el panel C: fila superior: cromosoma 3 completo (0-198,295,559 Mb); fila central: 186-196 Mb; fila inferior: 191.0-191.5 Mb. (C) Gráficos de cobertura para las regiones representadas en A. Las flechas negras muestran la cobertura más baja (%cis reads) para la reticulación solo de FA. Abreviaturas: FA = formaldehído; DSG = glutarato de disuccinimidilo; CHR = cromosoma. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
No todas las lecturas asignadas son útiles. Un segundo indicador de calidad es el número de duplicados de PCR. Es muy poco probable que las lecturas duplicadas exactas ocurran por casualidad después de la ligadura y la sonicación. Por lo tanto, tales lecturas probablemente resultaron de la amplificación de PCR y deben filtrarse. Los duplicados a menudo surgen cuando se requieren demasiados ciclos de PCR para amplificar las bibliotecas de baja complejidad. En general, para Hi-C, la mayoría de las bibliotecas solo necesitan de 5 a 8 ciclos de amplificación final de PCR, según lo determinado por la valoración PCR (ver paso 3.9; Figura 4). Sin embargo, se pueden obtener bibliotecas con suficiente complejidad incluso después de 14 ciclos de amplificación por PCR.
Otra categoría de lecturas duplicadas, los llamados duplicados ópticos, pueden surgir del proceso de amplificación en plataformas de secuenciación Illumina que utilizan celdas de flujo con patrones (como HiSeq4000). Los duplicados ópticos se encuentran ya sea por sobrecargar la celda de flujo, causando que los grupos (grandes) se llamen dos grupos separados, o por el reagrupamiento local de la molécula de extremo pareado original después de una primera ronda de PCR. Debido a que ambos tipos de duplicados ópticos son locales, se pueden identificar y distinguir de los duplicados de PCR por su ubicación en la celda de flujo. Mientras que las bibliotecas con >15% de duplicados de PCR necesitarían regeneración, las bibliotecas con duplicados ópticos se pueden volver a cargar después de optimizar el proceso de carga.
Cuadro complementario S1: Tampones y soluciones. Haga clic aquí para descargar esta tabla.
Discusión
Pasos críticos para el manejo celular
Aunque es posible utilizar un número menor de celdas de entrada, este protocolo se ha optimizado para ~ 5 × 106 celdas por carril de secuenciación (~ 400 M lecturas) para garantizar la complejidad adecuada después de la secuenciación profunda. Las células se cuentan mejor antes de la fijación. Para la generación de bibliotecas ultraprofundas, generalmente multiplicamos el número de carriles (y celdas) hasta que se alcanza la profundidad de lectura deseada. Para una fijación óptima, el medio que contiene suero debe ser reemplazado por PBS antes de la fijación de AF, y las soluciones fijadoras deben agregarse inmediatamente y sin gradientes de concentración15,22. Para la recolección celular, se prefiere el raspado a la tripsinización, porque la transición de una forma más plana a una forma esférica después de la tripsinización podría afectar la conformación nuclear. Después de la adición de DSG, los gránulos de células sueltas y grumosas se pierden fácilmente. Tenga cuidado al manipular células en esta etapa y agregue hasta 0.05% de BSA para disminuir la aglutinación.
Modificaciones al método
Este protocolo fue desarrollado utilizando células humanas17. Sin embargo, según la experiencia con la captura de la conformación cromosómica, este protocolo debería funcionar para la mayoría de las células eucariotas. Para una entrada significativamente menor (~ 1 × 106 celdas), recomendamos usar la mitad de los volúmenes para los procedimientos de captura de lisis y conformación [pasos 2.1-2.4]. Esto también permitiría realizar el aislamiento de ADN [paso 2.5] en una centrífuga de mesa con tubos de 1,7 ml, lo que podría mejorar la granulación para bajas concentraciones de ADN. La cuantificación del ADN (paso 2.6.6) indicará cómo proceder. Para cantidades bajas de ADN aislado (1-5 μg), sugerimos omitir la selección de tamaño (paso 3.3) y proceder con la eliminación de biotina después de reducir el volumen de 130 μL a ~ 45 μL con una UFC.
Este protocolo fue desarrollado específicamente para garantizar datos de alta calidad después de la posterior reticulación con FA y DSG y la digestión con DpnII y DdeI. Sin embargo, las estrategias alternativas de reticulación como FA seguida de EGS (etilenglicol bis (succinato de succinidil)), que también se utiliza en ChIP-seq23 y ChIA-PET24, podrían funcionar igualmente bien17. Del mismo modo, se pueden usar diferentes combinaciones de enzimas, como DpnII y HinfI18 o MboI, MseI y NlaIII19 para la digestión. Al adaptar combinaciones de enzimas, asegúrese de usar nucleótidos biotinilados que puedan llenar los voladizos específicos de 5' y use los tampones más óptimos para cada cóctel. DpnII viene con su propio tampón y el fabricante de enzimas recomienda un tampón específico para la digestión DdeI. Sin embargo, para la doble digestión con DpnII y DdeI en este protocolo, se recomienda el tampón de restricción porque está clasificado en 100% de actividad para ambas enzimas.
Solución de problemas de captura de conformación
Los tres pasos clave en la captura de la conformación cromosómica: reticulación, digestión y religación se han realizado antes de que los resultados se puedan visualizar en gel. Para determinar la calidad de cada uno de estos tres pasos y discernir dónde podrían haber surgido los problemas, se toman alícuotas antes (IC) y después de la digestión (DC) y se cargan en el gel junto con la muestra Hi-C ligada (Figura 2). Este gel se utiliza para determinar la calidad de la muestra Hi-C y si valdrá la pena continuar con el protocolo. Sin el IC y el DC, es difícil identificar posibles pasos subóptimos. Vale la pena señalar que la ligadura subóptima podría deberse a un problema en la ligadura en sí, el relleno o un problema con la reticulación. Para solucionar problemas de reticulación, asegúrese de no usar más de 1 × 107 celdas por biblioteca y comience con reactivos de reticulación nuevos y celdas limpias (es decir, enjuagadas con PBS). Para la ligadura, asegúrese de que las células y la mezcla de ligadura se mantengan en hielo. Añadir T4 DNA ligasa justo antes de la incubación de 4 h a 16 °C y mezclar bien.
Solución de problemas de preparación de la biblioteca
Si se necesitan más de 10 ciclos de PCR o no se puede ver ningún producto de PCR en el gel después de la titulación por PCR (Figura 4), hay algunas opciones para guardar la muestra Hi-C. Trabajando hacia atrás desde la titulación de PCR, la primera opción es probar la PCR nuevamente. Si todavía no hay suficiente producto, es posible intentar otra ronda de A-tailing y ligadura del adaptador (paso 3.6) después de lavar las perlas dos veces con 1x tampón TLE. Después de esta ligadura adicional de cola A y adaptador, se puede proceder a la titulación por PCR como antes. Si todavía no hay producto, la última opción es volver a sonizar la fracción 0.8x del paso 3.3 y proceder desde allí.
Limitaciones y ventajas de Hi-C3.0
Es importante darse cuenta de que Hi-C es un método basado en la población que captura la frecuencia promedio de interacciones entre pares de loci en la población celular. Algunos análisis computacionales están diseñados para desentrañar combinaciones de conformaciones de una población25, pero en principio, Hi-C es ciego a las diferencias entre células. Aunque es posible realizar Hi-C26,27 de una sola celda y se pueden hacer inferencias computacionales28, Hi-C de una sola celda no es adecuado para obtener información 3C de ultra alta resolución. Una limitación adicional de Hi-C es que solo detecta interacciones por pares. Para detectar interacciones multicontacto, se pueden usar cortadores frecuentes combinados con secuenciación de lectura corta (Illumina)16 o realizar multicontacto 3C29 o 4C30, utilizando secuenciación de lectura larga de plataformas PacBio u Oxford Nanopore. También se han desarrollado derivados de Hi-C para detectar específicamente contactos entre y a lo largo de cromátidas hermanas31,32.
Aunque Hi-C19 y Micro-C33 se pueden usar para generar mapas de contactos a resoluciones subkilobase, ambos requieren una gran cantidad de lecturas de secuenciación y esto puede convertirse en una tarea costosa. Para llegar a una resolución similar o incluso mayor sin los costos, se puede aplicar el enriquecimiento para regiones genómicas específicas (captura-C 34) o interacciones proteicas específicas (ChiA-PET 35, PLAC-seq36, Hi-ChIP37). La fuerza y la desventaja de estas aplicaciones de enriquecimiento es que solo se muestrea un número limitado de interacciones. Con tales enriquecimientos, el aspecto global de Hi-C (y la opción de normalización global) se pierde.
Importancia y aplicaciones potenciales de Hi-C3.0
Este protocolo fue diseñado para permitir 3C ultraprofundo de alta resolución y, al mismo tiempo, detectar características de plegado a gran escala, como TAD y compartimentos17 (Figura 6). Este protocolo comienza con 5 × 106 celdas por tubo para cada biblioteca Hi-C, que debería ser material más que suficiente para secuenciar uno o dos carriles en una celda de flujo para obtener hasta mil millones de lecturas de extremo emparejado. Para la secuenciación ultraprofunda, se deben preparar múltiples tubos de 5 × 106 células, dependiendo del número de lecturas mapeadas y duplicados de PCR. A la resolución más alta (<1 kb), las interacciones en bucle se encuentran principalmente entre los sitios CTCF, pero también se pueden detectar interacciones promotor-potenciador. Los lectores pueden consultar Akgol Oksuz et al.17 para una descripción detallada del análisis de datos.
Divulgaciones
Los autores no tienen conflictos de intereses que revelar.
Agradecimientos
Nos gustaría agradecer a Denis Lafontaine por el desarrollo del protocolo y a Sergey Venev por la asistencia bioinformática. Este trabajo fue apoyado por una subvención del Programa de Nucleomas 4D del Fondo Común de los Institutos Nacionales de Salud a J.D. (U54-DK107980, UM1-HG011536). J.D. es investigador del Instituto Médico Howard Hughes.
Este artículo está sujeto a la política de acceso abierto a publicaciones del HHMI. Los jefes de laboratorio de HHMI han otorgado previamente una licencia CC BY 4.0 no exclusiva al público y una licencia sublicenciable a HHMI en sus artículos de investigación. De conformidad con esas licencias, el manuscrito aceptado por el autor de este artículo puede estar disponible gratuitamente bajo una licencia CC BY 4.0 inmediatamente después de su publicación.
Materiales
| Name | Company | Catalog Number | Comments |
| 1 kb Ladder | New England Biolabs | N3232L | |
| Agarose | Invitrogen | 16500100 | |
| Agencourt AMPure XP magnetic beads , 60 mL | Beckman Coulter | A63881 | |
| Amicon Ultra-0.5 Centrifugal Filter Unit (CFU) | EMD Millipore | UFC500396 | |
| Annealing Buffer (5x) | See recipe in supplemental materials | ||
| ATP 10 mM | ThermoFisher | R0441 | |
| Avanti J-25i High Speed Refrigerated ultra-centrifuge | Beckman Coulter | ||
| beckman ultracentrifuge tube 35 mL | Beckman Coulter | 357002 | |
| Binding Buffer (2x) | See recipe in supplemental materials | ||
| biotin-14-dATP 0.4 mM | Invitrogen | 19524-016 | |
| BSA 10 mg/mL | New England Biolabs | B9000S | dilute from 20 mg/mL |
| Cell scraper | Falcon | 353089 | |
| Cell scraper | Corning | 3008 | |
| Conical polypropylene tubes 50 mL | Denville | C1062-P | |
| Conical tube 15 mL | Denville | C1017-P | |
| Covaris micro tube AFA fiber with snap-cap 130 µL | Covaris | 520045/520077 | |
| Covaris Sonicator | Covaris | E220/E220evolution/M220 | |
| Culture flask 175 cm2 | Falcon | 353112 | |
| Culture plates 150 mm x 25 mm | Corning | 430599 | |
| dATP 1 mM | Invitrogen | 56172 | |
| dATP 10 mM | Invitrogen | 56172 | |
| dCTP 10 mM | Invitrogen | 56173 | |
| DdeI | New England Biolabs | R0175L | |
| dGTP 10 mM | Invitrogen | 56174 | |
| DMSO | Sigma | D2650-5x10ML | |
| dNTP mix 25 mM | Invitrogen | 10297117 | |
| Dounce homogenizer | DWK Life Sciences | 8853010002/8853030002 | |
| DPBS | Gibco | 14190-144 | |
| DpnII | New England Biolabs | R0543M | |
| DSG | ThermoScientific | 20593 | |
| dTTP 10 mM | Invitrogen | 56175 | |
| Ethanol 70% | Fisher | A409-4 | Diluted from 100% |
| Ethidium Bromide | Fisher | BP1302-10 | |
| Formaldehyde (37%) | Fisher | BP531-500 | |
| Gel loading dye (6x ) | New England Biolabs | B7024S | |
| Glycine in ultrapure water 2.5 M | Sigma | G8898-1KG | |
| HBSS | Gibco | 14025-092 | |
| Igepal CA-630 detergent | MP Biomedicals | 198596 | |
| Klenow DNA polymerase 5 U/µL | New England Biolabs | M0210L | |
| Klenow Fragment 3-->5’ exo-, 5 U/µL | New England Biolabs | M0212L | |
| ligation buffer (10x) | New England Biolabs | B7203S | |
| Liquid nitrogen | |||
| LoBind microcentrifuge tube 1.7 mL | Eppendorf | 22431021 | |
| Low Molecular Weight DNA Ladder | New England Biolabs | N3233L | |
| Lysis buffer | See recipe in supplemental materials | ||
| Magnetic Particle separator | ThermoFisher | 12321D | |
| Microfuge tubes 1.7 mL | Axygen | MCT-175-C | |
| MyOne Streptavidin C1 beads | Invitrogen | 65001 | |
| NEBuffer 2.1 (10x) | New England Biolabs | B7002S | |
| NEBuffer 3.1 (10x) | New England Biolabs | B7203S | |
| PBS | Gibco | 70013-032 | |
| PCR (strip) tubes | Biorad | TBS0201/ TCS0803 | |
| PCR thermocycler | Biorad | T100 | |
| Pfu Ultra II Buffer (10x) | Agilent | Comes with Pfu Ultra | |
| PfuUltra II Fusion HS DNA Polymerase | Agilent | 600674 | |
| Phase lock tube 15 mL | Qiagen | 129065 | |
| Phase lock tubes 2 mL | Qiagen | 129056 | |
| Phenol:chloroform:isoamyl alcohol | Invitrogen | 15593-049 | |
| Protease inhibitor cocktail | ThermoFisher | 78440 | |
| Proteinase K in ultrapure water 10 mg/mL | Invitrogen | 25530-031 | |
| Refrigerated Centrifuge | Eppendorf | 5810R | |
| RNase A, DNase and protease-free 10 mg/mL | Thermo Scientific | EN0531 | |
| Rotator | Argos technologies | EW-04397-40 or rocking platform | |
| SDS 1% | Fisher | BP13111 | |
| Sodium acetate pH = 5.2, 3 M | Sigma | ||
| Sub-Cell GT Horizontal Electrophoresis System | Biorad | 1704401 | |
| T4 DNA ligase 1 U/µL | Invitrogen | 100004817 | |
| T4 DNA polymerase | New England Biolabs | M0203L | |
| T4 DNA polymerase | New England Biolabs | M0203L | |
| T4 DNA polymerase 3 U/µL | New England Biolabs | M0203L | |
| T4 ligation buffer (5x) | Invitrogen | Y90001 | |
| T4 polynucleotide kinase 10 U/µL | New England Biolabs | M0201L | |
| Tabletop centrifuge | Eppendorf | 5425 | |
| TBE buffer | See recipe in supplemental materials | ||
| Tris Low EDTA Buffer (TLE) | See recipe in supplemental materials | ||
| Triton X-100 (10%) | Sigma | 93443 | |
| Truseq adapter oligos | Integrated DNA Technologies (IDT)) | https://www.idtdna.com/site/order/oligoentry | 250 nmole and HPLC purified |
| Tween 20 detergent | Fisher | 9005-64-5 | |
| Tween Wash Buffer | See recipe in supplemental materials | ||
| Vortex | Scientific Industries | (G560)SI-0236 |
Referencias
- Dekker, J., Rippe, K., Dekker, M., Kleckner, N. Capturing chromosome conformation. Science. 295 (5558), 1306-1311 (2002).
- Simonis, M., et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nature Genetics. 38 (11), 1348-1354 (2006).
- Dostie, J., et al. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Research. 16 (10), 1299-1309 (2006).
- Lieberman-Aiden, E., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326 (5950), 289-293 (2009).
- Stadhouders, R., et al. Multiplexed chromosome conformation capture sequencing for rapid genome-scale high-resolution detection of long-range chromatin interactions. Nature Protocols. 8 (3), 509-524 (2013).
- Kalhor, R., Tjong, H., Jayathilaka, N., Alber, F., Chen, L. Genome architectures revealed by tethered chromosome conformation capture and population-based modeling. Nature Biotechnology. 30 (1), 90-98 (2012).
- Hsieh, T. H., et al. Mapping nucleosome resolution chromosome folding in yeast by micro-C. Cell. 162 (1), 108-119 (2015).
- Hsieh, T. -H. S., Fudenberg, G., Goloborodko, A., Rando, O. J. Micro-C XL: assaying chromosome conformation from the nucleosome to the entire genome. Nature Methods. 13 (12), 1009-1011 (2016).
- Denker, A., de Laat, W. The second decade of 3C technologies: detailed insights into nuclear organization. Genes & Development. 30 (12), 1357-1382 (2016).
- Rodley, C. D., Bertels, F., Jones, B., O’Sullivan, J. M. Global identification of yeast chromosome interactions using Genome conformation capture. Fungal Genetics and Biology. 46 (11), 879-886 (2009).
- Rao, S. S., et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 159 (7), 1665-1680 (2014).
- Alipour, E., Marko, J. F. Self-organization of domain structures by DNA-loop-extruding enzymes. Nucleic Acids Research. 40 (22), 11202-11212 (2012).
- Lajoie, B. R., Dekker, J., Kaplan, N. The Hitchhiker’s guide to Hi-C analysis: Practical guidelines. Methods. 72, 65-75 (2015).
- Belaghzal, H., Dekker, J., Gibcus, J. H. Hi-C 2.0: An optimized Hi-C procedure for high-resolution genome-wide mapping of chromosome conformation. Methods. 123, 56-65 (2017).
- Golloshi, R., Sanders, J. T., McCord, R. P. Iteratively improving Hi-C experiments one step at a time. Methods. 142, 47-58 (2018).
- Darrow, E. M., et al. Deletion of DXZ4 on the human inactive X chromosome alters higher-order genome architecture. Proceedings of the National Academy of Sciences of the United States of America. 113 (31), 4504-4512 (2016).
- Akgol Oksuz, B., et al. Systematic evaluation of chromosome conformation capture assays. Nature Methods. 18 (9), 1046-1055 (2021).
- Ghuryeid, J., et al. Integrating Hi-C links with assembly graphs for chromosome-scale assembly. PLoS Computational Biology. 15 (8), 1007273(2019).
- Gu, H., et al. Fine-mapping of nuclear compartments using ultra-deep Hi-C shows that active promoter and enhancer elements localize in the active A compartment even when adjacent sequences do not. bioRxiv. , (2021).
- Ramani, V., et al. Mapping 3D genome architecture through in situ DNase Hi-C. Nature Protocols. 11 (11), 2104-2121 (2016).
- Lafontaine, D. L., Yang, L., Dekker, J., Gibcus, J. H. Hi-C 3.0: Improved Protocol for Genome-Wide Chromosome Conformation Capture. Current Protocols. 1 (7), 198(2021).
- Belton, J. -M. M., et al. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods. 58 (3), 268-276 (2012).
- Truch, J., Telenius, J., Higgs, D. R., Gibbons, R. J. How to tackle challenging ChIP-Seq, with long-range cross-linking, Using ATRX as an example. Methods in Molecular Biology. 1832, Clifton, N.J. 105-130 (2018).
- Wang, P., et al. In situ chromatin interaction analysis using paired-end tag sequencing. Current Protocols. 1 (8), 174(2021).
- Tjong, H., et al. Population-based 3D genome structure analysis reveals driving forces in spatial genome organization. Proceedings of the National Academy of Sciences of the United States of America. 113 (12), 1663-1672 (2016).
- Nagano, T., et al. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature. 547 (7661), 61-67 (2017).
- Ramani, V., et al. Massively multiplex single-cell Hi-C. Nature Methods. 14 (3), 263-266 (2017).
- Meng, L., Wang, C., Shi, Y., Luo, Q. Si-C is a method for inferring super-resolution intact genome structure from single-cell Hi-C data. Nature Communications. 12 (1), 4369(2021).
- Tavares-Cadete, F., Norouzi, D., Dekker, B., Liu, Y., Dekker, J. Multi-contact 3C reveals that the human genome during interphase is largely not entangled. Nature Structural & Molecular Biology. 27 (12), 1105-1114 (2020).
- Vermeulen, C., et al. Multi-contact 4C: long-molecule sequencing of complex proximity ligation products to uncover local cooperative and competitive chromatin topologies. Nature Protocols. 15 (2), 364-397 (2020).
- Oomen, M. E., Hedger, A. K., Watts, J. K., Dekker, J. Detecting chromatin interactions between and along sister chromatids with SisterC. Nature Methods. 17 (10), 1002-1009 (2020).
- Mitter, M., et al. Conformation of sister chromatids in the replicated human genome. Nature. 586 (7827), 139-144 (2020).
- Krietenstein, N., et al. Ultrastructural Details of Mammalian Chromosome Architecture. Molecular Cell. 78 (3), 554-565 (2020).
- Hughes, J. R., et al. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nature Genetics. 46 (2), 205-212 (2014).
- Fullwood, M. J., et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 462 (7269), 58-64 (2009).
- Fang, R., et al. Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Research. 26 (12), 1345-1348 (2016).
- Mumbach, M. R., et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nature Methods. 13 (11), 919-922 (2016).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados