
Method Article
Capture de la conformation chromosomique à travers les échelles de longueur
Dans cet article
Résumé
Hi-C 3.0 est un protocole Hi-C amélioré qui combine des agents de réticulation du formaldéhyde et du glutarate de disuccinimidyl avec un cocktail d’enzymes de restriction DpnII et DdeI pour augmenter le rapport signal sur bruit et la résolution de la détection des interactions chromatines.
Résumé
La capture de conformation chromosomique (3C) est utilisée pour détecter les interactions tridimensionnelles de la chromatine. Typiquement, la réticulation chimique avec le formaldéhyde (FA) est utilisée pour fixer les interactions de la chromatine. Ensuite, la digestion de la chromatine avec une enzyme de restriction et la ligature subséquente des extrémités des fragments convertissent la proximité tridimensionnelle (3D) en produits de ligature uniques. Enfin, après l’inversion des réticulations, l’élimination des protéines et l’isolement de l’ADN, l’ADN est cisaillé et préparé pour le séquençage à haut débit. La fréquence de ligature de proximité des paires de loci est une mesure de la fréquence de leur colocalisation dans l’espace tridimensionnel dans une population cellulaire.
Une bibliothèque Hi-C séquencée fournit des informations à l’échelle du génome sur les fréquences d’interaction entre toutes les paires de loci. La résolution et la précision de Hi-C reposent sur une réticulation efficace qui maintient les contacts de la chromatine et la fragmentation fréquente et uniforme de la chromatine. Cet article décrit un protocole Hi-C in situ amélioré, Hi-C 3.0, qui augmente l’efficacité de la réticulation en combinant deux agents de réticulation (formaldéhyde [FA] et glutarate de disuccinimidyl [DSG]), suivi d’une digestion plus fine à l’aide de deux enzymes de restriction (DpnII et DdeI). Hi-C 3.0 est un protocole unique pour la quantification précise des caractéristiques de repliement du génome à des échelles plus petites telles que les boucles et les domaines d’association topologique (TAD), ainsi que des caractéristiques à des échelles plus grandes à l’échelle du noyau telles que les compartiments.
Introduction
La capture de conformation chromosomique est utilisée depuis 20021. Fondamentalement, chaque variante de capture de conformation repose sur la fixation des interactions ADN-protéine et protéine-protéine pour préserver l’organisation de la chromatine 3D. Ceci est suivi par la fragmentation de l’ADN, généralement par la digestion de restriction, et, enfin, la ligature des extrémités d’ADN proches pour convertir les loci spatialement proximaux en séquences d’ADN covalentes uniques. Les protocoles 3C initiaux utilisaient la PCR pour échantillonner des interactions spécifiques « un-à-un ». Les tests 4C ultérieurs ont permis de détecter les interactions « un-à-tous »2, tandis que 5C a détecté les interactions « plusieurs-à-plusieurs »3. La capture de la conformation chromosomique a porté ses fruits après la mise en œuvre du séquençage à haut débit (NGS) de nouvelle génération, qui a permis de détecter des interactions génomiques « tout-à-tous » en utilisant Hi-C4 à l’échelle du génome et des techniques comparables telles que 3C-seq5, TCC6 et Micro-C 7,8 (voir également l’examen de Denker et De Laat9).
Dans Hi-C, des nucléotides biotinylés sont utilisés pour marquer les surplombs 5' après digestion et avant ligature (Figure 1). Cela permet de sélectionner des fragments correctement digérés et religués à l’aide de billes enrobées de streptavidine, ce qui les distingue de GCC10. Une mise à jour importante du protocole Hi-C a été mise en œuvre par Rao et coll.11, qui ont effectué la digestion et la ligature dans des noyaux intacts (c.-à-d. in situ) afin de réduire les faux produits de ligature. De plus, le remplacement de la digestion HindIII par la digestion MboI (ou DpnII) a réduit la taille des fragments et augmenté le potentiel de résolution de l’Hi-C. Cette augmentation a permis la détection de structures à relativement petite échelle et une localisation génomique plus précise des points de contact, tels que les boucles d’ADN entre petits éléments cis, par exemple les boucles entre les sites liés à la CTCF générées par l’extrusion de boucles11,12. Cependant, ce potentiel a un coût. Premièrement, une multiplication par deux de la résolution nécessite une multiplication par quatre (22) des lectures de séquençage13. Deuxièmement, les petites tailles de fragments augmentent la possibilité de confondre les fragments voisins non digérés avec les fragments digérés et relidés14. Comme mentionné, dans Hi-C, les fragments digérés et religués diffèrent des fragments non digérés par la présence de biotine à la jonction de ligature. Cependant, une élimination appropriée de la biotine des extrémités non ligaturées est nécessaire pour s’assurer que seules les jonctions de ligature sont abaissées14,15.
Avec la diminution du coût du NGS, il devient possible d’étudier le repliement des chromosomes plus en détail. Pour diminuer la taille des fragments d’ADN, et ainsi augmenter la résolution, le protocole Hi-C peut être adapté pour utiliser plus fréquemment des enzymes de restriction de coupe16 ou pour utiliser des combinaisons d’enzymes de restriction17,18,19. Alternativement, MNase 7,8 dans Micro-C et DNase dans DNase Hi-C20 peuvent être titrés pour obtenir une digestion optimale.
Une évaluation systématique récente des principes fondamentaux des méthodes 3C a montré que la détection des caractéristiques de repliement des chromosomes à chaque échelle de longueur s’améliorait considérablement avec la réticulation séquentielle avec 1% FA suivi de 3 mM DSG17. De plus, Hi-C avec digestion HindIII était la meilleure option pour détecter les caractéristiques de pliage à grande échelle, telles que les compartiments, et que Micro-C était supérieur pour détecter les caractéristiques de repliement à petite échelle telles que les boucles d’ADN. Ces résultats ont conduit au développement d’une stratégie unique et haute résolution « Hi-C 3.0 », qui utilise la combinaison de réticulateurs FA et DSG suivie d’une double digestion avec les endonucléases DpnII et DdeI21. Hi-C 3.0 fournit une stratégie efficace pour une utilisation générale car il détecte avec précision les caractéristiques de pliage sur toutes les échelles de longueur17. La partie expérimentale du protocole Hi-C 3.0 est détaillée ici et les résultats typiques auxquels on peut s’attendre après le séquençage sont présentés.

Figure 1 : Procédure Hi-C en six étapes. Les cellules sont fixées d’abord avec FA, puis DSG (1). Ensuite, la lyse précède une double digestion avec DdeI et DpnII (2). La biotine est ajoutée par remplissage en surplomb et les extrémités émoussées proximales sont ligaturées (3) avant la purification de l’ADN (4). La biotine est retirée des extrémités non ligaturées avant la sonication et la sélection de la taille (5). Enfin, l’extraction de la biotine permet la ligature de l’adaptateur et l’amplification de la bibliothèque par PCR (6). Abréviations : FA = formaldéhyde; DSG = glutarate de disuccinimidyl; B = Biotine. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Protocole
1. Fixation par réticulation
- Fixation du formaldéhyde : à partir des cellules en monocouche
- Avoir des cellules ensemencées dans un milieu approprié pour récolter 5 × 106 cellules par plaque de 150 mm.
REMARQUE: Les utilisateurs peuvent choisir n’importe quel récipient préféré qui assure une croissance optimale de toute lignée cellulaire de mammifères. De plus, les cellules peuvent être isolées des tissus. - Aspirer le milieu avec une pipette Pasteur couplée à un piège à vide à partir d’une plaque de 150 mm, laver 2x avec ~10 mL HBSS.
- Immédiatement avant la réticulation, préparer une solution de réticulation à 1 % d’AF dans un tube de 50 mL en combinant 22,5 mL d’HBSS et 625 μL d’AF à 37 % jusqu’à une concentration finale de 1 %. Mélanger doucement en berçant.
ATTENTION : Utilisez une hotte; Le formaldéhyde est toxique. - Pour réticuler les cellules, verser 23,125 mL de la solution d’acide FA à 1 % sur chaque plaque de 15 cm.
- Incuber à température ambiante pendant 10 min et bercer doucement les assiettes à la main toutes les 2 minutes.
- Ajouter 1,25 mL de glycine 2,5 M (128 mM final) et agiter doucement la plaque pour éteindre la réaction de réticulation.
- Incuber à température ambiante pendant 5 min et poursuivre l’incubation sur glace pendant au moins 15 min pour arrêter la réticulation.
- Grattez les cellules des plaques avec un grattoir de cellule ou un policier en caoutchouc.
- Transférer la suspension cellulaire dans un tube conique de 50 ml à l’aide d’une pipette. Centrifuger à 1 000 × g pendant 10 min à température ambiante et éliminer le surnageant par aspiration.
- Lavez la pastille cellulaire une fois avec 10 mL de solution saline tamponnée au phosphate (DPBS) de Dulbecco, à l’aide d’une pipette pour la remettre en suspension. Ensuite, centrifuger à 1 000 × g pendant 10 min à température ambiante. Passez immédiatement à la réticulation DSG.
REMARQUE: Soyez prudent lorsque vous lavez la pastille de cellule car les pastilles de cellule peuvent être desserrées et les cellules peuvent être perdues.
- Avoir des cellules ensemencées dans un milieu approprié pour récolter 5 × 106 cellules par plaque de 150 mm.
- Fixation du formaldéhyde : à partir de cellules en suspension
- Avoir des cellules ensemencées dans un milieu approprié pour récolter 5 × 106 cellules par récipient.
REMARQUE: Les utilisateurs peuvent choisir n’importe quel récipient préféré qui assure une croissance cellulaire optimale de n’importe quelle lignée cellulaire de mammifère. - Immédiatement avant la récolte, compter les cellules et transférer 5 × 106 cellules dans un tube conique de 50 mL.
- Enduire doucement les cellules par centrifugation à 300 × g pendant 10 min à température ambiante.
- Préparer une solution de réticulation à 1 % d’AF en ajoutant 1,25 mL d’AF à 37 % à 45 mL d’HBSS et mélanger en retournant le tube plusieurs fois.
REMARQUE : Ajouter la totalité de 1,25 mL d’AF sans fractionner la quantité.
ATTENTION : Le formaldéhyde est hautement toxique. - Remettez en suspension la pastille de cellule dans la solution de réticulation à 1 % d’acide sulfureux à 1 % préparée à l’étape précédente en la tuytant de haut en bas.
- Incuber à température ambiante pendant exactement 10 minutes sur rotateur, bascule ou par inversion manuelle douce du tube toutes les 1-2 minutes.
- Éteindre la réaction de réticulation en ajoutant 2,5 mL de glycine 2,5 M (128 mM final) et bien mélanger en inversant le tube.
- Incuber pendant 5 min à température ambiante, puis sur la glace pendant au moins 15 min pour arrêter complètement la réticulation.
- Centrifuger à température ambiante pour enduire les cellules réticulées à 1 000 × g pendant 10 min et éliminer le surnageant par aspiration.
- Laver les cellules une fois avec 10 mL de DPBS, puis centrifuger à 1 000 × g pendant 10 min à température ambiante. Jetez complètement le surnageant à l’aide d’une pipette et procédez immédiatement à la réticulation DSG.
REMARQUE: Soyez prudent lorsque vous lavez la pastille de cellule car les pastilles de cellule peuvent être desserrées et les cellules peuvent être perdues.
- Avoir des cellules ensemencées dans un milieu approprié pour récolter 5 × 106 cellules par récipient.
- Réticulation avec le glutarate de disuccinimidyl
- Resuspendre les cellules granulées dans 9,9 mL de DPBS avant d’ajouter 100 μL de 300 mM DSG (3 mM final). Mélanger par inversion.
REMARQUE: DSG est sensible à l’humidité. Il est important de préparer un nouveau stock de 300 mM DSG en DMSO le jour de la réticulation.
ATTENTION : Le DSG dans le DMSO est très toxique. - Réticuler les cellules à température ambiante pendant 40 min sur un rotateur.
- Ajouter 1,925 mL de glycine 2,5 M (400 mM final), retourner pour mélanger et incuber à température ambiante pendant 5 min.
- Centrifuger les cellules à 2 000 × g pendant 15 min à température ambiante.
REMARQUE: Soyez prudent lorsque vous retirez le surnageant des pastilles de cellules en vrac. - Remettez la pastille en suspension dans 1 mL d’albumine sérique bovine (BSA)-DPBS à 0,05 % et transvaser dans un tube de 1,7 mL.
REMARQUE: L’ajout de BSA peut aider à réduire l’agglutination cellulaire. - Centrifuger les cellules à 2 000 × g pendant 15 min à 4 °C et retirer le surnageant à l’aide d’une pipette.
REMARQUE: Pour éviter de perdre la pastille, retirez rapidement et complètement le surnageant. - Congeler la pastille dans de l’azote liquide et la conserver à -80 °C ou passer immédiatement à l’étape suivante.
- Resuspendre les cellules granulées dans 9,9 mL de DPBS avant d’ajouter 100 μL de 300 mM DSG (3 mM final). Mélanger par inversion.
2. Capture de conformation chromosomique
- Lyse cellulaire et digestion de la chromatine
- Resuspendre (pipette) les aliquotes des cellules réticulées (~5 × 106 cellules) dans 1 mL de tampon de lyse glacée (recette du tableau supplémentaire S1) contenant 10 μL de cocktail d’inhibiteurs de protéase et transférer dans un homogénéisateur de rebond pour une incubation de 15 minutes sur glace.
REMARQUE: Ajouter des inhibiteurs de protéase au tampon de lyse immédiatement avant utilisation. - Déplacez lentement le pilon A de haut en bas 30 fois pour homogénéiser les cellules sur la glace et incuber sur la glace pendant 1 minute pour permettre aux cellules de refroidir, avant 30 autres coups.
- Transférer le lysat dans un tube microcentrifuge de 1,7 mL.
REMARQUE: Gardez la suspension en mouvement car parfois des cellules collent dans l’embout de la pipette. - Centrifuger la suspension lysée à 2 500 × g pendant 5 min à température ambiante.
- Jeter le surnageant et agiter ou vorter la pastille humide pour la remettre en suspension. Enlevez autant de surnageant que possible pour obtenir une substance semblable à celle du yaourt avec un minimum de touffes.
- Remettez la pastille en suspension dans 500 μL de tampon de restriction 1x glacé (à partir de 10x; voir la recette dans le tableau supplémentaire S1) et centrifugez pendant 5 min à 2 500 × g. Répétez cette étape pour un deuxième lavage.
REMARQUE: La pastille dans 1x tampon de restriction est plus granulaire que la pastille précédente dans le tampon de lyse. - Resuspendre les cellules dans un volume final de 360 μL de 1x tampon de restriction par pipetage après avoir ajouté ~340 μL au volume de transfert de la pastille, qui dépend de la taille de la cellule.
- Réserver 18 μL de chaque lysat pour tester l’intégrité de la chromatine (IC). Conserver les échantillons d’IC à 4 °C.
- Ajouter 38 μL de dodécylsulfate de sodium (SDS) à 1 % à chaque tube Hi-C (volume total de 380 μL) et mélanger soigneusement en pipetant sans introduire de bulles.
ATTENTION : Les FDS sont toxiques. - Incuber les échantillons à 65 °C sans les agiter pendant exactement 10 min pour ouvrir la chromatine.
- Placer immédiatement les tubes sur de la glace et préparer le mélange de digestion avec Triton X-100 pour éteindre la FDS comme décrit dans le tableau 1.
- Ajouter 107 μL du mélange de digestion dans le tube Hi-C (487 μL au total) pour digérer la chromatine pendant une nuit (~16 h) à 37 °C dans un thermomélangeur avec agitation par intervalle (par exemple, 900 tr/min, 30 s allumé, 4 min off).
NOTE: L’ajout de Triton à une concentration finale de 1% sert à éteindre la FDS.
- Resuspendre (pipette) les aliquotes des cellules réticulées (~5 × 106 cellules) dans 1 mL de tampon de lyse glacée (recette du tableau supplémentaire S1) contenant 10 μL de cocktail d’inhibiteurs de protéase et transférer dans un homogénéisateur de rebond pour une incubation de 15 minutes sur glace.
- Fin de la biotinylation de l’ADN
- Après digestion pendant une nuit, transférer les échantillons à 65 °C pendant 20 minutes pour désactiver l’activité endonucléase restante.
- Pendant l’incubation, préparer un mélange maître à remplir comme indiqué dans le tableau 2.
- Après l’incubation, placer les échantillons immédiatement sur la glace.
- Mettre de côté un contrôle de la digestion (CC) de 10 μL pour chaque échantillon et conserver à 4 °C.
- Retirez la condensation du couvercle à l’aide d’une pipette ou en faisant tourner. À chaque échantillon, ajouter 58 μL de mélange de remplissage de biotine (volume total de l’échantillon 535 μL) et pipeter doucement sans former de bulles.
- Incuber les échantillons à 23 °C pendant 4 h dans un thermomélangeur (p. ex., 900 tr/min, 30 s allumé, 4 min off).
- Ligature de fragments d’ADN proximaux
- Préparer le mélange de ligature comme indiqué dans le tableau 3 pendant l’incubation de la biotine.
- Ajouter 665 μL du mélange de ligature à chaque échantillon (volume total de l’échantillon 1 200 μL). Mélanger doucement par pipetage.
- Incuber les échantillons à 16 °C pendant 4 h dans un thermomélangeur avec agitation par intervalles (p. ex., 900 tr/min, 30 s allumé, 4 min d’arrêt). Conservez ces échantillons contenant de la chromatine liée par covalence à 4 °C pendant quelques jours.
- Inversion de la réticulation
- Porter les volumes des échantillons CI et DC à 50 μL avec 1x Tris Low EDTA (TLE; voir la recette dans le tableau supplémentaire S1).
- Ajouter 10 μL de 10 mg/mL de protéinase K aux échantillons d’IC et de DC.
- Incuber à 65 °C pendant une nuit avec agitation par intervalles (p. ex. 900 tr/min, 30 s allumé, 4 min d’arrêt). Vous pouvez également effectuer une inversion de réticulation de 30 minutes pour ces contrôles pendant la purification de l’ADN des échantillons Hi-C.
- À chaque échantillon de Hi-C, ajouter 50 μL de 10 mg/mL de protéinase K et incuber à 65 °C pendant au moins 2 h en agitant par intervalle (p. ex. 900 tr/min, 30 s allumé, 4 min off).
- Ajouter 50 μL supplémentaires de 10 mg/mL de protéinase K dans chaque tube Hi-C (volume total de l’échantillon 1 300 μL) et poursuivre l’incubation à 65 °C pendant la nuit. Conserver à 4 °C jusqu’à purification de l’ADN.
REMARQUE: La division des incubations de protéinase K assure une digestion totale des protéines.
- Purification de l’ADN
- Laisser refroidir les tubes de 65 °C jusqu’à la température ambiante.
- Transférer chaque échantillon dans un tube conique de 15 mL et ajouter 2,6 mL (2x volume) de phénol:chloroforme:alcool isoamylique dans chaque tube.
ATTENTION : Le phénol:chloroforme:alcool isoamylique est un irritant très toxique et potentiellement cancérigène. - Vortex chaque tube pendant 1 min, puis transférer son contenu dans un tube à verrouillage de phase de 15 mL.
- Centrifuger les échantillons pendant 5 min à la vitesse maximale (1 500-3 500 × g) dans une centrifugeuse de paillasse.
- Verser délicatement la phase aqueuse dans un tube à ultracentrifugation de 35 mL et ajouter de l’eau ultrapure jusqu’à un volume final de 1 250 μL.
REMARQUE: Utilisez des tubes pour adapter l’ultracentrifugeuse disponible ou divisez-les en plusieurs tubes de microcentrifugeuse. - Ajouterun volume 1 /10e (~125 μL) d’acétate de sodium 3 M et bien mélanger par inversion.
- Ajouter un volume 2,5x (~3,4 mL) d’éthanol 100% glacé à chaque échantillon, équilibrer les tubes pour l’ultracentrifugation en ajoutant de l’éthanol glacé à 100% et bien mélanger par inversion.
- Incuber les tubes sur de la glace sèche pendant ~15 min (éviter la solidification).
- Centrifuger les tubes à 18 000 × g pendant 30 min à 4 °C.
REMARQUE: Pour les rotors inclinés: marquez les tubes pour l’emplacement de la pastille. - À l’aide d’une pipette, retirer et jeter complètement le surnageant du côté non granulé.
REMARQUE: À ce stade, le granulé doit devenir visible et peut être marqué sur le tube, car il peut ne pas être clairement visible après séchage à l’étape suivante. - Sécher les échantillons à l’air pendant environ 10 minutes ou jusqu’à ce qu’ils soient visiblement secs.
- Solubiliser chaque pastille dans 450 μL de 1x TLE par pipetage ou tourbillonnement et transférer dans une unité de filtration centrifuge (UFC) de 0,5 mL avec une coupure de poids moléculaire de 3 kDa.
- Centrifuger le CFU à la vitesse maximale pendant 10 min et jeter le débit. Lavez chaque tube à ultracentrifuger avec 450 μL supplémentaires de 1x TLE et transférez-le dans son UFC pour un autre lavage.
REMARQUE: Le lavage de l’UFC de cette façon limite la perte d’ADN tout en réduisant la concentration de sel. - Centrifuger le CFU à la vitesse maximale pendant 10 min et jeter le débit.
- Ajouter 80 μL de 1x TLE à la colonne et tourner la colonne dans un nouveau tube de collecte avant de centrifuger pendant 2 min à la vitesse maximale pour obtenir un volume final de ~100 μL.
- Ajouter 1 μL de RnaseA (1 mg/mL; dilution 10 fois de 10 mg/mL de stock) à chaque échantillon et incuber à 37 °C sur un bloc de chaleur, au bain-marie ou dans un thermomélangeur pendant au moins 30 minutes.
- Après le traitement à la rnase, prélever les échantillons à 37 °C et conserver à 4 °C jusqu’à l’étape de contrôle de la qualité.
- Vérification de la qualité de la chromatine, de la digestion enzymatique et de la ligature des échantillons
- Refroidir les échantillons CI et DC à température ambiante après avoir inversé les réticulations à l’étape 2.4.5. Ensuite, transférer dans un tube à verrouillage de phase de 2 mL.
REMARQUE: Assurez-vous que le contenu du verrouillage de phase est centrifugé à une pastille (vitesse maximale pendant 2 min). - Ajouter 200 μL d’alcool phénol:chloroforme:isoamylique et mélanger les échantillons en tourbillonnant pendant 1 min.
- Centrifuger les tubes pendant 5 min à vitesse maximale.
- Transférer la phase aqueuse de chaque échantillon (~50 μL) dans un nouveau tube à microfuge de 1,7 mL.
- Ajouter 1 μL de Rnase A (à partir de 1 mg/mL) et incuber à 37 °C pendant au moins 30 min.
- Charger les échantillons sur un gel d’agarose à 0,8 %, comme le recommande le tableau 4.
REMARQUE : Les résultats attendus d’un contrôle de la qualité sont présentés à la figure 2. - Quantifier l’ADN par densitométrie à partir du gel ou en utilisant Qubit ou Nanodrop.
REMARQUE: Une quantification précise garantit le volume d’entrée correct dans la partie suivante du protocole. Utilisez plusieurs étalons avec une quantité connue pour construire une courbe standard.
- Refroidir les échantillons CI et DC à température ambiante après avoir inversé les réticulations à l’étape 2.4.5. Ensuite, transférer dans un tube à verrouillage de phase de 2 mL.
Tableau 1 : Réactifs de digestion. Veuillez cliquer ici pour télécharger ce tableau.
Tableau 2 : Réactifs de remplissage de biotine. *Notez que le changement d’enzymes peut nécessiter des tampons différents et des dNTP biotinylés. Veuillez cliquer ici pour télécharger ce tableau.
Tableau 3 : Réactifs de mélange de ligature. Abréviation : BSA = albumine sérique bovine. Veuillez cliquer ici pour télécharger ce tableau.
Tableau 4 : Paramètres de charge du gel pour l’évaluation de la qualité et de la sélection de la taille. Veuillez cliquer ici pour télécharger ce tableau.

Figure 2 : Gel d’agarose présentant les résultats typiques du contrôle de la qualité post-purification de l’ADN. (A) Le contrôle IC doit indiquer une bande d’ADN de poids moléculaire élevé. (B) Les échantillons DC et Hi-C montrent une gamme de tailles d’ADN. L’échantillon de Hi-C, ayant été combiné en fragments plus gros, doit avoir un poids moléculaire plus élevé que le DC. La plage de concentration des marqueurs permet de générer une courbe standard. Notez que dans cet exemple, l’IC a été chargé sur un gel séparé, mais il est conseillé de charger et d’exécuter tous les échantillons et les contrôles ensemble. Abréviations : IC = intégrité de la chromatine; DC = contrôle de la digestion; Hi-C = ligaturé de proximité. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
3. Préparation de la bibliothèque de séquençage Hi-C
- Élimination de la biotine des extrémités non ligaturées
- Préparer les réactions d’élimination de la biotine comme indiqué dans le tableau 5.
REMARQUE: En règle générale, 10 μg d’ADN sont suffisants, mais jusqu’à 30 μg peuvent être utilisés. - Répartir 2 aliquotes de 65 μL de chaque réaction de 130 μL dans deux tubes de PCR.
- Transférer dans un thermocycleur ou une machine de PCR et incuber comme décrit dans le tableau 5.
REMARQUE: Les échantillons peuvent être stockés ici à 4 ° C (jours à semaines), -20 ° C (à long terme) ou immédiatement transférés à la sonication.
- Préparer les réactions d’élimination de la biotine comme indiqué dans le tableau 5.
- Sonication
- Regroupez les doublons d’échantillon de l’étape d’élimination de la biotine (volume total d’échantillon de 130 μL) dans un tube sonicateur de 130 μL pour la sonication.
- Sonicer les échantillons en utilisant les paramètres donnés dans le tableau 6 pour obtenir une distribution étroite et étroite inférieure à 500 pb.
REMARQUE: Différents types de sonicateurs peuvent être utilisés, mais pour une distribution de fragments étroite (100-500 pb), les paramètres de sonicator peuvent nécessiter une optimisation.
- Sélection de taille avec perles magnétiques
- Pipeter l’ADN soniqué du ou des tubes sonicateurs dans un tube à faible liaison de 1,7 mL.
- Porter chaque échantillon à un volume total de 500 μL avec 1x TLE. Essayez d’obtenir le volume le plus proche possible de 500 μL, car le rapport entre l’échantillon et le mélange de billes magnétiques est essentiel pour le choix de la taille.
- Ajouter 400 μL de mélange de billes magnétiques à chaque tube pour obtenir un rapport de mélange de billes magnétiques au volume d’échantillon de 0,8.
REMARQUE: Dans ces conditions, les billes capturent des fragments d’ADN >300 pb, qui seront la fraction supérieure. Le surnageant contiendra des fragments <300 pb, qui sera la fraction inférieure. - Mélanger les tubes par vortex et incuber pendant 10 min à température ambiante sur un rotateur. Pour ce rapport et d’autres ratios de sélection de taille, assurez-vous que le volume complet de l’échantillon se mélange bien. Recherchez le « mode erratique » de certains rotors, qui fonctionne bien pour les petits volumes.
- Incuber pendant 5 min à température ambiante sur un séparateur de particules magnétiques (MPS).
- Pendant l’incubation, ajouter 500 μL du mélange de billes magnétiques dans un nouveau tube à faible liaison de 1,7 μL pour chaque échantillon.
NOTE: Ces tubes seront utilisés pour la prochaine sélection de taille de la fraction inférieure en générant un rapport mélange de billes magnétiques sur échantillon de 1,1: 1. - Laissez les tubes sur le MPS pendant 5 min.
- Retirer le surnageant des billes et remettre en suspension avec 150 μL du mélange de billes magnétiques.
NOTE: Cette étape évite la saturation des billes avec de l’ADN en augmentant le nombre de billes sans augmenter le volume. - Transférer le surnageant de l’étape 3.3.5 au tube étiqueté préparé pour sélectionner la fraction inférieure (étape 3.3.8).
REMARQUE : 150 μL de mélange de billes magnétiques + 400 μL de mélange de billes magnétiques 0,8x (550 μL au total) divisé par 500 μL d’échantillon initial = 1,1x le rapport mélange de billes magnétiques sur échantillon. - Mélanger les tubes de fraction inférieure par vortex et incuber pendant 10 min à température ambiante sur un rotateur.
REMARQUE: Les billes lieront des fragments d’ADN > 100 pb, ce qui donnera une fraction finale liée aux billes de 100 à 300 pb. - Placez les tubes de fraction inférieure sur le MPS pendant 5 min (température ambiante).
- Retirez le surnageant et centrifugez brièvement les tubes pour éliminer davantage le surnageant autant que possible.
- Lavez les billes des deux fractions en utilisant 200 μL d’éthanol à 70% et récupérez les billes pendant 5 minutes sur le MPS à chaque fois.
- Après un rapide essorage dans une centrifugeuse, retirez complètement l’éthanol et séchez davantage les billes sur le MPS.
NOTE: Sécher jusqu’à ce que l’alcool soit complètement évaporé. La pastille doit ressembler à du chocolat noir sans craquer (peut prendre ~10 min). - Resuspendre les deux fractions dans 50 μL de 1x tampon TLE. Incuber à température ambiante pendant 10 minutes et tapoter ou agiter les tubes toutes les deux minutes pour stimuler le mélange et l’élution.
- Séparer les billes du surnageant sur le MPS pendant 5 min pour les deux fractions.
- Conserver le surnageant de chaque échantillon. Introduire le surnageant dans un tube à faible liaison de 1,7 mL.
NOTE: Les échantillons peuvent être conservés à 4 ° C pendant quelques jours ou à -20 ° C à long terme. - Exécutez un gel d’agarose à 2 % comme indiqué dans le tableau 4 pour déterminer la qualité et la quantité de l’échantillon. Voir la figure 3 pour un exemple d’un tel gel.
REMARQUE: Si par expérience, la sonication est hautement reproductible, on peut sauter ce gel et procéder à la réparation immédiatement. Il est recommandé aux utilisateurs de conserver les fractions supérieures jusqu’à la PCR de titrage. Des quantités d’ADN sous-optimales qui nécessiteraient beaucoup d’amplification par PCR peuvent être sauvées du matériel de la fraction supérieure. - Quantifier la quantité d’ADN du gel directement après avoir généré une courbe standard à partir de l’entrée d’échelle d’ADN connue ou en utilisant un Qubit ou une Nanodrop.
- Fin de la réparation
- Préparer le mélange de réparation final comme indiqué dans le tableau 7 (quantité par réaction donnée).
- Transférer les 46 μL restants d’ADN élué de la fraction inférieure vers des tubes de PCR et ajouter 24 μL du mélange de réparation final. Incuber dans une machine de PCR, comme proposé dans le tableau 7.
- Une fois le programme terminé, conserver les échantillons à 4 °C jusqu’à ce qu’ils soient abaissés.
- Retrait de produits de ligature biotinylés avec des billes enrobées de streptavidine
- Déterminer la quantité de billes enrobées de streptavidine pour chaque bibliothèque à partir de la sélection de taille quantifiée (étape 3.3.19).
REMARQUE : Ces billes enrobées de streptavidine (solution à 10 mg/mL) peuvent se lier à 20 μg d’ADN double brin par billes mg (= 20 μg / 100 μL de billes). Utilisez 2 μL pour chaque 1 μg d’ADN Hi-C, mais pas moins de 10 μL. - Mélanger les billes enrobées de streptavidine et pipeter le volume de billes nécessaires pour chaque bibliothèque (calculé à l’étape précédente) dans des tubes individuels à faible liaison de 1,7 mL.
- Remettez les billes en suspension dans 400 μL de tampon de lavage Tween (TWB; voir la recette dans le tableau supplémentaire S1) et incuber pendant ~3 min à température ambiante sur un rotateur (voir les instructions à l’étape 3.3.4).
- Séparez les perles du surnageant sur le MPS pendant 1 min et retirez le surnageant.
- Lavez les billes en pipetant 400 μL de TWB supplémentaires.
- Séparez les perles du surnageant sur le MPS pendant 1 min et retirez le surnageant.
- Ajouter 400 μL de tampon de liaison 2x (recette du tableau supplémentaire S1) aux billes et remettre en suspension. De plus, ajoutez 330 μL de 1x TLE et la solution de End-repair (à partir de l’étape 3.4.3).
- Incuber les échantillons pendant 15 min à température ambiante tout en mélangeant sur un rotateur.
- Séparez les perles du surnageant sur le MPS pendant 1 min et retirez le surnageant.
- Ajouter 400 μL de 1x BB aux billes et remettre en suspension.
- Séparez les perles du surnageant sur le MPS pendant 1 min et retirez le surnageant.
- Ajouter 100 μL de 1x TLE pour laver les billes.
- Séparez les perles du surnageant sur le MPS pendant 1 min et retirez le surnageant.
- Enfin, ajoutez 41 μL de 1x TLE pour remettre les billes en suspension.
- Déterminer la quantité de billes enrobées de streptavidine pour chaque bibliothèque à partir de la sélection de taille quantifiée (étape 3.3.19).
- A-tailing
- Préparer le mélange A-tailing comme indiqué dans le tableau 8.
- Pipeter les réactions dans des tubes de PCR et incuber comme indiqué dans le tableau 8.
- Placez les tubes PCR sur de la glace immédiatement après avoir été retirés du thermocycleur et transférez le contenu dans des tubes à faible liaison de 1,7 mL.
- Séparez les perles du surnageant sur le MPS pendant 1 min et jetez le surnageant.
- Ajouter 400 μL de tampon de ligature 1x, dilué dans 5x tampon ADN ligase T4 avec de l’eau ultrapure.
- Séparez les perles du surnageant sur le MPS pendant 1 min, puis jetez le surnageant.
- Ajouter 1x tampon de ligature à un volume final de 40 μL.
- Oligos adaptateurs de recuit
- Préparer des stocks d’oligo adaptateurs de 100 μM (tableau 9).
NOTE: Commandez 250 nmoles d’oligos purifiés par HPLC. - Recuit les adaptateurs dans des tubes PCR comme décrit dans le tableau 9.
- Utilisez un thermocycleur PCR pour augmenter progressivement la température de 0,5 °C/s à 97,5 °C. Maintenir à 97,5 °C pendant 2,5 min.
- Utilisez un thermocycleur PCR pour augmenter progressivement la température à 0,1 °C/s pendant 775 cycles (atteignant 20 °C). Maintenez la température à 4 °C jusqu’à nouvel usage.
- Ajouter 83 μL de 1x tampon de recuit (recette du tableau supplémentaire S1) pour diluer les adaptateurs à 15 μM. Conserver les adaptateurs à -20 °C.
- Préparer des stocks d’oligo adaptateurs de 100 μM (tableau 9).
- Ligature de l’adaptateur de séquençage
- Préparer le mélange de ligature de l’adaptateur dans un tube à faible liaison de 1,7 ml (tableau 10).
- Ligate pendant 2 h à température ambiante.
- Séparez les perles du surnageant sur le MPS pendant 1 min et jetez le surnageant.
- Ajouter 400 μL de TWB et pipeter soigneusement les billes de haut en bas avant l’incubation sur le rotateur pendant 5 minutes à température ambiante. Séparez les perles du surnageant sur le MPS et répétez cette étape une fois de plus.
- Séparez les billes du surnageant sur le MPS (~1 min), jetez le surnageant et ajoutez 200 μL de 1x BB.
- Séparez les perles du surnageant sur le MPS (~1 min) et jetez le surnageant.
- Ajouter 200 μL de 1x tampon pré-PCR (à partir de 10x; recette dans le tableau supplémentaire S1) et transférer dans un nouveau tube à faible liaison de 1,7 mL.
- Séparez les perles du surnageant sur le MPS (~1 min) et jetez le surnageant.
- Ajouter 20 μL de 1x tampon pré-PCR et mélanger par pipetage.
- Conserver les tubes sur de la glace pendant leur utilisation ou les conserver à 4 °C.
- Optimisation du nombre de cycles PCR par titrage
- Mettre en place 30 μL de réactions du mélange maître par échantillon, comme indiqué au tableau 11 (quantité par réaction donnée).
- Exécutez le plus petit nombre de cycles et prenez une partie aliquote de 5 μL. Exécutez 2-3 cycles supplémentaires sur la réaction restante avant de prendre la partie aliquote suivante de 5 μL. Répétez l’opération pour recueillir quatre aliquotes.
- Utilisez les paramètres de PCR du tableau 11 pour chaque aliquote.
- Ajouter 5 μL d’eau et 2 μL de colorant 6x à chaque échantillon de 5 μL. Exécuter sur un gel d’agarose TBE à 2% (recette du tableau supplémentaire S1) avec 25-150 ng d’échelle de faible poids moléculaire. Voir la figure 4 pour les résultats attendus.
REMARQUE: Un nombre optimal de cycles pour l’amplification PCR finale de la bibliothèque [étape 3.10] est le plus petit nombre de cycles pour obtenir un produit visible sur le gel moins un cycle.
- Amplification PCR finale de la bibliothèque
- Configurez des réactions de 12 x 30 μL pour amplifier chaque bibliothèque finale pour le séquençage comme dans le tableau 11.
- Faire défiler les réactions de PCR conformément au tableau 11 après avoir déterminé le nombre de cycles après le titrage PCR (étape 3.9.3).
- Une fois la PCR terminée, regrouper les échantillons répliqués dans un tube à microfuge à faible liaison de 1,7 mL.
- Placez les tubes sur le MPS et transférez le surnageant dans un nouveau tube à microfuge à faible liaison de 1,7 mL.
- Remettez en suspension les billes restantes enrobées de streptavidine dans 20 μL de 1x tampon pré-PCR.
REMARQUE: Ce modèle peut être réutilisé lorsqu’il est stocké à 4 ° C pendant des jours ou des semaines ou à long terme à -20 ° C.
- Enlèvement des apprêts avec mélange de billes magnétiques
- Utilisez 1x tampon TLE (recette du tableau supplémentaire S1) pour ajuster le volume de l’étape 3.10.4 à exactement 360 μL.
- À chaque échantillon, ajouter 360 μL du mélange de billes magnétiques et pipeter de haut en bas pour mélanger.
- Sur un rotateur, mélanger les échantillons pendant 10 min à température ambiante.
- Séparer les billes du surnageant sur le MPS à température ambiante (3-5 min).
- Lavez les billes deux fois avec 200 μL d’éthanol à 70 % et récupérez les billes pendant 5 min sur le MPS à chaque fois.
- Essorage rapide dans une centrifugeuse et pipette complète de l’éthanol. Sécher les billes à l’air sur le MPS pour évaporer davantage l’éthanol.
REMARQUE: La pastille doit ressembler à du chocolat noir sans craquer (peut prendre ~ 10 min). - Ajouter 30 μL d’eau ultrapure et remettre en suspension pour éluer l’ADN pendant 10 min à température ambiante. Agitez les tubes toutes les 2 minutes pour faciliter le mélange.
- Séparez les perles du surnageant sur le MPS pendant 5 min.
- Prélever le surnageant de chaque échantillon dans un tube frais de 1,7 mL.
- Exécutez 1 μL de la bibliothèque sur un gel d’agarose TBE à 2 % (recette du tableau supplémentaire S1) pour obtenir une distribution granulométrique des fragments et quantifier la bibliothèque finale (figure 5).
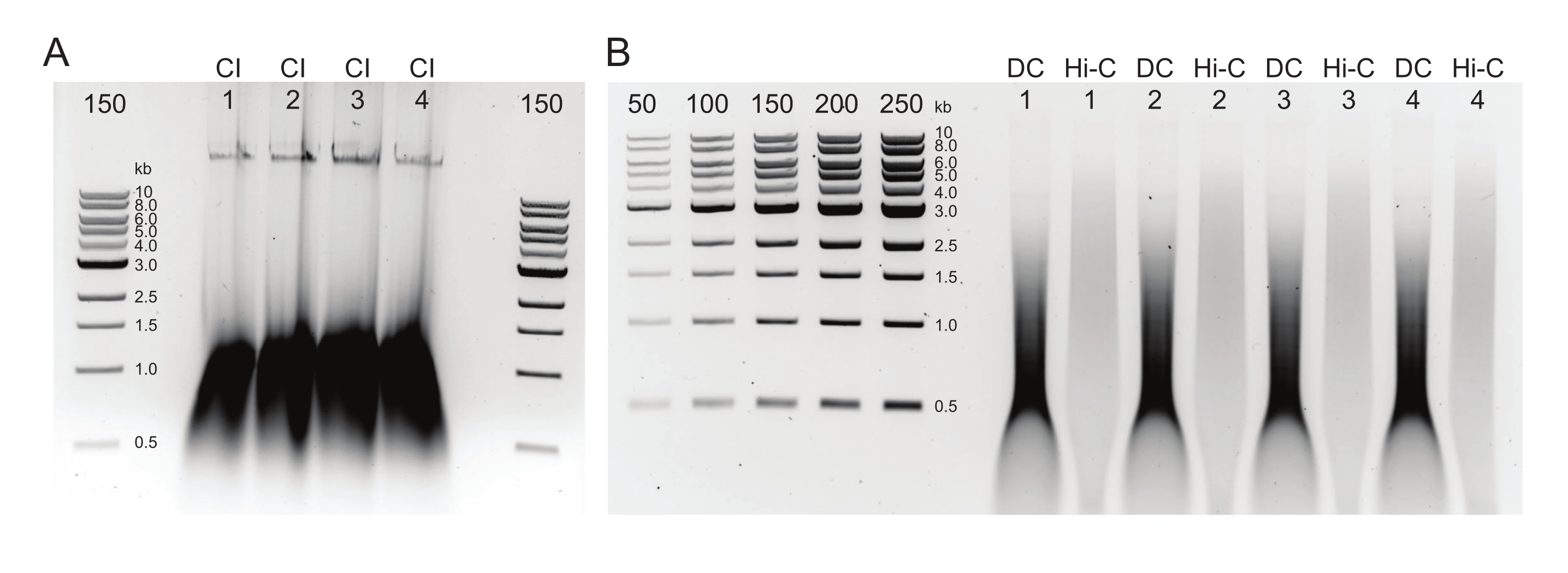
REMARQUE: La digestion ClaI ne peut se produire que pour les jonctions DpnII-DpnII et sert de contrôle de ligature positive qui devrait entraîner une distribution de taille de fragment plus faible pour la bibliothèque finale. Les bibliothèques finales peuvent être conservées pendant quelques jours à 4 °C, à long terme à -20 °C, ou immédiatement diluées et soumises au séquençage.
Tableau 5 : Réactifs d’élimination de la biotine et températures Veuillez cliquer ici pour télécharger ce tableau.
Tableau 6 : Paramètres de sonication. Veuillez cliquer ici pour télécharger ce tableau.
Tableau 7 : Réactifs de réparation finale et températures. Veuillez cliquer ici pour télécharger ce tableau.
Tableau 8 : Réactifs résiduels A et températures. Veuillez cliquer ici pour télécharger ce tableau.
Tableau 9 : Amorces PCR et oligos adaptateurs d’extrémité appariés avec recuit des réactifs pour le recuit. Abréviation : 5PHOS = 5' phosphate. Les astérisques indiquent des bases d’ADN phosphorothioées. # Combinez un oligo indexé avec l’oligo universel pour recuit dans un adaptateur indexé. Veuillez cliquer ici pour télécharger ce tableau.
Tableau 10 : Réactifs de ligature de l’adaptateur. Veuillez cliquer ici pour télécharger ce tableau.
Tableau 11 : Réactifs PCR et paramètres de cyclage. Veuillez cliquer ici pour télécharger ce tableau.

Figure 3 : Gel d’agarose montrant les résultats typiques de la sélection post-taille. Les fractions supérieure et inférieure de quatre échantillons (numérotés de 1 à 4) de DpnII-DdeI Hi-C sont indiquées. La première voie de chaque échantillon contient la fraction supérieure, dérivée d’un mélange de billes magnétiques 0,8x, et les deuxième et troisième voies contiennent une dilution de la fraction inférieure dérivée d’un mélange de billes magnétiques 1,1x. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 4 : Gel d’agarose avec résultats de titrage PCR. À partir de 5 cycles de PCR, des échantillons sont prélevés tous les 2 cycles (5, 7, 9 et 11 cycles) pour chacune des quatre bibliothèques. Sur la base de ce chiffre, 6 cycles ont été choisis comme cycle optimal pour chaque échantillon. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 5 : Produits finaux de PCR. Après le nettoyage et la sélection de la taille, les produits PCR (Hi-C) ont été chargés à côté d’une fraction digérée ClaI de la même bibliothèque (ClaI). Les fragments digérés par le claI indiquent la présence de ligatures DpnII-DpnII recherchées. Notez que ClaI ne digère pas les jonctions DpnII-DdeI et, par conséquent, toutes les ligatures ne contribueront pas à une réduction de taille de cette restriction. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Résultats
Les figures de ce manuscrit ont été générées à partir d’une expérience distincte et répliquée de celle publiée précédemment par Lafontaine et al.21. Après avoir obtenu des données de séquençage à haut débit, l’Open Chromatin Collective (Open2C: https://github.com/open2c) a été utilisé pour traiter les données Hi-C. Un pipeline similaire peut être trouvé sur le portail de données du projet 4D Nucleome (https://data.4dnucleome.org/resources/data-analysis/hi_c-processing-pipeline). En bref, le distillateur de pipeline Nextflow (https://github.com/open2c/distiller-nf) a été mis en œuvre pour (1) aligner les séquences de molécules Hi-C sur le génome de référence, (2) analyser l’alignement .sam et les fichiers de formulaire avec des paires Hi-C, (3) filtrer les doublons de PCR et (4) agréger les paires dans des matrices groupées d’interactions Hi-C. Ces matrices formatées HDF5, appelées refroidisseurs, peuvent ensuite être (1) visualisées sur un serveur HiGlass (https://higlass.io/) et (2) analysées à l’aide d’un grand ensemble d’outils de calcul open source présents dans la collection « cooltools » maintenue par l’Open Chromatin Collective (https://github.com/open2c/cooltools) pour extraire et quantifier les caractéristiques de pliage telles que les compartiments, les TAD et les boucles.
Certains indicateurs de qualité des bibliothèques Hi-C3.0 peuvent être évalués immédiatement après avoir mappé des paires de lecture à un génome de référence, à l’aide de quelques mesures/indicateurs simples. Tout d’abord, ~50% des paires de lecture séquencées peuvent être mappées de manière unique pour les cellules humaines. En raison de la nature polymérique des chromosomes, la plupart de ces lectures cartographiées (~60%-90%) représentent des interactions au sein d’un chromosome (cis), avec des fréquences d’interaction décroissant rapidement avec l’augmentation de la distance génomique (désintégration dépendante de la distance). La désintégration dépendante de la distance peut être mieux visualisée dans un « diagramme d’échelle », qui montre la probabilité de contact (par bras chromosomique) en fonction de la distance génomique. Nous avons constaté que l’utilisation de différents agents de réticulation et enzymes peut modifier la désintégration dépendante de la distance à des distances à longue et à courte distance17. L’ajout de la réticulation DSG augmente la détectabilité des interactions à courte distance lorsqu’il est combiné avec des enzymes telles que la Mnase et des combinaisons de DpnII-DdeI qui produisent des fragments plus petits (Figure 6A).
La décroissance dépendante de la distance peut également être observée directement à partir des matrices d’interaction 2D : les interactions deviennent plus rares lorsqu’elles sont situées plus loin de la diagonale centrale (Figure 6B). De plus, les caractéristiques de repliement génomique, telles que les compartiments, les TAD et les boucles, peuvent être identifiées à partir de matrices Hi-C et de diagrammes de mise à l’échelle comme des écarts par rapport à la désintégration moyenne dépendant de la distance à l’échelle du génome. Il est important de noter que la réticulation avec la DSG en plus de l’AF diminue les ligatures aléatoires, qui ne sont pas restreintes en raison de la nature polymère des chromosomes et, par conséquent, plus susceptibles de se produire entre les chromosomes (en trans) (Figure 6C). La réduction de la ligature aléatoire entraîne une augmentation des rapports signal/bruit, en particulier pour les interactions intrachromosomiques et intrachromosomiques à très longue portée (>10-50 Mb).

Figure 6 : Résultats représentatifs des bibliothèques Hi-C cartographiées et filtrées. (A) Tracés de mise à l’échelle avec probabilité de contact et son dérivé pour diverses enzymes, classés par longueur de fragment (en haut) et réticulation avec FA ou FA + DSG (en bas). La digestion avec la MNAse (microC) ou le DpnII-DdeI (Hi-C 3.0) augmente considérablement les contacts à courte portée (en haut), tout comme l’ajout de DSG à FA (en bas). (B) Les colonnes montrent les cartes thermiques Hi-C de la digestion DpnII après réticulation FA et de la digestion DpnII ou DdeI après réticulation FA + DSG. Les flèches blanches montrent une force croissante des « points » après la réticulation DSG et la digestion DdeI, ce qui implique une meilleure détection des boucles d’ADN. Les rangées montrent différentes parties du chromosome 3 à une résolution croissante, alignées sur le panneau C: rangée supérieure: chromosome 3 entier (0-198,295,559 Mb); rangée du milieu : 186-196 Mb; rangée du bas : 191,0-191,5 Mo. (C) Graphiques de couverture pour les régions représentées en A. Les flèches noires indiquent la couverture inférieure (%cis en lecture) pour la réticulation FA uniquement. Abréviations : FA = formaldéhyde; DSG = glutarate de disuccinimidyl; chr = chromosome. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Toutes les lectures mappées ne sont pas utiles. Un deuxième indicateur de qualité est le nombre de doublons PCR. Il est très peu probable que des lectures exactes en double se produisent par hasard après la ligature et la sonication. Ainsi, ces lectures résultent probablement de l’amplification de la PCR et doivent être filtrées. Les doublons surviennent souvent lorsque trop de cycles de PCR sont nécessaires pour amplifier les bibliothèques de faible complexité. En général, pour Hi-C, la plupart des bibliothèques n’ont besoin que de 5 à 8 cycles d’amplification finale de la PCR, comme déterminé par la PCR de titrage (voir étape 3.9 ; Graphique 4). Cependant, des bibliothèques suffisamment complexes peuvent être obtenues même après 14 cycles d’amplification PCR.
Une autre catégorie de lectures de doublons, appelées duplications optiques, peut découler du processus d’amplification sur les plates-formes de séquençage Illumina qui utilisent des cellules de flux à motifs (telles que HiSeq4000). Les doublons optiques sont trouvés soit en surchargeant la cellule d’écoulement, ce qui fait que les (grands) clusters sont appelés deux clusters distincts, soit en regroupant localement la molécule d’origine appariée après un premier cycle de PCR. Parce que les deux types de duplications optiques sont locaux, ils peuvent être identifiés et distingués des doublons PCR par leur emplacement sur la cellule d’écoulement. Alors que les bibliothèques avec >15% de duplications PCR auraient besoin d’être régénérées, les bibliothèques avec des doublons optiques peuvent être rechargées après avoir optimisé le processus de chargement.
Tableau supplémentaire S1 : Tampons et solutions. Veuillez cliquer ici pour télécharger ce tableau.
Discussion
Étapes critiques pour la manipulation des cellules
Bien qu’il soit possible d’utiliser un nombre inférieur de cellules d’entrée, ce protocole a été optimisé pour ~5 × 106 cellules par voie de séquençage (~400 M de lectures) pour assurer une complexité appropriée après un séquençage profond. Il est préférable de compter les cellules avant la fixation. Pour la génération de bibliothèques ultraprofondes, nous multiplions généralement le nombre de voies (et de cellules) jusqu’à ce que la profondeur de lecture souhaitée soit atteinte. Pour une fixation optimale, le milieu contenant du sérum doit être remplacé par du PBS avant la fixation par FA, et des solutions fixatrices doivent être ajoutées immédiatement et sans gradients de concentration15,22. Pour la récolte cellulaire, le grattage est préféré à la trypsinisation, car le passage d’une forme plus plate à une forme sphérique après trypsinisation pourrait affecter la conformation nucléaire. Après l’ajout de DSG, les granulés cellulaires lâches et grumeleux sont facilement perdus. Soyez prudent lorsque vous manipulez des cellules à ce stade et ajoutez jusqu’à 0,05% de BSA pour diminuer l’agglutination.
Modifications de la méthode
Ce protocole a été développé à partir de cellules humaines17. Pourtant, sur la base de l’expérience avec la capture de conformation chromosomique, ce protocole devrait fonctionner pour la plupart des cellules eucaryotes. Pour une entrée significativement plus faible (~1 × 106 cellules), nous conseillons d’utiliser la moitié des volumes pour les procédures de lyse et de capture de conformation [étapes 2.1-2.4]. Cela permettrait également d’isoler l’ADN [étape 2.5] dans une centrifugeuse de table avec des tubes de 1,7 mL, ce qui pourrait améliorer la granulation pour de faibles concentrations d’ADN. La quantification de l’ADN (étape 2.6.6) indiquera comment procéder. Pour de faibles quantités d’ADN isolé (1-5 μg), nous suggérons de sauter la sélection de la taille (étape 3.3) et de procéder à l’élimination de la biotine après avoir réduit le volume de 130 μL à ~45 μL avec une UFC.
Ce protocole a été développé spécifiquement pour garantir des données de haute qualité après réticulation ultérieure avec FA et DSG et digestion avec DpnII et DdeI. Cependant, d’autres stratégies de réticulation telles que l’AF suivie de l’EGS (éthylène glycol bis(succinimidyl succinate)), qui est également utilisé dans ChIP-seq23 et-PET24, pourraient fonctionner tout aussi bien17. De même, différentes combinaisons d’enzymes, telles que DpnII et HinfI18 ou MboI, MseI et NlaIII19 peuvent être utilisées pour la digestion. Lors de l’adaptation des combinaisons d’enzymes, assurez-vous d’utiliser des nucléotides biotinylés qui peuvent remplir les surplombs spécifiques de 5' et utilisez les tampons les plus optimaux pour chaque cocktail. DpnII est livré avec son propre tampon et le fabricant d’enzymes recommande un tampon spécifique pour la digestion DdeI. Pourtant, pour la double digestion avec DpnII et DdeI dans ce protocole, Restriction Buffer est recommandé car il est évalué à 100% d’activité pour les deux enzymes.
Résolution des problèmes liés à la capture de conformation
Les trois étapes clés de la capture de la conformation chromosomique: la réticulation, la digestion et la ligature ont toutes été effectuées avant que les résultats puissent être visualisés sur gel. Pour déterminer la qualité de chacune de ces trois étapes et discerner où des problèmes auraient pu survenir, des aliquotes avant (IC) et après digestion (DC) sont prélevées et chargées sur le gel avec l’échantillon de Hi-C ligaturé (Figure 2). Ce gel est utilisé pour déterminer la qualité de l’échantillon Hi-C et s’il vaut la peine de poursuivre le protocole. Sans l’IC et le CD, il est difficile d’identifier les étapes sous-optimales potentielles. Il convient de noter que la ligature sous-optimale peut être due à un problème de ligature elle-même, au remplissage ou à un problème de réticulation. Pour résoudre les problèmes de réticulation, veillez à ne pas utiliser plus de 1 × 10 à7 cellules par bibliothèque et commencez par de nouveaux réactifs de réticulation et des cellules propres (c’est-à-dire rincées avec du PBS). Pour la ligature, assurez-vous que les cellules et le mélange de ligature sont conservés sur la glace. Ajouter l’ADN ligase T4 juste avant l’incubation de 4 h à 16 °C et bien mélanger.
Dépannage de la préparation de la bibliothèque
Si plus de 10 cycles de PCR sont nécessaires ou si aucun produit PCR ne peut être vu sur le gel après titrage PCR (Figure 4), il existe quelques options pour enregistrer l’échantillon Hi-C. En revenant au titrage PCR, la première option consiste à réessayer la PCR. S’il n’y a toujours pas assez de produit, il est possible de tenter une autre série de ligature de queue A et d’adaptateur (étape 3.6) après avoir lavé les billes deux fois avec 1x tampon TLE. Après cette ligature supplémentaire de la queue A et de l’adaptateur, on peut procéder au titrage PCR comme précédemment. S’il n’y a toujours pas de produit, la dernière option consiste à résoniser la fraction 0,8x de l’étape 3.3 et à partir de là.
Limitations et avantages de Hi-C3.0
Il est important de réaliser que Hi-C est une méthode basée sur la population qui capture la fréquence moyenne des interactions entre les paires de loci dans la population cellulaire. Certaines analyses informatiques sont conçues pour démêler les combinaisons de conformations d’une population25, mais en principe, Hi-C est aveugle aux différences entre les cellules. Bien qu’il soit possible d’effectuer une Hi-C monocellulaire26,27 et que des inférences informatiques puissent être faites28, la Hi-C monocellulaire ne convient pas pour obtenir des informations 3C à ultrahaute résolution. Une limitation supplémentaire de Hi-C est qu’il ne détecte que les interactions par paires. Pour détecter les interactions multicontacts, on peut soit utiliser des couteaux fréquents combinés avec un séquençage à lecture courte (Illumina)16 ou effectuer des multicontacts 3C29 ou 4C30, en utilisant le séquençage à lecture longue des plateformes PacBio ou Oxford Nanopore. Des dérivés Hi-C pour détecter spécifiquement les contacts entre et le long des chromatides sœurs ont également été développés31,32.
Bien que Hi-C19 et Micro-C33 puissent être utilisés pour générer des cartes de contact à des résolutions inférieures à la kilobase, les deux nécessitent une grande quantité de lectures de séquençage, ce qui peut devenir une entreprise coûteuse. Pour obtenir une résolution similaire ou même supérieure sans les coûts, l’enrichissement pour des régions génomiques spécifiques (capture-C34) ou des interactions protéiques spécifiques (-PET 35, PLAC-seq 36, Hi-ChIP37) peuvent être appliqués. La force et l’inconvénient de ces applications d’enrichissement sont que seul un nombre limité d’interactions sont échantillonnées. Avec de tels enrichissements, l’aspect global de Hi-C (et l’option de normalisation globale) est perdu.
Importance et applications potentielles de Hi-C3.0
Ce protocole a été conçu pour permettre une 3C ultraprofonde haute résolution tout en détectant simultanément les caractéristiques de pliage à grande échelle telles que les TAD et les compartiments17 (Figure 6). Ce protocole commence avec 5 × 106 cellules par tube pour chaque bibliothèque Hi-C, ce qui devrait être plus que suffisant pour séquencer une ou deux voies sur une cellule d’écoulement pour obtenir jusqu’à 1 milliard de lectures d’extrémité appariée. Pour le séquençage ultraprofond, plusieurs tubes de 5 × 106 cellules doivent être préparés, en fonction du nombre de lectures cartographiées et de doublons PCR. À la résolution la plus élevée (<1 kb), les interactions en boucle se trouvent principalement entre les sites CTCF, mais les interactions promoteur-amplificateur peuvent également être détectées. Les lecteurs peuvent consulter Akgol Oksuz et coll.17 pour une description détaillée de l’analyse des données.
Déclarations de divulgation
Les auteurs n’ont aucun conflit d’intérêts à divulguer.
Remerciements
Nous tenons à remercier Denis Lafontaine pour le développement du protocole et Sergey Venev pour l’assistance bioinformatique. Ce travail a été soutenu par une subvention du National Institutes of Health Common Fund 4D Nucleome Program à J.D. (U54-DK107980, UM1-HG011536). J.D. est un chercheur du Howard Hughes Medical Institute.
Cet article est soumis à la politique de libre accès aux publications de HHMI. Les chefs de laboratoire HHMI ont déjà accordé une licence CC BY 4.0 non exclusive au public et une licence sous-licenciable à HHMI dans leurs articles de recherche. Conformément à ces licences, le manuscrit accepté par l’auteur de cet article peut être mis gratuitement à disposition sous une licence CC BY 4.0 dès sa publication.
matériels
| Name | Company | Catalog Number | Comments |
| 1 kb Ladder | New England Biolabs | N3232L | |
| Agarose | Invitrogen | 16500100 | |
| Agencourt AMPure XP magnetic beads , 60 mL | Beckman Coulter | A63881 | |
| Amicon Ultra-0.5 Centrifugal Filter Unit (CFU) | EMD Millipore | UFC500396 | |
| Annealing Buffer (5x) | See recipe in supplemental materials | ||
| ATP 10 mM | ThermoFisher | R0441 | |
| Avanti J-25i High Speed Refrigerated ultra-centrifuge | Beckman Coulter | ||
| beckman ultracentrifuge tube 35 mL | Beckman Coulter | 357002 | |
| Binding Buffer (2x) | See recipe in supplemental materials | ||
| biotin-14-dATP 0.4 mM | Invitrogen | 19524-016 | |
| BSA 10 mg/mL | New England Biolabs | B9000S | dilute from 20 mg/mL |
| Cell scraper | Falcon | 353089 | |
| Cell scraper | Corning | 3008 | |
| Conical polypropylene tubes 50 mL | Denville | C1062-P | |
| Conical tube 15 mL | Denville | C1017-P | |
| Covaris micro tube AFA fiber with snap-cap 130 µL | Covaris | 520045/520077 | |
| Covaris Sonicator | Covaris | E220/E220evolution/M220 | |
| Culture flask 175 cm2 | Falcon | 353112 | |
| Culture plates 150 mm x 25 mm | Corning | 430599 | |
| dATP 1 mM | Invitrogen | 56172 | |
| dATP 10 mM | Invitrogen | 56172 | |
| dCTP 10 mM | Invitrogen | 56173 | |
| DdeI | New England Biolabs | R0175L | |
| dGTP 10 mM | Invitrogen | 56174 | |
| DMSO | Sigma | D2650-5x10ML | |
| dNTP mix 25 mM | Invitrogen | 10297117 | |
| Dounce homogenizer | DWK Life Sciences | 8853010002/8853030002 | |
| DPBS | Gibco | 14190-144 | |
| DpnII | New England Biolabs | R0543M | |
| DSG | ThermoScientific | 20593 | |
| dTTP 10 mM | Invitrogen | 56175 | |
| Ethanol 70% | Fisher | A409-4 | Diluted from 100% |
| Ethidium Bromide | Fisher | BP1302-10 | |
| Formaldehyde (37%) | Fisher | BP531-500 | |
| Gel loading dye (6x ) | New England Biolabs | B7024S | |
| Glycine in ultrapure water 2.5 M | Sigma | G8898-1KG | |
| HBSS | Gibco | 14025-092 | |
| Igepal CA-630 detergent | MP Biomedicals | 198596 | |
| Klenow DNA polymerase 5 U/µL | New England Biolabs | M0210L | |
| Klenow Fragment 3-->5’ exo-, 5 U/µL | New England Biolabs | M0212L | |
| ligation buffer (10x) | New England Biolabs | B7203S | |
| Liquid nitrogen | |||
| LoBind microcentrifuge tube 1.7 mL | Eppendorf | 22431021 | |
| Low Molecular Weight DNA Ladder | New England Biolabs | N3233L | |
| Lysis buffer | See recipe in supplemental materials | ||
| Magnetic Particle separator | ThermoFisher | 12321D | |
| Microfuge tubes 1.7 mL | Axygen | MCT-175-C | |
| MyOne Streptavidin C1 beads | Invitrogen | 65001 | |
| NEBuffer 2.1 (10x) | New England Biolabs | B7002S | |
| NEBuffer 3.1 (10x) | New England Biolabs | B7203S | |
| PBS | Gibco | 70013-032 | |
| PCR (strip) tubes | Biorad | TBS0201/ TCS0803 | |
| PCR thermocycler | Biorad | T100 | |
| Pfu Ultra II Buffer (10x) | Agilent | Comes with Pfu Ultra | |
| PfuUltra II Fusion HS DNA Polymerase | Agilent | 600674 | |
| Phase lock tube 15 mL | Qiagen | 129065 | |
| Phase lock tubes 2 mL | Qiagen | 129056 | |
| Phenol:chloroform:isoamyl alcohol | Invitrogen | 15593-049 | |
| Protease inhibitor cocktail | ThermoFisher | 78440 | |
| Proteinase K in ultrapure water 10 mg/mL | Invitrogen | 25530-031 | |
| Refrigerated Centrifuge | Eppendorf | 5810R | |
| RNase A, DNase and protease-free 10 mg/mL | Thermo Scientific | EN0531 | |
| Rotator | Argos technologies | EW-04397-40 or rocking platform | |
| SDS 1% | Fisher | BP13111 | |
| Sodium acetate pH = 5.2, 3 M | Sigma | ||
| Sub-Cell GT Horizontal Electrophoresis System | Biorad | 1704401 | |
| T4 DNA ligase 1 U/µL | Invitrogen | 100004817 | |
| T4 DNA polymerase | New England Biolabs | M0203L | |
| T4 DNA polymerase | New England Biolabs | M0203L | |
| T4 DNA polymerase 3 U/µL | New England Biolabs | M0203L | |
| T4 ligation buffer (5x) | Invitrogen | Y90001 | |
| T4 polynucleotide kinase 10 U/µL | New England Biolabs | M0201L | |
| Tabletop centrifuge | Eppendorf | 5425 | |
| TBE buffer | See recipe in supplemental materials | ||
| Tris Low EDTA Buffer (TLE) | See recipe in supplemental materials | ||
| Triton X-100 (10%) | Sigma | 93443 | |
| Truseq adapter oligos | Integrated DNA Technologies (IDT)) | https://www.idtdna.com/site/order/oligoentry | 250 nmole and HPLC purified |
| Tween 20 detergent | Fisher | 9005-64-5 | |
| Tween Wash Buffer | See recipe in supplemental materials | ||
| Vortex | Scientific Industries | (G560)SI-0236 |
Références
- Dekker, J., Rippe, K., Dekker, M., Kleckner, N. Capturing chromosome conformation. Science. 295 (5558), 1306-1311 (2002).
- Simonis, M., et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nature Genetics. 38 (11), 1348-1354 (2006).
- Dostie, J., et al. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Research. 16 (10), 1299-1309 (2006).
- Lieberman-Aiden, E., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326 (5950), 289-293 (2009).
- Stadhouders, R., et al. Multiplexed chromosome conformation capture sequencing for rapid genome-scale high-resolution detection of long-range chromatin interactions. Nature Protocols. 8 (3), 509-524 (2013).
- Kalhor, R., Tjong, H., Jayathilaka, N., Alber, F., Chen, L. Genome architectures revealed by tethered chromosome conformation capture and population-based modeling. Nature Biotechnology. 30 (1), 90-98 (2012).
- Hsieh, T. H., et al. Mapping nucleosome resolution chromosome folding in yeast by micro-C. Cell. 162 (1), 108-119 (2015).
- Hsieh, T. -. H. S., Fudenberg, G., Goloborodko, A., Rando, O. J. Micro-C XL: assaying chromosome conformation from the nucleosome to the entire genome. Nature Methods. 13 (12), 1009-1011 (2016).
- Denker, A., de Laat, W. The second decade of 3C technologies: detailed insights into nuclear organization. Genes & Development. 30 (12), 1357-1382 (2016).
- Rodley, C. D., Bertels, F., Jones, B., O’Sullivan, J. M. Global identification of yeast chromosome interactions using Genome conformation capture. Fungal Genetics and Biology. 46 (11), 879-886 (2009).
- Rao, S. S., et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 159 (7), 1665-1680 (2014).
- Alipour, E., Marko, J. F. Self-organization of domain structures by DNA-loop-extruding enzymes. Nucleic Acids Research. 40 (22), 11202-11212 (2012).
- Lajoie, B. R., Dekker, J., Kaplan, N. The Hitchhiker’s guide to Hi-C analysis: Practical guidelines. Methods. 72, 65-75 (2015).
- Belaghzal, H., Dekker, J., Gibcus, J. H. Hi-C 2.0: An optimized Hi-C procedure for high-resolution genome-wide mapping of chromosome conformation. Methods. 123, 56-65 (2017).
- Golloshi, R., Sanders, J. T., McCord, R. P. Iteratively improving Hi-C experiments one step at a time. Methods. 142, 47-58 (2018).
- Darrow, E. M., et al. Deletion of DXZ4 on the human inactive X chromosome alters higher-order genome architecture. Proceedings of the National Academy of Sciences of the United States of America. 113 (31), 4504-4512 (2016).
- Akgol Oksuz, B., et al. Systematic evaluation of chromosome conformation capture assays. Nature Methods. 18 (9), 1046-1055 (2021).
- Ghuryeid, J., et al. Integrating Hi-C links with assembly graphs for chromosome-scale assembly. PLoS Computational Biology. 15 (8), 1007273 (2019).
- Gu, H., et al. Fine-mapping of nuclear compartments using ultra-deep Hi-C shows that active promoter and enhancer elements localize in the active A compartment even when adjacent sequences do not. bioRxiv. , (2021).
- Ramani, V., et al. Mapping 3D genome architecture through in situ DNase Hi-C. Nature Protocols. 11 (11), 2104-2121 (2016).
- Lafontaine, D. L., Yang, L., Dekker, J., Gibcus, J. H. Hi-C 3.0: Improved Protocol for Genome-Wide Chromosome Conformation Capture. Current Protocols. 1 (7), 198 (2021).
- Belton, J. -. M. M., et al. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods. 58 (3), 268-276 (2012).
- Truch, J., Telenius, J., Higgs, D. R., Gibbons, R. J. How to tackle challenging ChIP-Seq, with long-range cross-linking, Using ATRX as an example. Methods in Molecular Biology. 1832, 105-130 (2018).
- Wang, P., et al. In situ chromatin interaction analysis using paired-end tag sequencing. Current Protocols. 1 (8), 174 (2021).
- Tjong, H., et al. Population-based 3D genome structure analysis reveals driving forces in spatial genome organization. Proceedings of the National Academy of Sciences of the United States of America. 113 (12), 1663-1672 (2016).
- Nagano, T., et al. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature. 547 (7661), 61-67 (2017).
- Ramani, V., et al. Massively multiplex single-cell Hi-C. Nature Methods. 14 (3), 263-266 (2017).
- Meng, L., Wang, C., Shi, Y., Luo, Q. Si-C is a method for inferring super-resolution intact genome structure from single-cell Hi-C data. Nature Communications. 12 (1), 4369 (2021).
- Tavares-Cadete, F., Norouzi, D., Dekker, B., Liu, Y., Dekker, J. Multi-contact 3C reveals that the human genome during interphase is largely not entangled. Nature Structural & Molecular Biology. 27 (12), 1105-1114 (2020).
- Vermeulen, C., et al. Multi-contact 4C: long-molecule sequencing of complex proximity ligation products to uncover local cooperative and competitive chromatin topologies. Nature Protocols. 15 (2), 364-397 (2020).
- Oomen, M. E., Hedger, A. K., Watts, J. K., Dekker, J. Detecting chromatin interactions between and along sister chromatids with SisterC. Nature Methods. 17 (10), 1002-1009 (2020).
- Mitter, M., et al. Conformation of sister chromatids in the replicated human genome. Nature. 586 (7827), 139-144 (2020).
- Krietenstein, N., et al. Ultrastructural Details of Mammalian Chromosome Architecture. Molecular Cell. 78 (3), 554-565 (2020).
- Hughes, J. R., et al. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nature Genetics. 46 (2), 205-212 (2014).
- Fullwood, M. J., et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 462 (7269), 58-64 (2009).
- Fang, R., et al. Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Research. 26 (12), 1345-1348 (2016).
- Mumbach, M. R., et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nature Methods. 13 (11), 919-922 (2016).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.