
Method Article
Erfassung der Chromosomenkonformation über Längenskalen hinweg
In diesem Artikel
Zusammenfassung
Hi-C 3.0 ist ein verbessertes Hi-C-Protokoll, das Formaldehyd- und Disuccinimidylglutarat-Vernetzer mit einem Cocktail aus DpnII- und DdeI-Restriktionsenzymen kombiniert, um das Signal-Rausch-Verhältnis und die Auflösung der Chromatin-Interaktionsdetektion zu erhöhen.
Zusammenfassung
Chromosomenkonformationserfassung (3C) wird verwendet, um dreidimensionale Chromatin-Wechselwirkungen zu erkennen. Typischerweise wird chemische Vernetzung mit Formaldehyd (FA) verwendet, um Chromatinwechselwirkungen zu fixieren. Dann wandelt der Chromatinaufschluss mit einem Restriktionsenzym und die anschließende Religation der Fragmentenden dreidimensionale (3D) Nähe in einzigartige Ligationsprodukte um. Schließlich wird die DNA nach der Umkehrung der Vernetzungen, der Proteinentfernung und der DNA-Isolierung geschert und für die Hochdurchsatzsequenzierung vorbereitet. Die Häufigkeit der Annäherungsligation von Loci-Paaren ist ein Maß für die Häufigkeit ihrer Kolokalisation im dreidimensionalen Raum in einer Zellpopulation.
Eine sequenzierte Hi-C-Bibliothek liefert genomweite Informationen über Interaktionsfrequenzen zwischen allen Loci-Paaren. Die Auflösung und Präzision von Hi-C beruht auf einer effizienten Vernetzung, die Chromatinkontakte und eine häufige und gleichmäßige Fragmentierung des Chromatins aufrechterhält. Dieser Artikel beschreibt ein verbessertes In-situ-Hi-C-Protokoll, Hi-C 3.0, das die Effizienz der Vernetzung durch Kombination von zwei Vernetzern (Formaldehyd [FA] und Disuccinimidylglutarat [DSG]) erhöht, gefolgt von einem feineren Aufschluss mit zwei Restriktionsenzymen (DpnII und DdeI). Hi-C 3.0 ist ein einziges Protokoll für die genaue Quantifizierung von Genomfaltungsmerkmalen auf kleineren Skalen wie Schleifen und topologisch assoziierenden Domänen (TADs) sowie von Merkmalen auf größeren kernweiten Skalen wie Kompartimenten.
Einleitung
Chromosomenkonformationserfassung wird seit 2002 verwendet1. Grundsätzlich beruht jede Konformationserfassungsvariante auf der Fixierung von DNA-Protein- und Protein-Protein-Wechselwirkungen, um die 3D-Chromatinorganisation zu erhalten. Es folgt eine DNA-Fragmentierung, in der Regel durch Restriktionsverdauung, und schließlich endet die Religation der nahe gelegenen DNA, um räumlich proximale Loci in einzigartige kovalente DNA-Sequenzen umzuwandeln. Anfängliche 3C-Protokolle verwendeten PCR, um spezifische "Eins-zu-Eins"-Interaktionen abzutasten. Nachfolgende 4C-Assays ermöglichten den Nachweis von "One-to-all"-Interaktionen2, während 5C "many-to-many"-Interaktionen3 detektierten. Die Erfassung von Chromosomenkonformationen kam nach der Implementierung der Hochdurchsatzsequenzierung (NGS) der nächsten Generation, die den Nachweis von "all-to-all" genomischen Interaktionen mit genomweitem Hi-C4 und vergleichbaren Techniken wie 3C-seq5, TCC6 und Micro-C7,8 ermöglichte, vollständig zum Tragen (siehe auch Review von Denker und De Laat9).
In Hi-C werden biotinylierte Nukleotide verwendet, um 5′-Überhänge nach dem Verdauung und vor der Ligation zu markieren (Abbildung 1). Dies ermöglicht die Auswahl von richtig verdauten und religierten Fragmenten mit Streptavidin-beschichteten Perlen und unterscheidet sie von GCC10. Eine wichtige Aktualisierung des Hi-C-Protokolls wurde von Rao et al.11 implementiert, die die Verdauung und Religation in intakten Kernen (d.h. in situ) durchführten, um falsche Ligaturprodukte zu reduzieren. Darüber hinaus reduzierte der Ersatz des HindIII-Aufschlusses durch den MboI- (oder DpnII-) Aufschluss die Fragmentgröße und erhöhte das Auflösungspotenzial von Hi-C. Dieser Anstieg ermöglichte den Nachweis relativ kleiner Strukturen und eine genauere genomische Lokalisierung von Kontaktpunkten, wie z.B. DNA-Schleifen zwischen kleinen cis-Elementen, z.B. Schleifen zwischen CTCF-gebundenen Stellen, die durch Schleifenextrusionerzeugt werden 11,12. Dieses Potenzial hat jedoch seinen Preis. Erstens erfordert eine zweifache Erhöhung der Auflösung eine vierfache (22) Erhöhung der Sequenzierungslesevorgänge13. Zweitens erhöhen die kleinen Fragmentgrößen die Möglichkeit, unverdaute benachbarte Fragmente mit verdauten und religierten Fragmenten zu verwechseln14. Wie bereits erwähnt, unterscheiden sich bei Hi-C verdaute und religierte Fragmente von unverdauten Fragmenten durch die Anwesenheit von Biotin an der Ligationsverbindung. Es ist jedoch eine ordnungsgemäße Biotinentfernung von unligierten Enden erforderlich, um sicherzustellen, dass nur Ligationsverbindungen nach unten gezogenwerden 14,15.
Mit den sinkenden Kosten von NGS wird es möglich, die Chromosomenfaltung genauer zu untersuchen. Um die Größe von DNA-Fragmenten zu verringern und dadurch die Auflösung zu erhöhen, kann das Hi-C-Protokoll angepasst werden, um häufiger schneidende Restriktionsenzyme16 oder Kombinationen von Restriktionsenzymen 17,18,19 zu verwenden. Alternativ können MNase 7,8 in Micro-C und DNase in DNase Hi-C20 titriert werden, um eine optimale Verdauung zu erreichen.
Eine kürzlich durchgeführte systematische Auswertung der Grundlagen von 3C-Methoden zeigte, dass sich die Detektion von Chromosomenfaltungsmerkmalen auf jeder Längenskala mit sequentieller Vernetzung mit 1% FA gefolgt von 3 mM DSG17 stark verbesserte. Darüber hinaus war Hi-C mit HindIII-Aufschluss die beste Option zum Nachweis großräumiger Faltungsmerkmale wie Kompartimente, und Micro-C war bei der Erkennung kleiner Faltungsmerkmale wie DNA-Schleifen überlegen. Diese Ergebnisse führten zur Entwicklung einer einzigen, hochauflösenden "Hi-C 3.0"-Strategie, die die Kombination von FA- und DSG-Vernetzern gefolgt von einem Doppelaufschluss mit DpnII- und DdeI-Endonukleasen verwendet21. Hi-C 3.0 bietet eine effektive Strategie für den allgemeinen Gebrauch, da es Faltmerkmale über alle Längenskalen hinweg genau erkennt17. Der experimentelle Teil des Hi-C 3.0-Protokolls wird hier detailliert beschrieben und typische Ergebnisse, die nach der Sequenzierung zu erwarten sind, werden gezeigt.

Abbildung 1: Hi-C-Verfahren in sechs Schritten. Die Zellen werden zuerst mit FA und dann mit DSG fixiert (1). Dann geht die Lyse einer Doppelverdauung mit DdeI und DpnII voraus (2). Biotin wird durch Überhangfüllung hinzugefügt und proximal abgestumpfte Enden werden vor der DNA-Reinigung ligiert (3). Biotin wird vor der Beschallung und Größenauswahl von unligierten Enden entfernt (5). Schließlich ermöglicht das Pull-down von Biotin eine Adapterligation und Bibliotheksamplifikation durch PCR (6). Abkürzungen: FA = Formaldehyd; DSG = Disuccinimidylglutarat; B = Biotin. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
Protokoll
1. Fixierung durch Vernetzung
- Formaldehydfixierung: ausgehend von Zellen in Monolayer
- Die Zellen werden in geeignetem Medium ausgesät, um 5 × 106 Zellen pro 150 mm Platte zu ernten.
HINWEIS: Benutzer können jeden bevorzugten Behälter auswählen, der ein optimales Wachstum jeder Säugetierzelllinie gewährleistet. Zusätzlich können Zellen aus Gewebe isoliert werden. - Das Medium mit einer Pasteur-Pipette aus 150 mm Platte absaugen, 2x mit ~10 mL HBSS waschen.
- Unmittelbar vor der Vernetzung wird eine 1%ige FA-Vernetzungslösung in einem 50-ml-Röhrchen hergestellt, indem 22,5 ml HBSS und 625 μl 37% FA zu einer endgültigen Konzentration von 1% kombiniert werden. Sanft durch Schaukeln mischen.
VORSICHT: Verwenden Sie einen Abzug; Formaldehyd ist giftig. - Um die Zellen zu vernetzen, gießen Sie 23,125 ml der 1% igen FA-Lösung auf jede 15 cm große Platte.
- Bei Raumtemperatur 10 min inkubieren und die Platten alle 2 min vorsichtig von Hand schaukeln.
- Fügen Sie 1,25 ml 2,5 M Glycin (128 mM endgültig) hinzu und schwenken Sie die Platte vorsichtig, um die Vernetzungsreaktion zu löschen.
- Inkubieren Sie bei Raumtemperatur für 5 min und setzen Sie die Inkubation auf Eis für mindestens 15 Minuten fort, um die Vernetzung zu stoppen.
- Kratzen Sie die Zellen mit einem Zellschaber oder Gummipolizisten von den Platten.
- Die Zellsuspension wird mit einer Pipette in ein 50-ml-konisches Röhrchen überführt. Bei 1.000 × g 10 min bei Raumtemperatur zentrifugieren und den Überstand durch Absaugen verwerfen.
- Waschen Sie das Zellpellet einmal mit 10 ml Dulbeccos phosphatgepufferter Kochsalzlösung (DPBS) und verwenden Sie eine Pipette zum Resuspendieren. Dann bei 1.000 × g für 10 min bei Raumtemperatur zentrifugieren. Fahren Sie sofort mit der DSG-Vernetzung fort.
HINWEIS: Seien Sie vorsichtig beim Waschen des Zellpellets, da Zellpellets lose sein können und Zellen verloren gehen können.
- Die Zellen werden in geeignetem Medium ausgesät, um 5 × 106 Zellen pro 150 mm Platte zu ernten.
- Formaldehydfixierung: ausgehend von Zellen in Suspension
- Lassen Sie die Zellen in geeignetem Medium säen, um 5 × 106 Zellen pro Gefäß zu ernten.
HINWEIS: Benutzer können jeden bevorzugten Behälter auswählen, der ein optimales Zellwachstum jeder Säugetierzelllinie gewährleistet. - Unmittelbar vor der Ernte die Zellen zählen und 5 × 106 Zellen in ein 50 ml konisches Röhrchen überführen.
- Pelletieren Sie die Zellen vorsichtig durch Zentrifugieren bei 300 × g für 10 min bei Raumtemperatur.
- Bereiten Sie 1% FA-Vernetzungslösung vor, indem Sie 1,25 ml 37% FA zu 45 ml HBSS hinzufügen und durch mehrmaliges Invertieren des Röhrchens mischen.
HINWEIS: Fügen Sie die gesamten 1,25 ml FA hinzu, ohne die Menge aufzuteilen.
ACHTUNG: Formaldehyd ist hochgiftig. - Resuspendieren Sie das Zellpellet in der im vorherigen Schritt hergestellten 46,25 ml 1% FA-Vernetzungslösung durch Pipettieren auf und ab.
- Inkubieren Sie bei Raumtemperatur für genau 10 min auf Rotator, Wippe oder durch sanfte manuelle Inversion des Rohres alle 1-2 min.
- Löschen Sie die Vernetzungsreaktion durch Zugabe von 2,5 ml 2,5 M Glycin (128 mM endgültig) und mischen Sie gut durch Invertieren des Röhrchens.
- Inkubieren Sie für 5 min bei Raumtemperatur und dann mindestens 15 min auf Eis, um die Vernetzung vollständig zu beenden.
- Zentrifugieren Sie bei Raumtemperatur, um die vernetzten Zellen bei 1.000 × g für 10 min zu pelletieren und den Überstand durch Aspiration zu verwerfen.
- Waschen Sie die Zellen einmal mit 10 ml DPBS und zentrifugieren Sie dann bei 1.000 × g für 10 min bei Raumtemperatur. Entsorgen Sie den Überstand vollständig mit einer Pipette und fahren Sie sofort mit der DSG-Vernetzung fort.
HINWEIS: Seien Sie vorsichtig beim Waschen des Zellpellets, da Zellpellets lose sein können und Zellen verloren gehen können.
- Lassen Sie die Zellen in geeignetem Medium säen, um 5 × 106 Zellen pro Gefäß zu ernten.
- Vernetzung mit Disuccinimidylglutarat
- Resuspendieren Sie die pelletierten Zellen in 9,9 ml DPBS, bevor Sie 100 μL 300 mM DSG (3 mM endgültig) hinzufügen. Mischen durch Inversion.
HINWEIS: DSG ist feuchtigkeitsempfindlich. Es ist wichtig, am Tag der Vernetzung einen frischen Vorrat von 300 mM DSG in DMSO vorzubereiten.
ACHTUNG: DSG in DMSO ist hochgiftig. - Vernetzen Sie die Zellen bei Raumtemperatur für 40 min auf einem Rotator.
- 1,925 ml 2,5 M Glycin (400 mM endgültig) zugeben, zum Mischen umkehren und 5 min bei Raumtemperatur inkubieren.
- Zentrifugieren Sie die Zellen bei 2.000 × g für 15 min bei Raumtemperatur.
HINWEIS: Seien Sie vorsichtig, wenn Sie den Überstand von losen Zellpellets entfernen. - Das Pellet wird in 1 ml 0,05% Rinderserumalbumin (BSA)-DPBS resuspendiert und in ein 1,7 ml Röhrchen überführt.
HINWEIS: Die Zugabe von BSA kann helfen, die Zellverklumpung zu reduzieren. - Zentrifugieren Sie die Zellen bei 2.000 × g für 15 min bei 4 °C und entfernen Sie den Überstand per Pipette.
HINWEIS: Um den Verlust des Pellets zu vermeiden, entfernen Sie den Überstand schnell und vollständig. - Das Pellet in flüssigem Stickstoff einfrieren und bei -80 °C lagern oder sofort mit dem nächsten Schritt fortfahren.
- Resuspendieren Sie die pelletierten Zellen in 9,9 ml DPBS, bevor Sie 100 μL 300 mM DSG (3 mM endgültig) hinzufügen. Mischen durch Inversion.
2. Erfassung der Chromosomenkonformation
- Zelllyse und Chromatinverdauung
- Resuspendieren (Pipetieren) der vernetzten Zellaliquots (~5 × 106 Zellen) in 1 ml eiskaltem Lysepuffer (Rezeptur in Zusatztabelle S1), der 10 μL Proteaseinhibitorcocktail enthält, und Transfer in einen Dounce-Homogenisator für eine 15-minütige Inkubation auf Eis.
HINWEIS: Fügen Sie dem Lysepuffer unmittelbar vor Gebrauch Proteaseinhibitoren hinzu. - Bewegen Sie Stößel A langsam 30 Mal auf und ab, um die Zellen auf Eis zu homogenisieren, und inkubieren Sie auf Eis für 1 Minute, damit die Zellen abkühlen können, bevor weitere 30 Schläge ausgeführt werden.
- Das Lysat in ein 1,7 ml Mikrozentrifugenröhrchen überführen.
HINWEIS: Halten Sie die Suspension in Bewegung, da manchmal Zellen in der Pipettenspitze stecken bleiben. - Zentrifugieren Sie die lysierte Suspension bei 2.500 × g für 5 min bei Raumtemperatur.
- Verwerfen Sie den Überstand und streichen oder wirbeln Sie das nasse Pellet, um es zu resuspendieren. Entfernen Sie so viel wie möglich vom Überstand, um eine joghurtähnliche Substanz mit minimalen Klumpen zu erhalten.
- Das Pellet in 500 μL eiskaltem 1x Restriktionspuffer (ab 10x; siehe Rezept in Zusatztabelle S1) resuspendieren und 5 min bei 2.500 × g zentrifugieren. Wiederholen Sie diesen Schritt für eine zweite Wäsche.
HINWEIS: Das Pellet im 1x Restriktionspuffer ist körniger als das vorherige Pellet im Lysepuffer. - Resuspendieren Sie die Zellen in einem Endvolumen von 360 μL 1x Restriktionspuffer durch Pipettieren nach Zugabe von ~340 μL zum Verschleppungsvolumen des Pellets, das von der Zellgröße abhängt.
- Legen Sie 18 μL jedes Lysats beiseite, um die Chromatinintegrität (CI) zu testen. Lagern Sie die CI-Proben bei 4 °C.
- Fügen Sie 38 μL 1% Natriumdodecylsulfat (SDS) zu jedem Hi-C-Röhrchen hinzu (Gesamtvolumen von 380 μL) und mischen Sie vorsichtig durch Pipettieren ohne Blasenbildung.
ACHTUNG: SDS ist giftig. - Inkubieren Sie die Proben bei 65 °C ohne Schütteln für genau 10 min, um das Chromatin zu öffnen.
- Stellen Sie die Röhrchen sofort auf Eis und bereiten Sie die Aufschlussmischung mit Triton X-100 vor, um das SDB wie in Tabelle 1 beschrieben zu löschen.
- 107 μL der Aufschlussmischung in das Hi-C-Rohr (487 μL insgesamt) geben, um das Chromatin über Nacht (~16 h) bei 37 °C in einem Thermomixer mit Intervallschütteln (z. B. 900 U/min, 30 s an, 4 min aus) aufzuschlussen.
ANMERKUNG: Die Zugabe von Triton zu einer Endkonzentration von 1% dient dazu, das SDB zu löschen.
- Resuspendieren (Pipetieren) der vernetzten Zellaliquots (~5 × 106 Zellen) in 1 ml eiskaltem Lysepuffer (Rezeptur in Zusatztabelle S1), der 10 μL Proteaseinhibitorcocktail enthält, und Transfer in einen Dounce-Homogenisator für eine 15-minütige Inkubation auf Eis.
- Biotinylierung von DNA-Enden
- Nach dem nächtlichen Aufschluss werden die Proben für 20 min auf 65 °C überführt, um die verbleibende Endonukleaseaktivität zu deaktivieren.
- Bereiten Sie während der Inkubation eine Fill-in-Mastermischung vor, wie in Tabelle 2 dargestellt.
- Nach der Inkubation legen Sie die Proben sofort auf Eis.
- Für jede Probe eine 10 μL Aufschlusskontrolle (DC) beiseite legen und bei 4 °C lagern.
- Entfernen Sie das Kondenswasser mit einer Pipette oder durch Schleudern vom Deckel. Zu jeder Probe werden 58 μL Biotin-Fill-in-Mischung (Gesamtprobenvolumen 535 μL) hinzugefügt und vorsichtig pipettiert, ohne Blasen zu bilden.
- Inkubieren Sie die Proben bei 23 °C für 4 h in einem Thermomixer (z. B. 900 U/min, 30 s an, 4 min aus).
- Ligatur proximaler DNA-Fragmente
- Bereiten Sie die Ligationsmischung wie in Tabelle 3 gezeigt vor, während die Biotinfüllung inkubiert.
- Fügen Sie jeder Probe 665 μL der Ligationsmischung hinzu (Gesamtprobenvolumen 1.200 μL). Durch Pipettieren vorsichtig mischen.
- Die Proben werden bei 16 °C für 4 h in einem Thermomischer mit Intervallschütteln (z. B. 900 U/min, 30 s an, 4 min aus) inkubiert. Lagern Sie diese Proben, die kovalent verknüpftes Chromatin enthalten, einige Tage bei 4 °C.
- Umkehrung der Vernetzung
- Bringen Sie die Volumina der CI- und DC-Proben mit 1x Tris Low EDTA (TLE; siehe Rezept in Zusatztabelle S1) auf 50 μL.
- 10 μL 10 mg/ml Proteinase K zu CI- und DC-Proben hinzufügen.
- Bei 65 °C über Nacht unter Intervallschütteln (z. B. 900 U/min, 30 s an, 4 min aus) inkubieren. Alternativ können Sie eine 30-minütige Umkehrung der Vernetzung für diese Kontrollen während der DNA-Reinigung der Hi-C-Proben durchführen.
- Zu jeder Hi-C-Probe werden 50 μL 10 mg/ml Proteinase K hinzugefügt und bei 65 °C für mindestens 2 h mit Intervallschütteln (z. B. 900 U/min, 30 s an, 4 min aus) inkubiert.
- Zu jedem Hi-C-Röhrchen weitere 50 μL 10 mg/ml Proteinase K geben (Gesamtprobenvolumen 1.300 μL) und über Nacht bei 65 °C weiter inkubieren. Bei 4 °C bis zur DNA-Reinigung lagern.
HINWEIS: Die Aufteilung der Proteinase-K-Inkubationen stellt die gesamte Proteinverdauung sicher.
- DNA-Reinigung
- Lassen Sie die Röhrchen von 65 °C auf Raumtemperatur abkühlen.
- Jede Probe wird in ein 15-ml-konisches Röhrchen überführt und jedem Röhrchen 2,6 ml (2x Volumen) Phenol:Chloroform:Isoamylalkohol hinzugefügt.
ACHTUNG: Phenol:Chloroform:Isoamylalkohol ist ein sehr giftiger Reizstoff und potentiell krebserregend. - Wirbeln Sie jedes Röhrchen für 1 min ab und übertragen Sie dann seinen Inhalt in ein 15-ml-Phasenverschlussrohr.
- Zentrifugieren Sie die Proben für 5 min bei maximaler Geschwindigkeit (1.500-3.500 × g) in einer Tischzentrifuge.
- Gießen Sie die wässrige Phase vorsichtig in ein 35-ml-Ultrazentrifugenröhrchen und fügen Sie Reinstwasser zu einem Endvolumen von 1.250 μL hinzu.
HINWEIS: Verwenden Sie Röhrchen, um die verfügbare Ultrazentrifuge anzupassen, oder teilen Sie sie in mehrere Mikrozentrifugenröhrchen auf. - Fügen Sie ein 1/10-Volumen (~ 125 μL) von 3 M Natriumacetat hinzu und mischen Sie gut durch Inversion.
- Fügen Sie jeder Probe ein 2,5-faches Volumen (~ 3,4 ml) eiskaltes 100% Ethanol hinzu, balancieren Sie die Röhrchen für die Ultrazentrifugation aus, indem Sie eiskaltes 100% Ethanol hinzufügen und gut durch Inversion mischen.
- Inkubieren Sie die Röhrchen auf Trockeneis für ~15 min (Verfestigung vermeiden).
- Zentrifugieren Sie die Röhrchen bei 18.000 × g für 30 min bei 4 °C.
HINWEIS: Für abgewinkelte Rotoren: Markieren Sie die Rohre, wo sich das Pellet befinden wird. - Entfernen und entsorgen Sie den Überstand mit einer Pipette vollständig von der Nichtpelletseite.
HINWEIS: An dieser Stelle sollte das Pellet sichtbar werden und kann auf dem Röhrchen markiert werden, da es nach dem Trocknen im nächsten Schritt möglicherweise nicht mehr deutlich sichtbar ist. - Trocknen Sie die Proben ca. 10 min an der Luft oder bis sie sichtbar trocken sind.
- Jedes Pellet wird in 450 μL 1x TLE durch Pipettieren oder Wirbeln gelöst und in eine 0,5 mL Zentrifugalfiltereinheit (CFU) mit einer Molekulargewichtsgrenze von 3 kDa überführt.
- Zentrifugieren Sie die KBE 10 min lang mit maximaler Geschwindigkeit und verwerfen Sie den Durchfluss. Waschen Sie jedes Ultrazentrifugenröhrchen mit zusätzlichen 450 μL 1x TLE und geben Sie es für eine weitere Wäsche in die CFU.
HINWEIS: Das Waschen der KBE auf diese Weise begrenzt den DNA-Verlust und reduziert gleichzeitig die Salzkonzentration. - Zentrifugieren Sie die KBE 10 min lang mit maximaler Geschwindigkeit und verwerfen Sie den Durchfluss.
- Fügen Sie 80 μL 1x TLE zur Säule hinzu und drehen Sie die Säule in ein neues Sammelröhrchen, bevor Sie 2 Minuten lang mit maximaler Geschwindigkeit zentrifugieren, um ein Endvolumen von ~100 μL zu erhalten.
- Zu jeder Probe werden 1 μL RnaseA (1 mg/ml; 10-fache Verdünnung von 10 mg/ml Brühe) gegeben und bei 37 °C auf einem Hitzeblock, in einem Wasserbad oder einem Thermomixer für mindestens 30 min inkubiert.
- Nach der Rnase-Behandlung die Proben bei 37 °C entnehmen und bei 4 °C bis zur Qualitätskontrolle lagern.
- Überprüfung der Chromatinqualität, des Enzymaufschlusses und der Probenligatur
- Kühlen Sie die CI- und DC-Proben auf Raumtemperatur ab, nachdem Sie die Vernetzungen in Schritt 2.4.5 umgekehrt haben. Dann in ein vorgesponnenes 2-ml-Phasenverschlussrohr überführen.
HINWEIS: Stellen Sie sicher, dass der Inhalt der Phasensperre zu einem Pellet zentrifugiert ist (maximale Geschwindigkeit für 2 Minuten). - Fügen Sie 200 μL Phenol:Chloroform:Isoamylalkohol hinzu und mischen Sie die Proben durch Wirbeln für 1 min.
- Zentrifugieren Sie die Röhrchen für 5 min bei maximaler Geschwindigkeit.
- Die wässrige Phase von jeder Probe (~50 μL) wird in ein neues 1,7 ml Mikrofugenröhrchen überführt.
- 1 μL Rnase A (ab 1 mg/ml) zugeben und bei 37 °C für mindestens 30 min inkubieren.
- Laden Sie die Proben auf ein 0,8%iges Agarosegel, wie in Tabelle 4 empfohlen.
HINWEIS: Die von einer Qualitätskontrolle erwarteten Ergebnisse sind in Abbildung 2 dargestellt. - Quantifizieren Sie die DNA durch Densitometrie aus dem Gel oder mit Qubit oder Nanodrop.
HINWEIS: Eine genaue Quantifizierung stellt das korrekte Eingangsvolumen im nächsten Teil des Protokolls sicher. Verwenden Sie mehrere Standards mit einer bekannten Größe, um eine Standardkurve zu erstellen.
- Kühlen Sie die CI- und DC-Proben auf Raumtemperatur ab, nachdem Sie die Vernetzungen in Schritt 2.4.5 umgekehrt haben. Dann in ein vorgesponnenes 2-ml-Phasenverschlussrohr überführen.
Tabelle 1: Verdauungsreagenzien. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Tabelle 2: Biotin-Fill-in-Reagenzien. *Beachten Sie, dass wechselnde Enzyme unterschiedliche Puffer und biotinylierte dNTPs erfordern können. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Tabelle 3: Reagenzien der Ligationsmischung. Abkürzung: BSA = Rinderserumalbumin. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Tabelle 4: Gelbelastungsparameter für die Qualitäts- und Größenauswahl. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.

Abbildung 2: Agarose-Gel mit typischen Qualitätskontrollergebnissen nach der DNA-Reinigung . (A) Die CI-Kontrolle sollte eine Bande hochmolekularer DNA angeben. (B) Die DC- und Hi-C-Proben zeigen eine Reihe von DNA-Größen. Die Hi-C-Probe, die zu größeren Fragmenten zusammengefasst wurde, sollte ein höheres Molekulargewicht als die DC-Probe haben. Der Konzentrationsbereich der Marker ermöglicht die Erzeugung einer Standardkurve. Beachten Sie, dass in diesem Beispiel das CI auf ein separates Gel geladen wurde, es wird jedoch empfohlen, alle Proben und Kontrollen zusammen zu laden und auszuführen. Abkürzungen: CI = Chromatinintegrität; DC = Verdauungskontrolle; Hi-C = proximity-ligated. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
3. Vorbereitung der Hi-C-Sequenzierungsbibliothek
- Entfernung von Biotin aus unligierten Enden
- Bereiten Sie Biotinentfernungsreaktionen vor, wie in Tabelle 5 gezeigt.
HINWEIS: Typischerweise sind 10 μg DNA ausreichend, aber es können bis zu 30 μg verwendet werden. - 2 x 65 μL Aliquots aus jeder 130 μL Reaktion in zwei PCR-Röhrchen verteilen.
- Transfer zu einem Thermocycler oder PCR-Gerät und Inkubation wie in Tabelle 5 beschrieben.
HINWEIS: Die Proben können hier bei 4 °C (Tage bis Wochen), -20 °C (Langzeit) gelagert oder sofort in die Ultraschalluntersuchung übertragen werden.
- Bereiten Sie Biotinentfernungsreaktionen vor, wie in Tabelle 5 gezeigt.
- Sonorisierung
- Poolen Sie die Probenduplikate aus dem Biotinentfernungsschritt (130 μL Gesamtprobenvolumen) in ein 130 μL Ultraschallröhrchen zur Beschallung.
- Die Proben werden mit den in Tabelle 6 angegebenen Parametern beschallt, um eine enge Verteilung unter 500 bp zu erreichen.
HINWEIS: Es können verschiedene Sonicator-Typen verwendet werden, aber für eine enge Fragmentverteilung (100-500 bp) müssen die Sonicator-Einstellungen möglicherweise optimiert werden.
- Größenauswahl mit Magnetperlen
- Pipeten Sie die beschallte DNA aus dem/den Ultraschallröhrchen(n) in ein 1,7 ml niedrig bindendes Röhrchen.
- Jede Probe mit 1x TLE auf ein Gesamtvolumen von 500 μL bringen. Versuchen Sie, das Volumen so nah wie möglich an 500 μL zu bringen, da das Verhältnis von Proben- zu Magnetperlenmischung für die Größenauswahl entscheidend ist.
- Fügen Sie 400 μL magnetische Kügelmischung in jedes Röhrchen hinzu, um ein Verhältnis der magnetischen Beads-Mischung zum Probenvolumen von 0,8 zu erhalten.
HINWEIS: Unter diesen Bedingungen erfassen Perlen DNA-Fragmente >300 bp, was die obere Fraktion ist. Der Überstand wird Fragmente <300 bp enthalten, was die untere Fraktion sein wird. - Mischen Sie die Röhrchen durch Wirbeln und inkubieren Sie sie für 10 min bei Raumtemperatur auf einem Rotator. Stellen Sie bei diesem und anderen Größenauswahlverhältnissen sicher, dass sich das gesamte Volumen der Probe gut mischt. Suchen Sie nach dem "erratischen Modus", den einige Rotoren haben, der für kleinere Volumina gut funktioniert.
- Inkubieren Sie für 5 min bei Raumtemperatur auf einem Magnetpartikelabscheider (MPS).
- Während der Inkubation 500 μL der magnetischen Perlenmischung in ein frisches 1,7 μL niedrigbindendes Röhrchen für jede Probe geben.
HINWEIS: Diese Röhrchen werden für die nächste Größenauswahl der unteren Fraktion verwendet, indem ein magnetisches Kügelmischungs-Proben-Verhältnis von 1,1: 1 erzeugt wird. - Lassen Sie die Röhrchen 5 min auf dem MPS.
- Entfernen Sie den Überstand von den Perlen und resuspendieren Sie mit 150 μL der magnetischen Perlenmischung.
HINWEIS: Dieser Schritt vermeidet die Sättigung der Perlen mit DNA, indem die Anzahl der Perlen erhöht wird, ohne das Volumen zu erhöhen. - Der Überstand von Schritt 3.3.5 wird in das beschriftete Röhrchen überführt, das zur Auswahl der unteren Fraktion vorbereitet ist (Schritt 3.3.8).
HINWEIS: 150 μL magnetische Perlenmischung + 400 μL 0,8x magnetische Perlenmischung (550 μL insgesamt) geteilt durch 500 μL der ursprünglichen Probe = 1,1x magnetische Beads Mischung zu Probenverhältnis. - Mischen Sie die unteren Fraktionsröhrchen durch Wirbeln und inkubieren Sie für 10 min bei Raumtemperatur auf einem Rotator.
HINWEIS: Die Perlen binden DNA-Fragmente >100 bp, was zu einer endgültigen perlengebundenen Fraktion von 100-300 bp führt. - Legen Sie die unteren Fraktionsröhrchen für 5 min (Raumtemperatur) auf das MPS.
- Entfernen Sie den Überstand und zentrifugieren Sie die Röhrchen kurz, um den Überstand so weit wie möglich weiter zu entfernen.
- Waschen Sie die Perlen beider Fraktionen zweimal mit 200 μL 70% Ethanol und fordern Sie die Perlen jedes Mal für 5 Minuten auf dem MPS zurück.
- Nach einer schnellen Drehung in einer Zentrifuge das Ethanol vollständig entfernen und die Perlen auf dem MPS weiter trocknen.
HINWEIS: Trocknen, bis der Alkohol vollständig verdampft ist. Das Pellet sollte wie dunkle Schokolade aussehen, ohne zu knacken (kann ~ 10 min dauern). - Resuspendieren Sie beide Fraktionen in 50 μL 1x TLE-Puffer. Bei Raumtemperatur 10 Minuten inkubieren und alle zwei Minuten auf die Röhrchen klopfen oder streichen, um das Mischen und Elution anzuregen.
- Trennen Sie die Perlen vom Überstand auf dem MPS für 5 min für beide Fraktionen.
- Bewahren Sie den Überstand jeder Probe auf. Pipeten Sie den Überstand in ein 1,7 ml niedrig bindendes Rohr.
HINWEIS: Die Proben können einige Tage bei 4 °C oder langfristig bei -20 °C aufbewahrt werden. - Lassen Sie ein 2%iges Agarosegel wie in Tabelle 4 durchführen, um die Probenqualität und -quantität zu bestimmen. Siehe Abbildung 3 für ein Beispiel für ein solches Gel.
HINWEIS: Wenn die Ultraschallbehandlung erfahrungsgemäß sehr reproduzierbar ist, kann man dieses Gel überspringen und die Reparatur sofort beenden. Es wird empfohlen, dass Anwender die oberen Fraktionen bis nach der Titrations-PCR beibehalten. Suboptimale DNA-Mengen, die viel PCR-Amplifikation erfordern würden, können aus Material in der oberen Fraktion gerettet werden. - Quantifizieren Sie die DNA-Menge aus dem Gel direkt nach der Erzeugung einer Standardkurve aus dem bekannten DNA-Leitereingang oder mit einem Qubit oder Nanodrop.
- Reparatur beenden
- Bereiten Sie die Endreparaturmischung wie in Tabelle 7 beschrieben vor (Menge pro Reaktion angegeben).
- Die restlichen 46 μL eluierter DNA aus der unteren Fraktion werden in PCR-Röhrchen überführt und 24 μL der vorbereiteten Endreparaturmischung hinzugefügt. Inkubation in einer PCR-Maschine, wie in Tabelle 7 vorgeschlagen.
- Sobald das Programm abgeschlossen ist, halten Sie die Proben bis zum Pulldown bei 4 °C.
- Pull-down von biotinylierten Ligationsprodukten mit Streptavidin-beschichteten Perlen
- Bestimmen Sie die Menge der mit Streptavidin beschichteten Perlen für jede Bibliothek aus der quantifizierten Größenauswahl (Schritt 3.3.19).
HINWEIS: Diese mit Streptavidin beschichteten Beads (10 mg/ml Lösung) können 20 μg doppelsträngige DNA pro mg Beads (= 20 μg / 100 μL Beads) binden. Verwenden Sie 2 μL für jeweils 1 μg Hi-C-DNA, aber nicht weniger als 10 μL. - Mischen Sie die mit Streptavidin beschichteten Perlen und pipeten Sie das Volumen der für jede Bibliothek benötigten Perlen (berechnet im vorherigen Schritt) in einzelne 1,7-ml-Rohre mit niedriger Bindung.
- Die Perlen werden in 400 μL Tween-Waschpuffer (TWB; siehe Rezept in Zusatztabelle S1) resuspendiert und ~3 min bei Raumtemperatur auf einem Rotator inkubiert (siehe Anweisungen in Schritt 3.3.4).
- Trennen Sie die Perlen für 1 min vom Überstand auf dem MPS und entfernen Sie den Überstand.
- Waschen Sie die Perlen, indem Sie weitere 400 μL TWB pipettieren.
- Trennen Sie die Perlen für 1 min vom Überstand auf dem MPS und entfernen Sie den Überstand.
- 400 μL 2x Binding Buffer (BB) (Rezept in Zusatztabelle S1) zu den Perlen geben und resuspendieren. Fügen Sie zusätzlich 330 μL 1x TLE und die Lösung aus End-repair (ab Schritt 3.4.3) hinzu.
- Inkubieren Sie die Proben für 15 min bei Raumtemperatur, während Sie auf einem Rotator mischen.
- Trennen Sie die Perlen für 1 min vom Überstand auf dem MPS und entfernen Sie den Überstand.
- 400 μL 1x BB zu den Perlen geben und resuspendieren.
- Trennen Sie die Perlen für 1 min vom Überstand auf dem MPS und entfernen Sie den Überstand.
- Fügen Sie 100 μL 1x TLE hinzu, um die Perlen zu waschen.
- Trennen Sie die Perlen für 1 min vom Überstand auf dem MPS und entfernen Sie den Überstand.
- Zum Schluss fügen Sie 41 μL 1x TLE hinzu, um die Perlen zu resuspendieren.
- Bestimmen Sie die Menge der mit Streptavidin beschichteten Perlen für jede Bibliothek aus der quantifizierten Größenauswahl (Schritt 3.3.19).
- A-Tailing
- Bereiten Sie die A-Tailing-Mischung wie in Tabelle 8 beschrieben vor.
- Die Reaktionen werden in PCR-Röhrchen pipettiert und inkubiert wie in Tabelle 8.
- Legen Sie die PCR-Röhrchen unmittelbar nach der Entnahme aus dem Thermocycler auf Eis und überführen Sie den Inhalt in 1,7 ml Low-Binding-Röhrchen.
- Trennen Sie die Perlen für 1 min vom Überstand auf dem MPS und verwerfen Sie den Überstand.
- Fügen Sie 400 μL 1x Ligationspuffer hinzu, verdünnt aus 5x T4-DNA-Ligasepuffer mit Reinstwasser.
- Trennen Sie die Perlen für 1 Minute vom Überstand auf dem MPS und verwerfen Sie dann den Überstand.
- Fügen Sie 1x Ligationspuffer zu einem Endvolumen von 40 μL hinzu.
- Glühadapter-Oligos
- Adapter-Oligo-Bestände von 100 μM vorbereiten (Tabelle 9).
HINWEIS: Bestellen Sie 250 nmol HPLC-gereinigte Oligos. - Glühen Sie die Adapter in PCR-Röhrchen wie in Tabelle 9 beschrieben.
- Verwenden Sie einen PCR-Thermocycler, um die Temperatur bei 0,5 °C/s schrittweise auf 97,5 °C zu erhöhen. Bei 97,5 °C 2,5 min halten.
- Verwenden Sie einen PCR-Thermocycler, um die Temperatur für 775 Zyklen schrittweise auf 0,1 ° C / s zu erhöhen (20 ° C zu erreichen). Halten Sie die Temperatur bis zur weiteren Verwendung bei 4 °C.
- Fügen Sie 83 μL 1x Glühpuffer (Rezept in Zusatztabelle S1) hinzu, um die Adapter auf 15 μM zu verdünnen. Lagern Sie die Adapter bei -20 °C.
- Adapter-Oligo-Bestände von 100 μM vorbereiten (Tabelle 9).
- Sequenzierungsadapterligatur
- Bereiten Sie die Adapterligationsmischung in einem 1,7-ml-Schlauch mit niedriger Bindung vor (Tabelle 10).
- Ligate für 2 h bei Raumtemperatur.
- Trennen Sie die Perlen für 1 min vom Überstand auf dem MPS und verwerfen Sie den Überstand.
- Fügen Sie 400 μL TWB hinzu und pipepern Sie die Perlen vorsichtig auf und ab, bevor sie 5 Minuten bei Raumtemperatur auf dem Rotator inkubiert werden. Trennen Sie die Perlen vom Überstand auf dem MPS und wiederholen Sie diesen Schritt noch einmal.
- Trennen Sie die Perlen vom Überstand auf dem MPS (~1 min), verwerfen Sie den Überstand und fügen Sie 200 μL 1x BB hinzu.
- Trennen Sie die Perlen vom Überstand auf dem MPS (~1 min) und verwerfen Sie den Überstand.
- Fügen Sie 200 μL 1x Pre-PCR-Puffer hinzu (ab 10x; Rezept in Zusatztabelle S1) und überführen Sie es in ein neues 1,7 mL Low Binding Röhrchen.
- Trennen Sie die Perlen vom Überstand auf dem MPS (~1 min) und verwerfen Sie den Überstand.
- Fügen Sie 20 μL 1x pre-PCR Buffer hinzu und mischen Sie durch Pipettieren.
- Halten Sie die Röhrchen während des Gebrauchs auf Eis oder lagern Sie sie bei 4 °C.
- Optimierung der PCR-Zykluszahl durch Titration
- Richten Sie 30 μL Master-Mix-Reaktionen pro Probe ein, wie in Tabelle 11 angegeben (Menge pro Reaktion angegeben).
- Führen Sie die niedrigste Anzahl von Zyklen aus und nehmen Sie ein Aliquot von 5 μL. Führen Sie weitere 2-3 Zyklen für die verbleibende Reaktion durch, bevor Sie die nächsten 5 μL aliquot einnehmen. Wiederholen Sie diesen Vorgang, um vier Aliquots zu sammeln.
- Verwenden Sie für jedes Aliquot PCR-Parameter aus Tabelle 11 .
- Zu jeder 5 μL Probe werden 5 μL Wasser und 2 μL 6x Farbstoff hinzugefügt. Laufen Sie auf einem 2% igen Agarose-FSME-Gel (Rezept in Zusatztabelle S1) mit 25-150 ng niedermolekularer Leiter. Die erwarteten Ergebnisse finden Sie in Abbildung 4 .
HINWEIS: Eine optimale Anzahl von Zyklen für die endgültige Bibliotheks-PCR-Amplifikation [Schritt 3.10] ist die niedrigste Anzahl von Zyklen, um sichtbares Produkt auf dem Gel zu erhalten, abzüglich eines Zyklus.
- Finale Bibliothek PCR-Amplifikation
- Richten Sie 12 x 30 μL-Reaktionen ein, um jede endgültige Bibliothek für die Sequenzierung zu amplifizieren, wie in Tabelle 11 beschrieben.
- Zyklus der PCR-Reaktionen gemäß Tabelle 11 nach Bestimmung der Anzahl der Zyklen nach PCR-Titration (Schritt 3.9.3).
- Sobald die PCR abgeschlossen ist, poolen Sie die Replikatproben in einem 1,7 ml Mikrofugenröhrchen mit niedriger Bindung.
- Legen Sie die Röhrchen auf das MPS und überführen Sie den Überstand in ein neues 1,7-ml-Mikrofugenröhrchen mit niedriger Bindung.
- Resuspendieren Sie die übrig gebliebenen Streptavidin-beschichteten Beads in 20 μL 1x pre-PCR Buffer.
HINWEIS: Diese Vorlage kann wiederverwendet werden, wenn sie bei 4 °C für Tage bis Wochen oder langfristig bei -20 °C gelagert wird.
- Entfernen von Primern mit Magnetperlenmischung
- Verwenden Sie 1x TLE-Puffer (Rezept in Zusatztabelle S1), um das Volumen von Schritt 3.10.4 auf genau 360 μL einzustellen.
- Zu jeder Probe 360 μL der magnetischen Perlenmischung hinzufügen und zum Mischen auf und ab pipettieren.
- Auf einem Rotator die Proben 10 min bei Raumtemperatur mischen.
- Trennen Sie die Perlen vom Überstand auf dem MPS bei Raumtemperatur (3-5 min).
- Waschen Sie die Perlen zweimal mit 200 μL 70% Ethanol und gewinnen Sie die Perlen jedes Mal für 5 Minuten auf dem MPS zurück.
- In einer Zentrifuge schnell umdrehen und das Ethanol vollständig pipetten. Trocknen Sie die Perlen an der Luft auf dem MPS, um das Ethanol weiter zu verdampfen.
HINWEIS: Das Pellet sollte wie dunkle Schokolade aussehen, ohne zu knacken (kann ~ 10 Minuten dauern). - Fügen Sie 30 μL Reinstwasser hinzu und resuspendieren Sie, um die DNA für 10 Minuten bei Raumtemperatur zu eluieren. Streichen Sie die Röhrchen alle 2 Minuten, um das Mischen zu erleichtern.
- Trennen Sie die Perlen vom Überstand auf dem MPS für 5 min.
- Sammeln Sie den Überstand von jeder Probe in einem frischen 1,7-ml-Röhrchen.
- Lassen Sie 1 μL der Bibliothek auf ein 2% Agarose-FSME-Gel (Rezept in Zusatztabelle S1) laufen, um eine Fragmentgrößenverteilung zu erhalten und die endgültige Bibliothek zu quantifizieren (Abbildung 5).
HINWEIS: Der ClaI-Aufschluss kann nur für DpnII-DpnII-Übergänge auftreten und dient als positive Ligationskontrolle, die zu einer geringeren Fragmentgrößenverteilung für die endgültige Bibliothek führen sollte. Endbibliotheken können einige Tage bei 4 °C, langfristig bei -20 °C gelagert oder sofort verdünnt und zur Sequenzierung eingereicht werden.
Tabelle 5: Reagenzien und Temperaturen zur Entfernung von Biotin Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Tabelle 6: Parameter für die Beschallung. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Tabelle 7: Endreparaturreagenzien und Temperaturen. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Tabelle 8: A-Tailing-Reagenzien und Temperaturen. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Tabelle 9: PCR-Primer und Paired-End-Adapter-Oligos mit Reagenzienglühen zum Glühen. Abkürzung: 5PHOS = 5' Phosphat. Sternchen weisen auf phosphorothioierte DNA-Basen hin. # Kombinieren Sie einen indizierten Oligo mit dem Universal-Oligo zum Glühen zu einem indizierten Adapter. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Tabelle 10: Adapterligationsreagenzien. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Tabelle 11: PCR-Reagenzien und Zyklusparameter. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.

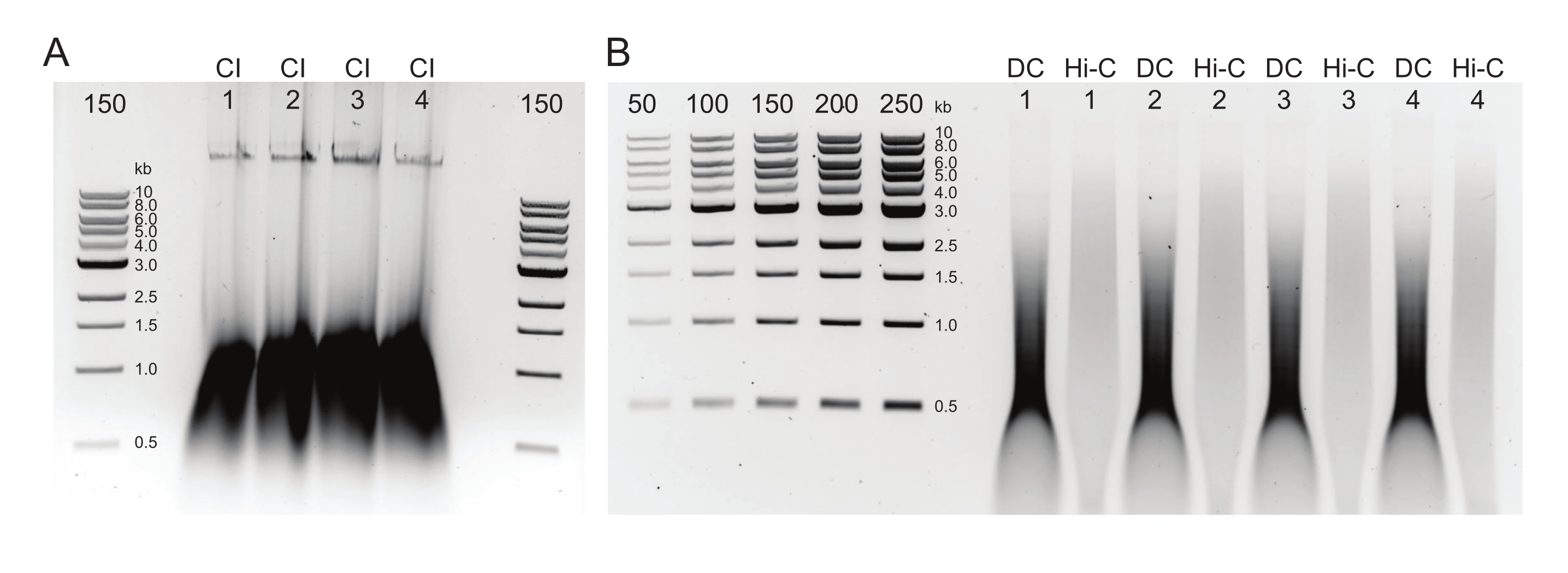
Abbildung 3: Agarose-Gel mit typischen Ergebnissen bei der Auswahl nach der Größe. Die oberen und unteren Fraktionen für vier Proben (nummeriert 1-4) von DpnII-DdeI Hi-C sind dargestellt. Die erste Spur für jede Probe enthält die obere Fraktion, die aus einer 0,8-fachen magnetischen Perlenmischung abgeleitet ist, und die zweite und dritte Spur enthalten eine Verdünnung der unteren Fraktion, die von einer 1,1-fachen magnetischen Perlenmischung abgeleitet ist. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 4: Agarose-Gel mit PCR-Titrationsergebnissen. Ausgehend von 5 PCR-Zyklen werden alle 2 Zyklen (5, 7, 9 und 11 Zyklen) für jede der vier Bibliotheken Proben entnommen. Basierend auf dieser Abbildung wurden 6 Zyklen als optimaler Zyklus für jede Probe ausgewählt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 5: Finale PCR-Produkte. Nach Reinigung und Größenauswahl wurden PCR-Produkte (Hi-C) neben einer ClaI-verdauten Fraktion derselben Bibliothek (ClaI) geladen. ClaI-verdaute Fragmente weisen auf das Vorhandensein begehrter DpnII-DpnII-Ligaturen hin. Beachten Sie, dass ClaI keine DpnII-DdeI-Verbindungen verdaut und daher nicht alle Ligaturen zu einer Größenreduktion dieser Einschränkung beitragen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
Ergebnisse
Die Abbildungen in diesem Manuskript stammen aus einem separaten, replizierten Experiment des zuvor von Lafontaine et al.21 veröffentlichten. Nach der Gewinnung von Hochdurchsatz-Sequenzierungsdaten wurde das Open Chromatin Collective (Open2C: https://github.com/open2c) zur Verarbeitung der Hi-C-Daten verwendet. Eine ähnliche Pipeline findet sich auf dem Datenportal des Projekts 4D Nucleome (https://data.4dnucleome.org/resources/data-analysis/hi_c-processing-pipeline). Kurz gesagt, der Nextflow Pipeline Distiller (https://github.com/open2c/distiller-nf) wurde implementiert, um (1) die Sequenzen von Hi-C-Molekülen auf das Referenzgenom auszurichten, (2) .sam Alignment und Formulardateien mit Hi-C-Paaren zu analysieren, (3) PCR-Duplikate zu filtern und (4) Paare in binned Matrices von Hi-C-Interaktionen zu aggregieren. Diese HDF5-formatierten Matrizen, die als Kühler bezeichnet werden, können dann (1) auf einem HiGlass-Server (https://higlass.io/) angezeigt und (2) mit einem großen Satz von Open-Source-Berechnungswerkzeugen analysiert werden, die in der vom Open Chromatin Collective (https://github.com/open2c/cooltools) verwalteten Sammlung "cooltools" vorhanden sind, um Faltmerkmale wie Fächer, TADs und Schleifen zu extrahieren und zu quantifizieren.
Einige Qualitätsindikatoren der Hi-C3.0-Bibliotheken können unmittelbar nach der Zuordnung von Lesepaaren zu einem Referenzgenom mit wenigen einfachen Metriken/Indikatoren bewertet werden. Erstens können typischerweise ~50% der sequenzierten Lesepaare eindeutig für menschliche Zellen abgebildet werden. Aufgrund der polymeren Natur von Chromosomen stellen die meisten dieser kartierten Lesevorgänge (~60%-90%) Interaktionen innerhalb eines Chromosoms (cis) dar, wobei die Interaktionsfrequenzen mit zunehmender genomischer Entfernung (entfernungsabhängiger Zerfall) schnell abnehmen. Der entfernungsabhängige Zerfall lässt sich am besten in einem "Skalierungsdiagramm" visualisieren, das die Kontaktwahrscheinlichkeit (pro Chromosomenarm) als Funktion der genomischen Distanz zeigt. Wir fanden heraus, dass die Verwendung verschiedener Vernetzer und Enzyme den entfernungsabhängigen Zerfall auf Lang- und Kurzstreckenentfernungen verändern kann17. Die Zugabe von DSG-Vernetzung erhöht die Detektierbarkeit von Wechselwirkungen auf kurze Distanzen, wenn sie mit Enzymen wie Mnase und Kombinationen von DpnII-DdeI kombiniert wird, die kleinere Fragmente erzeugen (Abbildung 6A).
Entfernungsabhängiger Zerfall kann auch direkt aus 2D-Interaktionsmatrizen beobachtet werden: Wechselwirkungen werden seltener, wenn sie weiter von der zentralen Diagonale entfernt sind (Abbildung 6B). Darüber hinaus können genomische Faltungsmerkmale wie Kompartimente, TADs und Schleifen aus Hi-C-Matrizen und Skalierungsdiagrammen als Abweichungen vom allgemeinen genomweiten durchschnittlichen entfernungsabhängigen Zerfall identifiziert werden. Wichtig ist, dass die Vernetzung mit DSG zusätzlich zu FA die zufälligen Ligaturen verringert, die aufgrund der Polymernatur der Chromosomen uneingeschränkt sind und daher eher zwischen Chromosomen (in trans) auftreten (Abbildung 6C). Die Reduzierung der zufälligen Ligatur führt zu erhöhten Signal-Rausch-Verhältnissen, insbesondere bei interchromosomalen und intrachromosomalen Interaktionen mit sehr großer Reichweite (>10-50 Mb).

Abbildung 6: Repräsentative Ergebnisse von kartierten und gefilterten Hi-C-Bibliotheken. (A) Skalierungsdiagramme mit Kontaktwahrscheinlichkeit und deren Ableitung für verschiedene Enzyme, geordnet nach Fragmentlänge (oben) und Vernetzung mit FA oder FA + DSG (unten). Der Aufschluss mit MNAse (microC) oder DpnII-DdeI (Hi-C 3.0) erhöht die Kurzstreckenkontakte (oben) signifikant, ebenso wie die Zugabe von DSG zu FA (unten). (B) Spalten zeigen Hi-C-Heatmaps der DpnII-Vergärung nach FA-Vernetzung und der DpnII- oder DdeI-Verdauung nach FA+DSG-Vernetzung. Weiße Pfeile zeigen eine zunehmende Stärke der "Punkte" nach DSG-Vernetzung und DdeI-Verdauung, was eine bessere Erkennung von DNA-Schleifen impliziert. Die Zeilen zeigen verschiedene Teile von Chromosom 3 mit zunehmender Auflösung, ausgerichtet mit Panel C: obere Reihe: gesamtes Chromosom 3 (0-198,295,559 Mb); mittlere Reihe: 186-196 Mb; untere Reihe: 191.0-191.5 Mb. (C) Erfassungsdiagramme für die in A dargestellten Regionen. Schwarze Pfeile zeigen die geringere Abdeckung (%cis-Lesevorgänge) für die reine FA-Vernetzung. Abkürzungen: FA = Formaldehyd; DSG = Disuccinimidylglutarat; CHR = Chromosom. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
Nicht alle zugeordneten Lesevorgänge sind nützlich. Ein zweiter Qualitätsindikator ist die Anzahl der PCR-Duplikate. Es ist sehr unwahrscheinlich, dass exakte doppelte Lesevorgänge zufällig nach Ligatur und Beschallung auftreten. Daher resultierten solche Messwerte wahrscheinlich aus der PCR-Amplifikation und müssen herausgefiltert werden. Duplikate entstehen oft, wenn zu viele PCR-Zyklen erforderlich sind, um Bibliotheken mit geringer Komplexität zu amplifizieren. Im Allgemeinen benötigen die meisten Bibliotheken für Hi-C nur 5-8 Zyklen der endgültigen PCR-Amplifikation, wie durch Titrations-PCR bestimmt (siehe Schritt 3.9; Abbildung 4). Bibliotheken mit ausreichender Komplexität können jedoch auch nach 14 Zyklen der PCR-Amplifikation erhalten werden.
Eine weitere Kategorie von Duplicate Reads, sogenannte optische Duplikate, kann durch den Verstärkungsprozess auf Illumina-Sequenzierungsplattformen entstehen, die strukturierte Flusszellen verwenden (wie HiSeq4000). Optische Duplikate werden entweder durch Überlastung der Flusszelle gefunden, wodurch (große) Cluster als zwei separate Cluster bezeichnet werden, oder durch lokale Reclusterung des ursprünglichen gepaarten Moleküls nach einer ersten Runde der PCR. Da beide Arten von optischen Duplikaten lokal sind, können sie identifiziert und von PCR-Duplikaten durch ihre Position auf der Flusszelle unterschieden werden. Während Bibliotheken mit >15% PCR-Duplikaten regeneriert werden müssten, können Bibliotheken mit optischen Duplikaten nach Optimierung des Ladevorgangs neu geladen werden.
Zusatztabelle S1: Puffer und Lösungen. Bitte klicken Sie hier, um diese Tabelle herunterzuladen.
Diskussion
Kritische Schritte für das Zellhandling
Obwohl es möglich ist, eine geringere Anzahl von Eingangszellen zu verwenden, wurde dieses Protokoll für ~5 × 106 Zellen pro Sequenzierungsspur (~400 M Lesevorgänge) optimiert, um eine angemessene Komplexität nach der tiefen Sequenzierung zu gewährleisten. Zellen werden am besten vor der Fixierung gezählt. Für die Generierung von Ultradeep-Bibliotheken multiplizieren wir in der Regel die Anzahl der Lanes (und Zellen), bis die gewünschte Lesetiefe erreicht ist. Für eine optimale Fixierung sollte das serumhaltige Medium vor der FA-Fixierung durch PBS ersetzt werden, und Fixierlösungen sollten sofort und ohne Konzentrationsgradienten15,22 hinzugefügt werden. Für die Zellernte wird das Schaben der Trypsinisierung vorgezogen, da der Übergang von einer flacheren zu einer kugelförmigen Form nach der Trypsinisierung die Kernkonformation beeinflussen könnte. Nach der Zugabe von DSG gehen lose und klumpige Zellpellets leicht verloren. Seien Sie vorsichtig beim Umgang mit Zellen in diesem Stadium und fügen Sie bis zu 0,05% BSA hinzu, um die Verklumpung zu verringern.
Änderungen an der Methode
Dieses Protokoll wurde unter Verwendung menschlicher Zellenentwickelt 17. Basierend auf Erfahrungen mit der Erfassung von Chromosomen sollte dieses Protokoll jedoch für die meisten eukaryotischen Zellen funktionieren. Für einen signifikant geringeren Input (~1 × 106 Zellen) empfehlen wir, die Hälfte der Volumina für die Lyse- und Exterieurerfassungsverfahren zu verwenden [Schritte 2.1-2.4]. Dies würde auch die DNA-Isolierung [Schritt 2.5] in einer Tischzentrifuge mit 1,7-ml-Röhrchen ermöglichen, was die Pelletierung bei niedrigen DNA-Konzentrationen verbessern könnte. Die Quantifizierung der DNA (Schritt 2.6.6) zeigt das weitere Vorgehen an. Für geringe Mengen isolierter DNA (1-5 μg) empfehlen wir, die Größenauswahl (Schritt 3.3) zu überspringen und mit der Biotinentfernung fortzufahren, nachdem das Volumen mit einer KBE von 130 μL auf ~45 μL reduziert wurde.
Dieses Protokoll wurde speziell entwickelt, um qualitativ hochwertige Daten nach anschließender Vernetzung mit FA und DSG und Aufschluss mit DpnII und DdeI zu gewährleisten. Alternative Vernetzungsstrategien wie FA gefolgt von EGS (Ethylenglykolbis(succinimidylsuccinat)), das auch in ChIP-seq23 und ChIA-PET24 verwendet wird, könnten jedoch ebenso gut funktionieren17. Ebenso können verschiedene Enzymkombinationen wie DpnII und HinfI18 oder MboI, MseI und NlaIII19 für die Verdauung verwendet werden. Achten Sie bei der Anpassung von Enzymkombinationen darauf, biotinylierte Nukleotide zu verwenden, die die spezifischen 5'-Überhänge ausfüllen können, und verwenden Sie die optimalen Puffer für jeden Cocktail. DpnII wird mit einem eigenen Puffer geliefert und der Enzymhersteller empfiehlt einen speziellen Puffer für die DdeI-Verdauung. Für die Doppelverdauung mit DpnII und DdeI in diesem Protokoll wird jedoch Restriction Buffer empfohlen, da es für beide Enzyme mit 100% Aktivität bewertet wird.
Fehlerbehebung bei der Konformationserfassung
Die drei wichtigsten Schritte bei der Erfassung der Chromosomenkonformation: Crosslinking, Verdauung und Religation wurden alle durchgeführt, bevor die Ergebnisse auf Gel visualisiert werden können. Um die Qualität jedes dieser drei Schritte zu bestimmen und zu erkennen, wo Probleme aufgetreten sein könnten, werden Aliquots vor (CI) und nach dem Aufschluss (DC) entnommen und zusammen mit der ligierten Hi-C-Probe auf das Gel geladen (Abbildung 2). Dieses Gel wird verwendet, um die Qualität der Hi-C-Probe zu bestimmen und ob es sich lohnt, das Protokoll fortzusetzen. Ohne CI und DC ist es schwierig, potenzielle suboptimale Schritte zu lokalisieren. Es ist erwähnenswert, dass eine suboptimale Ligatur auf ein Problem in der Ligatur selbst, das Ausfüllen oder ein Problem mit der Vernetzung zurückzuführen sein kann. Um Probleme bei der Vernetzung zu beheben, achten Sie darauf, nicht mehr als 1 × 107 Zellen pro Bibliothek zu verwenden, und beginnen Sie mit frischen Vernetzungsreagenzien und sauberen Zellen (d. h. mit PBS gespült). Stellen Sie für die Ligatur sicher, dass Zellen und Ligationsmischung auf Eis gehalten werden. T4-DNA-Ligase kurz vor der 4-stündigen Inkubation bei 16 °C hinzufügen und gut mischen.
Problembehandlung bei der Bibliotheksvorbereitung
Wenn mehr als 10 PCR-Zyklen erforderlich sind oder nach der PCR-Titration kein PCR-Produkt auf Gel zu sehen ist (Abbildung 4), gibt es einige Möglichkeiten, die Hi-C-Probe zu speichern. Nach der PCR-Titration besteht die erste Möglichkeit darin, die PCR erneut zu versuchen. Wenn immer noch nicht genügend Produkt vorhanden ist, ist es möglich, nach zweimaligem Waschen der Perlen mit 1x TLE-Puffer eine weitere Runde A-Tailing und Adapterligation (Schritt 3.6) zu versuchen. Nach dieser zusätzlichen A-Tailing und Adapterligatur kann man wie bisher zur PCR-Titration übergehen. Wenn immer noch kein Produkt vorhanden ist, besteht die letzte Option darin, den 0,8x-Anteil aus Schritt 3.3 zu wiederholen und von dort aus fortzufahren.
Einschränkungen und Vorteile von Hi-C3.0
Es ist wichtig zu wissen, dass Hi-C eine populationsbasierte Methode ist, die die durchschnittliche Häufigkeit von Interaktionen zwischen Loci-Paaren in der Zellpopulation erfasst. Einige Computeranalysen sind darauf ausgelegt, Kombinationen von Konformationen aus einer Population25 zu entwirren, aber im Prinzip ist Hi-C blind für Unterschiede zwischen Zellen. Obwohl es möglich ist, Einzelzell-Hi-C26,27 durchzuführen und rechnerische Schlussfolgerungen28 zu ziehen, ist Einzelzell-Hi-C nicht geeignet, ultrahochauflösende 3C-Informationen zu erhalten. Eine weitere Einschränkung von Hi-C besteht darin, dass es nur paarweise Wechselwirkungen erkennt. Um Multikontakt-Interaktionen zu erkennen, kann man entweder häufige Cutter in Kombination mit Short-Read-Sequenzierung (Illumina)16 verwenden oder Multicontact 3C29 oder 4C30 mit Long-Read-Sequenzierung von PacBio- oder Oxford Nanopore-Plattformen durchführen. Hi-C-Derivate zur spezifischen Detektion von Kontakten zwischen und entlang von Schwesterchromatiden wurden ebenfalls entwickelt31,32.
Obwohl Hi-C19 und Micro-C33 verwendet werden können, um Kontaktkarten mit Subkilobasenauflösungen zu erstellen, erfordern beide eine große Menge an Sequenzierungslesevorgängen, was zu einem kostspieligen Unterfangen werden kann. Um ohne Kosten eine ähnliche oder sogar höhere Auflösung zu erreichen, kann eine Anreicherung für bestimmte genomische Regionen (capture-C 34) oder spezifische Proteininteraktionen (ChiA-PET 35, PLAC-seq36, Hi-ChIP37) angewendet werden. Die Stärke und der Nachteil dieser Anreicherungsanwendungen besteht darin, dass nur eine begrenzte Anzahl von Interaktionen untersucht wird. Mit solchen Anreicherungen geht der globale Aspekt von Hi-C (und die Option der globalen Normalisierung) verloren.
Bedeutung und Einsatzmöglichkeiten von Hi-C3.0
Dieses Protokoll wurde entwickelt, um hochauflösendes, ultratiefes 3C zu ermöglichen und gleichzeitig großflächige Faltmerkmale wie TADs und Fächer17 zu erkennen (Abbildung 6). Dieses Protokoll beginnt mit 5 × 106 Zellen pro Röhre für jede Hi-C-Bibliothek, was mehr als genug Material sein sollte, um eine oder zwei Bahnen auf einer Flusszelle zu sequenzieren, um bis zu 1 Milliarde Paired-End-Lesevorgänge zu erhalten. Für die ultratiefe Sequenzierung sollten mehrere Röhrchen mit 5 × 106 Zellen vorbereitet werden, abhängig von der Anzahl der kartierten Reads und PCR-Duplikate. Bei der höchsten Auflösung (<1 kb) werden Looping-Interaktionen meist zwischen CTCF-Stellen gefunden, aber auch Promotor-Enhancer-Interaktionen können nachgewiesen werden. Leser können sich auf Akgol Oksuz et al.17 für eine detaillierte Beschreibung der Datenanalyse beziehen.
Offenlegungen
Die Autoren haben keine Interessenkonflikte offenzulegen.
Danksagungen
Wir danken Denis Lafontaine für die Protokollentwicklung und Sergey Venev für die bioinformatische Unterstützung. Diese Arbeit wurde durch einen Zuschuss des National Institutes of Health Common Fund 4D Nucleome Program an J.D. (U54-DK107980, UM1-HG011536) unterstützt. J.D. ist ein Forscher des Howard Hughes Medical Institute.
Dieser Artikel unterliegt der HHMI-Richtlinie Open Access to Publications. HHMI-Laborleiter haben HHMI in ihren Forschungsartikeln zuvor eine nicht-exklusive CC BY 4.0-Lizenz und HHMI eine unterlizenzierbare Lizenz gewährt. Gemäß diesen Lizenzen kann das vom Autor akzeptierte Manuskript dieses Artikels unmittelbar nach der Veröffentlichung unter einer CC BY 4.0-Lizenz frei verfügbar gemacht werden.
Materialien
| Name | Company | Catalog Number | Comments |
| 1 kb Ladder | New England Biolabs | N3232L | |
| Agarose | Invitrogen | 16500100 | |
| Agencourt AMPure XP magnetic beads , 60 mL | Beckman Coulter | A63881 | |
| Amicon Ultra-0.5 Centrifugal Filter Unit (CFU) | EMD Millipore | UFC500396 | |
| Annealing Buffer (5x) | See recipe in supplemental materials | ||
| ATP 10 mM | ThermoFisher | R0441 | |
| Avanti J-25i High Speed Refrigerated ultra-centrifuge | Beckman Coulter | ||
| beckman ultracentrifuge tube 35 mL | Beckman Coulter | 357002 | |
| Binding Buffer (2x) | See recipe in supplemental materials | ||
| biotin-14-dATP 0.4 mM | Invitrogen | 19524-016 | |
| BSA 10 mg/mL | New England Biolabs | B9000S | dilute from 20 mg/mL |
| Cell scraper | Falcon | 353089 | |
| Cell scraper | Corning | 3008 | |
| Conical polypropylene tubes 50 mL | Denville | C1062-P | |
| Conical tube 15 mL | Denville | C1017-P | |
| Covaris micro tube AFA fiber with snap-cap 130 µL | Covaris | 520045/520077 | |
| Covaris Sonicator | Covaris | E220/E220evolution/M220 | |
| Culture flask 175 cm2 | Falcon | 353112 | |
| Culture plates 150 mm x 25 mm | Corning | 430599 | |
| dATP 1 mM | Invitrogen | 56172 | |
| dATP 10 mM | Invitrogen | 56172 | |
| dCTP 10 mM | Invitrogen | 56173 | |
| DdeI | New England Biolabs | R0175L | |
| dGTP 10 mM | Invitrogen | 56174 | |
| DMSO | Sigma | D2650-5x10ML | |
| dNTP mix 25 mM | Invitrogen | 10297117 | |
| Dounce homogenizer | DWK Life Sciences | 8853010002/8853030002 | |
| DPBS | Gibco | 14190-144 | |
| DpnII | New England Biolabs | R0543M | |
| DSG | ThermoScientific | 20593 | |
| dTTP 10 mM | Invitrogen | 56175 | |
| Ethanol 70% | Fisher | A409-4 | Diluted from 100% |
| Ethidium Bromide | Fisher | BP1302-10 | |
| Formaldehyde (37%) | Fisher | BP531-500 | |
| Gel loading dye (6x ) | New England Biolabs | B7024S | |
| Glycine in ultrapure water 2.5 M | Sigma | G8898-1KG | |
| HBSS | Gibco | 14025-092 | |
| Igepal CA-630 detergent | MP Biomedicals | 198596 | |
| Klenow DNA polymerase 5 U/µL | New England Biolabs | M0210L | |
| Klenow Fragment 3-->5’ exo-, 5 U/µL | New England Biolabs | M0212L | |
| ligation buffer (10x) | New England Biolabs | B7203S | |
| Liquid nitrogen | |||
| LoBind microcentrifuge tube 1.7 mL | Eppendorf | 22431021 | |
| Low Molecular Weight DNA Ladder | New England Biolabs | N3233L | |
| Lysis buffer | See recipe in supplemental materials | ||
| Magnetic Particle separator | ThermoFisher | 12321D | |
| Microfuge tubes 1.7 mL | Axygen | MCT-175-C | |
| MyOne Streptavidin C1 beads | Invitrogen | 65001 | |
| NEBuffer 2.1 (10x) | New England Biolabs | B7002S | |
| NEBuffer 3.1 (10x) | New England Biolabs | B7203S | |
| PBS | Gibco | 70013-032 | |
| PCR (strip) tubes | Biorad | TBS0201/ TCS0803 | |
| PCR thermocycler | Biorad | T100 | |
| Pfu Ultra II Buffer (10x) | Agilent | Comes with Pfu Ultra | |
| PfuUltra II Fusion HS DNA Polymerase | Agilent | 600674 | |
| Phase lock tube 15 mL | Qiagen | 129065 | |
| Phase lock tubes 2 mL | Qiagen | 129056 | |
| Phenol:chloroform:isoamyl alcohol | Invitrogen | 15593-049 | |
| Protease inhibitor cocktail | ThermoFisher | 78440 | |
| Proteinase K in ultrapure water 10 mg/mL | Invitrogen | 25530-031 | |
| Refrigerated Centrifuge | Eppendorf | 5810R | |
| RNase A, DNase and protease-free 10 mg/mL | Thermo Scientific | EN0531 | |
| Rotator | Argos technologies | EW-04397-40 or rocking platform | |
| SDS 1% | Fisher | BP13111 | |
| Sodium acetate pH = 5.2, 3 M | Sigma | ||
| Sub-Cell GT Horizontal Electrophoresis System | Biorad | 1704401 | |
| T4 DNA ligase 1 U/µL | Invitrogen | 100004817 | |
| T4 DNA polymerase | New England Biolabs | M0203L | |
| T4 DNA polymerase | New England Biolabs | M0203L | |
| T4 DNA polymerase 3 U/µL | New England Biolabs | M0203L | |
| T4 ligation buffer (5x) | Invitrogen | Y90001 | |
| T4 polynucleotide kinase 10 U/µL | New England Biolabs | M0201L | |
| Tabletop centrifuge | Eppendorf | 5425 | |
| TBE buffer | See recipe in supplemental materials | ||
| Tris Low EDTA Buffer (TLE) | See recipe in supplemental materials | ||
| Triton X-100 (10%) | Sigma | 93443 | |
| Truseq adapter oligos | Integrated DNA Technologies (IDT)) | https://www.idtdna.com/site/order/oligoentry | 250 nmole and HPLC purified |
| Tween 20 detergent | Fisher | 9005-64-5 | |
| Tween Wash Buffer | See recipe in supplemental materials | ||
| Vortex | Scientific Industries | (G560)SI-0236 |
Referenzen
- Dekker, J., Rippe, K., Dekker, M., Kleckner, N. Capturing chromosome conformation. Science. 295 (5558), 1306-1311 (2002).
- Simonis, M., et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nature Genetics. 38 (11), 1348-1354 (2006).
- Dostie, J., et al. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Research. 16 (10), 1299-1309 (2006).
- Lieberman-Aiden, E., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 326 (5950), 289-293 (2009).
- Stadhouders, R., et al. Multiplexed chromosome conformation capture sequencing for rapid genome-scale high-resolution detection of long-range chromatin interactions. Nature Protocols. 8 (3), 509-524 (2013).
- Kalhor, R., Tjong, H., Jayathilaka, N., Alber, F., Chen, L. Genome architectures revealed by tethered chromosome conformation capture and population-based modeling. Nature Biotechnology. 30 (1), 90-98 (2012).
- Hsieh, T. H., et al. Mapping nucleosome resolution chromosome folding in yeast by micro-C. Cell. 162 (1), 108-119 (2015).
- Hsieh, T. -H. S., Fudenberg, G., Goloborodko, A., Rando, O. J. Micro-C XL: assaying chromosome conformation from the nucleosome to the entire genome. Nature Methods. 13 (12), 1009-1011 (2016).
- Denker, A., de Laat, W. The second decade of 3C technologies: detailed insights into nuclear organization. Genes & Development. 30 (12), 1357-1382 (2016).
- Rodley, C. D., Bertels, F., Jones, B., O’Sullivan, J. M. Global identification of yeast chromosome interactions using Genome conformation capture. Fungal Genetics and Biology. 46 (11), 879-886 (2009).
- Rao, S. S., et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 159 (7), 1665-1680 (2014).
- Alipour, E., Marko, J. F. Self-organization of domain structures by DNA-loop-extruding enzymes. Nucleic Acids Research. 40 (22), 11202-11212 (2012).
- Lajoie, B. R., Dekker, J., Kaplan, N. The Hitchhiker’s guide to Hi-C analysis: Practical guidelines. Methods. 72, 65-75 (2015).
- Belaghzal, H., Dekker, J., Gibcus, J. H. Hi-C 2.0: An optimized Hi-C procedure for high-resolution genome-wide mapping of chromosome conformation. Methods. 123, 56-65 (2017).
- Golloshi, R., Sanders, J. T., McCord, R. P. Iteratively improving Hi-C experiments one step at a time. Methods. 142, 47-58 (2018).
- Darrow, E. M., et al. Deletion of DXZ4 on the human inactive X chromosome alters higher-order genome architecture. Proceedings of the National Academy of Sciences of the United States of America. 113 (31), 4504-4512 (2016).
- Akgol Oksuz, B., et al. Systematic evaluation of chromosome conformation capture assays. Nature Methods. 18 (9), 1046-1055 (2021).
- Ghuryeid, J., et al. Integrating Hi-C links with assembly graphs for chromosome-scale assembly. PLoS Computational Biology. 15 (8), 1007273(2019).
- Gu, H., et al. Fine-mapping of nuclear compartments using ultra-deep Hi-C shows that active promoter and enhancer elements localize in the active A compartment even when adjacent sequences do not. bioRxiv. , (2021).
- Ramani, V., et al. Mapping 3D genome architecture through in situ DNase Hi-C. Nature Protocols. 11 (11), 2104-2121 (2016).
- Lafontaine, D. L., Yang, L., Dekker, J., Gibcus, J. H. Hi-C 3.0: Improved Protocol for Genome-Wide Chromosome Conformation Capture. Current Protocols. 1 (7), 198(2021).
- Belton, J. -M. M., et al. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods. 58 (3), 268-276 (2012).
- Truch, J., Telenius, J., Higgs, D. R., Gibbons, R. J. How to tackle challenging ChIP-Seq, with long-range cross-linking, Using ATRX as an example. Methods in Molecular Biology. 1832, Clifton, N.J. 105-130 (2018).
- Wang, P., et al. In situ chromatin interaction analysis using paired-end tag sequencing. Current Protocols. 1 (8), 174(2021).
- Tjong, H., et al. Population-based 3D genome structure analysis reveals driving forces in spatial genome organization. Proceedings of the National Academy of Sciences of the United States of America. 113 (12), 1663-1672 (2016).
- Nagano, T., et al. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature. 547 (7661), 61-67 (2017).
- Ramani, V., et al. Massively multiplex single-cell Hi-C. Nature Methods. 14 (3), 263-266 (2017).
- Meng, L., Wang, C., Shi, Y., Luo, Q. Si-C is a method for inferring super-resolution intact genome structure from single-cell Hi-C data. Nature Communications. 12 (1), 4369(2021).
- Tavares-Cadete, F., Norouzi, D., Dekker, B., Liu, Y., Dekker, J. Multi-contact 3C reveals that the human genome during interphase is largely not entangled. Nature Structural & Molecular Biology. 27 (12), 1105-1114 (2020).
- Vermeulen, C., et al. Multi-contact 4C: long-molecule sequencing of complex proximity ligation products to uncover local cooperative and competitive chromatin topologies. Nature Protocols. 15 (2), 364-397 (2020).
- Oomen, M. E., Hedger, A. K., Watts, J. K., Dekker, J. Detecting chromatin interactions between and along sister chromatids with SisterC. Nature Methods. 17 (10), 1002-1009 (2020).
- Mitter, M., et al. Conformation of sister chromatids in the replicated human genome. Nature. 586 (7827), 139-144 (2020).
- Krietenstein, N., et al. Ultrastructural Details of Mammalian Chromosome Architecture. Molecular Cell. 78 (3), 554-565 (2020).
- Hughes, J. R., et al. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nature Genetics. 46 (2), 205-212 (2014).
- Fullwood, M. J., et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 462 (7269), 58-64 (2009).
- Fang, R., et al. Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Research. 26 (12), 1345-1348 (2016).
- Mumbach, M. R., et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nature Methods. 13 (11), 919-922 (2016).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten