
Method Article
Захват конформации хромосом по длинным шкалам
В этой статье
Резюме
Hi-C 3.0 представляет собой улучшенный протокол Hi-C, который сочетает в себе формальдегидные и дисукцинимидиллутаратные сшивки с коктейлем ферментов dpnII и DdeI для увеличения отношения сигнал/шум и разрешения обнаружения взаимодействия хроматина.
Аннотация
Захват конформации хромосом (3C) используется для обнаружения трехмерных взаимодействий хроматина. Как правило, химическое сшивание с формальдегидом (ФА) используется для фиксации взаимодействий хроматина. Затем переваривание хроматина ферментом рестрикции и последующее религирование концов фрагмента превращает трехмерную (3D) близость в уникальные продукты лигирования. Наконец, после разворота сшивок, удаления белка и выделения ДНК ДНК срезается и подготавливается к высокопроизводительному секвенированию. Частота бесконтактного лигирования пар локусов является мерой частоты их колокализации в трехмерном пространстве в клеточной популяции.
Секвенированная библиотека Hi-C предоставляет общегеномную информацию о частотах взаимодействия между всеми парами локусов. Разрешение и точность Hi-C зависит от эффективного сшивания, которое поддерживает контакты хроматина и частую и равномерную фрагментацию хроматина. В этой статье описывается улучшенный in situ протокол Hi-C, Hi-C 3.0, который повышает эффективность сшивки путем объединения двух сшивающих (формальдегид [FA] и дисукцинимидиллутарат [DSG]), с последующим более тонким пищеварением с использованием двух ферментов рестрикции (DpnII и DdeI). Hi-C 3.0 - это единый протокол для точной количественной оценки признаков складывания генома в меньших масштабах, таких как петли и топологически ассоциирующие домены (TADs), а также особенностей в более крупных масштабах ядра, таких как компартменты.
Введение
Захват конформации хромосом используется с 2002 года1. По сути, каждый вариант захвата конформации зависит от фиксации взаимодействий ДНК-белок и белок-белок для сохранения 3D-организации хроматина. За этим следует фрагментация ДНК, обычно путем рестрикции пищеварения, и, наконец, религирование близлежащих концов ДНК для преобразования пространственно проксимальных локусов в уникальные ковалентные последовательности ДНК. Первоначальные протоколы 3C использовали ПЦР для выборки конкретных взаимодействий «один к одному». Последующие анализы 4C позволили обнаружить взаимодействия «один ко всем»2, в то время как 5C обнаружил взаимодействия «многие ко многим»3. Захват конформации хромосом полностью осуществился после внедрения высокопроизводительного секвенирования следующего поколения (NGS), которое позволило обнаружить геномные взаимодействия «все ко всем» с использованием геномных Hi-C4 и сопоставимых методов, таких как 3C-seq5, TCC6 и Micro-C 7,8 (см. также обзор Denker и De Laat9).
В Hi-C биотинилированные нуклеотиды используются для обозначения 5'-свесов после пищеварения и перед лигированием (рисунок 1). Это позволяет выбирать правильно переваренные и религированные фрагменты с использованием бусин, покрытых стрептавидином, что отличает их от GCC10. Важное обновление протокола Hi-C было реализовано Rao et al.11, которые выполнили переваривание и религирование в интактных ядрах (т.е. in situ) для уменьшения ложных продуктов лигирования. Кроме того, замена пищеварения HindIII перевариванием MboI (или DpnII) уменьшала размер фрагмента и увеличивала потенциал разрешения Hi-C. Это увеличение позволило обнаружить относительно небольшие структуры и более точную геномную локализацию точек контакта, таких как петли ДНК между мелкими цис-элементами, например, петли между CTCF-связанными сайтами, генерируемыми экструзией петли11,12. Однако этот потенциал имеет свою цену. Во-первых, двукратное увеличение разрешения требует четырехкратного (22) увеличения последовательности чтения13. Во-вторых, небольшие размеры фрагментов увеличивают возможность ошибочного принятия непереваренных соседних фрагментов за переваренные и лигированные фрагменты14. Как уже упоминалось, в Hi-C переваренные и религированные фрагменты отличаются от непереваренных фрагментов наличием биотина в лигативном соединении. Тем не менее, требуется правильное удаление биотина с нелигированных концов, чтобы гарантировать, что только лигационные соединения вытягиваются вниз14,15.
С уменьшением стоимости NGS становится возможным более детально изучить сворачивание хромосом. Чтобы уменьшить размер фрагментов ДНК и тем самым увеличить разрешение, протокол Hi-C может быть адаптирован для более частого использования редукционных ферментов16 или для использования комбинаций ферментоврестрикции 17,18,19. Альтернативно,MNase 7,8 в Micro-C и DNase в DNase Hi-C20 могут быть титрованы для достижения оптимального пищеварения.
Недавняя систематическая оценка основ методов 3C показала, что обнаружение особенностей сворачивания хромосом на каждой шкале длины значительно улучшилось благодаря последовательному сшиванию с 1% FA, за которым следует 3 мМ DSG17. Кроме того, Hi-C с пищеварением HindIII был лучшим вариантом для обнаружения крупномасштабных складных функций, таких как компартменты, и что Micro-C превосходил в обнаружении мелкомасштабных складных функций, таких как петли ДНК. Эти результаты привели к разработке единой стратегии высокого разрешения «Hi-C 3.0», которая использует комбинацию сшивающих устройств FA и DSG с последующим двойным перевариванием с эндонуклеазами DpnII и DdeI21. Hi-C 3.0 обеспечивает эффективную стратегию для общего использования, поскольку он точно обнаруживает складные элементы на всех масштабах длины17. Экспериментальная часть протокола Hi-C 3.0 подробно описана здесь и показаны типичные результаты, которые можно ожидать после секвенирования.

Рисунок 1: Процедура Hi-C в шесть этапов. Клетки фиксируются сначала ФА, а затем DSG (1). Затем лизис предшествует двойному пищеварению с DdeI и DpnII (2). Биотин добавляют путем заполнения свеса, а проксимальные притупленные концы перевязывают (3) перед очисткой ДНК (4). Биотин удаляется с нелигированных концов перед обработкой ультразвуком и подбором размера (5). Наконец, вытягивание биотина позволяет осуществлять лигирование адаптера и библиотечное усиление с помощью ПЦР (6). Сокращения: FA = формальдегид; DSG = дисукцинимидиллутарат; B = Биотин. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
протокол
1. Фиксация с помощью сшивки
- Фиксация формальдегида: начиная с клеток в монослое
- Посеять клетки в соответствующей среде для сбора урожая 5 × 106 клеток на пластину 150 мм.
ПРИМЕЧАНИЕ: Пользователи могут выбрать любой предпочтительный контейнер, который обеспечивает оптимальный рост любой клеточной линии млекопитающих. Кроме того, клетки могут быть выделены из ткани. - Аспирируйте среду пипеткой Пастера, соединенной с вакуумной ловушкой из пластины 150 мм, промывайте 2x с ~10 мл HBSS.
- Непосредственно перед сшиванием приготовьте 1% раствор для сшивания FA в пробирке объемом 50 мл, объединив 22,5 мл HBSS и 625 мкл 37% FA до конечной 1% концентрации. Мягко перемешайте, покачивая.
ВНИМАНИЕ: Используйте вытяжной капюшон; формальдегид токсичен. - Чтобы сшить ячейки, налейте 23,125 мл 1% раствора FA на каждую пластину размером 15 см.
- Инкубировать при комнатной температуре в течение 10 мин и аккуратно раскачивать пластины вручную каждые 2 мин.
- Добавьте 1,25 мл 2,5 М глицина (128 мМ конечного) и осторожно закрутите пластину, чтобы погасить реакцию сшивания.
- Инкубировать при комнатной температуре в течение 5 мин и продолжать инкубацию на льду не менее 15 мин, чтобы остановить сшивание.
- Соскоблите ячейки с пластин с помощью скребка или резины полицейского.
- Переложите клеточную суспензию на коническую трубку объемом 50 мл с пипеткой. Центрифугу при 1000 × г в течение 10 мин при комнатной температуре и выбросьте супернатант путем аспирации.
- Вымойте гранулу ячейки один раз 10 мл фосфатно-буферного физиологического раствора Dulbecco (DPBS), используя пипетку для повторного суспендирования. Затем центрифугу при 1000 × г в течение 10 мин при комнатной температуре. Немедленно перейдите к сшиванию DSG.
ПРИМЕЧАНИЕ: Будьте осторожны при промывке гранул клетки, так как гранулы клеток могут быть рыхлыми, и ячейки могут быть потеряны.
- Посеять клетки в соответствующей среде для сбора урожая 5 × 106 клеток на пластину 150 мм.
- Фиксация формальдегида: начиная с клеток в суспензии
- Посеять клетки в соответствующей среде, чтобы собрать 5 × 106 клеток на сосуд.
ПРИМЕЧАНИЕ: Пользователи могут выбрать любой предпочтительный контейнер, который обеспечивает оптимальный рост клеток любой клеточной линии млекопитающих. - Непосредственно перед сбором урожая подсчитайте клетки и переведите 5 × 106 клеток в коническую трубку объемом 50 мл.
- Аккуратно гранулируют клетки центрифугированием при 300 × г в течение 10 мин при комнатной температуре.
- Приготовьте 1% раствор для сшивания FA, добавив 1,25 мл 37% FA к 45 мл HBSS и перемешайте, перевернув трубку несколько раз.
ПРИМЕЧАНИЕ: Добавьте все 1,25 мл FA без разделения количества.
ВНИМАНИЕ: Формальдегид очень токсичен. - Повторно суспендируют ячейку гранулы в 46,25 мл 1% раствора для сшивания FA, приготовленного на предыдущем этапе путем пипетки вверх и вниз.
- Инкубировать при комнатной температуре ровно 10 мин на ротаторе, коромысле или путем мягкой ручной инверсии трубки каждые 1-2 мин.
- Погасите реакцию сшивания, добавив 2,5 мл 2,5 М глицина (128 мМ финала) и хорошо перемешайте, перевернув трубку.
- Инкубировать в течение 5 мин при комнатной температуре, а затем на льду не менее 15 мин, чтобы полностью прекратить сшивание.
- Центрифугу при комнатной температуре гранулируют сшитые ячейки при 1000 × г в течение 10 мин и выбрасывают супернатант путем аспирации.
- Промыть ячейки один раз 10 мл DPBS, а затем центрифугу при 1000 × г в течение 10 мин при комнатной температуре. Полностью отбросьте супернатант с помощью пипетки и сразу же переходите к сшиванию DSG.
ПРИМЕЧАНИЕ: Будьте осторожны при промывке гранул клетки, так как гранулы клеток могут быть рыхлыми, и ячейки могут быть потеряны.
- Посеять клетки в соответствующей среде, чтобы собрать 5 × 106 клеток на сосуд.
- Сшивание с дисукцинимидиллутаратом
- Повторно суспендировать гранулированные ячейки в 9,9 мл DPBS перед добавлением 100 мкл 300 мМ DSG (3 мМ конечного). Смешивать путем инверсии.
ПРИМЕЧАНИЕ: DSG чувствительна к влаге. Важно подготовить свежий запас 300 мМ DSG в DMSO в день сшивания.
ВНИМАНИЕ: DSG в ДМСО очень токсичен. - Сшивайте ячейки при комнатной температуре в течение 40 мин на ротаторе.
- Добавьте 1,925 мл 2,5 М глицина (400 мМ конечного), инвертируйте для смешивания и инкубируйте при комнатной температуре в течение 5 мин.
- Центрифугирование клеток при 2000 × г в течение 15 мин при комнатной температуре.
ПРИМЕЧАНИЕ: Будьте осторожны при удалении супернатанта из гранул сыпучих клеток. - Повторно суспендируют гранулы в 1 мл 0,05% бычьего сывороточного альбумина (BSA)-DPBS и перекладывают в пробирку объемом 1,7 мл.
ПРИМЕЧАНИЕ: Добавление BSA может помочь уменьшить слипание клеток. - Центрифугируют клетки при 2000 × г в течение 15 мин при 4 °C и удаляют супернатант пипеткой.
ПРИМЕЧАНИЕ: Чтобы избежать потери гранулы, быстро и полностью удалите супернатант. - Заморозьте гранулы в жидком азоте и храните при -80 °C или сразу переходите к следующему этапу.
- Повторно суспендировать гранулированные ячейки в 9,9 мл DPBS перед добавлением 100 мкл 300 мМ DSG (3 мМ конечного). Смешивать путем инверсии.
2. Захват конформации хромосом
- Лизис клеток и переваривание хроматина
- Повторное суспендирование (пипет) сшитых клеточных аликвот (~5 × 106 клеток) в 1 мл буфера ледяного лизиса (рецепт в Дополнительной таблице S1), содержащего 10 мкл коктейля ингибитора протеазы, и перенос в гомогенизатор доунса для 15-минутной инкубации на льду.
ПРИМЕЧАНИЕ: Добавьте ингибиторы протеазы в буфер лизиса непосредственно перед использованием. - Медленно перемещайте пестик А вверх и вниз 30 раз, чтобы гомогенизировать клетки на льду и инкубировать на льду в течение 1 мин, чтобы дать клеткам остыть, еще до 30 ударов.
- Перенесите лизат в микроцентрифужную трубку объемом 1,7 мл.
ПРИМЕЧАНИЕ: Держите подвеску в движении, потому что иногда клетки застревают в кончике пипетки. - Центрифугируют лизированную суспензию при 2 500 × г в течение 5 мин при комнатной температуре.
- Выбросьте супернатант и щелкните или вихрьте влажную гранулу для повторного суспендирования. Удалите как можно больше супернатанта, чтобы получить йогуртоподобное вещество с минимальным количеством сгустков.
- Повторно суспендировать гранулу в 500 мкл ледяного 1x рестрикционного буфера (от 10x; см. рецепт в Дополнительной таблице S1) и центрифугу в течение 5 мин при 2 500 × г. Повторите этот шаг для второй стирки.
ПРИМЕЧАНИЕ: Гранула в 1x Restriction Buffer является более гранулированной, чем предыдущая гранула в буфере лизиса. - Повторное суспендирование клеток в конечном объеме 360 мкл 1x Restriction Buffer путем пипетирования после добавления ~340 мкл к переходящего объему гранулы, который зависит от размера ячейки.
- Отложите 18 мкл каждого лизата для проверки целостности хроматина (CI). Храните образцы CI при температуре 4 °C.
- Добавьте 38 мкл 1% додецилсульфата натрия (SDS) в каждую трубку Hi-C (общий объем 380 мкл) и тщательно перемешайте путем пипетирования без введения пузырьков.
ВНИМАНИЕ: SDS токсичен. - Инкубируйте образцы при 65 °C без встряхивания в течение ровно 10 минут, чтобы открыть хроматин.
- Немедленно поместите трубки на лед и приготовьте смесь для пищеварения с тритоном X-100 для закалки SDS, как описано в таблице 1.
- Добавьте 107 мкл смеси для пищеварения в Hi-C-трубку (всего 487 мкл), чтобы переварить хроматин в течение ночи (~16 ч) при 37 °C в термомиксоре с интервальным встряхиванием (например, 900 об/мин, 30 с включено, 4 мин выключение).
ПРИМЕЧАНИЕ: Добавление Тритона к конечной концентрации 1% служит для гашения SDS.
- Повторное суспендирование (пипет) сшитых клеточных аликвот (~5 × 106 клеток) в 1 мл буфера ледяного лизиса (рецепт в Дополнительной таблице S1), содержащего 10 мкл коктейля ингибитора протеазы, и перенос в гомогенизатор доунса для 15-минутной инкубации на льду.
- Биотинилирование концов ДНК
- После ночного пищеварения переведите образцы до 65 °C в течение 20 мин, чтобы деактивировать оставшуюся активность эндонуклеазы.
- Во время инкубации приготовьте восполняющую мастер-смесь, как показано в таблице 2.
- После инкубации поместите образцы сразу на лед.
- Отложите 10 мкл контроля пищеварения (DC) для каждого образца и храните при 4 °C.
- Снимите конденсат с крышки пипеткой или спиннингом. К каждому образцу добавляют 58 мкл биотиновой смеси (общий объем образца 535 мкл) и аккуратно пипетируют без образования пузырьков.
- Инкубируйте образцы при 23 °C в течение 4 ч в термомиксоре (например, 900 об/мин, 30 с вкл., 4 мин выключение).
- Перевязка проксимальных фрагментов ДНК
- Приготовьте лигированную смесь, как показано в таблице 3 , пока биотиновая добавка инкубируется.
- Добавьте 665 мкл лигационной смеси к каждому образцу (общий объем образца 1 200 мкл). Аккуратно перемешайте путем пипетки.
- Инкубировать образцы при 16 °C в течение 4 ч в термомиксаторе с интервальным встряхиванием (например, 900 об/мин, 30 с вкл., 4 мин выключение). Храните эти образцы, содержащие ковалентно связанный хроматин при 4 °C, в течение нескольких дней.
- Разворот сшивки
- Доведите объемы образцов CI и DC до 50 мкл с 1x Tris Low EDTA (TLE; см. рецепт в Дополнительной таблице S1).
- Добавьте 10 мкл 10 мг/мл протеиназы K к образцам CI и DC.
- Инкубировать при 65 °C в течение ночи с интервальным встряхиванием (например, 900 об/мин, 30 с вкл., 4 мин выключение). В качестве альтернативы, выполните 30-минутное изменение сшивки для этих элементов управления во время очистки ДНК образцов Hi-C.
- К каждому образцу Hi-C добавляют 50 мкл 10 мг/мл протеиназы K и инкубируют при 65 °C в течение не менее 2 ч с интервалом встряхивания (например, 900 об/мин, 30 с вкл., 4 мин выключения).
- Добавьте еще 50 мкл 10 мг/мл протеиназы K в каждую пробирку Hi-C (общий объем образца 1 300 мкл) и продолжайте инкубацию при 65 °C в течение ночи. Хранить при температуре 4 °C до очистки ДНК.
ПРИМЕЧАНИЕ: Расщепление инкубаций протеиназы К обеспечивает общее переваривание белка.
- Очистка ДНК
- Дайте трубам остыть от 65 °C до комнатной температуры.
- Переложите каждый образец в коническую пробирку объемом 15 мл и добавьте 2,6 мл (объем 2x) фенола:хлороформа:изоамилового спирта в каждую пробирку.
ВНИМАНИЕ: Фенол: хлороформ: изоамиловый спирт является очень токсичным раздражителем и потенциально канцерогенным. - Вращайте каждую трубку в течение 1 мин, а затем перенесите ее содержимое в 15 мл фазовой запирающей трубки.
- Центрифугирование образцов в течение 5 мин на максимальной скорости (1 500-3 500 × g) в настольной центрифуге.
- Осторожно налейте водную фазу в ультрацентрифужную трубку объемом 35 мл и добавьте сверхчистую воду к конечному объему 1 250 мкл.
ПРИМЕЧАНИЕ: Используйте трубки, чтобы установить доступную ультрацентрифугу или разделить на несколько микроцентрифужных трубок. - Добавьте 1/10-й объем (~125 мкл) 3 М ацетата натрия и хорошо перемешайте путем инверсии.
- Добавьте в каждый образец 2,5-кратный объем (~3,4 мл) ледяного 100% этанола, сбалансируйте пробирки для ультрацентрифугирования, добавив ледяной 100% этанол, и хорошо перемешайте путем инверсии.
- Инкубируйте трубки на сухом льду в течение ~15 мин (избегайте затвердевания).
- Центрифугируйте пробирки по 18 000 × г в течение 30 мин при 4 °C.
ПРИМЕЧАНИЕ: Для угловых роторов: отметьте трубы, в которых будет находиться гранула. - Используя пипетку, удалите и выбросьте супернатант полностью со стороны без гранул.
ПРИМЕЧАНИЕ: В этот момент гранула должна стать видимой и может быть отмечена на трубке, потому что она может быть не хорошо видна после сушки на следующем этапе. - Высушите образцы на воздухе в течение приблизительно 10 минут или до тех пор, пока они не станут заметно сухими.
- Солюбилизируйте каждую гранулу в 450 мкл 1x TLE путем пипетирования или закручивания и переведите на центробежный фильтрующий блок (КОЕ) объемом 0,5 мл с отсечкой молекулярной массы 3 кДа.
- Центрифугируйте КОЕ на максимальной скорости в течение 10 мин и отбросьте проточное прохождение. Вымойте каждую ультрацентрифужную трубку дополнительными 450 мкл 1x TLE и переложите на ее КОЕ для другой стирки.
ПРИМЕЧАНИЕ: Промывка КОЕ таким образом ограничивает потерю ДНК при одновременном снижении концентрации соли. - Центрифугируйте КОЕ на максимальной скорости в течение 10 мин и отбросьте проточное прохождение.
- Добавьте 80 мкл 1x TLE в колонну и превратите колонну в новую коллекторную трубку перед центрифугированием в течение 2 мин на максимальной скорости, чтобы получить конечный объем ~100 мкл.
- Добавьте 1 мкл Рназы А (1 мг/мл; 10-кратное разведение 10 мг/мл) к каждому образцу и инкубируйте при 37 °C на тепловом блоке, на водяной бане или термомикшере в течение не менее 30 мин.
- После обработки Rnase удалите образцы с температуры 37 °C и храните при температуре 4 °C до этапа контроля качества.
- Проверка качества хроматина, переваривания ферментов и лигирования образцов
- Охладите образцы CI и DC до комнатной температуры после разворота сшивок на этапе 2.4.5. Затем переведите на предвращенную 2 мл фазовую запирающую трубку.
ПРИМЕЧАНИЕ: Убедитесь, что содержимое фазовой блокировки центрифугировано до гранулы (максимальная скорость в течение 2 мин). - Добавьте 200 мкл фенола:хлороформа:изоамилового спирта и перемешайте образцы путем вихря в течение 1 мин.
- Центрифугируйте трубки в течение 5 мин на максимальной скорости.
- Перенесите водную фазу из каждого образца (~50 мкл) в новую микрофрагменную трубку объемом 1,7 мл.
- Добавляют 1 мкл Рназы А (от 1 мг/мл) и инкубируют при 37 °C в течение не менее 30 мин.
- Загрузите образцы на 0,8% агарозный гель, как рекомендовано в таблице 4.
ПРИМЕЧАНИЕ: Результаты, ожидаемые от контроля качества, показаны на рисунке 2. - Количественно оценить ДНК с помощью денситометрии из геля или с помощью кубита или нанодропа.
ПРИМЕЧАНИЕ: Точная количественная оценка обеспечивает правильный входной объем в следующей части протокола. Используйте несколько стандартов с известной величиной, чтобы построить стандартную кривую.
- Охладите образцы CI и DC до комнатной температуры после разворота сшивок на этапе 2.4.5. Затем переведите на предвращенную 2 мл фазовую запирающую трубку.
Таблица 1: Реагенты для пищеварения. Пожалуйста, нажмите здесь, чтобы загрузить эту таблицу.
Таблица 2: Биотиновые реагенты. *Обратите внимание, что для изменения ферментов могут потребоваться различные буферы и биотинилированные дНТП. Пожалуйста, нажмите здесь, чтобы загрузить эту таблицу.
Таблица 3: Лигационная смесь реагентов. Аббревиатура: BSA = бычий сывороточный альбумин. Пожалуйста, нажмите здесь, чтобы загрузить эту таблицу.
Таблица 4: Параметры загрузки геля для оценки качества и выбора размера. Пожалуйста, нажмите здесь, чтобы загрузить эту таблицу.

Рисунок 2: Агарозный гель, показывающий типичные результаты контроля качества очистки постДНК. (A) Контроль CI должен указывать на полосу высокомолекулярной ДНК. (B) Образцы DC и Hi-C показывают диапазон размеров ДНК. Образец Hi-C, объединенный в более крупные фрагменты, должен иметь более высокую молекулярную массу, чем DC. Диапазон концентрации маркеров позволяет генерировать стандартную кривую. Обратите внимание, что в этом примере CI был загружен на отдельный гель, но рекомендуется загружать и запускать все образцы и элементы управления вместе. Сокращения: CI = целостность хроматина; DC = контроль пищеварения; Hi-C = бесконтактное лигирование. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
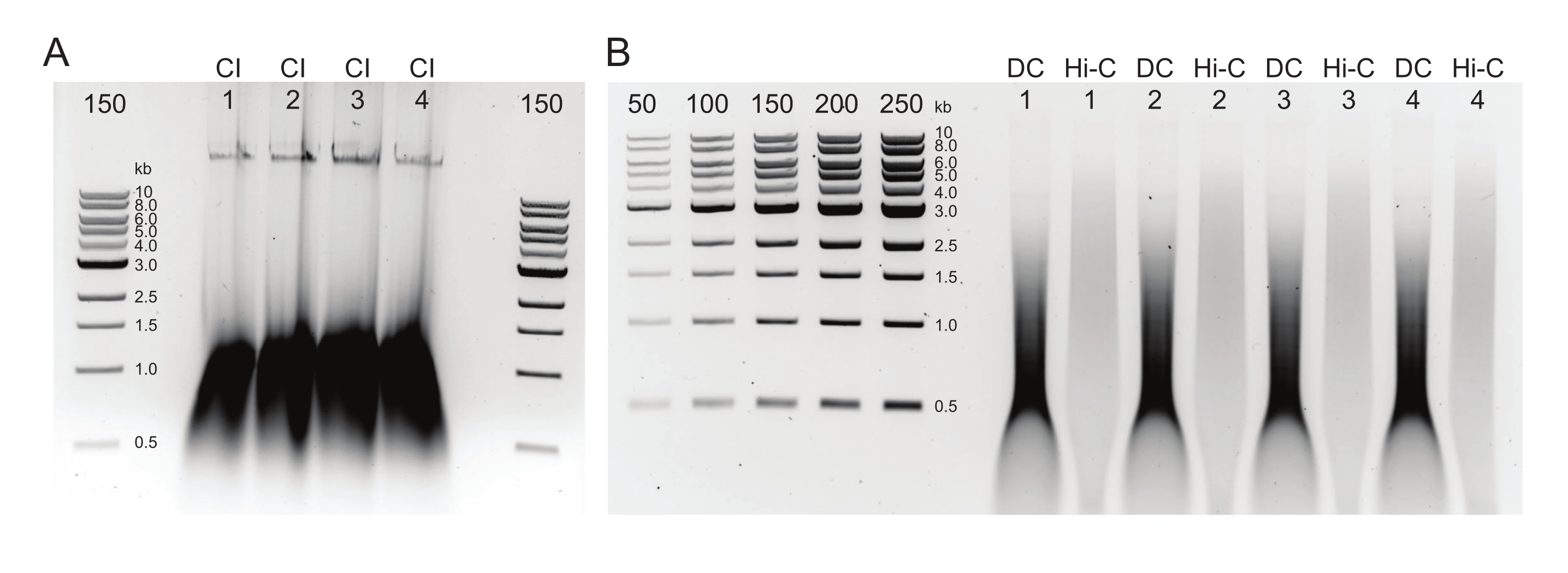{kind=link}
3. Подготовка библиотеки секвенирования Hi-C
- Удаление биотина с нелигированных концов
- Готовят реакции удаления биотина, как показано в таблице 5.
ПРИМЕЧАНИЕ: Как правило, достаточно 10 мкг ДНК, но можно использовать до 30 мкг. - Распределите 2 х 65 мкл аликвот из каждой реакции 130 мкл в две трубки ПЦР.
- Переложить в термоциклер или ПЦР-машину и инкубировать, как описано в таблице 5.
ПРИМЕЧАНИЕ: Образцы могут храниться здесь при температуре от 4 °C (от дней до недель), -20 °C (в долгосрочной перспективе) или немедленно переноситься на обработку ультразвуком.
- Готовят реакции удаления биотина, как показано в таблице 5.
- Обработка ультразвуком
- Объедините дубликаты образца со стадии удаления биотина (общий объем образца 130 мкл) в трубку ультразвукового аппарата объемом 130 мкл для обработки ультразвуком.
- Обработка образцов ультразвуком с использованием параметров, приведенных в таблице 6 , для достижения плотного узкого распределения ниже 500 bp.
ПРИМЕЧАНИЕ: Могут использоваться различные типы ультразвуковых аппаратов, но для узкого распределения фрагментов (100-500 bp) настройки ультразвуковика могут потребовать оптимизации.
- Подбор размера с помощью магнитных шариков
- Пипетируйте ультразвуковую ДНК из пробирки (пробирок) ультразвукового аппарата в трубку с низким связыванием объемом 1,7 мл.
- Доведите каждый образец до общего объема 500 мкл с 1x TLE. Постарайтесь получить объем как можно ближе к 500 мкл, так как отношение образца к смеси магнитных шариков имеет важное значение для подбора размера.
- Добавьте 400 мкл смеси магнитных шариков в каждую трубку, чтобы получить отношение смеси магнитных шариков к объему образца 0,8.
ПРИМЕЧАНИЕ: В этих условиях бусины захватывают фрагменты ДНК >300 bp, которые будут верхней фракцией. Супернатант будет содержать фрагменты <300 bp, которые будут нижней фракцией. - Перемешайте трубки путем вихря и инкубируйте в течение 10 мин при комнатной температуре на ротаторе. Для этого и других соотношений выбора размера убедитесь, что весь объем образца хорошо перемешивается. Ищите «неустойчивый режим», который есть у некоторых роторов, который хорошо работает для небольших объемов.
- Инкубировать в течение 5 мин при комнатной температуре на магнитопорошковом сепараторе (МПС).
- Во время инкубации добавляют 500 мкл смеси магнитных шариков в свежую трубку с низким связыванием 1,7 мкл для каждого образца.
ПРИМЕЧАНИЕ: Эти трубки будут использоваться для выбора следующего размера нижней фракции путем создания смеси магнитных шариков к соотношению образца 1,1:1. - Оставьте трубки на MPS на 5 мин.
- Удалите супернатант из шариков и повторно суспендируйте 150 мкл смеси магнитных шариков.
ПРИМЕЧАНИЕ: Этот шаг позволяет избежать насыщения бусин ДНК за счет увеличения количества бусин без увеличения объема. - Перенесите супернатант со стадии 3.3.5 в маркированную трубку, подготовленную для выбора нижней фракции (стадия 3.3.8).
ПРИМЕЧАНИЕ: 150 мкл смеси магнитных шариков + 400 мкл смеси 0,8x магнитных шариков (всего 550 мкл), деленной на 500 мкл исходного образца = 1,1x смесь магнитных шариков к образцу. - Смешайте трубки нижней фракции путем вихря и инкубируйте в течение 10 мин при комнатной температуре на ротаторе.
ПРИМЕЧАНИЕ: Бусины будут связывать фрагменты ДНК >100 bp, в результате чего конечная фракция, связанная с бисером, составляет 100-300 bp. - Поместите трубки Нижней Фракции на MPS на 5 мин (комнатная температура).
- Удалите супернатант и ненадолго центрифугируйте трубки, чтобы в дальнейшем удалить супернатант как можно больше.
- Дважды вымойте бусины из обеих фракций, используя 200 мкл 70% этанола, и каждый раз восстанавливайте шарики в течение 5 минут на MPS.
- После быстрого отжима в центрифуге полностью удалите этанол и дополнительно высушите шарики на MPS.
ПРИМЕЧАНИЕ: Высушите до полного испарения спирта. Гранула должна выглядеть как темный шоколад без растрескивания (может занять ~10 мин). - Повторное суспендирование обеих фракций в 50 мкл 1x TLE буфера. Инкубировать при комнатной температуре в течение 10 мин и постукивать или щелкать по тюбикам каждые две минуты, чтобы стимулировать смешивание и элюирование.
- Отделите бусины от супернатанта на МПС в течение 5 мин для обеих фракций.
- Сохраните супернатант из каждого образца. Пипетка супернатанта в трубку с низким связыванием объемом 1,7 мл.
ПРИМЕЧАНИЕ: Образцы могут храниться при температуре 4 °C в течение нескольких дней или при -20 °C в течение длительного времени. - Запустите 2% агарозный гель, как показано в таблице 4 , чтобы определить качество и количество образца. Пример такого геля приведен на рисунке 3 .
ПРИМЕЧАНИЕ: Если по опыту обработка ультразвуком очень воспроизводима, можно пропустить этот гель и немедленно приступить к завершению ремонта. Рекомендуется, чтобы пользователи держались за верхние фракции до тех пор, пока не закончится титрование ПЦР. Неоптимальные количества ДНК, которые потребовали бы большой амплификации ПЦР, могут быть спасены из материала в верхней фракции. - Количественно оценить количество ДНК из геля непосредственно после генерации стандартной кривой из известного входа лестницы ДНК или с помощью кубита или нанодропа.
- Окончательный ремонт
- Подготовьте конечную ремонтную смесь, как показано в таблице 7 (количество на реакцию).
- Переложите оставшиеся 46 мкл элюированной ДНК из Нижней фракции в пробирки ПЦР и добавьте 24 мкл подготовленной концевой репарационной смеси. Инкубировать в машине ПЦР, как предложено в таблице 7.
- После завершения программы держите образцы при температуре 4 °C до вытягивания.
- Вытягивание биотинилированных продуктов лигирования с шариками со стрептавидиновым покрытием
- Определите количество бусин со стрептавидиновым покрытием для каждой библиотеки из количественного выбора размера (шаг 3.3.19).
ПРИМЕЧАНИЕ: Эти бусины, покрытые стрептавидином (раствор 10 мг/мл), могут связывать 20 мкг двухцепочечной ДНК на мг бусин (= 20 мкг / 100 мкл бусин). Используйте 2 мкл на каждый 1 мкг ДНК Hi-C, но не менее 10 мкл. - Смешайте бусины, покрытые стрептавидином, и испелите объем бусин, необходимый для каждой библиотеки (рассчитанный на предыдущем этапе), в отдельные трубки с низким связыванием объемом 1,7 мл.
- Повторно суспендировать шарики в 400 мкл буфера для промывки Tween (TWB; см. рецепт в дополнительной таблице S1) и инкубировать в течение ~3 мин при комнатной температуре на ротаторе (см. инструкции в шаге 3.3.4)..
- Отделите бусины от супернатанта на MPS в течение 1 мин и удалите супернатант.
- Вымойте шарики, пипеткой еще 400 мкл TWB.
- Отделите бусины от супернатанта на MPS в течение 1 мин и удалите супернатант.
- Добавьте 400 мкл 2x Binding Buffer (BB) (рецепт в дополнительной таблице S1) к шарикам и повторно суспендируйте. Кроме того, добавьте 330 мкл 1x TLE и раствор из End-repair (из шага 3.4.3).
- Инкубируйте образцы в течение 15 мин при комнатной температуре при смешивании на ротаторе.
- Отделите бусины от супернатанта на MPS в течение 1 мин и удалите супернатант.
- Добавьте 400 мкл 1x BB к бусинам и повторно суспендируйте.
- Отделите бусины от супернатанта на MPS в течение 1 мин и удалите супернатант.
- Добавьте 100 мкл 1x TLE для мытья бусин.
- Отделите бусины от супернатанта на MPS в течение 1 мин и удалите супернатант.
- Наконец, добавьте 41 мкл 1x TLE, чтобы повторно суспендировать бусины.
- Определите количество бусин со стрептавидиновым покрытием для каждой библиотеки из количественного выбора размера (шаг 3.3.19).
- Хвостатый хвост
- Приготовьте смесь А-хвоста, как показано в таблице 8.
- Пипетируйте реакции в ПЦР-трубки и инкубируйте, как показано в таблице 8.
- Поместите трубки ПЦР на лед сразу после извлечения из термоциклера и перенесите содержимое в трубки с низким связыванием 1,7 мл.
- Отделите бусины от супернатанта на MPS в течение 1 мин и выбросьте супернатант.
- Добавьте 400 мкл 1x лигационного буфера, разбавленного из 5x T4 ДНК-лигазного буфера сверхчистой водой.
- Отделите бусины от супернатанта на MPS в течение 1 мин, а затем выбросьте супернатант.
- Добавьте 1x лигационный буфер к конечному объему 40 мкл.
- Адаптер для отжига олиго
- Готовят адаптер олиго запасов 100 мкМ (табл. 9).
ПРИМЕЧАНИЕ: Закажите 250 нмолей олиго, очищенных ВЭЖХ. - Отжиг адаптеров в ПЦР-трубках, как описано в таблице 9.
- Используйте термоциклер ПЦР для постепенного повышения температуры от 0,5 ° C / с до 97,5 ° C. Удерживать при температуре 97,5 °C в течение 2,5 мин.
- Используйте термоциклер ПЦР для постепенного повышения температуры при 0,1 °C/с в течение 775 циклов (достигая 20 °C). Держите температуру на уровне 4 °C до дальнейшего использования.
- Добавьте 83 мкл 1x буфера отжига (рецепт в дополнительной таблице S1), чтобы разбавить адаптеры до 15 мкМ. Храните адаптеры при -20 °C.
- Готовят адаптер олиго запасов 100 мкМ (табл. 9).
- Лигирование адаптера секвенирования
- Приготовьте переходную лигатационную смесь в трубке с низким связыванием объемом 1,7 мл (таблица 10)..
- Лигат в течение 2 ч при комнатной температуре.
- Отделите бусины от супернатанта на MPS в течение 1 мин и выбросьте супернатант.
- Добавьте 400 мкл TWB и тщательно пипетируйте шарики вверх и вниз перед инкубацией на ротаторе в течение 5 мин при комнатной температуре. Отделите бусины от супернатанта на MPS и повторите этот шаг еще раз.
- Отделите бусины от супернатанта на MPS (~1 мин), выбросьте супернатант и добавьте 200 мкл 1x BB.
- Отделите бусины от супернатанта на MPS (~1 мин) и выбросьте супернатант.
- Добавьте 200 мкл 1x буфера предварительной ПЦР (из 10x; рецепт в дополнительной таблице S1) и переложите в новую трубку с низким связыванием объемом 1,7 мл.
- Отделите бусины от супернатанта на MPS (~1 мин) и выбросьте супернатант.
- Добавьте 20 мкл 1x буфера предварительной ПЦР и перемешайте путем пипетирования.
- Держите трубки на льду во время использования или храните при температуре 4 °C.
- Оптимизация числа циклов ПЦР путем титрования
- Установите 30 мкл реакций основной смеси на образец, как показано в таблице 11 (количество на реакцию).
- Выполните наименьшее количество циклов и возьмите аликвоту 5 мкл. Запустите дополнительные 2-3 цикла по оставшейся реакции, прежде чем принимать следующие 5 мкл аликвоты. Повторите, чтобы собрать четыре аликвоты.
- Используйте параметры ПЦР из таблицы 11 для каждой аликвоты.
- Добавьте 5 мкл воды и 2 мкл 6-кратного красителя к каждому образцу размером 5 мкл. Запускайте на 2% агарозном геле TBE (рецепт в дополнительной таблице S1) с 25-150 нг низкомолекулярной лестницы. Ожидаемые результаты см. на рисунке 4 .
ПРИМЕЧАНИЕ: Оптимальное количество циклов для окончательной библиотеки ПЦР-амплификации [шаг 3.10] является наименьшим числом циклов для получения видимого продукта на геле минус один цикл.
- Итоговая библиотека ПЦР-амплификации
- Настройте реакции 12 x 30 мкл для усиления каждой конечной библиотеки для секвенирования, как показано в таблице 11.
- Цикл ПЦР-реакций согласно таблице 11 после определения числа циклов после титрования ПЦР (этап 3.9.3).
- Как только ПЦР будет завершена, объедините реплицированные образцы в 1,7 мл низкосвязанной микрофьюжной трубки.
- Поместите трубки на MPS и перенесите супернатант в новую микрофрагменную трубку с низким связыванием объемом 1,7 мл.
- Повторное суспендирование оставшихся шариков, покрытых стрептавидином, в 20 мкл 1x буфера до ПЦР.
ПРИМЕЧАНИЕ: Этот шаблон можно повторно использовать при хранении при температуре от 4 °C в течение нескольких дней до недель или при длительном хранении при -20 °C.
- Снятие грунтовок с магнитной смесью бусин
- Используйте 1-кратный буфер TLE (рецепт в дополнительной таблице S1) для регулировки громкости с шага 3.10.4 до ровно 360 мкл.
- К каждому образцу добавьте 360 мкл смеси магнитных шариков и пипетку вверх и вниз, чтобы перемешать.
- На ротаторе перемешайте образцы в течение 10 мин при комнатной температуре.
- Отделите шарики от надосадочного вещества на МПС при комнатной температуре (3-5 мин).
- Дважды вымойте бусины, используя 200 мкл 70% этанола, и каждый раз восстанавливайте шарики в течение 5 минут на MPS.
- Быстрое отжим в центрифуге и полностью отключите этанол. Высушите шарики на воздухе на MPS для дальнейшего испарения этанола.
ПРИМЕЧАНИЕ: Гранула должна выглядеть как темный шоколад без растрескивания (может занять ~ 10 мин). - Добавьте 30 мкл сверхчистой воды и повторно суспендируйте для элюирования ДНК в течение 10 минут при комнатной температуре. Проводите по тюбикам каждые 2 минуты, чтобы помочь перемешать.
- Отделите бусины от супернатанта на MPS в течение 5 минут.
- Соберите супернатант из каждого образца в свежую пробирку объемом 1,7 мл.
- Запустите 1 мкл библиотеки на 2% агарозном TBE (рецепт в дополнительной таблице S1) геля для получения распределения фрагментов по размерам и количественной оценки конечной библиотеки (рисунок 5).
ПРИМЕЧАНИЕ: Переваривание ClaI может происходить только для переходов DpnII-DpnII и служит в качестве положительного контроля лигирования, что должно привести к более низкому распределению размеров фрагментов для конечной библиотеки. Конечные библиотеки могут храниться в течение нескольких дней при 4 °C, в течение длительного времени при -20 °C или немедленно разбавляться и подаваться на секвенирование.
Таблица 5: Реагенты для удаления биотина и температура Пожалуйста, нажмите здесь, чтобы загрузить эту таблицу.
Таблица 6: Параметры обработки ультразвуком. Пожалуйста, нажмите здесь, чтобы загрузить эту таблицу.
Таблица 7: Конечные реагенты и температуры. Пожалуйста, нажмите здесь, чтобы загрузить эту таблицу.
Таблица 8: Реагенты хвостохранилища и температуры. Пожалуйста, нажмите здесь, чтобы загрузить эту таблицу.
Таблица 9: Грунтовки ПЦР и парно-концевые адаптерные олиго с отжигом реагентов для отжига. Аббревиатура: 5ФОС = 5' фосфат. Звездочки указывают на фосфоротиоированные основания ДНК. # Объедините индексированное олиго с универсальным олиго для отжига в индексированный адаптер. Пожалуйста, нажмите здесь, чтобы загрузить эту таблицу.
Таблица 10: Адаптерные реагенты для лигирования. Пожалуйста, нажмите здесь, чтобы загрузить эту таблицу.
Таблица 11: Реагенты ПЦР и параметры цикличности. Пожалуйста, нажмите здесь, чтобы загрузить эту таблицу.

Рисунок 3: Агарозный гель показывает типичные результаты выбора после размера. Показаны Верхняя и Нижняя фракции для четырех образцов (пронумерованных 1-4) DpnII-DdeI Hi-C. Первая полоса для каждого образца содержит верхнюю фракцию, полученную из 0,8-кратной смеси магнитных шариков, а вторая и третья полосы содержат разбавление нижней фракции, полученной из 1,1-кратной смеси магнитных шариков. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}

Рисунок 4: Агарозный гель с результатами титрования ПЦР. Начиная с 5 циклов ПЦР, образцы берутся после каждых 2 циклов (5, 7, 9 и 11 циклов) для каждой из четырех библиотек. Исходя из этого рисунка, в качестве оптимального цикла для каждого образца было выбрано 6 циклов. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}

Рисунок 5: Конечные продукты ПЦР. После очистки и выбора размера продукты ПЦР (Hi-C) загружались рядом с переваренной ClaI фракцией той же библиотеки (ClaI). ClaI-переваренные фрагменты указывают на наличие востребованных DpnII-DpnII лигирования. Обратите внимание, что ClaI не переваривает соединения DpnII-DdeI и, следовательно, не все лигации будут способствовать уменьшению размера от этого ограничения. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
Результаты
Рисунки в этой рукописи были получены из отдельного, повторяющего эксперимента, опубликованного ранее Lafontaine et al.21. После получения высокопроизводительных данных секвенирования для обработки данных Hi-C был использован Open Chromatin Collective (Open2C: https://github.com/open2c). Аналогичный конвейер можно найти на портале данных проекта 4D Nucleome (https://data.4dnucleome.org/resources/data-analysis/hi_c-processing-pipeline). Вкратце, конвейерный дистиллятор Nextflow (https://github.com/open2c/distiller-nf) был реализован для (1) выравнивания последовательностей молекул Hi-C с эталонным геномом, (2) разбора выравнивания .sam и файлов форм с парами Hi-C, (3) фильтрации дубликатов ПЦР и (4) агрегирования пар в объединенные матрицы взаимодействий Hi-C. Эти матрицы в формате HDF5, называемые кулерами, затем можно (1) просмотреть на сервере HiGlass (https://higlass.io/) и (2) проанализировать с использованием большого набора вычислительных инструментов с открытым исходным кодом, присутствующих в коллекции «cooltools», поддерживаемой Open Chromatin Collective (https://github.com/open2c/cooltools), для извлечения и количественной оценки складных функций, таких как отсеки, TAD и петли.
Некоторые показатели качества библиотек Hi-C3.0 могут быть оценены сразу после сопоставления считываемых пар с эталонным геномом, используя несколько простых метрик / индикаторов. Во-первых, как правило, ~ 50% секвенированных пар чтения могут быть уникально отображены для клеток человека. Из-за полимерной природы хромосом большинство из этих нанесенных на карту считываний (~ 60%-90%) представляют собой взаимодействия внутри хромосомы (цис), причем частоты взаимодействия быстро распадаются с увеличением геномного расстояния (зависящий от расстояния распад). Зависящий от расстояния распад может быть лучше всего визуализирован на «графике масштабирования», который показывает вероятность контакта (на хромосомное плечо) как функцию геномного расстояния. Мы обнаружили, что использование различных сшивающих веществ и ферментов может изменить зависящий от расстояния распад на больших и ближних расстояниях17. Добавление сшивки DSG повышает обнаруживаемость взаимодействий на коротких расстояниях в сочетании с ферментами, такими как Mnase и комбинациями DpnII-DdeI, которые производят меньшие фрагменты (рисунок 6A).
Зависящий от расстояния распад также можно наблюдать непосредственно из 2D-матриц взаимодействия: взаимодействия становятся более редкими, когда они расположены дальше от центральной диагонали (рисунок 6B). Кроме того, геномные особенности сворачивания, такие как компартменты, TAD и петли, могут быть идентифицированы из матриц Hi-C и графиков масштабирования как отклонения от общего среднего расстояния геномного распада. Важно отметить, что сшивание с DSG в дополнение к FA уменьшает случайные лигации, которые не ограничены из-за полимерной природы хромосом и, следовательно, более вероятны между хромосомами (у транс) (рисунок 6C). Уменьшение случайного лигирования приводит к увеличению отношения сигнал/шум, особенно для межхромосомных и очень дальних (>10-50 Мб) внутрихромосомных взаимодействий.

Рисунок 6: Репрезентативные результаты картографированных и отфильтрованных библиотек Hi-C. (A) Графики масштабирования с вероятностью контакта и его производными для различных ферментов, упорядоченные по длине фрагмента (вверху) и сшивки с FA или FA + DSG (внизу). Пищеварение с помощью MNAse (microC) или DpnII-DdeI (Hi-C 3.0) значительно увеличивает контакты ближнего действия (сверху), как и добавление DSG к FA (внизу). (B) Столбцы показывают тепловые карты Hi-C пищеварения DpnII после сшивки FA и dpnII или DdeI после сшивки FA + DSG. Белые стрелки показывают увеличение силы «точек» после сшивания DSG и переваривания DdeI, что подразумевает лучшее обнаружение петель ДНК. Ряды показывают различные части хромосомы 3 с возрастающим разрешением, выравниваясь с панелью C: верхний ряд: вся хромосома 3 (0-198,295,559 Mb); средний ряд: 186-196 Мб; нижний ряд: 191.0-191.5 Mb. (C) Графики покрытия для регионов, изображенных в A. Черные стрелки показывают более низкое покрытие (%cis reads) для сшивки только FA. Сокращения: FA = формальдегид; DSG = дисукцинимидиллутарат; chr = хромосома. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
Не все сопоставленные чтения полезны. Вторым показателем качества является количество дубликатов ПЦР. Точные дублирующие показания вряд ли произойдут случайно после перевязки и обработки ультразвуком. Таким образом, такие показания, вероятно, являются результатом амплификации ПЦР и должны быть отфильтрованы. Дубликаты часто возникают, когда требуется слишком много циклов ПЦР для усиления библиотек низкой сложности. Как правило, для Hi-C большинству библиотек требуется только 5-8 циклов конечной амплификации ПЦР, определяемой титрованием ПЦР (см. шаг 3.9; Рисунок 4). Однако библиотеки достаточной сложности можно получить даже после 14 циклов ПЦР-амплификации.
Другая категория дублированных считываний, так называемые оптические дубликаты, может возникнуть в результате процесса усиления на платформах секвенирования Illumina, которые используют узорчатые проточные ячейки (такие как HiSeq4000). Оптические дубликаты обнаруживаются либо от перегрузки проточной ячейки, в результате чего (большие) кластеры называются двумя отдельными кластерами, либо от локального повторного повторения исходной парной концевой молекулы после первого раунда ПЦР. Поскольку оба типа оптических дубликатов являются локальными, их можно идентифицировать и отличить от дубликатов ПЦР по их расположению на проточной ячейке. В то время как библиотеки с >15% дубликатами ПЦР потребуют регенерации, библиотеки с оптическими дубликатами могут быть перезагружены после оптимизации процесса загрузки.
Дополнительная таблица S1: Буферы и растворы. Пожалуйста, нажмите здесь, чтобы загрузить эту таблицу.
Обсуждение
Критические шаги для обработки ячеек
Хотя можно использовать меньшее количество входных ячеек, этот протокол был оптимизирован для ~ 5 × 106 ячеек на линию секвенирования (~ 400 M чтения), чтобы обеспечить надлежащую сложность после глубокого секвенирования. Клетки лучше всего подсчитывать перед фиксацией. Для генерации сверхглубоких библиотек мы обычно умножаем количество полос (и ячеек) до тех пор, пока не будет достигнута желаемая глубина чтения. Для оптимальной фиксации сывороточную среду следует заменить ПБС до фиксации ФА, а фиксирующие растворы добавлять немедленно и без градиентов концентрации 15,22. Для сбора клеток соскоб предпочтительнее трипсинизации, потому что переход от более плоской к сферической форме после трипсинизации может повлиять на ядерную конформацию. После добавления DSG гранулы сыпучих и комковатых клеток легко теряются. Будьте осторожны при работе с клетками на этом этапе и добавьте до 0,05% BSA, чтобы уменьшить слипание.
Изменения в методе
Этот протокол был разработан с использованием клеток человека17. Тем не менее, основываясь на опыте захвата конформации хромосом, этот протокол должен работать для большинства эукариотических клеток. Для значительно более низкого ввода (~1 × 106 ячеек) мы советуем использовать половину объемов для процедур лизиса и конформационного захвата [этапы 2.1-2.4]. Это также позволит выполнить изоляцию ДНК [этап 2.5] в настольной центрифуге с пробирками объемом 1,7 мл, что может улучшить гранулирование для низких концентраций ДНК. Количественная оценка ДНК (этап 2.6.6) укажет, как действовать дальше. Для небольших количеств выделенной ДНК (1-5 мкг) мы предлагаем пропустить выбор размера (этап 3.3) и приступить к удалению биотина после уменьшения объема со 130 мкл до ~45 мкл с помощью КОЕ.
Этот протокол был разработан специально для обеспечения высокого качества данных после последующего сшивания с FA и DSG и пищеварения с DpnII и DdeI. Однако альтернативные стратегии сшивания, такие как FA, за которым следует EGS (этиленгликоль-бис (сукцинимидилсукцинат)), который также используется в ChIP-seq23 и ChIA-PET24, могут работать одинаково хорошо17. Аналогичным образом, различные комбинации ферментов, такие как DpnII и HinfI18 или MboI, MseI и NlaIII19 , могут быть использованы для пищеварения. При адаптации комбинаций ферментов обязательно используйте биотинилированные нуклеотиды, которые могут заполнять определенные 5'-свесы и использовать наиболее оптимальные буферы для каждого коктейля. DpnII поставляется с собственным буфером, и производитель ферментов рекомендует специальный буфер для пищеварения DdeI. Тем не менее, для двойного пищеварения с DpnII и DdeI в этом протоколе рекомендуется рестрикционный буфер, потому что он рассчитан на 100% активность для обоих ферментов.
Поиск и устранение неисправностей при захвате соответствия
Три ключевых шага в захвате конформации хромосом: сшивание, пищеварение и религирование были выполнены до того, как результаты могут быть визуализированы на геле. Чтобы определить качество каждого из этих трех этапов и определить, где могли возникнуть проблемы, аликвоты до (CI) и после пищеварения (DC) берутся и загружаются на гель вместе с лигированным образцом Hi-C (рисунок 2). Этот гель используется для определения качества образца Hi-C и того, стоит ли продолжать протокол. Без CI и DC трудно точно определить потенциальные неоптимальные шаги. Стоит отметить, что неоптимальная перевязка может быть вызвана проблемой в самой лигации, заполнении или проблемой с сшивки. Для устранения неполадок сшивки убедитесь, что вы не используете более 1 × 107 ячеек на библиотеку и начните со свежих реагентов для сшивания и чистых ячеек (т. Е. Промытых PBS). Для перевязки убедитесь, что клетки и лигационная смесь хранятся на льду. Добавьте Т4 ДНК-лигазу непосредственно перед 4-часовой инкубацией при 16 °C и хорошо перемешайте.
Устранение неполадок при подготовке библиотеки
Если требуется более 10 циклов ПЦР или продукт ПЦР не виден на геле после титрования ПЦР (рисунок 4), есть несколько вариантов сохранения образца Hi-C. Возвращаясь к титрованию ПЦР, первый вариант заключается в том, чтобы попробовать ПЦР еще раз. Если продукта все еще недостаточно, можно попробовать еще один раунд перевязки A-хвоста и адаптера (шаг 3.6) после промывки бусин дважды с буфером 1x TLE. После этого дополнительного А-хвоста и перевязки адаптера можно приступать к титрованию ПЦР, как и раньше. Если продукта по-прежнему нет, последний вариант состоит в том, чтобы повторно обработать фракцию 0,8x с шага 3.3 и продолжить оттуда.
Ограничения и преимущества Hi-C3.0
Важно понимать, что Hi-C является популяционным методом, который фиксирует среднюю частоту взаимодействий между парами локусов в клеточной популяции. Некоторые вычислительные анализы предназначены для разделения комбинаций конформаций из популяции25, но в принципе Hi-C слеп к различиям между клетками. Хотя можно выполнить одноэлементный Hi-C26,27 и вычислительные выводы могут быть сделаны28, одноэлементный Hi-C не подходит для получения информации 3C сверхвысокого разрешения. Дополнительным ограничением Hi-C является то, что он обнаруживает только парные взаимодействия. Для обнаружения многоконтактных взаимодействий можно либо использовать частые фрезы в сочетании с секвенированием короткого считывания (Illumina)16, либо выполнять многоконтактные 3C29 или 4C30, используя секвенирование с длинным чтением с платформ PacBio или Oxford Nanopore. Производные Hi-C для конкретного обнаружения контактов между и вдоль сестринских хроматид также были разработаны31,32.
Хотя Hi-C19 и Micro-C33 могут использоваться для создания карт контактов с субкилобазовыми разрешениями, оба требуют большого количества последовательных считываний, и это может стать дорогостоящим мероприятием. Чтобы получить аналогичное или даже более высокое разрешение без затрат, может быть применено обогащение для конкретных геномных областей (захват-C34) или специфических белковых взаимодействий (ChiA-PET35, PLAC-seq36, Hi-ChIP37). Сила и недостаток этих применений обогащения заключается в том, что отбирается только ограниченное количество взаимодействий. При таких обогащениях теряется глобальный аспект Hi-C (и возможность глобальной нормализации).
Важность и потенциальные возможности применения Hi-C3.0
Этот протокол был разработан для обеспечения сверхглубокого 3C с высоким разрешением при одновременном обнаружении крупномасштабных складных функций, таких как TAD и отсеки17 (рисунок 6). Этот протокол начинается с 5 × 106 ячеек на трубку для каждой библиотеки Hi-C, что должно быть более чем достаточным материалом для секвенирования одной или двух полос на проточной ячейке для получения до 1 миллиарда парных считываний. Для сверхглубокого секвенирования следует подготовить несколько пробирок по 5 × 106 клеток, в зависимости от количества отображаемых считываний и дубликатов ПЦР. При самом высоком разрешении (<1 кб) зацикленные взаимодействия в основном обнаруживаются между сайтами CTCF, но также могут быть обнаружены взаимодействия промотор-энхансер. Читатели могут обратиться к Akgol Oksuz et al.17 для подробного описания анализа данных.
Раскрытие информации
У авторов нет конфликта интересов для раскрытия.
Благодарности
Мы хотели бы поблагодарить Дениса Лафонтена за разработку протокола и Сергея Венева за биоинформационную помощь. Эта работа была поддержана грантом Национального института здравоохранения Common Fund 4D Nucleome Program для J.D. (U54-DK107980, UM1-HG011536). Джей Ди является исследователем Медицинского института Говарда Хьюза.
Эта статья подпадает под действие политики открытого доступа к публикациям HHMI. Руководители лабораторий HHMI ранее предоставили неисключительную лицензию CC BY 4.0 для общественности и сублицензируемую лицензию HHMI в своих исследовательских статьях. В соответствии с этими лицензиями принятая автором рукопись этой статьи может быть предоставлена в свободный доступ по лицензии CC BY 4.0 сразу после публикации.
Материалы
| Name | Company | Catalog Number | Comments |
| 1 kb Ladder | New England Biolabs | N3232L | |
| Agarose | Invitrogen | 16500100 | |
| Agencourt AMPure XP magnetic beads , 60 mL | Beckman Coulter | A63881 | |
| Amicon Ultra-0.5 Centrifugal Filter Unit (CFU) | EMD Millipore | UFC500396 | |
| Annealing Buffer (5x) | See recipe in supplemental materials | ||
| ATP 10 mM | ThermoFisher | R0441 | |
| Avanti J-25i High Speed Refrigerated ultra-centrifuge | Beckman Coulter | ||
| beckman ultracentrifuge tube 35 mL | Beckman Coulter | 357002 | |
| Binding Buffer (2x) | See recipe in supplemental materials | ||
| biotin-14-dATP 0.4 mM | Invitrogen | 19524-016 | |
| BSA 10 mg/mL | New England Biolabs | B9000S | dilute from 20 mg/mL |
| Cell scraper | Falcon | 353089 | |
| Cell scraper | Corning | 3008 | |
| Conical polypropylene tubes 50 mL | Denville | C1062-P | |
| Conical tube 15 mL | Denville | C1017-P | |
| Covaris micro tube AFA fiber with snap-cap 130 µL | Covaris | 520045/520077 | |
| Covaris Sonicator | Covaris | E220/E220evolution/M220 | |
| Culture flask 175 cm2 | Falcon | 353112 | |
| Culture plates 150 mm x 25 mm | Corning | 430599 | |
| dATP 1 mM | Invitrogen | 56172 | |
| dATP 10 mM | Invitrogen | 56172 | |
| dCTP 10 mM | Invitrogen | 56173 | |
| DdeI | New England Biolabs | R0175L | |
| dGTP 10 mM | Invitrogen | 56174 | |
| DMSO | Sigma | D2650-5x10ML | |
| dNTP mix 25 mM | Invitrogen | 10297117 | |
| Dounce homogenizer | DWK Life Sciences | 8853010002/8853030002 | |
| DPBS | Gibco | 14190-144 | |
| DpnII | New England Biolabs | R0543M | |
| DSG | ThermoScientific | 20593 | |
| dTTP 10 mM | Invitrogen | 56175 | |
| Ethanol 70% | Fisher | A409-4 | Diluted from 100% |
| Ethidium Bromide | Fisher | BP1302-10 | |
| Formaldehyde (37%) | Fisher | BP531-500 | |
| Gel loading dye (6x ) | New England Biolabs | B7024S | |
| Glycine in ultrapure water 2.5 M | Sigma | G8898-1KG | |
| HBSS | Gibco | 14025-092 | |
| Igepal CA-630 detergent | MP Biomedicals | 198596 | |
| Klenow DNA polymerase 5 U/µL | New England Biolabs | M0210L | |
| Klenow Fragment 3-->5’ exo-, 5 U/µL | New England Biolabs | M0212L | |
| ligation buffer (10x) | New England Biolabs | B7203S | |
| Liquid nitrogen | |||
| LoBind microcentrifuge tube 1.7 mL | Eppendorf | 22431021 | |
| Low Molecular Weight DNA Ladder | New England Biolabs | N3233L | |
| Lysis buffer | See recipe in supplemental materials | ||
| Magnetic Particle separator | ThermoFisher | 12321D | |
| Microfuge tubes 1.7 mL | Axygen | MCT-175-C | |
| MyOne Streptavidin C1 beads | Invitrogen | 65001 | |
| NEBuffer 2.1 (10x) | New England Biolabs | B7002S | |
| NEBuffer 3.1 (10x) | New England Biolabs | B7203S | |
| PBS | Gibco | 70013-032 | |
| PCR (strip) tubes | Biorad | TBS0201/ TCS0803 | |
| PCR thermocycler | Biorad | T100 | |
| Pfu Ultra II Buffer (10x) | Agilent | Comes with Pfu Ultra | |
| PfuUltra II Fusion HS DNA Polymerase | Agilent | 600674 | |
| Phase lock tube 15 mL | Qiagen | 129065 | |
| Phase lock tubes 2 mL | Qiagen | 129056 | |
| Phenol:chloroform:isoamyl alcohol | Invitrogen | 15593-049 | |
| Protease inhibitor cocktail | ThermoFisher | 78440 | |
| Proteinase K in ultrapure water 10 mg/mL | Invitrogen | 25530-031 | |
| Refrigerated Centrifuge | Eppendorf | 5810R | |
| RNase A, DNase and protease-free 10 mg/mL | Thermo Scientific | EN0531 | |
| Rotator | Argos technologies | EW-04397-40 or rocking platform | |
| SDS 1% | Fisher | BP13111 | |
| Sodium acetate pH = 5.2, 3 M | Sigma | ||
| Sub-Cell GT Horizontal Electrophoresis System | Biorad | 1704401 | |
| T4 DNA ligase 1 U/µL | Invitrogen | 100004817 | |
| T4 DNA polymerase | New England Biolabs | M0203L | |
| T4 DNA polymerase | New England Biolabs | M0203L | |
| T4 DNA polymerase 3 U/µL | New England Biolabs | M0203L | |
| T4 ligation buffer (5x) | Invitrogen | Y90001 | |
| T4 polynucleotide kinase 10 U/µL | New England Biolabs | M0201L | |
| Tabletop centrifuge | Eppendorf | 5425 | |
| TBE buffer | See recipe in supplemental materials | ||
| Tris Low EDTA Buffer (TLE) | See recipe in supplemental materials | ||
| Triton X-100 (10%) | Sigma | 93443 | |
| Truseq adapter oligos | Integrated DNA Technologies (IDT)) | https://www.idtdna.com/site/order/oligoentry | 250 nmole and HPLC purified |
| Tween 20 detergent | Fisher | 9005-64-5 | |
| Tween Wash Buffer | See recipe in supplemental materials | ||
| Vortex | Scientific Industries | (G560)SI-0236 |
Ссылки
- Dekker, J., Rippe, K., Dekker, M., Kleckner, N. Capturing chromosome conformation. Science. 295 (5558), 1306-1311 (2002).
- Simonis, M., et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nature Genetics. 38 (11), 1348-1354 (2006).
- Dostie, J., et al. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Research. 16 (10), 1299-1309 (2006).
- Lieberman-Aiden, E., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326 (5950), 289-293 (2009).
- Stadhouders, R., et al. Multiplexed chromosome conformation capture sequencing for rapid genome-scale high-resolution detection of long-range chromatin interactions. Nature Protocols. 8 (3), 509-524 (2013).
- Kalhor, R., Tjong, H., Jayathilaka, N., Alber, F., Chen, L. Genome architectures revealed by tethered chromosome conformation capture and population-based modeling. Nature Biotechnology. 30 (1), 90-98 (2012).
- Hsieh, T. H., et al. Mapping nucleosome resolution chromosome folding in yeast by micro-C. Cell. 162 (1), 108-119 (2015).
- Hsieh, T. -H. S., Fudenberg, G., Goloborodko, A., Rando, O. J. Micro-C XL: assaying chromosome conformation from the nucleosome to the entire genome. Nature Methods. 13 (12), 1009-1011 (2016).
- Denker, A., de Laat, W. The second decade of 3C technologies: detailed insights into nuclear organization. Genes & Development. 30 (12), 1357-1382 (2016).
- Rodley, C. D., Bertels, F., Jones, B., O’Sullivan, J. M. Global identification of yeast chromosome interactions using Genome conformation capture. Fungal Genetics and Biology. 46 (11), 879-886 (2009).
- Rao, S. S., et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 159 (7), 1665-1680 (2014).
- Alipour, E., Marko, J. F. Self-organization of domain structures by DNA-loop-extruding enzymes. Nucleic Acids Research. 40 (22), 11202-11212 (2012).
- Lajoie, B. R., Dekker, J., Kaplan, N. The Hitchhiker’s guide to Hi-C analysis: Practical guidelines. Methods. 72, 65-75 (2015).
- Belaghzal, H., Dekker, J., Gibcus, J. H. Hi-C 2.0: An optimized Hi-C procedure for high-resolution genome-wide mapping of chromosome conformation. Methods. 123, 56-65 (2017).
- Golloshi, R., Sanders, J. T., McCord, R. P. Iteratively improving Hi-C experiments one step at a time. Methods. 142, 47-58 (2018).
- Darrow, E. M., et al. Deletion of DXZ4 on the human inactive X chromosome alters higher-order genome architecture. Proceedings of the National Academy of Sciences of the United States of America. 113 (31), 4504-4512 (2016).
- Akgol Oksuz, B., et al. Systematic evaluation of chromosome conformation capture assays. Nature Methods. 18 (9), 1046-1055 (2021).
- Ghuryeid, J., et al. Integrating Hi-C links with assembly graphs for chromosome-scale assembly. PLoS Computational Biology. 15 (8), 1007273(2019).
- Gu, H., et al. Fine-mapping of nuclear compartments using ultra-deep Hi-C shows that active promoter and enhancer elements localize in the active A compartment even when adjacent sequences do not. bioRxiv. , (2021).
- Ramani, V., et al. Mapping 3D genome architecture through in situ DNase Hi-C. Nature Protocols. 11 (11), 2104-2121 (2016).
- Lafontaine, D. L., Yang, L., Dekker, J., Gibcus, J. H. Hi-C 3.0: Improved Protocol for Genome-Wide Chromosome Conformation Capture. Current Protocols. 1 (7), 198(2021).
- Belton, J. -M. M., et al. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods. 58 (3), 268-276 (2012).
- Truch, J., Telenius, J., Higgs, D. R., Gibbons, R. J. How to tackle challenging ChIP-Seq, with long-range cross-linking, Using ATRX as an example. Methods in Molecular Biology. 1832, Clifton, N.J. 105-130 (2018).
- Wang, P., et al. In situ chromatin interaction analysis using paired-end tag sequencing. Current Protocols. 1 (8), 174(2021).
- Tjong, H., et al. Population-based 3D genome structure analysis reveals driving forces in spatial genome organization. Proceedings of the National Academy of Sciences of the United States of America. 113 (12), 1663-1672 (2016).
- Nagano, T., et al. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature. 547 (7661), 61-67 (2017).
- Ramani, V., et al. Massively multiplex single-cell Hi-C. Nature Methods. 14 (3), 263-266 (2017).
- Meng, L., Wang, C., Shi, Y., Luo, Q. Si-C is a method for inferring super-resolution intact genome structure from single-cell Hi-C data. Nature Communications. 12 (1), 4369(2021).
- Tavares-Cadete, F., Norouzi, D., Dekker, B., Liu, Y., Dekker, J. Multi-contact 3C reveals that the human genome during interphase is largely not entangled. Nature Structural & Molecular Biology. 27 (12), 1105-1114 (2020).
- Vermeulen, C., et al. Multi-contact 4C: long-molecule sequencing of complex proximity ligation products to uncover local cooperative and competitive chromatin topologies. Nature Protocols. 15 (2), 364-397 (2020).
- Oomen, M. E., Hedger, A. K., Watts, J. K., Dekker, J. Detecting chromatin interactions between and along sister chromatids with SisterC. Nature Methods. 17 (10), 1002-1009 (2020).
- Mitter, M., et al. Conformation of sister chromatids in the replicated human genome. Nature. 586 (7827), 139-144 (2020).
- Krietenstein, N., et al. Ultrastructural Details of Mammalian Chromosome Architecture. Molecular Cell. 78 (3), 554-565 (2020).
- Hughes, J. R., et al. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nature Genetics. 46 (2), 205-212 (2014).
- Fullwood, M. J., et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 462 (7269), 58-64 (2009).
- Fang, R., et al. Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Research. 26 (12), 1345-1348 (2016).
- Mumbach, M. R., et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nature Methods. 13 (11), 919-922 (2016).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеСмотреть дополнительные статьи
This article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены