
Method Article
Acquisizione della conformazione cromosomica su scale di lunghezza
In questo articolo
Riepilogo
Hi-C 3.0 è un protocollo Hi-C migliorato che combina crosslinker di formaldeide e disuccinimidil glutarato con un cocktail di enzimi di restrizione DpnII e DdeI per aumentare il rapporto segnale-rumore e la risoluzione del rilevamento dell'interazione cromatina.
Abstract
La cattura della conformazione cromosomica (3C) viene utilizzata per rilevare interazioni tridimensionali della cromatina. Tipicamente, la reticolazione chimica con la formaldeide (FA) viene utilizzata per fissare le interazioni della cromatina. Quindi, la digestione della cromatina con un enzima di restrizione e la successiva legatura delle estremità del frammento converte la prossimità tridimensionale (3D) in prodotti di legatura unici. Infine, dopo l'inversione dei legami incrociati, la rimozione delle proteine e l'isolamento del DNA, il DNA viene tagliato e preparato per il sequenziamento ad alto rendimento. La frequenza di legatura di prossimità di coppie di loci è una misura della frequenza della loro colocalizzazione nello spazio tridimensionale in una popolazione cellulare.
Una libreria Hi-C sequenziata fornisce informazioni sull'intero genoma sulle frequenze di interazione tra tutte le coppie di loci. La risoluzione e la precisione di Hi-C si basano su una reticolazione efficiente che mantiene i contatti della cromatina e la frammentazione frequente e uniforme della cromatina. Questo articolo descrive un protocollo Hi-C in situ migliorato, Hi-C 3.0, che aumenta l'efficienza della reticolazione combinando due reticolanti (formaldeide [FA] e disuccinimidil glutarato [DSG]), seguiti da una digestione più fine utilizzando due enzimi di restrizione (DpnII e DdeI). Hi-C 3.0 è un singolo protocollo per la quantificazione accurata delle caratteristiche di ripiegamento del genoma su scale più piccole come loop e domini topologicamente associati (TAD), nonché caratteristiche su scale più grandi a livello di nucleo come i compartimenti.
Introduzione
La cattura della conformazione cromosomica è stata utilizzata dal 20021. Fondamentalmente, ogni variante di cattura della conformazione si basa sulla fissazione delle interazioni DNA-proteina e proteina-proteina per preservare l'organizzazione della cromatina 3D. Questo è seguito dalla frammentazione del DNA, di solito dalla digestione di restrizione e, infine, dalla religenza delle estremità del DNA vicine per convertire loci spazialmente prossimali in sequenze di DNA covalenti uniche. I protocolli 3C iniziali utilizzavano la PCR per campionare specifiche interazioni "one-to-one". I successivi saggi 4C hanno permesso di rilevare interazioni "uno-a-tutti"2, mentre 5C ha rilevato interazioni "molti-a-molti"3. La cattura della conformazione cromosomica è arrivata a pieno compimento dopo aver implementato il sequenziamento ad alto rendimento (NGS) di nuova generazione, che ha permesso di rilevare interazioni genomiche "all-to-all" utilizzando Hi-C 4 a livello di genoma e tecniche comparabili come 3C-seq5, TCC6 eMicro-C 7,8 (vedi anche revisione di Denker e De Laat9).
In Hi-C, i nucleotidi biotinilati sono usati per marcare le sporgenze di 5' dopo la digestione e prima della legatura (Figura 1). Ciò consente la selezione di frammenti correttamente digeriti e religati utilizzando perle rivestite di streptavidina, distinguendolo dal GCC10. Un importante aggiornamento del protocollo Hi-C è stato implementato da Rao et al.11, che hanno eseguito la digestione e la religazione in nuclei intatti (cioè in situ) per ridurre i prodotti di legatura spuria. Inoltre, la sostituzione della digestione HindIII con la digestione MboI (o DpnII) ha ridotto la dimensione del frammento e aumentato il potenziale di risoluzione di Hi-C. Questo aumento ha permesso la rilevazione di strutture relativamente piccole e una localizzazione genomica più precisa dei punti di contatto, come i loop di DNA tra piccoli cis-elementi, ad esempio loop tra siti legati al CTCF generati dall'estrusione di loop11,12. Tuttavia, questo potenziale ha un costo. In primo luogo, un aumento di due volte della risoluzione richiede un aumento di quattro volte (22) delle letture di sequenziamento13. In secondo luogo, le piccole dimensioni dei frammenti aumentano la possibilità di confondere i frammenti vicini non digeriti con frammenti digeriti e relegati14. Come accennato, in Hi-C, i frammenti digeriti e relegati differiscono dai frammenti non digeriti per la presenza di biotina alla giunzione della legatura. Tuttavia, è necessaria una corretta rimozione della biotina dalle estremità non legate per assicurare che solo le giunzioni di legatura vengano abbassate14,15.
Con la diminuzione del costo di NGS, diventa possibile studiare il ripiegamento cromosomico in modo più dettagliato. Per ridurre le dimensioni dei frammenti di DNA, e quindi aumentare la risoluzione, il protocollo Hi-C può essere adattato per utilizzare più frequentemente gli enzimi di restrizione di taglio16 o per utilizzare combinazioni di enzimi di restrizione17,18,19. In alternativa, MNasi 7,8 in Micro-C e DNasi in DNasi Hi-C20 possono essere titolate per ottenere una digestione ottimale.
Una recente valutazione sistematica dei fondamenti dei metodi 3C ha mostrato che la rilevazione delle caratteristiche di ripiegamento cromosomico su ogni scala di lunghezza è notevolmente migliorata con la reticolazione sequenziale con 1% FA seguito da 3 mM DSG17. Inoltre, Hi-C con digestione HindIII era l'opzione migliore per rilevare caratteristiche di piegatura su larga scala, come i compartimenti, e che Micro-C era superiore nel rilevare caratteristiche di ripiegamento su piccola scala come i loop di DNA. Questi risultati hanno portato allo sviluppo di un'unica strategia "Hi-C 3.0" ad alta risoluzione, che utilizza la combinazione di reticolanti FA e DSG seguita da una doppia digestione con endonucleasi DpnII e DdeI21. Hi-C 3.0 fornisce una strategia efficace per l'uso generale perché rileva con precisione le caratteristiche di piegatura su tutte le scale di lunghezza17. La parte sperimentale del protocollo Hi-C 3.0 è dettagliata qui e vengono mostrati i risultati tipici che ci si può aspettare dopo il sequenziamento.

Figura 1: Procedura Hi-C in sei fasi. Le celle vengono fissate prima con FA e poi DSG (1). Quindi, la lisi precede una doppia digestione con DdeI e DpnII (2). La biotina viene aggiunta per riempimento sporgente e le estremità smussate prossimali vengono legate (3) prima della purificazione del DNA (4). La biotina viene rimossa dalle estremità non legate prima della sonicazione e della selezione delle dimensioni (5). Infine, il pull-down della biotina consente la legatura dell'adattatore e l'amplificazione della libreria mediante PCR (6). Abbreviazioni: FA = formaldeide; DSG = glutarato di disuccinimidile; B = Biotina. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}
Protocollo
1. Fissazione mediante reticolazione
- Fissazione della formaldeide: a partire da cellule in monostrato
- Avere cellule seminate in terreno appropriato per raccogliere 5 × 106 cellule per piastra di 150 mm.
NOTA: Gli utenti possono scegliere qualsiasi contenitore preferito che garantisca una crescita ottimale di qualsiasi linea cellulare di mammifero. Inoltre, le cellule possono essere isolate dal tessuto. - Aspirare il fluido con una pipetta Pasteur accoppiata a una trappola sottovuoto da piastra da 150 mm, lavare 2x con ~10 mL HBSS.
- Immediatamente prima della reticolazione, preparare una soluzione di reticolazione all'1% di FA in una provetta da 50 mL combinando 22,5 mL di HBSS e 625 μL di FA al 37% fino a una concentrazione finale dell'1%. Mescolare delicatamente dondolando.
ATTENZIONE: Utilizzare cappa aspirante; La formaldeide è tossica. - Per reticolare le cellule, versare 23,125 ml della soluzione FA all'1% su ciascuna piastra da 15 cm.
- Incubare a temperatura ambiente per 10 minuti e dondolare delicatamente le piastre a mano ogni 2 minuti.
- Aggiungere 1,25 mL di glicina 2,5 M (128 mM finale) e ruotare delicatamente la piastra per estinguere la reazione di reticolazione.
- Incubare a temperatura ambiente per 5 minuti e continuare l'incubazione su ghiaccio per almeno 15 minuti per interrompere la reticolazione.
- Raschia le cellule dalle piastre con un raschietto cellulare o un poliziotto di gomma.
- Trasferire la sospensione cellulare in un tubo conico da 50 mL con un pipet. Centrifugare a 1.000 × g per 10 minuti a temperatura ambiente ed eliminare il surnatante mediante aspirazione.
- Lavare il pellet cellulare una volta con 10 ml di soluzione salina tamponata fosfato di Dulbecco (DPBS), utilizzando un pipet per risospendere. Quindi, centrifugare a 1.000 × g per 10 minuti a temperatura ambiente. Procedere immediatamente alla reticolazione DSG.
NOTA: Fare attenzione quando si lava il pellet cellulare poiché i pellet cellulari possono essere sciolti e le cellule potrebbero essere perse.
- Avere cellule seminate in terreno appropriato per raccogliere 5 × 106 cellule per piastra di 150 mm.
- Fissazione della formaldeide: a partire dalle cellule in sospensione
- Avere cellule seminate in terreno appropriato per raccogliere 5 × 106 cellule per vaso.
NOTA: Gli utenti possono scegliere qualsiasi contenitore preferito che garantisca una crescita cellulare ottimale di qualsiasi linea cellulare di mammifero. - Immediatamente prima del raccolto, contare le cellule e trasferire 5 × 106 cellule in un tubo conico da 50 ml.
- Pellettare delicatamente le celle centrifugando a 300 × g per 10 minuti a temperatura ambiente.
- Preparare la soluzione di reticolazione all'1% di FA aggiungendo 1,25 ml di FA al 37% a 45 ml di HBSS e mescolare capovolgendo il tubo più volte.
NOTA: Aggiungere l'intero 1,25 ml di FA senza dividere la quantità.
ATTENZIONE: La formaldeide è altamente tossica. - Risospendere il pellet cellulare nella soluzione di reticolazione FA da 46,25 mL all'1% preparata nella fase precedente mediante pipettaggio su e giù.
- Incubare a temperatura ambiente per esattamente 10 minuti su rotatore, bilanciere o inversione manuale delicata del tubo ogni 1-2 minuti.
- Estinguere la reazione di reticolazione aggiungendo 2,5 ml di glicina 2,5 M (128 mM finale) e mescolare bene capovolgendo il tubo.
- Incubare per 5 minuti a temperatura ambiente, quindi su ghiaccio per almeno 15 minuti per interrompere completamente la reticolazione.
- Centrifugare a temperatura ambiente per pellettare le cellule reticolate a 1.000 × g per 10 minuti ed eliminare il surnatante mediante aspirazione.
- Lavare le celle una volta con 10 ml di DPBS, quindi centrifugare a 1.000 × g per 10 minuti a temperatura ambiente. Scartare completamente il surnatante usando un pipet e procedere immediatamente alla reticolazione DSG.
NOTA: Fare attenzione quando si lava il pellet cellulare poiché i pellet cellulari possono essere sciolti e le cellule potrebbero essere perse.
- Avere cellule seminate in terreno appropriato per raccogliere 5 × 106 cellule per vaso.
- Crosslinking con glutarato di disuccinimidil
- Risospendere le celle pellettate in 9,9 mL di DPBS prima di aggiungere 100 μL di 300 mM DSG (3 mM finale). Mescolare per inversione.
NOTA: DSG è sensibile all'umidità. È importante preparare un nuovo stock di 300 mM DSG in DMSO il giorno della reticolazione.
ATTENZIONE: DSG in DMSO è altamente tossico. - Crosslink le celle a temperatura ambiente per 40 minuti su un rotatore.
- Aggiungere 1,925 ml di glicina 2,5 M (400 mM finale), capovolgere per miscelare e incubare a temperatura ambiente per 5 minuti.
- Centrifugare le celle a 2.000 × g per 15 minuti a temperatura ambiente.
NOTA: Prestare attenzione quando si rimuove il surnatante dai pellet a cellule sciolte. - Risospendere il pellet in 1 mL di albumina sierica bovina allo 0,05% (BSA)-DPBS e trasferirlo in una provetta da 1,7 ml.
NOTA: L'aggiunta di BSA può aiutare a ridurre l'aggregazione cellulare. - Centrifugare le cellule a 2.000 × g per 15 minuti a 4 °C e rimuovere il surnatante mediante pipetta.
NOTA: Per evitare di perdere il pellet, rimuovere rapidamente e completamente il surnatante. - Congelare a scatto il pellet in azoto liquido e conservare a -80 °C o procedere immediatamente alla fase successiva.
- Risospendere le celle pellettate in 9,9 mL di DPBS prima di aggiungere 100 μL di 300 mM DSG (3 mM finale). Mescolare per inversione.
2. Cattura della conformazione cromosomica
- Lisi cellulare e digestione della cromatina
- Risospendere (pipettare) le aliquote delle cellule reticolate (~5 × 106 cellule) in 1 mL di tampone di lisi ghiacciato (ricetta nella Tabella supplementare S1) contenente 10 μL di cocktail inibitore della proteasi e trasferire in un omogeneizzatore di dounce per un'incubazione di 15 minuti su ghiaccio.
NOTA: Aggiungere inibitori della proteasi al tampone di lisi immediatamente prima dell'uso. - Spostare lentamente il pestello A su e giù 30 volte per omogeneizzare le cellule sul ghiaccio e incubare sul ghiaccio per 1 minuto per consentire alle cellule di raffreddarsi, prima di altri 30 colpi.
- Trasferire il lisato in una provetta da microcentrifuga da 1,7 ml.
NOTA: Mantenere la sospensione in movimento perché a volte le cellule si attaccano alla punta della pipetta. - Centrifugare la sospensione lisata a 2.500 × g per 5 minuti a temperatura ambiente.
- Scartare il surnatante e far scorrere o vortice il pellet bagnato per risospendere. Rimuovere il più possibile il surnatante per ottenere una sostanza simile allo yogurt con grumi minimi.
- Risospendere il pellet in 500 μL di tampone di restrizione 1x ghiacciato (da 10x; vedi ricetta nella tabella supplementare S1) e centrifugare per 5 minuti a 2.500 × g. Ripetere questo passaggio per un secondo lavaggio.
NOTA: Il pellet in 1x Restriction Buffer è più granulare del pellet precedente in tampone di lisi. - Risospendere le celle in un volume finale di 360 μL di 1x Restriction Buffer mediante pipettaggio dopo aver aggiunto ~340 μL al volume di trascinamento del pellet, che dipende dalla dimensione della cella.
- Mettere da parte 18 μL di ciascun lisato per testare l'integrità della cromatina (CI). Conservare i campioni di IC a 4 °C.
- Aggiungere 38 μL di sodio dodecilsolfato (SDS) all'1% a ciascun tubo Hi-C (volume totale di 380 μL) e mescolare accuratamente mediante pipettaggio senza introdurre bolle.
ATTENZIONE: SDS è tossico. - Incubare i campioni a 65 °C senza agitare per esattamente 10 minuti per aprire la cromatina.
- Posizionare immediatamente i tubi sul ghiaccio e preparare la miscela di digestione con Triton X-100 per estinguere la SDS come descritto nella Tabella 1.
- Aggiungere 107 μL della miscela di digestione al tubo Hi-C (487 μL totali) per digerire la cromatina durante la notte (~16 h) a 37 °C in un termomiscelatore con agitazione a intervalli (ad esempio, 900 giri / min, 30 s accesi, 4 minuti spenti).
NOTA: L'aggiunta di Triton ad una concentrazione finale dell'1% serve a spegnere la SDS.
- Risospendere (pipettare) le aliquote delle cellule reticolate (~5 × 106 cellule) in 1 mL di tampone di lisi ghiacciato (ricetta nella Tabella supplementare S1) contenente 10 μL di cocktail inibitore della proteasi e trasferire in un omogeneizzatore di dounce per un'incubazione di 15 minuti su ghiaccio.
- Biotinilazione delle estremità del DNA
- Dopo la digestione durante la notte, trasferire i campioni a 65 °C per 20 minuti per disattivare l'attività endonucleasi rimanente.
- Durante l'incubazione, preparare una miscela master di riempimento come mostrato nella Tabella 2.
- Dopo l'incubazione, posizionare immediatamente i campioni sul ghiaccio.
- Mettere da parte un controllo della digestione (DC) da 10 μL per ciascun campione e conservare a 4 °C.
- Rimuovere la condensa dal coperchio con un pipet o ruotando. A ciascun campione, aggiungere 58 μL di miscela di riempimento di biotina (volume totale del campione 535 μL) e pipettare delicatamente senza formare bolle.
- Incubare i campioni a 23 °C per 4 ore in un termomiscelatore (ad esempio, 900 giri/min, 30 s acceso, 4 min spento).
- Legatura di frammenti di DNA prossimale
- Preparare la miscela di legatura come mostrato nella Tabella 3 mentre il riempimento della biotina è in incubazione.
- Aggiungere 665 μL della miscela di legatura a ciascun campione (volume totale del campione 1.200 μL). Mescolare delicatamente mediante pipettaggio.
- Incubare i campioni a 16 °C per 4 ore in un termomiscelatore con agitazione a intervalli (ad esempio, 900 giri/min, 30 s accesi, 4 minuti spenti). Conservare questi campioni contenenti cromatina legata covalentemente a 4 °C per alcuni giorni.
- Inversione della reticolazione
- Portare i volumi dei campioni CI e DC a 50 μL con 1x Tris Low EDTA (TLE; vedere la ricetta nella Tabella supplementare S1).
- Aggiungere 10 μL di 10 mg/mL di proteinasi K ai campioni di CI e DC.
- Incubare a 65 °C durante la notte con agitazione a intervalli (ad esempio, 900 giri/min, 30 s accesi, 4 minuti spenti). In alternativa, eseguire un'inversione di 30 minuti di reticolazione per questi controlli durante la purificazione del DNA dei campioni Hi-C.
- A ciascun campione Hi-C, aggiungere 50 μL di 10 mg/mL di proteinasi K e incubare a 65 °C per almeno 2 ore con agitazione a intervalli (ad esempio, 900 giri/min, 30 s accesi, 4 minuti di spegnimento).
- Aggiungere altri 50 μL di 10 mg/mL di proteinasi K a ciascuna provetta Hi-C (volume totale del campione 1.300 μL) e continuare l'incubazione a 65 °C per tutta la notte. Conservare a 4 °C fino alla purificazione del DNA.
NOTA: La suddivisione delle incubazioni della proteinasi K garantisce la digestione totale delle proteine.
- Purificazione del DNA
- Lasciare raffreddare i tubi da 65 °C fino alla temperatura ambiente.
- Trasferire ciascun campione in una provetta conica da 15 mL e aggiungere 2,6 mL (2x volume) di fenolo:cloroformio:alcool isoamilico a ciascuna provetta.
ATTENZIONE: Fenolo: cloroformio: l'alcol isoamilico è un irritante molto tossico ed è potenzialmente cancerogeno. - Vortice ogni tubo per 1 minuto e poi trasferire il suo contenuto in un tubo phase-lock da 15 ml.
- Centrifugare i campioni per 5 minuti alla massima velocità (1.500-3.500 × g) in una centrifuga da banco.
- Versare con cautela la fase acquosa in un tubo ultracentrifugo da 35 ml e aggiungere acqua ultrapura a un volume finale di 1.250 μL.
NOTA: Utilizzare tubi per adattarsi all'ultracentrifuga disponibile o dividere in più tubi di microcentrifuga. - Aggiungere un 1/10° volume (~125 μL) di acetato di sodio 3 M e mescolare bene per inversione.
- Aggiungere un volume 2,5x (~ 3,4 ml) di etanolo ghiacciato al 100% a ciascun campione, bilanciare i tubi per l'ultracentrifugazione aggiungendo etanolo ghiacciato al 100% e mescolare bene per inversione.
- Incubare i tubi su ghiaccio secco per ~ 15 minuti (evitare la solidificazione).
- Centrifugare i tubi a 18.000 × g per 30 minuti a 4 °C.
NOTA: Per i rotori angolati: contrassegnare i tubi per dove sarà il pellet. - Utilizzando una pipetta, rimuovere e scartare completamente il surnatante dal lato non pellet.
NOTA: A questo punto, il pellet dovrebbe diventare visibile e può essere marcato sul tubo, perché potrebbe non essere chiaramente visibile dopo l'essiccazione nella fase successiva. - Asciugare all'aria i campioni per circa 10 minuti o fino a quando non sono visibilmente asciutti.
- Solubilizzare ogni pellet in 450 μL di 1x TLE mediante pipettaggio o vortice e trasferire in un'unità filtrante centrifuga (CFU) da 0,5 mL con un cut-off di peso molecolare di 3 kDa.
- Centrifugare il CFU alla massima velocità per 10 minuti ed eliminare il flowthrough. Lavare ogni tubo ultracentrifuga con ulteriori 450 μL di 1x TLE e trasferire al suo CFU per un altro lavaggio.
NOTA: Lavare il CFU in questo modo limita la perdita di DNA riducendo la concentrazione di sale. - Centrifugare il CFU alla massima velocità per 10 minuti ed eliminare il flowthrough.
- Aggiungere 80 μL di 1x TLE alla colonna e girare la colonna in un nuovo tubo di raccolta prima di centrifugare per 2 minuti alla massima velocità per ottenere un volume finale di ~100 μL.
- Aggiungere 1 μL di RnaseA (1 mg/ml; diluizione 10 volte di 10 mg/mL di brodo) a ciascun campione e incubare a 37 °C su un blocco termico, a bagnomaria o in un termomiscelatore per almeno 30 minuti.
- Dopo il trattamento con RNASI, prelevare i campioni da 37 °C e conservarli a 4 °C fino alla fase di controllo qualità.
- Controllo della qualità della cromatina, digestione enzimatica e legatura del campione
- Raffreddare i campioni CI e DC a temperatura ambiente dopo aver invertito i reticoli al punto 2.4.5. Quindi, trasferire in un tubo di blocco di fase prefilato da 2 ml.
NOTA: Assicurarsi che il contenuto del blocco di fase sia centrifugato a pellet (velocità massima per 2 minuti). - Aggiungere 200 μL di fenolo:cloroformio:alcool isoamilico e mescolare i campioni a vortice per 1 minuto.
- Centrifugare i tubi per 5 minuti alla massima velocità.
- Trasferire la fase acquosa da ciascun campione (~50 μL) in una nuova provetta microfughe da 1,7 ml.
- Aggiungere 1 μL di Rnase A (da 1 mg/ml) e incubare a 37 °C per almeno 30 minuti.
- Caricare i campioni su un gel di agarosio allo 0,8% come raccomandato nella Tabella 4.
NOTA: i risultati attesi da un controllo di qualità sono illustrati nella Figura 2. - Quantificare il DNA mediante densitometria dal gel o utilizzando Qubit o Nanodrop.
NOTA: una quantificazione accurata garantisce il corretto volume di input nella parte successiva del protocollo. Utilizzare diversi standard con una quantità nota per costruire una curva standard.
- Raffreddare i campioni CI e DC a temperatura ambiente dopo aver invertito i reticoli al punto 2.4.5. Quindi, trasferire in un tubo di blocco di fase prefilato da 2 ml.
Tabella 1: Reagenti di digestione. Clicca qui per scaricare questa tabella.
Tabella 2: Reagenti fill-in della biotina. *Si noti che la modifica degli enzimi può richiedere diversi tamponi e dNTP biotinilati. Clicca qui per scaricare questa tabella.
Tabella 3: Reagenti della miscela di legatura. Abbreviazione: BSA = albumina sierica bovina. Clicca qui per scaricare questa tabella.
Tabella 4: Parametri di carico del gel per la valutazione della qualità e della dimensione della selezione. Clicca qui per scaricare questa tabella.

Figura 2: Gel di agarosio che mostra i risultati tipici del controllo di qualità della purificazione post-DNA . (A) Il controllo CI deve indicare una banda di DNA ad alto peso molecolare. (B) I campioni DC e Hi-C mostrano una gamma di dimensioni del DNA. Il campione Hi-C, essendo stato combinato in frammenti più grandi, dovrebbe avere un peso molecolare superiore rispetto al DC. L'intervallo di concentrazione dei marcatori consente di generare una curva standard. Si noti che, in questo esempio, l'IC è stato caricato su un gel separato, ma si consiglia di caricare ed eseguire tutti i campioni e i controlli insieme. Abbreviazioni: CI = integrità della cromatina; DC = controllo della digestione; Hi-C = legato alla prossimità. Fare clic qui per visualizzare una versione ingrandita di questa figura.
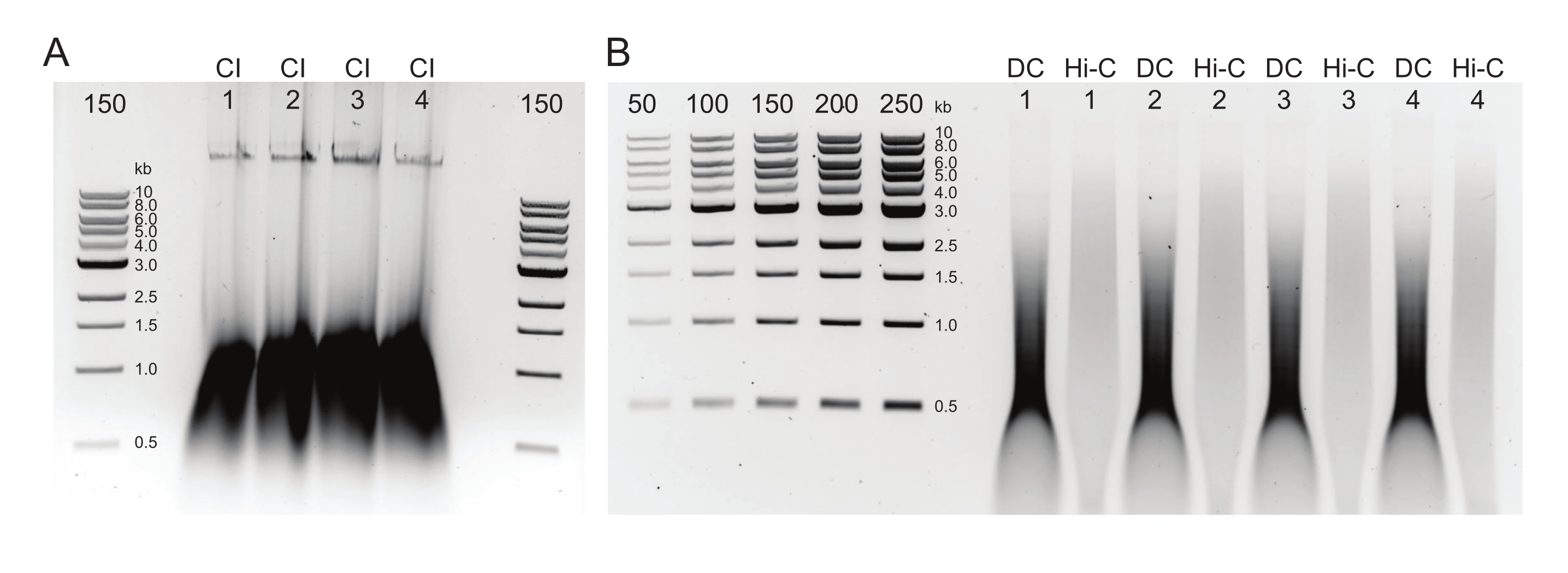{kind=link}
3. Preparazione della libreria di sequenziamento Hi-C
- Rimozione della biotina dalle estremità non legate
- Preparare le reazioni di rimozione della biotina come mostrato nella Tabella 5.
NOTA: In genere, 10 μg di DNA sono sufficienti, ma possono essere utilizzati fino a 30 μg. - Distribuire 2 aliquote da 65 μL da ogni reazione da 130 μL in due provette PCR.
- Trasferire in una macchina termociclatrice o PCR e incubare come descritto nella Tabella 5.
NOTA: I campioni possono essere conservati qui a 4 °C (giorni o settimane), -20 °C (a lungo termine) o immediatamente trasferiti alla sonicazione.
- Preparare le reazioni di rimozione della biotina come mostrato nella Tabella 5.
- Sonicazione
- Raggruppare i duplicati del campione dalla fase di rimozione della biotina (volume totale del campione di 130 μL) in una provetta da sonicatore da 130 μL per la sonicazione.
- Sonicare i campioni utilizzando i parametri indicati nella Tabella 6 per ottenere una distribuzione stretta e stretta al di sotto di 500 bp.
NOTA: è possibile utilizzare diversi tipi di sonicatore, ma per una distribuzione di frammenti stretta (100-500 bp), le impostazioni del sonicatore potrebbero richiedere l'ottimizzazione.
- Selezione delle taglie con perline magnetiche
- Pipet il DNA sonicato dai tubi sonicatori in un tubo a basso legame da 1,7 ml.
- Portare ogni campione a un volume totale di 500 μL con 1x TLE. Cerca di avvicinare il volume il più possibile a 500 μL, poiché il rapporto tra campione e miscela di sfere magnetiche è essenziale per la selezione delle dimensioni.
- Aggiungere 400 μL di miscela di sfere magnetiche a ciascun tubo per ottenere un rapporto tra miscela di sfere magnetiche e volume del campione di 0,8.
NOTA: In queste condizioni, le perle catturano frammenti di DNA >300 bp, che sarà la frazione superiore. Il surnatante conterrà frammenti <300 bp, che sarà la frazione inferiore. - Mescolare i tubi a vortice e incubare per 10 minuti a temperatura ambiente su un rotatore. Per questo e altri rapporti di selezione delle dimensioni, assicurarsi che l'intero volume del campione si mescoli bene. Cerca la "modalità irregolare" che hanno alcuni rotori, che funziona bene per volumi più piccoli.
- Incubare per 5 minuti a temperatura ambiente su un separatore di particelle magnetico (MPS).
- Durante l'incubazione, aggiungere 500 μL della miscela di sfere magnetiche a un tubo fresco a basso legame da 1,7 μL per ciascun campione.
NOTA: Questi tubi verranno utilizzati per la successiva selezione dimensionale della frazione inferiore generando una miscela di sfere magnetiche per il rapporto campione di 1,1: 1. - Lasciare i tubi sull'MPS per 5 minuti.
- Rimuovere il surnatante dalle perle e risospendere con 150 μL della miscela di sfere magnetiche.
NOTA: Questo passaggio evita la saturazione delle perle con DNA aumentando il numero di perle senza aumentare il volume. - Trasferire il surnatante dal punto 3.3.5 al tubo marcato preparato per selezionare la frazione inferiore (punto 3.3.8).
NOTA: 150 μL di miscela di sfere magnetiche + 400 μL di miscela di sfere magnetiche 0,8x (550 μL totali) diviso per 500 μL di campione iniziale = 1,1x rapporto sfere magnetiche/campione. - Mescolare i tubi della frazione inferiore mediante vortice e incubare per 10 minuti a temperatura ambiente su un rotatore.
NOTA: Le perle legheranno frammenti di DNA >100 bp, risultando in una frazione finale legata alle perline di 100-300 bp. - Posizionare i tubi della frazione inferiore sull'MPS per 5 minuti (temperatura ambiente).
- Rimuovere il surnatante e centrifugare brevemente le provette per rimuovere ulteriormente il surnatante il più possibile.
- Lavare le perle da entrambe le frazioni due volte utilizzando 200 μL di etanolo al 70% e recuperare le perle per 5 minuti sull'MPS ogni volta.
- Dopo una rapida centrifuga, rimuovere completamente l'etanolo e asciugare ulteriormente le perle sull'MPS.
NOTA: Asciugare fino a completa evaporazione dell'alcool. Il pellet dovrebbe apparire come cioccolato fondente senza cracking (può richiedere ~ 10 min). - Risospendere entrambe le frazioni in 50 μL di tampone 1x TLE. Incubare a temperatura ambiente per 10 minuti e picchiettare o sfiorare i tubi ogni due minuti per stimolare la miscelazione e l'eluizione.
- Separare le perle dal surnatante sull'MPS per 5 minuti per entrambe le frazioni.
- Conservare il surnatante da ogni campione. Pipet il surnatante in un tubo a basso legame da 1,7 ml.
NOTA: I campioni possono essere conservati a 4 °C per alcuni giorni o a -20 °C per un lungo periodo. - Eseguire un gel di agarosio al 2% come nella Tabella 4 per determinare la qualità e la quantità del campione. Vedere la Figura 3 per un esempio di tale gel.
NOTA: Se per esperienza, la sonicazione è altamente riproducibile, si può saltare questo gel e procedere immediatamente alla riparazione. Si raccomanda agli utenti di conservare le frazioni superiori fino a dopo la titolazione PCR. Quantità di DNA subottimali che richiederebbero molta amplificazione PCR possono essere salvate dal materiale nella frazione superiore. - Quantificare la quantità di DNA dal gel direttamente dopo aver generato una curva standard dall'input della scala di DNA noto o utilizzando un Qubit o Nanodrop.
- Fine riparazione
- Preparare la miscela di riparazione finale come nella tabella 7 (quantità per reazione data).
- Trasferire i restanti 46 μL di DNA eluito dalla frazione inferiore alle provette PCR e aggiungere 24 μL della miscela di riparazione finale preparata. Incubare in una macchina PCR, come proposto nella Tabella 7.
- Una volta completato il programma, mantenere i campioni a 4 °C fino al pull-down.
- Pull-down di prodotti di legatura biotinilati con perle rivestite di streptavidina
- Determinare la quantità di perle rivestite di streptavidina per ciascuna libreria dalla selezione delle dimensioni quantificate (passo 3.3.19).
NOTA: Queste sfere rivestite di streptavidina (soluzione da 10 mg/ml) possono legare 20 μg di DNA a doppio filamento per mg di perle (= 20 μg / 100 μL di perle). Utilizzare 2 μL per ogni 1 μg di DNA Hi-C ma non meno di 10 μL. - Mescolare le perle rivestite di streptavidina e pipetare il volume di perline necessario per ciascuna libreria (calcolato nel passaggio precedente) in singoli tubi a bassa legatura da 1,7 ml.
- Risospendere le perle in 400 μL di tampone di lavaggio Tween (TWB; vedere la ricetta nella tabella supplementare S1) e incubare per ~3 minuti a temperatura ambiente su un rotatore (vedere le istruzioni al punto 3.3.4).
- Separare le perline dal surnatante sull'MPS per 1 minuto e rimuovere il surnatante.
- Lavare le perline pipettando altri 400 μL di TWB.
- Separare le perline dal surnatante sull'MPS per 1 minuto e rimuovere il surnatante.
- Aggiungere 400 μL di 2x Binding Buffer (BB) (ricetta nella tabella supplementare S1) alle perline e risospendere. Inoltre, aggiungere 330 μL di 1x TLE e la soluzione da End-repair (dal punto 3.4.3).
- Incubare i campioni per 15 minuti a temperatura ambiente mentre si mescola su un rotatore.
- Separare le perline dal surnatante sull'MPS per 1 minuto e rimuovere il surnatante.
- Aggiungere 400 μL di 1x BB alle perline e risospendere.
- Separare le perline dal surnatante sull'MPS per 1 minuto e rimuovere il surnatante.
- Aggiungere 100 μL di 1x TLE per lavare le perline.
- Separare le perline dal surnatante sull'MPS per 1 minuto e rimuovere il surnatante.
- Infine, aggiungere 41 μL di 1x TLE per risospendere le perline.
- Determinare la quantità di perle rivestite di streptavidina per ciascuna libreria dalla selezione delle dimensioni quantificate (passo 3.3.19).
- A-tailing
- Preparare la miscela A-tailing come nella tabella 8.
- Pipet le reazioni in provette PCR e incubare come nella Tabella 8.
- Posizionare i tubi PCR sul ghiaccio immediatamente dopo la rimozione dal termociclatore e trasferire il contenuto in tubi a basso legame da 1,7 ml.
- Separare le perline dal surnatante sull'MPS per 1 minuto ed eliminare il surnatante.
- Aggiungere 400 μL di tampone di legatura 1x, diluito da 5x tampone DNA ligasi T4 con acqua ultrapura.
- Separare le perline dal surnatante sull'MPS per 1 minuto, quindi eliminare il surnatante.
- Aggiungere 1x tampone di legatura a un volume finale di 40 μL.
- Oligo adattatori di ricottura
- Preparare scorte di oligo adattatori da 100 μM (Tabella 9).
NOTA: Ordinare 250 nmoles di oligo purificati HPLC. - Ricottura degli adattatori in provette per PCR come descritto nella Tabella 9.
- Utilizzare un termociclatore PCR per aumentare gradualmente la temperatura da 0,5 °C / s a 97,5 °C. Tenere a 97,5 °C per 2,5 minuti.
- Utilizzare un termociclatore PCR per aumentare gradualmente la temperatura a 0,1 °C/s per 775 cicli (raggiungendo i 20 °C). Mantenere la temperatura a 4 °C fino a un ulteriore utilizzo.
- Aggiungere 83 μL di 1x tampone di ricottura (ricetta nella tabella supplementare S1) per diluire gli adattatori a 15 μM. Conservare gli adattatori a -20 °C.
- Preparare scorte di oligo adattatori da 100 μM (Tabella 9).
- Legatura dell'adattatore di sequenziamento
- Preparare la miscela di legatura adattatrice in un tubo di legame basso da 1,7 mL (Tabella 10).
- Ligate per 2 ore a temperatura ambiente.
- Separare le perline dal surnatante sull'MPS per 1 minuto ed eliminare il surnatante.
- Aggiungere 400 μL di TWB e pipettare le perline su e giù con attenzione prima di incubare sul rotatore per 5 minuti a temperatura ambiente. Separare le perline dal surnatante sull'MPS e ripetere ancora una volta questo passaggio.
- Separare le perle dal surnatante sull'MPS (~1 min), scartare il surnatante e aggiungere 200 μL di 1x BB.
- Separare le perle dal surnatante sull'MPS (~1 min) ed eliminare il surnatante.
- Aggiungere 200 μL di 1x tampone pre-PCR (da 10x; ricetta nella tabella supplementare S1) e trasferire in un nuovo tubo a basso legame da 1,7 ml.
- Separare le perle dal surnatante sull'MPS (~1 min) ed eliminare il surnatante.
- Aggiungere 20 μL di 1x tampone pre-PCR e miscelare mediante pipettaggio.
- Conservare i tubi in ghiaccio durante l'uso o conservare a 4 °C.
- Ottimizzazione del numero del ciclo PCR mediante titolazione
- Impostare 30 μL di reazioni master mix per campione come nella Tabella 11 (quantità per reazione data).
- Eseguire il minor numero di cicli e prendere un'aliquota di 5 μL. Eseguire ulteriori 2-3 cicli sulla reazione rimanente prima di prendere la successiva aliquota di 5 μL. Ripeti per raccogliere quattro aliquote.
- Utilizzare i parametri PCR della Tabella 11 per ciascuna aliquota.
- Aggiungere 5 μL di acqua e 2 μL di colorante 6x a ciascun campione da 5 μL. Eseguire su un gel TBE di agarosio al 2% (ricetta nella tabella supplementare S1) con 25-150 ng di scala a basso peso molecolare. Vedere la Figura 4 per i risultati attesi.
NOTA: Un numero ottimale di cicli per l'amplificazione finale della PCR della libreria [passo 3.10] è il numero più basso di cicli per ottenere un prodotto visibile sul gel meno un ciclo.
- Amplificazione PCR della libreria finale
- Impostare reazioni 12 x 30 μL per amplificare ogni libreria finale per il sequenziamento come nella Tabella 11.
- Ciclizzare le reazioni PCR secondo la Tabella 11 dopo aver determinato il numero di cicli dopo la titolazione della PCR (fase 3.9.3).
- Una volta completata la PCR, raggruppare i campioni replicati in una provetta microfuge a basso legame da 1,7 ml.
- Posizionare i tubi sull'MPS e trasferire il surnatante in un nuovo tubo microfuge a basso legame da 1,7 ml.
- Risospendere le rimanenti perle rivestite di streptavidina in 20 μL di 1x tampone pre-PCR.
NOTA: Questo modello può essere riutilizzato se conservato a 4 °C per giorni o settimane o a lungo termine a -20 °C.
- Rimozione dei primer con miscela di perline magnetiche
- Utilizzare 1x tampone TLE (ricetta nella tabella supplementare S1) per regolare il volume dal punto 3.10.4 a esattamente 360 μL.
- A ciascun campione, aggiungere 360 μL della miscela di sfere magnetiche e pipettare su e giù per mescolare.
- Su un rotatore, mescolare i campioni per 10 minuti a temperatura ambiente.
- Separare le perline dal surnatante sull'MPS a temperatura ambiente (3-5 min).
- Lavare le perle due volte utilizzando 200 μL di etanolo al 70% e recuperare le perle per 5 minuti sull'MPS ogni volta.
- Centrifugare rapidamente in una centrifuga e pipettare completamente l'etanolo. Asciugare le perle in aria sull'MPS per far evaporare ulteriormente l'etanolo.
NOTA: Il pellet dovrebbe apparire come cioccolato fondente senza cracking (può richiedere ~ 10 minuti). - Aggiungere 30 μL di acqua ultrapura e risospendere per eluire il DNA per 10 minuti a temperatura ambiente. Scorri i tubi ogni 2 minuti per facilitare la miscelazione.
- Separare le perline dal surnatante sull'MPS per 5 minuti.
- Raccogliere il surnatante da ciascun campione in una provetta fresca da 1,7 ml.
- Eseguire 1 μL della libreria su un gel TBE di agarosio al 2% (ricetta nella Tabella supplementare S1) per ottenere una distribuzione dimensionale del frammento e quantificare la libreria finale (Figura 5).
NOTA: la digestione ClaI può avvenire solo per le giunzioni DpnII-DpnII e serve come controllo di legatura positiva che dovrebbe comportare una distribuzione delle dimensioni dei frammenti inferiore per la libreria finale. Le librerie finali possono essere conservate per alcuni giorni a 4 °C, a lungo termine a -20 °C o immediatamente diluite e sottoposte a sequenziamento.
Tabella 5: Reagenti e temperature per la rimozione della biotina Fare clic qui per scaricare questa tabella.
Tabella 6: Parametri per la sonicazione. Clicca qui per scaricare questa tabella.
Tabella 7: Reagenti di riparazione finale e temperature. Clicca qui per scaricare questa tabella.
Tabella 8: Reagenti e temperature di coda A. Clicca qui per scaricare questa tabella.
Tabella 9: Primer PCR e oligo accoppiati-end-adapter con ricottura di reagenti per ricottura. Abbreviazione: 5PHOS = 5' fosfato. Gli asterischi indicano basi di DNA fosforotioato. # Combinare un oligo indicizzato con l'oligo universale per ricottura in un adattatore indicizzato. Clicca qui per scaricare questa tabella.
Tabella 10: Reagenti di legatura adattatori. Clicca qui per scaricare questa tabella.
Tabella 11: reagenti PCR e parametri ciclici. Clicca qui per scaricare questa tabella.

Figura 3: Gel di agarosio che mostra i tipici risultati di selezione post-size. Vengono mostrate le frazioni superiore e inferiore per quattro campioni (numerati 1-4) di DpnII-DdeI Hi-C. La prima corsia per ciascun campione contiene la frazione superiore, derivata da una miscela di sfere magnetiche 0,8x, e la seconda e la terza corsia contengono una diluizione della frazione inferiore derivata da una miscela di sfere magnetiche 1,1x. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

Figura 4: Gel di agarosio con risultati della titolazione PCR. A partire da 5 cicli di PCR, i campioni vengono prelevati dopo ogni 2 cicli (5, 7, 9 e 11 cicli) per ciascuna delle quattro librerie. Sulla base di questa figura, sono stati scelti 6 cicli come ciclo ottimale per ciascun campione. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

Figura 5: Prodotti PCR finali. Dopo la pulizia e la selezione delle dimensioni, i prodotti PCR (Hi-C) sono stati caricati accanto a una frazione digerita ClaI della stessa libreria (ClaI). I frammenti digeriti da ClaI indicano la presenza di legature DpnII-DpnII ricercate. Si noti che ClaI non digerisce le giunzioni DpnII-DdeI e, pertanto, non tutte le legature contribuiranno a una riduzione delle dimensioni di questa restrizione. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}
Risultati
Le figure in questo manoscritto sono state generate da un esperimento separato e replicato di quello pubblicato in precedenza da Lafontaine et al.21. Dopo aver ottenuto dati di sequenziamento ad alto throughput, è stato utilizzato l'Open Chromatin Collective (Open2C: https://github.com/open2c) per elaborare i dati Hi-C. Una pipeline simile può essere trovata sul portale dati del progetto 4D Nucleome (https://data.4dnucleome.org/resources/data-analysis/hi_c-processing-pipeline). In breve, il distillatore di pipeline Nextflow (https://github.com/open2c/distiller-nf) è stato implementato per (1) allineare le sequenze di molecole Hi-C al genoma di riferimento, (2) analizzare l'allineamento .sam e formare i file con coppie Hi-C, (3) filtrare i duplicati della PCR e (4) aggregare coppie in matrici binnate di interazioni Hi-C. Queste matrici formattate HDF5, chiamate cooler, possono quindi essere (1) visualizzate su un server HiGlass (https://higlass.io/) e (2) analizzate utilizzando un ampio set di strumenti computazionali open source presenti nella collezione "cooltools" gestita dall'Open Chromatin Collective (https://github.com/open2c/cooltools) per estrarre e quantificare caratteristiche di piegatura come scomparti, TAD e loop.
Alcuni indicatori di qualità delle librerie Hi-C3.0 possono essere valutati immediatamente dopo aver mappato le coppie di lettura a un genoma di riferimento, utilizzando alcune semplici metriche/indicatori. In primo luogo, in genere ~ 50% delle coppie di lettura sequenziate può essere mappato in modo univoco per le cellule umane. A causa della natura polimerica dei cromosomi, la maggior parte di queste letture mappate (~ 60% -90%) rappresentano interazioni all'interno di un cromosoma (cis), con frequenze di interazione che decadono rapidamente con l'aumentare della distanza genomica (decadimento dipendente dalla distanza). Il decadimento dipendente dalla distanza può essere visualizzato meglio in un "grafico di scala", che mostra la probabilità di contatto (per braccio cromosomico) in funzione della distanza genomica. Abbiamo scoperto che l'uso di diversi reticolanti ed enzimi può alterare il decadimento dipendente dalla distanza a lunghe e corte distanze17. L'aggiunta di reticolazione DSG aumenta la rilevabilità delle interazioni a brevi distanze quando combinata con enzimi come Mnase e combinazioni di DpnII-DdeI che producono frammenti più piccoli (Figura 6A).
Il decadimento dipendente dalla distanza può anche essere osservato direttamente dalle matrici di interazione 2D: le interazioni diventano più rare quando si trovano più lontano dalla diagonale centrale (Figura 6B). Inoltre, le caratteristiche di ripiegamento genomico, come compartimenti, TAD e loop possono essere identificate da matrici Hi-C e grafici di scala come deviazioni dal decadimento medio generale dipendente dalla distanza dell'intero genoma. È importante sottolineare che il crosslinking con DSG in aggiunta a FA diminuisce le legature casuali, che sono illimitate a causa della natura polimerica dei cromosomi e, quindi, è più probabile che si verifichino tra i cromosomi (in trans) (Figura 6C). La riduzione della legatura casuale porta ad un aumento dei rapporti segnale-rumore, specialmente per le interazioni intracromosomiche intercromosomiche e a lunghissimo raggio (>10-50 Mb).

Figura 6: Risultati rappresentativi delle librerie Hi-C mappate e filtrate. (A) Grafici di scala con probabilità di contatto e sua derivata per vari enzimi, ordinati per lunghezza del frammento (in alto) e reticolazione con FA o FA + DSG (in basso). La digestione con MNAse (microC) o DpnII-DdeI (Hi-C 3.0) aumenta significativamente i contatti a corto raggio (in alto) così come l'aggiunta di DSG a FA (in basso). (B) Le colonne mostrano le mappe di calore Hi-C della digestione DpnII dopo la sola reticolazione FA e la digestione DpnII o DdeI dopo la reticolazione FA + DSG. Le frecce bianche mostrano una crescente forza dei "punti" dopo la reticolazione DSG e la digestione DdeI, il che implica una migliore rilevazione dei loop di DNA. Le righe mostrano diverse parti del cromosoma 3 ad alta risoluzione, allineandosi con il pannello C: riga superiore: cromosoma intero 3 (0-198.295.559 Mb); fila centrale: 186-196 Mb; riga inferiore: 191,0-191,5 Mb. (C) Grafici di copertura per le regioni rappresentate in A. Le frecce nere mostrano la copertura inferiore (%cis legge) per la reticolazione solo FA. Abbreviazioni: FA = formaldeide; DSG = glutarato di disuccinimidile; CHR = cromosoma. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}
Non tutte le letture mappate sono utili. Un secondo indicatore di qualità è il numero di duplicati PCR. È altamente improbabile che le letture duplicate esatte si verifichino per caso dopo la legatura e la sonicazione. Pertanto, tali letture probabilmente derivano dall'amplificazione PCR e devono essere filtrate. I duplicati spesso si verificano quando sono necessari troppi cicli di PCR per amplificare librerie a bassa complessità. Generalmente, per Hi-C, la maggior parte delle librerie ha bisogno solo di 5-8 cicli di amplificazione finale della PCR, come determinato dalla titolazione PCR (vedere passo 3.9; Figura 4). Tuttavia, librerie con sufficiente complessità possono essere ottenute anche dopo 14 cicli di amplificazione PCR.
Un'altra categoria di letture duplicate, i cosiddetti duplicati ottici, può derivare dal processo di amplificazione su piattaforme di sequenziamento Illumina che utilizzano celle di flusso modellate (come HiSeq4000). I duplicati ottici si trovano sia dal sovraccarico della cella di flusso, causando cluster (grandi) chiamati due cluster separati, sia dal riclustering locale della molecola originale accoppiata dopo un primo ciclo di PCR. Poiché entrambi i tipi di duplicati ottici sono locali, possono essere identificati e distinti dai duplicati PCR in base alla loro posizione sulla cella di flusso. Mentre le librerie con duplicati PCR del >15% avrebbero bisogno di rigenerazione, le librerie con duplicati ottici possono essere ricaricate dopo aver ottimizzato il processo di caricamento.
Tabella supplementare S1: Buffer e soluzioni. Clicca qui per scaricare questa tabella.
Discussione
Passaggi critici per la gestione delle celle
Sebbene sia possibile utilizzare un numero inferiore di celle di input, questo protocollo è stato ottimizzato per ~ 5 × 106 celle per corsia di sequenziamento (~ 400 M letture) per garantire la corretta complessità dopo il sequenziamento profondo. Le cellule sono meglio contate prima della fissazione. Per la generazione di librerie ultraprofonde, generalmente moltiplichiamo il numero di corsie (e celle) fino a raggiungere la profondità di lettura desiderata. Per una fissazione ottimale, il mezzo contenente siero deve essere sostituito con PBS prima della fissazione FA e le soluzioni fissative devono essere aggiunte immediatamente e senza gradienti di concentrazione15,22. Per la raccolta cellulare, la raschiatura è preferita alla tripsinizzazione, perché il passaggio da una forma più piatta a una sferica dopo la tripsinizzazione potrebbe influenzare la conformazione nucleare. Dopo l'aggiunta di DSG, i pellet di cellule sciolte e grumose vengono facilmente persi. Fare attenzione quando si maneggiano le cellule in questa fase e aggiungere fino allo 0,05% di BSA per ridurre l'aggregazione.
Modifiche al metodo
Questo protocollo è stato sviluppato utilizzando cellule umane17. Tuttavia, sulla base dell'esperienza con la cattura della conformazione cromosomica, questo protocollo dovrebbe funzionare per la maggior parte delle cellule eucariotiche. Per un input significativamente inferiore (~1 × 106 celle), si consiglia di utilizzare metà dei volumi per le procedure di lisi e cattura della conformazione [passi 2.1-2.4]. Ciò consentirebbe anche di eseguire l'isolamento del DNA [fase 2.5] in una centrifuga da tavolo con tubi da 1,7 ml, che potrebbe migliorare la pellettizzazione per basse concentrazioni di DNA. La quantificazione del DNA (fase 2.6.6) indicherà come procedere. Per basse quantità di DNA isolato (1-5 μg), suggeriamo di saltare la selezione delle dimensioni (fase 3.3) e procedere con la rimozione della biotina dopo aver ridotto il volume da 130 μL a ~45 μL con un CFU.
Questo protocollo è stato sviluppato appositamente per garantire dati di alta qualità dopo la successiva reticolazione con FA e DSG e la digestione con DpnII e DdeI. Tuttavia, strategie di reticolazione alternative come la FA seguita da EGS (glicole etilenico bis (succinimidil succinato)), utilizzata anche in ChIP-seq23 e ChIA-PET24, potrebbero funzionare altrettanto bene17. Allo stesso modo, diverse combinazioni enzimatiche, come DpnII e HinfI18 o MboI, MseI e NlaIII19 possono essere utilizzate per la digestione. Quando si adattano le combinazioni enzimatiche, assicurarsi di utilizzare nucleotidi biotinilati in grado di riempire le sporgenze specifiche di 5' e utilizzare i tamponi più ottimali per ogni cocktail. DpnII viene fornito con il proprio tampone e il produttore dell'enzima raccomanda un tampone specifico per la digestione DdeI. Tuttavia, per la doppia digestione con DpnII e DdeI in questo protocollo, il Restriction Buffer è raccomandato perché è valutato al 100% di attività per entrambi gli enzimi.
Risoluzione dei problemi relativi all'acquisizione delle conformazioni
I tre passaggi chiave nella cattura della conformazione cromosomica: reticolazione, digestione e religenza sono stati tutti eseguiti prima che i risultati possano essere visualizzati su gel. Per determinare la qualità di ciascuno di questi tre passaggi e discernere dove potrebbero essere sorti problemi, le aliquote prima (CI) e dopo la digestione (DC) vengono prelevate e caricate sul gel insieme al campione Hi-C legato (Figura 2). Questo gel viene utilizzato per determinare la qualità del campione Hi-C e se varrà la pena continuare il protocollo. Senza CI e DC, è difficile individuare potenziali passaggi subottimali. Vale la pena notare che la legatura subottimale potrebbe essere dovuta a un problema nella legatura stessa, il fill-in o un problema con la reticolazione. Per risolvere i problemi di reticolazione, assicurarsi di non utilizzare più di 1 × 107 cellule per libreria e iniziare con reagenti di reticolazione freschi e cellule pulite (cioè risciacquate con PBS). Per la legatura, assicurarsi che le cellule e la miscela di legatura siano mantenute sul ghiaccio. Aggiungere T4 DNA ligasi poco prima dell'incubazione di 4 ore a 16 °C e mescolare bene.
Risoluzione dei problemi relativi alla preparazione della libreria
Se sono necessari più di 10 cicli di PCR o nessun prodotto PCR può essere visto sul gel dopo la titolazione PCR (Figura 4), ci sono alcune opzioni per salvare il campione Hi-C. Lavorando a ritroso dalla titolazione della PCR, la prima opzione è quella di provare di nuovo la PCR. Se non c'è ancora abbastanza prodotto, è possibile tentare un altro giro di coda A e legatura dell'adattatore (passaggio 3.6) dopo aver lavato le perline due volte con 1x tampone TLE. Dopo questa ulteriore legatura della coda A e dell'adattatore, si può procedere alla titolazione PCR come prima. Se non c'è ancora alcun prodotto, l'ultima opzione è quella di risonicare la frazione 0,8x dal passaggio 3.3 e procedere da lì.
Limitazioni e vantaggi di Hi-C3.0
È importante rendersi conto che Hi-C è un metodo basato sulla popolazione che cattura la frequenza media delle interazioni tra coppie di loci nella popolazione cellulare. Alcune analisi computazionali sono progettate per districare combinazioni di conformazioni da una popolazione25, ma in linea di principio, Hi-C è cieco alle differenze tra le cellule. Sebbene sia possibile eseguire Hi-C26,27 a cella singola e possano essere effettuate inferenze computazionali28, Hi-C a cella singola non è adatto per ottenere informazioni 3C ad altissima risoluzione. Un'ulteriore limitazione di Hi-C è che rileva solo interazioni a coppie. Per rilevare interazioni multicontatto, è possibile utilizzare frese frequenti combinate con sequenziamento a lettura breve (Illumina)16 o eseguire il sequenziamento multicontatto 3C29 o 4C30, utilizzando il sequenziamento a lunga lettura dalle piattaforme PacBio o Oxford Nanopore. Sono stati sviluppati anche derivati Hi-C per rilevare specificamente i contatti tra e lungo i cromatidi fratelli31,32.
Sebbene Hi-C19 e Micro-C33 possano essere utilizzati per generare mappe di contatto a risoluzioni subkilobase, entrambi richiedono una grande quantità di letture di sequenziamento e questo può diventare un'impresa costosa. Per ottenere una risoluzione simile o addirittura superiore senza i costi, è possibile applicare l'arricchimento per specifiche regioni genomiche (capture-C 34) o specifiche interazioni proteiche (ChiA-PET 35, PLAC-seq36, Hi-ChIP37). Il punto di forza e lo svantaggio di queste applicazioni di arricchimento è che viene campionato solo un numero limitato di interazioni. Con tali arricchimenti, l'aspetto globale di Hi-C (e l'opzione della normalizzazione globale) è perso.
Importanza e potenziali applicazioni di Hi-C3.0
Questo protocollo è stato progettato per consentire 3C ultraprofondi ad alta risoluzione e allo stesso tempo rilevare caratteristiche di piegatura su larga scala come TAD e scomparti17 (Figura 6). Questo protocollo inizia con 5 × 106 celle per tubo per ogni libreria Hi-C, che dovrebbe essere materiale più che sufficiente per sequenziare una o due corsie su una cella di flusso per ottenere fino a 1 miliardo di letture accoppiate. Per il sequenziamento ultraprofondo, devono essere preparate più provette da 5 × 106 cellule, a seconda del numero di letture mappate e duplicati PCR. Alla massima risoluzione (<1 kb), le interazioni cicliche si trovano principalmente tra i siti CTCF, ma possono anche essere rilevate interazioni promotore-potenziatore. I lettori possono fare riferimento ad Akgol Oksuz et al.17 per una descrizione dettagliata dell'analisi dei dati.
Divulgazioni
Gli autori non hanno conflitti di interesse da rivelare.
Riconoscimenti
Vorremmo ringraziare Denis Lafontaine per lo sviluppo del protocollo e Sergey Venev per l'assistenza bioinformatica. Questo lavoro è stato supportato da una sovvenzione del National Institutes of Health Common Fund 4D Nucleome Program a J.D. (U54-DK107980, UM1-HG011536). J.D. è un investigatore dell'Howard Hughes Medical Institute.
Questo articolo è soggetto alla politica Open Access to Publications di HHMI. I responsabili dei laboratori HHMI hanno precedentemente concesso una licenza CC BY 4.0 non esclusiva al pubblico e una licenza sublicenziabile a HHMI nei loro articoli di ricerca. In base a tali licenze, il manoscritto accettato dall'autore di questo articolo può essere reso liberamente disponibile sotto una licenza CC BY 4.0 immediatamente dopo la pubblicazione.
Materiali
| Name | Company | Catalog Number | Comments |
| 1 kb Ladder | New England Biolabs | N3232L | |
| Agarose | Invitrogen | 16500100 | |
| Agencourt AMPure XP magnetic beads , 60 mL | Beckman Coulter | A63881 | |
| Amicon Ultra-0.5 Centrifugal Filter Unit (CFU) | EMD Millipore | UFC500396 | |
| Annealing Buffer (5x) | See recipe in supplemental materials | ||
| ATP 10 mM | ThermoFisher | R0441 | |
| Avanti J-25i High Speed Refrigerated ultra-centrifuge | Beckman Coulter | ||
| beckman ultracentrifuge tube 35 mL | Beckman Coulter | 357002 | |
| Binding Buffer (2x) | See recipe in supplemental materials | ||
| biotin-14-dATP 0.4 mM | Invitrogen | 19524-016 | |
| BSA 10 mg/mL | New England Biolabs | B9000S | dilute from 20 mg/mL |
| Cell scraper | Falcon | 353089 | |
| Cell scraper | Corning | 3008 | |
| Conical polypropylene tubes 50 mL | Denville | C1062-P | |
| Conical tube 15 mL | Denville | C1017-P | |
| Covaris micro tube AFA fiber with snap-cap 130 µL | Covaris | 520045/520077 | |
| Covaris Sonicator | Covaris | E220/E220evolution/M220 | |
| Culture flask 175 cm2 | Falcon | 353112 | |
| Culture plates 150 mm x 25 mm | Corning | 430599 | |
| dATP 1 mM | Invitrogen | 56172 | |
| dATP 10 mM | Invitrogen | 56172 | |
| dCTP 10 mM | Invitrogen | 56173 | |
| DdeI | New England Biolabs | R0175L | |
| dGTP 10 mM | Invitrogen | 56174 | |
| DMSO | Sigma | D2650-5x10ML | |
| dNTP mix 25 mM | Invitrogen | 10297117 | |
| Dounce homogenizer | DWK Life Sciences | 8853010002/8853030002 | |
| DPBS | Gibco | 14190-144 | |
| DpnII | New England Biolabs | R0543M | |
| DSG | ThermoScientific | 20593 | |
| dTTP 10 mM | Invitrogen | 56175 | |
| Ethanol 70% | Fisher | A409-4 | Diluted from 100% |
| Ethidium Bromide | Fisher | BP1302-10 | |
| Formaldehyde (37%) | Fisher | BP531-500 | |
| Gel loading dye (6x ) | New England Biolabs | B7024S | |
| Glycine in ultrapure water 2.5 M | Sigma | G8898-1KG | |
| HBSS | Gibco | 14025-092 | |
| Igepal CA-630 detergent | MP Biomedicals | 198596 | |
| Klenow DNA polymerase 5 U/µL | New England Biolabs | M0210L | |
| Klenow Fragment 3-->5’ exo-, 5 U/µL | New England Biolabs | M0212L | |
| ligation buffer (10x) | New England Biolabs | B7203S | |
| Liquid nitrogen | |||
| LoBind microcentrifuge tube 1.7 mL | Eppendorf | 22431021 | |
| Low Molecular Weight DNA Ladder | New England Biolabs | N3233L | |
| Lysis buffer | See recipe in supplemental materials | ||
| Magnetic Particle separator | ThermoFisher | 12321D | |
| Microfuge tubes 1.7 mL | Axygen | MCT-175-C | |
| MyOne Streptavidin C1 beads | Invitrogen | 65001 | |
| NEBuffer 2.1 (10x) | New England Biolabs | B7002S | |
| NEBuffer 3.1 (10x) | New England Biolabs | B7203S | |
| PBS | Gibco | 70013-032 | |
| PCR (strip) tubes | Biorad | TBS0201/ TCS0803 | |
| PCR thermocycler | Biorad | T100 | |
| Pfu Ultra II Buffer (10x) | Agilent | Comes with Pfu Ultra | |
| PfuUltra II Fusion HS DNA Polymerase | Agilent | 600674 | |
| Phase lock tube 15 mL | Qiagen | 129065 | |
| Phase lock tubes 2 mL | Qiagen | 129056 | |
| Phenol:chloroform:isoamyl alcohol | Invitrogen | 15593-049 | |
| Protease inhibitor cocktail | ThermoFisher | 78440 | |
| Proteinase K in ultrapure water 10 mg/mL | Invitrogen | 25530-031 | |
| Refrigerated Centrifuge | Eppendorf | 5810R | |
| RNase A, DNase and protease-free 10 mg/mL | Thermo Scientific | EN0531 | |
| Rotator | Argos technologies | EW-04397-40 or rocking platform | |
| SDS 1% | Fisher | BP13111 | |
| Sodium acetate pH = 5.2, 3 M | Sigma | ||
| Sub-Cell GT Horizontal Electrophoresis System | Biorad | 1704401 | |
| T4 DNA ligase 1 U/µL | Invitrogen | 100004817 | |
| T4 DNA polymerase | New England Biolabs | M0203L | |
| T4 DNA polymerase | New England Biolabs | M0203L | |
| T4 DNA polymerase 3 U/µL | New England Biolabs | M0203L | |
| T4 ligation buffer (5x) | Invitrogen | Y90001 | |
| T4 polynucleotide kinase 10 U/µL | New England Biolabs | M0201L | |
| Tabletop centrifuge | Eppendorf | 5425 | |
| TBE buffer | See recipe in supplemental materials | ||
| Tris Low EDTA Buffer (TLE) | See recipe in supplemental materials | ||
| Triton X-100 (10%) | Sigma | 93443 | |
| Truseq adapter oligos | Integrated DNA Technologies (IDT)) | https://www.idtdna.com/site/order/oligoentry | 250 nmole and HPLC purified |
| Tween 20 detergent | Fisher | 9005-64-5 | |
| Tween Wash Buffer | See recipe in supplemental materials | ||
| Vortex | Scientific Industries | (G560)SI-0236 |
Riferimenti
- Dekker, J., Rippe, K., Dekker, M., Kleckner, N. Capturing chromosome conformation. Science. 295 (5558), 1306-1311 (2002).
- Simonis, M., et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nature Genetics. 38 (11), 1348-1354 (2006).
- Dostie, J., et al. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Research. 16 (10), 1299-1309 (2006).
- Lieberman-Aiden, E., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326 (5950), 289-293 (2009).
- Stadhouders, R., et al. Multiplexed chromosome conformation capture sequencing for rapid genome-scale high-resolution detection of long-range chromatin interactions. Nature Protocols. 8 (3), 509-524 (2013).
- Kalhor, R., Tjong, H., Jayathilaka, N., Alber, F., Chen, L. Genome architectures revealed by tethered chromosome conformation capture and population-based modeling. Nature Biotechnology. 30 (1), 90-98 (2012).
- Hsieh, T. H., et al. Mapping nucleosome resolution chromosome folding in yeast by micro-C. Cell. 162 (1), 108-119 (2015).
- Hsieh, T. -H. S., Fudenberg, G., Goloborodko, A., Rando, O. J. Micro-C XL: assaying chromosome conformation from the nucleosome to the entire genome. Nature Methods. 13 (12), 1009-1011 (2016).
- Denker, A., de Laat, W. The second decade of 3C technologies: detailed insights into nuclear organization. Genes & Development. 30 (12), 1357-1382 (2016).
- Rodley, C. D., Bertels, F., Jones, B., O’Sullivan, J. M. Global identification of yeast chromosome interactions using Genome conformation capture. Fungal Genetics and Biology. 46 (11), 879-886 (2009).
- Rao, S. S., et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 159 (7), 1665-1680 (2014).
- Alipour, E., Marko, J. F. Self-organization of domain structures by DNA-loop-extruding enzymes. Nucleic Acids Research. 40 (22), 11202-11212 (2012).
- Lajoie, B. R., Dekker, J., Kaplan, N. The Hitchhiker’s guide to Hi-C analysis: Practical guidelines. Methods. 72, 65-75 (2015).
- Belaghzal, H., Dekker, J., Gibcus, J. H. Hi-C 2.0: An optimized Hi-C procedure for high-resolution genome-wide mapping of chromosome conformation. Methods. 123, 56-65 (2017).
- Golloshi, R., Sanders, J. T., McCord, R. P. Iteratively improving Hi-C experiments one step at a time. Methods. 142, 47-58 (2018).
- Darrow, E. M., et al. Deletion of DXZ4 on the human inactive X chromosome alters higher-order genome architecture. Proceedings of the National Academy of Sciences of the United States of America. 113 (31), 4504-4512 (2016).
- Akgol Oksuz, B., et al. Systematic evaluation of chromosome conformation capture assays. Nature Methods. 18 (9), 1046-1055 (2021).
- Ghuryeid, J., et al. Integrating Hi-C links with assembly graphs for chromosome-scale assembly. PLoS Computational Biology. 15 (8), 1007273(2019).
- Gu, H., et al. Fine-mapping of nuclear compartments using ultra-deep Hi-C shows that active promoter and enhancer elements localize in the active A compartment even when adjacent sequences do not. bioRxiv. , (2021).
- Ramani, V., et al. Mapping 3D genome architecture through in situ DNase Hi-C. Nature Protocols. 11 (11), 2104-2121 (2016).
- Lafontaine, D. L., Yang, L., Dekker, J., Gibcus, J. H. Hi-C 3.0: Improved Protocol for Genome-Wide Chromosome Conformation Capture. Current Protocols. 1 (7), 198(2021).
- Belton, J. -M. M., et al. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods. 58 (3), 268-276 (2012).
- Truch, J., Telenius, J., Higgs, D. R., Gibbons, R. J. How to tackle challenging ChIP-Seq, with long-range cross-linking, Using ATRX as an example. Methods in Molecular Biology. 1832, Clifton, N.J. 105-130 (2018).
- Wang, P., et al. In situ chromatin interaction analysis using paired-end tag sequencing. Current Protocols. 1 (8), 174(2021).
- Tjong, H., et al. Population-based 3D genome structure analysis reveals driving forces in spatial genome organization. Proceedings of the National Academy of Sciences of the United States of America. 113 (12), 1663-1672 (2016).
- Nagano, T., et al. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature. 547 (7661), 61-67 (2017).
- Ramani, V., et al. Massively multiplex single-cell Hi-C. Nature Methods. 14 (3), 263-266 (2017).
- Meng, L., Wang, C., Shi, Y., Luo, Q. Si-C is a method for inferring super-resolution intact genome structure from single-cell Hi-C data. Nature Communications. 12 (1), 4369(2021).
- Tavares-Cadete, F., Norouzi, D., Dekker, B., Liu, Y., Dekker, J. Multi-contact 3C reveals that the human genome during interphase is largely not entangled. Nature Structural & Molecular Biology. 27 (12), 1105-1114 (2020).
- Vermeulen, C., et al. Multi-contact 4C: long-molecule sequencing of complex proximity ligation products to uncover local cooperative and competitive chromatin topologies. Nature Protocols. 15 (2), 364-397 (2020).
- Oomen, M. E., Hedger, A. K., Watts, J. K., Dekker, J. Detecting chromatin interactions between and along sister chromatids with SisterC. Nature Methods. 17 (10), 1002-1009 (2020).
- Mitter, M., et al. Conformation of sister chromatids in the replicated human genome. Nature. 586 (7827), 139-144 (2020).
- Krietenstein, N., et al. Ultrastructural Details of Mammalian Chromosome Architecture. Molecular Cell. 78 (3), 554-565 (2020).
- Hughes, J. R., et al. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nature Genetics. 46 (2), 205-212 (2014).
- Fullwood, M. J., et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 462 (7269), 58-64 (2009).
- Fang, R., et al. Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Research. 26 (12), 1345-1348 (2016).
- Mumbach, M. R., et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nature Methods. 13 (11), 919-922 (2016).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati