
Method Article
Perfil de epítopos de proteínas de superficie en partículas virales mediante la estrategia Multiplex Dual-Reporter
En este artículo
Resumen
Aquí, describimos un inmunoensayo fluorescente multiplex recientemente desarrollado que utiliza un sistema citométrico de flujo de doble reportero para detectar simultáneamente dos epítopos únicos de proteína de pico en partículas virales intactas del coronavirus 2 del síndrome respiratorio agudo severo (SARS-CoV-2) que habían sido capturadas por microesferas magnéticas acopladas a la enzima convertidora de angiotensina-2.
Resumen
Las proteínas de membrana de los virus con envoltura desempeñan un papel importante en muchas funciones biológicas relacionadas con la unión del virus a los receptores de las células diana, la fusión de partículas virales a las células huésped, las interacciones huésped-virus y la patogénesis de la enfermedad. Además, las proteínas de membrana viral en las partículas del virus y presentes en las superficies de las células huésped han demostrado ser excelentes objetivos para los antivirales y las vacunas. Aquí, describimos un protocolo para investigar proteínas de superficie en partículas intactas del coronavirus 2 del síndrome respiratorio agudo severo (SARS-CoV-2) utilizando el sistema de citometría de flujo de doble reportero. El ensayo aprovecha la tecnología multiplex para obtener una triple detección de partículas virales mediante tres reacciones de afinidad independientes. Se utilizaron perlas magnéticas conjugadas con la enzima convertidora de angiotensina-2 humana recombinante (ACE2) para capturar partículas virales del sobrenadante de células infectadas con SARS-CoV-2. A continuación, se aplicaron simultáneamente dos reactivos de detección marcados con R-ficoeritrina (PE) o Violeta Brillante 421 (BV421). Como prueba de concepto, se utilizaron fragmentos de anticuerpos dirigidos a diferentes epítopos de la proteína de superficie Spike (S1) del SARS-CoV-2. La detección de partículas virales mediante tres reacciones de afinidad independientes proporciona una fuerte especificidad y confirma la captura de partículas virales intactas. Se generaron curvas de dosis-dependencia del sobrenadante celular infectado con SARS-CoV-2 con varianzas de coeficiente replicado (media/DE) ˂14%. El buen rendimiento de los ensayos en ambos canales confirmó que dos epítopos de proteínas diana de la superficie del virus son detectables en paralelo. El protocolo descrito aquí podría aplicarse para (i) la creación de perfiles de alta multiplexación y alto rendimiento de proteínas de superficie expresadas en virus con envoltura; ii) detección de partículas virales activas intactas; y iii) evaluación de la especificidad y afinidad de los anticuerpos y los fármacos antivirales para los epítopos de superficie de los antígenos virales. La aplicación puede extenderse potencialmente a cualquier tipo de vesículas extracelulares y biopartículas, exponiendo antígenos de superficie en fluidos corporales u otras matrices líquidas.
Introducción
Los virus patógenos más comunes, como la gripe, el VIH, el citomegalovirus humano y las cepas del SARS-CoV, son virus con envoltura. La infección celular por virus con envoltura requiere la fusión de las membranas de la célula viral y del huésped, lo que resulta en la liberación del genoma viral en el citoplasma. A continuación, el ARN viral se replicará antes de ser empaquetado en una nueva partícula viral 1,2. Durante estos procesos, no solo las proteínas virales, sino también las proteínas de la membrana del huésped pueden incorporarse a la envoltura, convirtiéndose en una parte integral de la nueva partícula viral. Las proteínas de la membrana de la célula huésped incorporadas en la envoltura del virus pueden facilitar la entrada del virus en una nueva célula huésped, explotando los mecanismos de las interacciones célula-célula, la localización y el escape del sistema inmunitario 3,4.
A pesar de la importancia de investigar las proteínas asociadas a los virus, la mayoría de las técnicas disponibles actualmente para el análisis de virus5 no admiten la caracterización de alto rendimiento y alta multiplexación del antígeno de superficie del virus. Tampoco son capaces de detectar partículas virales individuales ni de discriminar entre partículas virales intactas infecciosas, ARN no infeccioso, proteínas virales y subpoblaciones de virus que expresan diferentes antígenos. Recientemente, la citometría de flujo ha sido modificada y adaptada a un método novedoso para el análisis de partículas virales, a saber, la virometría de flujo. La virometría de flujo permite la investigación de partículas virales individuales y sus antígenos de superficie. Sin embargo, siguen existiendo limitaciones como el bajo rendimiento, la baja capacidad de multiplexación, la complicada configuración experimental y el análisis de datos, y la limitada detectabilidad de partículas virales de pequeño tamaño 6,7.
La cuantificación multiplexada de proteínas y ácidos nucleicos basada en microesferas es una tecnología bien establecida con numerosas aplicaciones que van desde la cuantificación de proteínas en fluidos corporales, estudios de interacción proteína-proteína y diagnóstico de infecciones virales 8,9,10,11,12,13 . Un instrumento de análisis de flujo recientemente introducido cuenta con un canal reportero dual, lo que permite la medición de dos moléculas reporteras fluorescentes en el mismo pozo de reacción. Esta nueva capacidad ha demostrado ser particularmente útil para el perfil paralelo de diferentes isotipos de inmunoglobulinas14. Aquí, se describe cómo se puede utilizar el sistema de reportero dual para detectar partículas virales intactas, dirigiéndose a múltiples antígenos de superficie en paralelo.
Como prueba de concepto, este informe detalla el desarrollo de un sistema de triple detección de partículas del virus SARS-CoV-2. El SARS-CoV-2 consta de cuatro proteínas principales, una es la proteína de pico (S), que consta de dos subunidades. La primera subunidad, S1, realiza la unión primaria a la ACE2 expresada en las membranas celulares humanas. La segunda subunidad, S2, facilita la entrada en la célula diana por un péptido de fusión, creando un poro en la membrana de la célula diana por el que el virión puede entrar a través de15. Los tres componentes restantes del SARS-CoV-2 son la nucleocápside (N), la proteína de membrana (M) y la proteína de la envoltura (E). La nucleocápside es responsable del empaquetamiento del genoma viral mediante la formación de estructuras de ribonucleoproteínas con ARN, mientras que las proteínas de la membrana y la envoltura desempeñan un papel central en el ensamblaje del virus.
El ensayo descrito aquí se dirige a tres epítopos independientes de la subunidad S1 expresados en la superficie de la envoltura del SARS-CoV-2. Se utilizan diluciones seriadas de sobrenadantes celulares infectados y no infectados por SARS-CoV-2. Las partículas virales se capturan a través de microesferas conjugadas con ACE2 que se unen a la subunidad S1 en el virus. A continuación, la proteína S del virus de superficie se detecta en paralelo con un fragmento variable de cadena única (scFv) de inmunoglobulina marcada comercialmente y un anticuerpo monoclonal humano anti-S1 (Hu-anti-S1) junto con un scFv marcado con FLAG desarrollado internamente. El Hu-anti-S1 es detectado por el primer canal (RP1) en el sistema de doble reportero con el anticuerpo secundario anti-IgG-Fc humano conjugado con R-ficoeritrina (PE) naranja, y el scFv es detectado por el segundo canal (RP2) con un anticuerpo secundario anti-FLAG conjugado con Brilliant Violet 421 (BV421) azul. El ensayo de partículas de virus se representa en la Figura 1.
Protocolo
1. Conjugación de neutravidina y anticuerpos de control contra microesferas magnéticas
NOTA: Las perlas magnéticas teñidas con fluorescencia (microesferas de poliestireno de 6,5 μm de diámetro con magnetita incrustada) con diferentes etiquetas fluorescentes, enumeradas en la Tabla de Materiales , se utilizan para generar los siguientes conjugados y controles de perlas: (1) ACE2 humano recombinante biotinilado unido a perlas acoplado con un enlazador de neutravidina; (2) Biotina unida a perlas acopladas con un enlazador de neutravidina; (3) IgG de cabra acoplada directamente a las cuentas; y (4) Cuentas no conjugadas. La proteína que se va a acoplar a las perlas debe estar libre de azida de sodio, albúmina sérica bovina (BSA), glicina, tris(hidroxi-metil)aminometano (Tris), glicerol o aditivos que contengan aminas. El tampón de activación es de 0,1 M: fosfato de sodio monobásico, anhidro (NaH2, PO4), pH 6. El tampón de pH 5 se utiliza ácido 2-morfolinoetanosulfónico (MES; 50 mM) para diluir conjugados. El tampón de lavado es PBS-T (1x PBS [solución salina tamponada con fosfato], pH 7,4 + 0,05 % (v/v) Tween-20). El tampón de almacenamiento es un reactivo bloqueante de 2,7 mg/ml para ELISA (BRE) + 0,1% de antibióticos (aquí, ProClin 300).
- Retire el polvo de sulfo-N-hidroxisulfosuccinimida (NHS) del refrigerador y 65 mg de clorhidrato de 1-etil-3-[3-dimetilaminopropil]carbodiimida (EDC) prealícuota del congelador y deje que alcance la temperatura ambiente (RT; 18-22 °C) durante 30 min. Almacene tanto el NHS como el EDC en un sobre que contenga perlas de sílice durante este paso para evitar la hidrólisis de la humedad atmosférica.
- Preparar las microesferas para su activación y acoplamiento.
NOTA: Los tintes fluorescentes dentro de las microesferas son sensibles a la luz, y las perlas deben mantenerse en la oscuridad y a temperaturas de refrigerador (4-8 ° C) cuando no estén en uso activo.- Vuelva a suspender 4 existencias diferentes de las microesferas codificadas por color ID (12,5 x 106/mL) (Tabla de materiales) mediante un vórtice, sonicación o rotación breves (15 min a 15-30 rpm), de acuerdo con la hoja de información del producto.
- Transfiera 40 μL de cada suspensión de perlas (5 x 105 microesferas) a los pocillos asignados de una placa de microtitulación de medio pocillo, fondo plano y 96 pocillos (Tabla de materiales).
- Lava las cuentas magnéticas.
NOTA: Los pasos de lavado se pueden realizar manualmente o con una lavadora de placas automática.- Añada 80 μL/pocillo de tampón de activación a las perlas e inmovilice las perlas en un separador de placas magnéticas durante 30 s. Aspire el sobrenadante de las microesferas mientras las perlas se inmovilizan en el separador de placas magnéticas.
- Retire la placa de microtitulación del separador de placas magnéticas y vuelva a suspender las perlas en 50 μL de tampón de activación.
- Activa las perlas con Sulfo-NHS y EDC.
- Prepare la solución de trabajo Sulfo-NHS a 50 mg/mL en tampón de activación en un tubo de microfuga de 1,5 mL. Vuelva a colocar el polvo NHS en el frigorífico (4-8 °C), protegido de la humedad.
- Prepare la solución de trabajo EDC a 50 mg/mL en tampón de activación en su tubo de microfuga de 1,5 mL. Disuelva las alícuotas prefabricadas de 65 mg de polvo de EDC en 1,3 ml de tampón de activación.
NOTA: Sulfo-NHS y EDC comienzan a hidrolizarse y a perder actividad al disolverse. Evite interrumpir el procedimiento de acoplamiento hasta que NHS y EDC se hayan agregado a las cuentas. No guarde las soluciones disueltas del NHS o EDC para su uso posterior. - Prepare la solución de activación para la activación del cordón combinando volumétricamente una solución madre de Sulfo-NHS al 20 % (50 mg/ml), una solución madre de EDC al 20 % (50 mg/ml) y un tampón de activación al 60 %. Se requiere una solución de activación de 50 μL para cada reacción de activación del cordón (utilizando 5 × 105 perlas por reacción), además de un volumen adicional suficiente para acomodar las pérdidas de pipeteo.
- Añada 50 μL de Solución de Activación completa a cada pocillo que contenga perlas lavadas. Con el volumen de suspensión de perlas preexistente de 50 μL en el tampón de activación, por pocillo, la concentración final de Sulfo-NHS será de 5 mg/mL, y la concentración final de EDC también será de 5 mg/mL.
- Selle la placa de reacción de microesferas con un sellador de placas de plástico adhesivo desechable o de aluminio e incube durante 20 minutos en un agitador orbital (650 rpm) a temperatura ambiente (18-22 °C) en la oscuridad.
- Lave el exceso de Solución de Activación de las perlas.
- Centrifugar la placa de microtitulación a 233 × g durante 1 min.
- Inmovilizar las perlas activadas en un separador de placas magnéticas durante 30 s. Retire el sellador de placas y aspire el sobrenadante de las perlas inmovilizadas por imanes con la placa de microtitulación aún colocada en el separador magnético.
- Retire la placa de microtitulación del separador magnético y agregue 100 μL de tampón MES a cada pocillo.
- Repita los pasos 1.4.2-1.4.3 una vez más para un total de dos lavados.
- Acople la neutravidina y la IgG de cabra (control) para crear conjuntos de cuentas adecuados. Prepare suficientes soluciones de trabajo de neutravidina e IgG en cabras, planificando 100 μL/reacción y suficientes soluciones adicionales para acomodar las pérdidas de pipeteo de la siguiente manera:
NOTA: La proteína de neutravidina en polvo se reconstituye con agua ultrapura y luego se diluye a una solución madre de 1 mg / mL con PBS antes de alicuota para almacenamiento / uso (la proteína de neutravidina no es directamente soluble en PBS, pero es soluble en agua a ~ 10 mg / mL).- Prepare la solución de trabajo de neutravidina a una concentración de 125 μg/mL en tampón MES en un tubo de microfuga de baja unión a proteínas de 1,5 mL.
- Prepare la solución de trabajo de anticuerpos de control IgG de cabra a una concentración de 17,5 μg/mL en tampón MES.
- Prepare la placa de microtitulación que contiene las perlas activadas. Inmovilice las perlas en un separador de placas magnéticas durante 30 s. Con la placa de microtitulación todavía colocada en el separador magnético, aspire el sobrenadante de las perlas inmovilizadas por el imán.
- Añadir 100 μL de solución de trabajo de neutravidina (125 μg/mL) a los pocillos apropiados que contengan perlas (para el acoplamiento neutravidina-biotina y neutravidina-ACE2).
- Agregue 100 μL de solución de trabajo de IgG para cabras (17,5 μg/mL) al pocillo que contiene perlas asignadas como controles de IgG solo para cabras.
- Agregue 100 μL de tampón MES al pozo asignado como control de perlas no conjugadas.
- Sellar la placa de microtitulación e incubar durante 2 h en un agitador orbital (650 rpm) a RT (18-22 °C) en la oscuridad. Agite la placa brevemente después de 1 h de incubación para asegurarse de que las perlas permanezcan en suspensión.
- Lave las perlas con PBS-T.
- Centrifugar la placa de microtitulación a 233 × g durante 1 min.
- Inmovilice las perlas acopladas en un separador de placas magnéticas durante 30 s. Con la placa de microtitulación todavía colocada en el separador magnético, aspire el sobrenadante de las perlas inmovilizadas por el imán.
- Retire la placa de microtitulación del separador magnético.
- Agregue 100 μL de PBS-T a cada pocillo que contenga perlas.
- Repita los pasos de lavado 1.6.2-1.6.4 una vez para un total de dos lavados con PBS-T.
- Prepare las cuentas conjugadas para el almacenamiento.
- Inmovilice las perlas acopladas en un separador de placas magnéticas durante 30 s. Con la placa de microtitulación todavía colocada en el separador magnético, aspire el sobrenadante de las perlas inmovilizadas por el imán. Retire la placa de microtitulación del separador magnético.
- Agregue 50 μL de tampón de almacenamiento a cada ID de microesfera para apagar la actividad restante del cordón.
- Incubar la placa de microtitulación a temperatura frigorífica (4-8 °C) en la oscuridad durante la noche (16-22 h).
- Transfiera suspensiones de perlas no conjugadas y conjugadas con IgG de cabra (50 μL) a tubos de microfuga de baja unión a proteínas de 1,5 mL debidamente marcados, combinados con dos enjuagues de tampón de almacenamiento de 100 μL de los pocillos para garantizar la máxima recuperación de las perlas.
NOTA: Las perlas no conjugadas y las conjugadas con IgG de cabra numerarán 5 × 105 en un volumen final de 250 μL (es decir, 2 × 103 perlas/μL). Guarde los tubos de microfuga a temperaturas de refrigerador (4-8 °C) hasta su uso.
- Se une a la ACE2 biotinilada y a la biotina a las perlas conjugadas con neutravidina.
- Prepare la solución de trabajo ACE2 biotinilada humana recombinante a 18 μg/mL de ACE2 en 10 mM de PBS. Por reacción, se necesitarán 100 μL. Prepare la solución de trabajo de biotina a 2,4 mg/mL de biotina en 10 mM de PBS. Por reacción, se necesitarán 100 μL.
- Prepare la placa de microtitulación que contiene microesferas conjugadas con neutravidina.
- Inmovilice las microesferas en un separador de placas magnéticas durante 30 s. Con la placa de microtitulación aún colocada en el separador magnético, retire el sellador de placas y aspire el sobrenadante de las microesferas inmovilizadas por imanes.
- Retire la placa de microtitulación del separador magnético y agregue 50 μL de PBS de 10 mM / pocillo.
- Repita los pasos 1.8.2.1-1.8.2.2 una vez.
- Añadir 100 μL de la solución de trabajo biotinilada-ACE2 a los pocillos apropiados que contengan microesferas conjugadas con neutravidina. Añadir 100 μL de la solución de trabajo de biotina a los pocillos apropiados que contengan microesferas conjugadas con neutravidina.
- Selle la placa de microtitulación e incube durante 1 h en un agitador orbital (650 rpm) a RT (18-22 °C) en la oscuridad.
- Lave las microesferas como se describe en los pasos 1.6.1-1.6.5.
- Prepare y almacene las microesferas conjugadas con ACE2 y biotina como se describe en los pasos 1.7.1-1.7.4.
NOTA: Las perlas biotiniladas con ACE2 y conjugadas con biotina numerarán 5 × 105 en un volumen final de 250 μL (es decir, 2 × 103 perlas/μL).
2. Prueba de conjugación
- Prepare una mezcla de perlas combinando los cuatro tipos de microesferas creadas en la sección 1 (es decir, ACE2 biotinilada conjugada con neutravidina, biotina conjugada con neutravidina, conjugada con IgG de cabra y no conjugada).
NOTA: Las microesferas de stock se almacenaron a 2 × 103 perlas/μL y se combinaron de tal manera que la concentración final de perlas en la mezcla de perlas de trabajo es de 40 perlas de cada conjunto/μL.- Calcule el volumen de la mezcla de perlas de trabajo necesaria para la prueba (5 μL/reacción), permitiendo que el volumen adicional se adapte a las pérdidas de pipeteo. Agite brevemente cada tubo y combine volúmenes calculados iguales de cada suspensión de perlas en un nuevo tubo de microfuga de baja unión a proteínas. La concentración de perlas es ahora de 400 perlas de cada conjunto/μL.
- Cree la mezcla de cordones de trabajo diluyendo la suspensión de cordones combinada 10 veces más con el tampón de almacenamiento (40 de cada set/μL de concentración de trabajo).
NOTA: Primero haga una pequeña cantidad de mezcla de cuentas de trabajo para estimar el número de microesferas/μL para cada ID.
- Incubar las microesferas con el anticuerpo anti-ACE2 de cabra.
- Pipetee 5 μL de mezcla de perlas de trabajo en 3 pocillos de una placa de microtitulación de 96 pocillos de fondo plano.
- Añadir 50 μL de anti-ACE2 de cabra (0,4 μg/mL diluido en PBS-T, Tabla de Materiales) a cada uno de los 3 pocillos que contienen perlas en la placa de microtitulación. Selle la placa de microtitulación, el vórtice e incube en un agitador orbital (650 rpm) a RT (18-22 °C) durante 1 h en la oscuridad.
- Pulsar la placa de microtitulación a 233 × g durante 1 min y lavar las microesferas tres veces con PBS-T como se describe en 1.6.2-1.6.4.
- Incubar microesferas con anticuerpos de detección.
- Pipetear 5 μL de mezcla de perlas de trabajo en 6 pocillos nuevos de la placa de microtitulación.
- Prepare 1 μg/mL de cada una de las mezclas de detección de trabajo: IgG PE anti-cabra, IgG PE anti-ratón e IgG PE anti-conejo en 3 tubos separados de 1,5 mL, utilizando PBS-T como diluyente.
- Agregue 50 μL de las mezclas de detección a 3 pocillos cada uno, y la IgG anti-cabra se agrega a los mismos pocillos que el anti-ACE2 del paso 2.2.
- Sellar, vórtice e incubar en un agitador orbital (650 rpm) a RT (18-22 °C) durante 30 min.
- Pulse la placa a 233 × g durante 1 min y lave las microesferas tres veces con PBS-T como se describe en 1.6.2-1.6.4.
- Agregue 100 μL de PBS-T y ejecute en el instrumento de análisis de flujo de doble reportero con los siguientes ajustes:
Modo: Reportero dual; Tiempo muerto: 45 s; DD-gating: 7500-17500; Recuento mínimo de microesferas: 100 microesferas/conjunto (corte de control de calidad más bajo: 35 microesferas/conjunto).
3. Producción de sobrenadante de células infectadas por SARS-CoV-2
El virus SARS-CoV-2 se propaga en las células Vero E6 del huésped (línea celular epitelial de riñón de mono; ATCC; Tabla de Materiales). Las células Vero E6 se cultivan en medio Modified Eagles (MEM) a 37 °C en una atmósfera con 5% de CO2 y 95% de humedad relativa. Cada litro de MEM se complementa con 10 mL de L-glutamina (200 mM), 38 mL de NaHCO3 (7,5%), 5 mL de solución de penicilina/estreptomicina y 50 mL de suero fetal bovino (FCS); Tabla de Materiales.
PRECAUCIÓN: Utilice los procedimientos y equipos de bioseguridad adecuados cuando manipule el SARS-CoV-2.
- Las células Vero E6 se cultivan hasta la confluencia en dos matraces de cultivo de tejidos de150 cm2 . Infectar un matraz con el virus SARS-CoV-2 y usar el otro infectado falso como control.
- Mezcle aproximadamente 100,000 partículas infecciosas de SARS-CoV-2 de tipo salvaje (WT) con 5 mL de medio Eagles MEM.
- Aspire el medio de un matraz de150 cm2 y agregue 55 mL de MEM completo para generar sobrenadantes de control no infectados. Aspire el medio del otro matraz de150 cm2 y añada la mezcla de virus a las células. Incubar las células durante 1 h a 37 °C. Agite suavemente el matraz cada 15 minutos para distribuir el virus.
- Añadir 50 mL de medio MEM completo al matraz con SARS-CoV-2 añadido e incubar las células hasta observar efectos citopáticos, evaluando visualmente los matraces cada 24 h.
NOTA: Deben pasar aproximadamente 3-4 días después de la infección para que ocurra la citopatía. Los efectos citopáticos sobre la estructura monocapa de la célula Vero E6 (p. ej., retracción celular, crenación, redondeo, desadherencia, pérdida de granularidad intracitoplasmática, lisis manifiesta) se evalúan cualitativamente mediante la observación de las células utilizando un microscopio óptico invertido, según las directrices de la Organización Internacional de Normalización para las pruebas de citotoxicidad in vitro 16. - Recoja el sobrenadante celular de ambos matraces y centricé durante 6 min a 253 × g para sedimentar los restos celulares.
- Virus SARS-CoV-2 inactivado por los rayos UV en sobrenadante celular
- Pipetear 0,5 mL de sobrenadante por pocillo en 12 pocillos en una placa de microtitulación de 24 pocillos. Irradiar UV la placa de microtitulación, sin tapa, durante 30 s bajo una lámpara ultravioleta adecuada (Tabla de Materiales).
NOTA: La inactivación viral en el sobrenadante celular debe determinarse intentando la propagación del virus en cultivos celulares Vero E6.
- Pipetear 0,5 mL de sobrenadante por pocillo en 12 pocillos en una placa de microtitulación de 24 pocillos. Irradiar UV la placa de microtitulación, sin tapa, durante 30 s bajo una lámpara ultravioleta adecuada (Tabla de Materiales).
- Alicucuota el sobrenadante celular en tubos de 1,5 mL y almacene a -20 °C hasta su uso posterior.
NOTA: El sobrenadante celular se puede almacenar a -80 °C.
4. Ensayo: Detección de partículas virales del SARS-CoV-2 en sobrenadante celular
- Prepare el tampón de ensayos mezclando 0,1 % de caseína, 0,5 % de alcohol polivinílico, 0,8 % de polivinilpirrolidona y 1 % de BSA (todo p/v) (pH 7). Prepare el tampón de dilución de la muestra preparando un 10% de IgG de conejo en el tampón de ensayo.
- Calcule y prepare el volumen de la mezcla de perlas de trabajo (paso 2.1) necesario para la prueba (5 μL/reacción), permitiendo que el exceso de volumen se adapte a las pérdidas de pipeteo.
- Prepare la serie de diluciones del sobrenadante. Calcular los volúmenes de sobrenadantes necesarios. Ensaye cada punto de dilución en pocillos triplicados para cada uno de los cinco scFvs, lo que da como resultado 15 pocillos por punto de dilución y tipo de muestra. Utilice 45 μL de sobrenadantes diluidos en cada pocillo, para un total de 675 μL requeridos; un solo tubo de microfuga de 1,5 mL para cada uno de los sobrenadantes del SARS-CoV-2 y de control es suficiente.
- Descongele los sobrenadantes del SARS-CoV-2 y controle los sobrenadantes a 4 °C durante al menos 1 h. Mantener los sobrenadantes y sus diluciones en frío (2-8 °C) hasta su uso. Etiquete y coloque ocho tubos de microfuga de 1,5 ml cada uno para el SARS-CoV-2 y los sobrenadantes de control.
NOTA: La concentración más alta ensayada será una dilución de sobrenadante 1:1 (2 veces), a partir de la cual se realizarán una serie de diluciones 1:2 (3 veces) utilizando tampón de dilución de muestra, siendo la dilución más alta una dilución 1:1458. Los blancos que contienen tampón de dilución de muestra solo sirven como controles sin sobrenadante. Por lo tanto, las diluciones probadas de cada tipo de sobrenadante (SARS-CoV-2 o control) serán de 2, 6, 18, 54, 162, 486 y 1458 veces, con un control solo tampón. - Agregue tampón de dilución de muestra a los tubos de microfuga etiquetados. Los tubos de dilución 1:1 (2 veces) requieren 600 μL de tampón y los tubos restantes requieren 800 μL de tampón. Cree la dilución más alta (1:1; 2 veces) de cada sobrenadante combinando 600 μL del sobrenadante con 600 μL de tampón de dilución de muestra en tubos debidamente etiquetados, seguido de un breve vórtice del tubo para mezclar.
- Continúe la serie transfiriendo secuencialmente 400 μL de los sobrenadantes diluidos 1:1 (2 veces) al siguiente tubo de dilución (es decir, dilución 6 veces) y continúe con diluciones 3 veces hasta que se haya creado la dilución más baja (1458 veces). Agite brevemente cada sobrenadante diluido antes de proceder con la siguiente dilución.
- Descongele los sobrenadantes del SARS-CoV-2 y controle los sobrenadantes a 4 °C durante al menos 1 h. Mantener los sobrenadantes y sus diluciones en frío (2-8 °C) hasta su uso. Etiquete y coloque ocho tubos de microfuga de 1,5 ml cada uno para el SARS-CoV-2 y los sobrenadantes de control.
- Incubar las microesferas con el sobrenadante.
- Agite la mezcla de perlas de trabajo preparada previamente durante 30 s para resuspender las microesferas y agregue 5 μL de la mezcla de perlas a cada pocillo asignado de una placa de microtitulación de fondo plano de 384 pocillos.
NOTA: También se pueden utilizar placas de 96 pocillos. - Agregue 45 μL de las diluciones de sobrenadante preparadas a los pocillos asignados que contengan microesferas en la placa de 384 pocillos. Sellar la placa e incubar durante la noche (16-22 h) en un agitador orbital (650 rpm) a RT (18-22°C) en la oscuridad.
- Agite la mezcla de perlas de trabajo preparada previamente durante 30 s para resuspender las microesferas y agregue 5 μL de la mezcla de perlas a cada pocillo asignado de una placa de microtitulación de fondo plano de 384 pocillos.
- Lave el exceso de sobrenadante de las perlas.
- Retire la placa de microtitulación del agitador orbital y retire el sellador de placas. Centrifugar la placa a 931 x g durante 1 min.
- Inmovilice las perlas colocando la placa de microtitulación en un separador de placas magnéticas durante 30 s. Con la placa de microtitulación aún colocada en el separador magnético, retire el sellador de placas y aspire el sobrenadante de las perlas inmovilizadas por el imán.
- Retire la placa de microtitulación del separador magnético.
- Agregue 60 μL de PBS-T a cada pocillo que contenga perlas.
- Repita los pasos de lavado 4.5.2-4.5.4 dos veces para un total de tres lavados PBS-T.
- Prepare las diferentes mezclas de detección en tubos separados de 1,5 mL. Cada mezcla de detección consta de un anticuerpo monoclonal humano comercial anti-S1 (Hu-anti-S1) (1 μg/mL) y uno de los cinco scFvs (1 μg/mL) marcados con FLAG (Archivo complementario 1, Tabla complementaria 1 y Tabla complementaria 2) dirigidos a la proteína de la espícula en la partícula del SARS-CoV-2, diluida en tampón de ensayo (paso 4.1), lo que da como resultado un total de cinco mezclas de detección diferentes.
- Repita los pasos 4.5.2-4.5.3. Vuelva a suspender las microesferas lavadas en 50 μL/pocillo de la mezcla de detección de picos específica de scFv adecuada. Selle la placa de microtitulación e incube durante 1 h en un agitador orbital (650 rpm) a RT (18-22 °C) en la oscuridad.
- Centrifugar la placa de microtitulación a 931 × g durante 1 min. Lave el exceso de reactivo de detección de picos de las perlas. Realice tres pasos de lavado con 60 μL de PBS-T de acuerdo con los pasos 4.5.2-4.5.5.
- Incubar microesferas con una mezcla de anticuerpos de detección fluorescente.
- Prepare una mezcla de solución fluorescente que consista en IgG antihumana conjugada con PE disponible en el mercado junto con un anticuerpo anti-FLAG conjugado con BV421 diluido en el tampón de ensayo, con concentraciones de trabajo de 0,2 μg/mL y 1 μg/mL, respectivamente. Por reacción, se necesitan 50 μL de reactivo de detección fluorescente.
- Repita los pasos 4.5.2-4.5.3. Vuelva a suspender las microesferas en 50 μL/pocillo de mezcla de solución fluorescente. Selle la placa de microtitulación e incube durante 30 minutos en un agitador orbital (650 rpm) a RT (18-22 °C) en la oscuridad.
- Gire la placa de microtitulación a 931 × g durante 1 min. Lave el exceso de mezcla de solución fluorescente de las microesferas. Realice tres pasos de lavado con 60 μL de PBS-T de acuerdo con los pasos 4.5.2-4.5.5.
- Suspender las microesferas en 60 μL de PBS-T desde el último paso de lavado. Analice la placa en un sistema de análisis de flujo de doble reportero con la configuración descrita en el paso 2.6.
Resultados
Prueba de conjugación
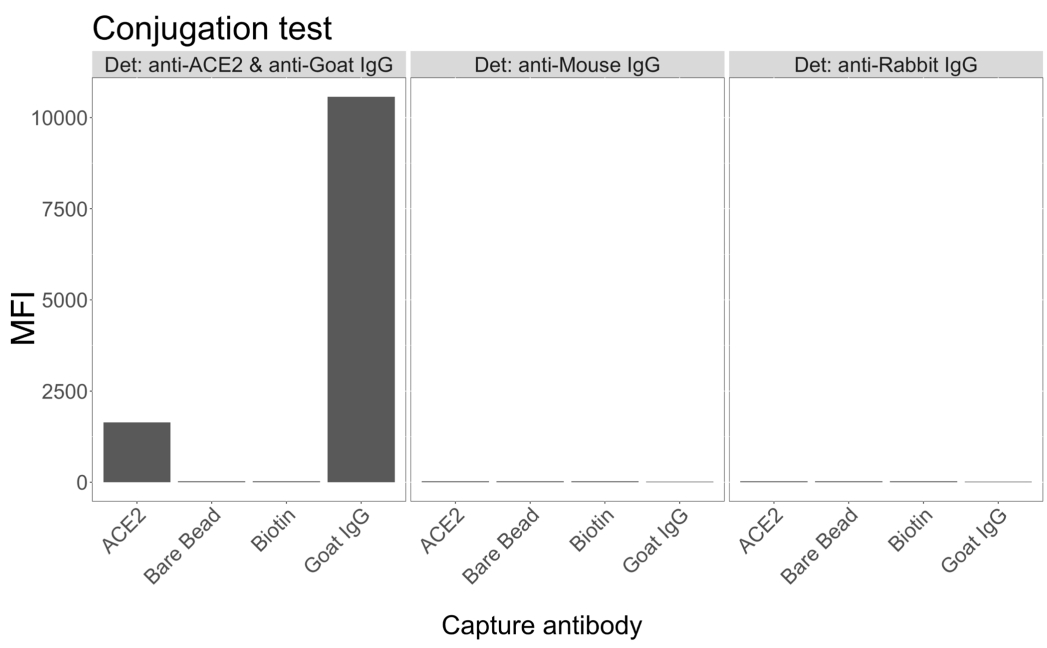
La prueba de conjugación mostró que la IgG de cabra y la ACE2 biotinilada con neutravidina se conjugaron con éxito con las microesferas. La especificidad de la detección del ensayo se confirmó mediante el sondeo de microesferas conjugadas con ACE2 con anticuerpos secundarios marcados con PE generados en diferentes especies animales (Figura 2). No se observó reactividad cruzada entre los diferentes anticuerpos de detección. Cuando las mezclas de perlas se sondearon con IgG PE anti-ACE2 + anti-cabra, se detectó un valor medio de intensidad de fluorescencia (MFI; unidades arbitrarias) por encima del fondo para las microesferas conjugadas con ACE2 e IgG de cabra, pero no para la microesfera no conjugada (desnuda) o para las microesferas recubiertas de biotina. Se utilizaron IgG PE anti-ratón y IgG PE anti-conejo como controles negativos para comprobar si había señales falsas positivas. Se generó una señal de fluorescencia insignificante durante la incubación con las microesferas, lo que indica que las señales positivas para la ACE2 y la IgG de cabra eran específicas.
Detectabilidad de partículas virales en sobrenadantes celulares
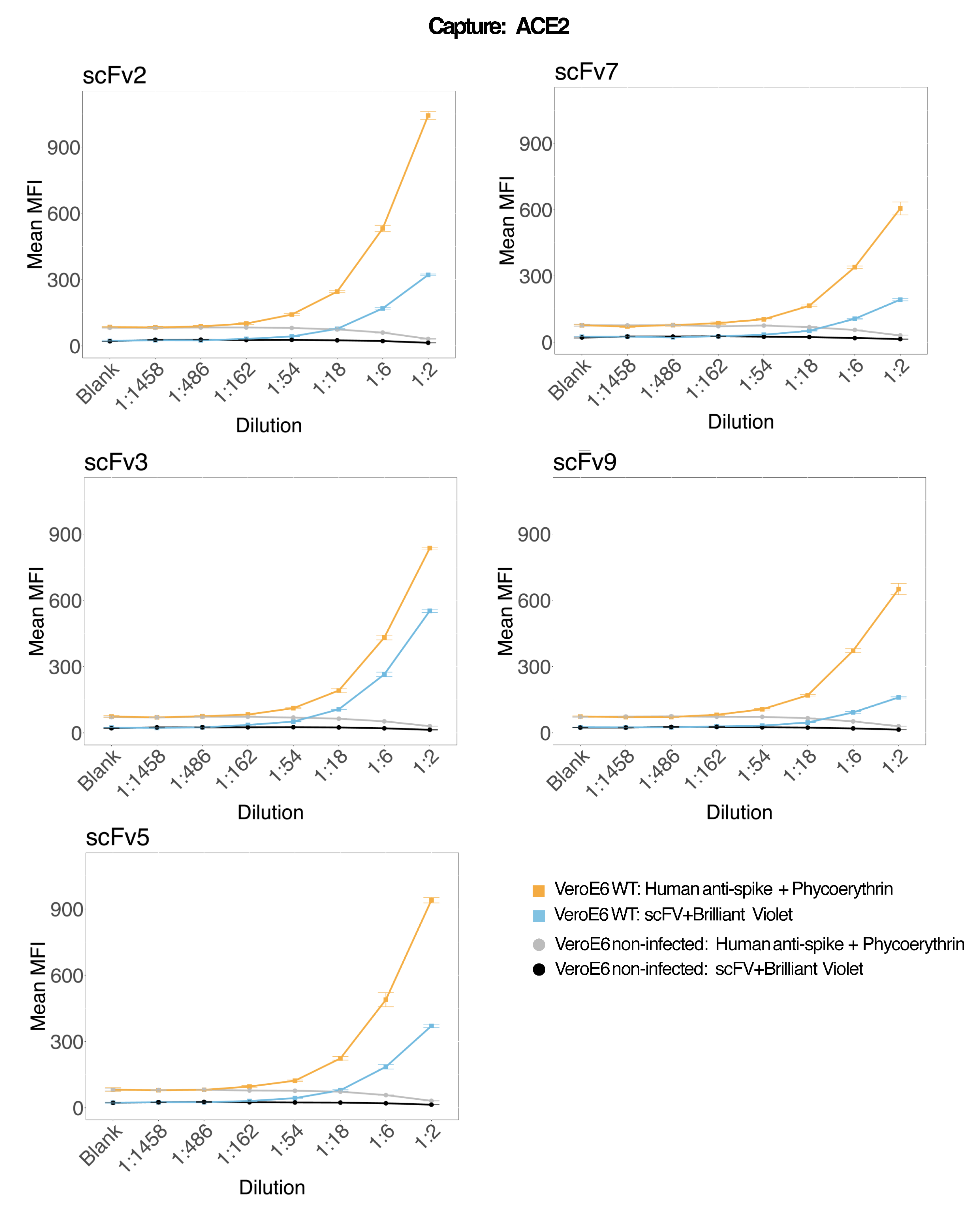
Se utilizaron perlas magnéticas acopladas a ACE2 humano recombinante para capturar partículas virales del SARS-CoV-2 de sobrenadantes de cultivo celular VeroE6 infectados y de control (sin virus) y luego se sondearon simultáneamente para dos regiones de pico viral distintas utilizando un anticuerpo monoclonal y uno de los cinco scFvs distintos. Se observó una señal dependiente de la concentración en las diluciones de sobrenadantes celulares infectados por SARS-Cov-2 en ambos canales reporteros (RP1 y RP2) (Figura 3), lo que indica que tanto el anticuerpo comercial Hu-anti-S1 como los diferentes scFvs detectaron la partícula viral unida a la microesfera conjugada con ACE2. Con tres de cada cinco scFvs, el virus es detectable en diluciones de hasta 1:18 (scFv2, scFv3, scFv5); para los dos scFvs restantes (scFv7 y scFv9), es detectable hasta diluciones 1:6. Esto podría atribuirse a una afinidad diferente con el objetivo. Como se muestra en la Figura 3 y la Tabla 1, scFv3 proporciona la intensidad de MFI más alta, seguido de scFv5, scFv2, scFv7 y scFv9, respectivamente.
A nivel mundial, la detección de scFvs da como resultado un MFI más bajo en comparación con el Hu-anti-S1. Esto podría indicar una menor afinidad, pero también podría ser un artefacto debido al marcaje con diferentes tintes fluorescentes (PE y BV421). Otra tendencia que se puede ver para scFv7 y scFv9 es que los valores de MFI son ligeramente más bajos para el canal RP1 (anti-pico) en comparación con las otras tres configuraciones. Esto podría indicar que los scFvs están reaccionando de forma cruzada o interfiriendo de otra manera con la interacción ACE2-Hu-anti-S1, lo que también podría explicar la menor señal en el canal RP2. No se detectaron partículas virales en el sobrenadante de las células Vero E6 no infectadas en el canal RP1 ni en el canal RP2.
La microesfera conjugada con neutravidina-biotina, la microesfera de cabra-IgG y las microesferas no conjugadas se utilizan como perlas de control negativo. Las partículas virales se capturaron con microesferas magnéticas acopladas a ACE2 y se probaron con anti-spike humano comercial en el canal reportero RP1 y con diferentes scFvs en el canal reportero RP2 (scFv se indica en la parte superior izquierda de cada panel). No se detectaron partículas de virus en ninguna de las muestras infectadas y no infectadas.
Precisión y robustez de los ensayos
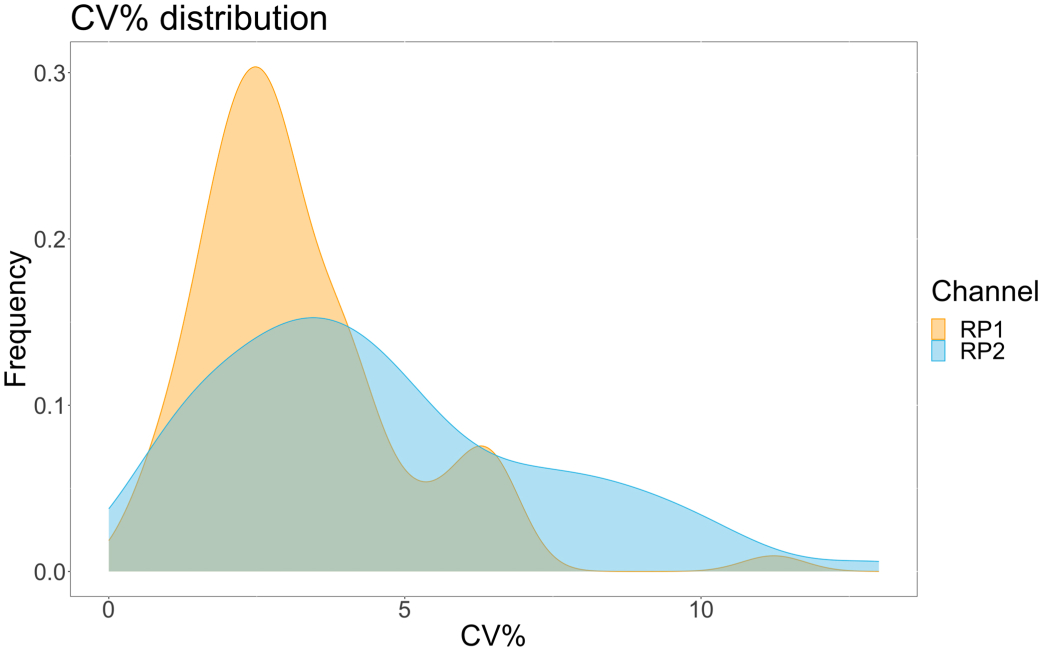
Para evaluar la precisión del ensayo, todas las condiciones se ejecutaron por triplicado. Se calculó un coeficiente de varianza (CV) para la microesfera ACE2 para cada punto de dilución. Todos los CV calculados para el ensayo estaban por debajo del 15%, donde el CV más alto medido fue del 13% y el CV más bajo fue del 1% (Tabla 2). Como se puede observar en el gráfico de densidad (Figura 4) del canal RP1, la detección de PE del Hu-anti-S1 comercial muestra una mayor precisión, concentrada principalmente en torno a un CV del 3%. El canal RP2, detección de BV de scFVs, muestra CVs más altos. Sin embargo, como se puede observar en la Tabla 2, el rango más alto de CV está impulsado por las muestras con bajas concentraciones de partículas virales, como el blanco. Para probar la robustez del protocolo, el ensayo se repitió dos veces por diferentes operadores, utilizando mezclas de perlas generadas en diferentes días y un volumen de muestra más bajo (72% menor). Se observó una muy buena correlación de Pearson, entre 0,98 y 1, tanto para el canal RP1 como para el RP2 (valor p < 0,01), lo que confirma la robustez del ensayo y la posibilidad de aplicar el ensayo cuando se dispone de menos muestra (Figura 5). Esta tecnología de análisis de flujo sigue la "teoría del analito ambiental"17, lo que hace que el ensayo sea sensible a la concentración pero no al volumen.

Figura 1: Ensayo de partículas de virus. (A) El sobrenadante celular de células Vero E6 infectadas y no infectadas se agrega en una dilución en serie a una placa de 96 pocillos o 384 pocillos, junto con microesferas magnéticas conjugadas con neutravidina, y luego se acopla a ACE2 humano biotinilado o biotina. Las microesferas no conjugadas junto con IgG de cabra y las microesferas desnudas se utilizan como controles negativos junto con la microesfera conjugada con neutravidina y biotina. (B) Los complejos de partículas de microesfera-virus que se han formado se detectan con un cóctel de detección que consiste en Hu-anti-S1 y uno de los diferentes scFvs con etiqueta FLAG. A continuación, se añade una mezcla fluorescente con IgG PE antihumana dirigida a Hu-anti-S1 y Brilliant Violet 421 anti-FLAG dirigida a las scFvs. (C) El sistema de detección dual de tres láseres emite un láser rojo, verde y violeta para detectar el complejo de micropartículas. El láser rojo detecta la etiqueta de tinte de microesfera, mientras que los láseres verde y violeta detectan el anti-S1 y el scFvs, respectivamente. A continuación, se analizan los datos generados. Haga clic aquí para ver una versión más grande de esta figura.
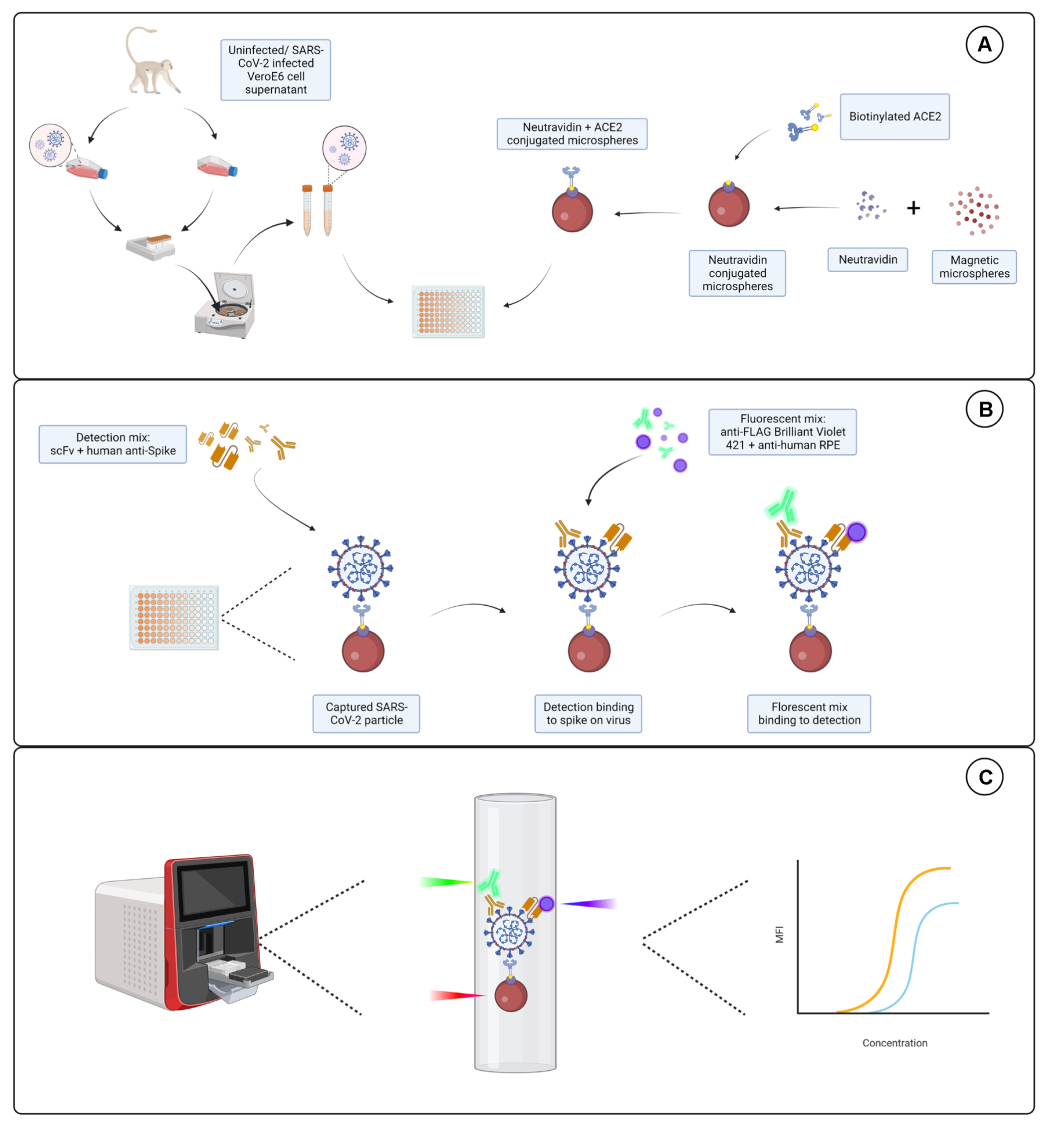{kind=link}

Figura 2: Gráfico de confirmación de conjugación. Las mezclas de perlas consistieron en cuatro identificaciones de microesferas diferentes, cada una conjugada con una proteína diferente: neutravidina-biotina-ACE2 (ACE2), microesfera no conjugada (Bare Bead), neutravidina-biotina (Biotina) e IgG de cabra (IgG de cabra). En la prueba de conjugación se utilizaron tres configuraciones diferentes de fluoróforos de detección. A saber, cabra anti-ACE2 + anti-cabra IgG PE, anti-ratón IgG PE y anti-conejo IgG PE. El eje Y muestra la señal MFI (intensidad de fluorescencia mediana; unidades arbitrarias) media medida de cada microesfera con las tres condiciones diferentes. El eje X muestra los diferentes anticuerpos de captura aplicados. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: Detección multiplexada de proteínas de superficie. Eje Y: MFI medio (mediana de intensidad de fluorescencia; unidades arbitrarias ± desviación estándar) para cada muestra, analizado en pocillos triplicados por condición. Eje X: Puntos de dilución en serie del sobrenadante celular. Naranja: Partículas virales en sobrenadante de Vero E6 infectadas con SARS-CoV-2 WT detectadas con anti-spike + anti-PE humano (ficoeritrina). Azul: Sobrenadante de Vero E6 infectado con SARS-CoV-2 WT detectado con los diferentes scFvs + anti-FLAG Brilliant Violet 421. Gris: Sobrenadante de células no infectadas detectado con anti-spike + anti-PE humano. Negro: Sobrenadante celular no infectado detectado con los cinco scFvs diferentes + anti-FLAG Brilliant Violet 421. Las partículas virales se capturaron con microesferas magnéticas acopladas a ACE2 y se probaron con anticuerpos anti-spike humanos comerciales en el canal reportero RP1 y con diferentes scFvs en el canal reportero RP2 (scFv se indica en la parte superior izquierda de cada panel). No se detectaron partículas de virus en ninguna de las muestras no infectadas. El epítopo al que se dirige scFv3 tenía la mayor afinidad. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4: Gráfico de dispersión de variación. El eje Y es la frecuencia de eventos, y el eje X muestra el coeficiente de varianza (CV) en porcentaje para cada réplica de las diferentes muestras. RP1 y RP2 son el primer y segundo canal reportero que detectan la fluorescencia asociada con la ficoeritrina y el violeta brillante 421, respectivamente. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5: Matriz de correlación de carreras. (A,B) Eje Y: Matriz de correlación de Pearson en escala log10 entre tres corridas separadas, ejecutadas por tres operadores diferentes y con diferentes mezclas de cuentas. En la tercera corrida se aplicó un volumen de muestra menor. Los histogramas muestran la distribución de los diferentes grupos de variables en función de la MFI medida. (A) Correlación para el canal reportero RP1 entre las diferentes corridas. (B) Correlación para el canal reportero RP2 entre las diferentes corridas. MFI=intensidad media de fluorescencia en unidades arbitrarias. p < 0,001. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
| Detección | Reactividad |
| scFv2 | ++ |
| scFv3 | +++ |
| scFv5 | ++ |
| scFv7 | + |
| scFv9 | + |
| IgG anti-Spike humana | ++++ |
Tabla 1: Clasificación de scFvs en detección en base a la intensidad MFI obtenida en las curvas estándar.
| RP1 (PE) | RP2 (BV421) | |
| Dilución de la muestra | Rango CV [%] | Rango CV [%] |
| Espacio en blanco | 3–11 | 2–13 |
| 1:1458 | 1–7 | 2–7 |
| 1:456 | 4–6 | 3–8 |
| 1:162 | 3–6 | 3–7 |
| 1:54 | 2–4 | 2–4 |
| 1:18 | 2–4 | 1–4 |
| 1:6 | 2–6 | 1–6 |
| 1:2 | 1–5 | 1–3 |
Tabla 2: Rango de CV% (media/desviación estándar × 100) de cada punto de dilución del sobrenadante infectado por SARS-CoV-2 para los canales reporteros RP1 y RP2.
Ficha complementaria 1: Generación de fragmentos variables de cadena simple (scFv) de inmunoglobulinas. Haga clic aquí para descargar este archivo.
Tabla complementaria 1: Cribado de scFvs en pares con Fabs frente a la dilución seriada de Spike recombinante (RBD). Para evaluar el rendimiento de diferentes péptidos de detección, se utilizaron 12 combinaciones de la proteína de pico, Fab, como captura en tampón enriquecido con RBD recombinante. Se aplicaron diez (10) scFvs dirigidos a diferentes epítopos de la proteína de la espícula como detección. En función del rendimiento del par de captura y detección, se marcaron como fallidos (-) o correctos (+). Haga clic aquí para descargar este archivo.
Tabla complementaria 2: Cribado de scFvs en pares con Fabs contra la dilución en serie del sobrenadante de células Calu-3 infectadas por SARS-Cov-2. Para evaluar el rendimiento de diferentes péptidos de detección, se utilizaron 12 combinaciones de la proteína de pico, Fab, como captura en el sobrenadante de células Calu-3 infectadas con SARS-Cov-2. Se aplicaron diez (10) scFvs dirigidos a diferentes epítopos de la proteína de la espícula como detección. En función del rendimiento del par de captura y detección, se marcaron como fallidos (-) o correctos (+). Haga clic aquí para descargar este archivo.
Discusión
Se ha demostrado que la tecnología multiplex basada en perlas es una plataforma valiosa para la detección de patógenos de alto rendimiento en una serie de aplicaciones clínicas. La alta flexibilidad de la plataforma, basada en los principios de la citometría de flujo, permite dirigirse a anticuerpos, proteínas y ácidos nucleicos 18,19,20,21,22, multiplexando cientos de analitos simultáneamente. Sin embargo, hasta donde sabemos, esta tecnología no se ha aplicado anteriormente para detectar partículas virales intactas. En este informe, la tecnología se aplicó para la detección de partículas virales intactas dirigiéndose a tres epítopos de superficie independientes del SARS-CoV-2.
Los virus de ARN con envoltura muestran una alta similitud estructural con las vesículas extracelulares (VE), pequeñas membranas de fosfolípidos que transportan ARN y proteínas virales junto con proteínas del huésped23. Los inmunoensayos sándwich se han aplicado previamente a la detección de VE, utilizando un par de anticuerpos dirigidos a dos proteínas de superficie distintas24,25. La limitación de los ensayos sándwich para detectar simultáneamente solo dos proteínas se elimina con enfoques multiplex que permiten la detección simultánea de más de dos proteínas por reacción.
El sistema de detección de doble reportero de tres láseres descrito aquí es el instrumento de análisis de flujo basado en perlas más avanzado hasta la fecha. Con respecto a los sistemas de lectura de un solo reportero, el doble reportero (canales RP1 y RP2) permite la detección de tres proteínas/epítopos de superficie en paralelo. Dirigirse a múltiples proteínas de superficie viral y epítopos proporciona una representación más precisa de la carga de proteínas virales, lo que, más allá de confirmar que el virus está, de hecho, intacto, también abre la oportunidad de investigar más a fondo los antígenos de superficie viral y los mecanismos de las interacciones de las proteínas virales y del huésped.
Durante la pandemia de COVID-19, la importancia de identificar rápidamente a las personas portadoras de partículas virales activas fue importante en los esfuerzos por contener la propagación del virus. El ARN genómico se detecta por RT-PCR cuantitativa independientemente de su origen (partículas virales intactas o libres). Sin embargo, solo una envoltura intacta con proteína S accesible puede mediar la entrada celular y la posterior replicación del virus. Estudios previos con chips microfluídicos en muestras de pacientes han demostrado cómo la detección de partículas virales intactas combinada con pruebas en el punto de atención permitiría realizar pruebas frecuentes y mejorar la vigilancia de la propagación de la enfermedad, incluida una elección más informada de las personas que deben ser puestas en cuarentena26. La aplicación de un ensayo basado en microesferas multiplexadas permitiría el diseño de ensayos destinados al cribado de múltiples virus y sus variantes de antígenos de superficie, obteniendo una imagen más precisa de la propagación del virus en la población.
La virometría de flujo es un desarrollo reciente de la citometría de flujo que tiene como objetivo el análisis de partículas virales. A pesar de ser capaz de detectar partículas virales discretas, el análisis de virus pequeños plantea un problema actual para la virometría de flujo27,28. De manera similar al método descrito aquí, la virometría de flujo implica la captura de viriones intactos por nanopartículas de oro acopladas a anticuerpos. Las limitaciones de ambos métodos incluyen (i) la dependencia de reactivos de captura y detección de alta afinidad para el antígeno expresado en superficie dirigido por las microesferas o nanopartículas, (ii) la capacidad limitada para discriminar entre partículas de virus y vesículas extracelulares, y (iii) falta de estándares para la cuantificación adecuada de partículas.
Las células secretan VE en su entorno, y cuando son infectadas por un virus, también pueden secretar viriones que tienen un tamaño similar al de las VE y pueden llegar a expresar los mismos antígenos29. Debido a que los VE tendrán composiciones de membrana similares a las del virus, podría ser difícil distinguirlos entre sí utilizando solo métodos basados en la afinidad, como el enfoque de láser dual de un solo reportero. Sin embargo, las estrategias descritas aquí presentan una mayor capacidad multiplex, lo que permite una investigación más amplia y profunda de la composición proteica de las partículas. Los métodos basados en flujo permiten el seguimiento de partículas discretas, lo que brinda oportunidades para la cuantificación digital. Una estrategia para abordar el problema de la cuantificación en nuestro método sería utilizar vesículas sintéticas bien caracterizadas que expresen antígenos de interés como partículas similares a virus (VLP) para preparar curvas estándar.
Una vía común de entrada y salida del SARS-CoV-2 de las células huésped es a través de la interacción del virus y la membrana de la célula huésped 2,15. En este proceso, la probabilidad de que las proteínas de la membrana del huésped se incorporen a la superficie del virus es alta. Mediante el cribado de las proteínas incorporadas del huésped, se puede rastrear la vía de la infección y potencialmente predecir el curso de la enfermedad para diferentes pacientes de riesgo, lo que permite tomar decisiones de tratamiento más tempranas. También permite la caracterización de los virus en diferentes lotes de muestras en laboratorios de investigación. Esto se puede explorar más a fondo probando si diferentes características están relacionadas con diferentes niveles de infectividad viral y para el cribado de anticuerpos y moléculas de fármacos que se dirigen a las proteínas de la superficie viral.
Un aspecto importante del método descrito es que se basa en la afinidad de los reactivos de captura y detección frente a sus proteínas diana en el virus. La elección de los reactivos de afinidad es, por lo tanto, un factor determinante en el rendimiento del ensayo. Posiblemente, los reactivos de afinidad múltiple deben ser examinados y probados para su captura y detección para seleccionar aquellos con la mayor afinidad. Aquí, se evaluó preliminarmente el rendimiento de diez scFvs y doce fragmentos de Fab utilizando RBD recombinante y en partículas virales del sobrenadante de células epiteliales pulmonares Calu-3 infectadas con SARS-Cov-2 (las células VeroE6 se utilizaron para cultivar/evaluar la citotoxicidad en todos los estudios posteriores). Se utilizó PE anti-FLAG para detectar los scFvs marcados con FLAG (Tabla Suplementaria 1 y Tabla Suplementaria 2). A continuación, se seleccionaron los cinco scFvs de mejor rendimiento para ser aplicados en el ensayo de doble reportero, junto con Hu-anti-S1 comercial (Tabla 1), en sobrenadantes de células epiteliales de riñón de mono verde africano VeroE6 infectadas.
Otro factor crítico para el éxito del protocolo es el procedimiento seleccionado para el acoplamiento de microesferas. El método de acoplamiento debe ser eficiente y, al mismo tiempo, mantener intactos y sin modificar los epítopos conformacionales o los residuos de aminoácidos involucrados en la unión a proteínas. Aquí, se aplicó la reacción EDC-NHS para acoplar neutravidina directamente a las microesferas, adaptando un protocolo previamente descrito30 y un sistema de neutravidina + biotina para unir ACE2 recombinante a las microesferas acopladas. Los métodos de acoplamiento alternativos y su eficiencia pueden probarse y compararse. Por último, se observó que diferentes reactivos de detección marcados con fluorescencia (por ejemplo, anti-FLAG PE (ficoeritrina) y anti-FLAG Brilliant Violet 421) pueden dar lugar a diferentes niveles de MFI que pueden afectar a la sensibilidad del ensayo.
En conclusión, el método descrito permite la detección de partículas virales intactas en solución, aplicando una estrategia de doble reportero. El análisis de tres determinantes de superficie en paralelo proporciona una herramienta más específica para caracterizar las partículas virales y, finalmente, discriminarlas de otros VE (por ejemplo, que no contienen antígenos virales). Esta estrategia es una alternativa a la virometría de flujo. Aunque el enfoque actual no discrimina los tamaños de partícula, las estrategias de perlas magnéticas que utilizan microesferas codificadas por colores ofrecen una capacidad más amplia en el perfil de antígenos de superficie y el diseño experimental mediante análisis de alta multiplexación y alto rendimiento. El ensayo muestra una alta precisión y robustez y puede extenderse al análisis de cualquier tipo de vesícula extracelular y cualquier otro tipo de biopartícula que exponga antígenos de superficie en fluidos corporales u otras matrices líquidas. Se trata de un estudio de prueba de concepto que demostró la utilidad de utilizar scFvs como reactivo de detección en un análisis multiplexado de múltiples epítopos de proteínas en partículas virales. Son necesarios estudios futuros para determinar las características específicas de las scFvs (p. ej., afinidades de unión, reactividad cruzada con otros reactivos y dianas) si se van a utilizar con fines cuantitativos o clínicos.
Divulgaciones
Los autores declaran no tener ningún conflicto de intereses.
Agradecimientos
Agradecemos en SciLifeLab, Suecia, al equipo de la Unidad Scilifelab de Affinity Proteomics-Stockholm por desarrollar y aplicar el método aquí descrito, a la Unidad de Terapéutica de Anticuerpos Humanos por proporcionar reactivos scFvs y Fab, y a Jonas Klingström por las células VeroE6 infectadas con aislados de SARS-CoV-2 procedentes de muestras clínicas. Los autores agradecen a Sherry Dunbar, PhD, MBA de Luminex Corporation (Austin, TX), por su apoyo a la investigación, y a Matt Silverman MSci, PhD de Biomedical Publishing Solutions (Ciudad de Panamá, FL; mattsilver@yahoo.com) por su asistencia científica y de redacción. Este trabajo contó con el apoyo de fondos de la Fundación Knut y Alice Wallenberg y el Laboratorio Ciencia para la Vida (SciLifeLab) (VC2020-0015 a Claudia Fredolini y Francesca Chiodi y VC-2022-0028 a Claudia Fredolini).
Materiales
| Name | Company | Catalog Number | Comments |
| ACE2-Biotin | Acro Biosystems (Newark, DE) | AC2-H82E6-25 ug | Conc: 340 µg/mL, LOT#BV35376-203HFI-2128 |
| Anti-Goat IgG, PE-conjugated | Jackson ImmunoResearch (West Grove, PA) | 705-116-147 | Host species: Donkey |
| Anti-Human IgG R-PE | Life Technologies/Thermo Fisher (Waltham, MA) | H10104 | Conc: 0.15 mg/mL, LOT#2079224, Host species: Goat |
| Anti-Mouse IgG, PE-conjugated | Jackson ImmunoResearch (West Grove, PA) | 115-116-146 | Host species: Goat |
| Anti-Rabbit IgG, PE-conjugated | Jackson ImmunoResearch (West Grove, PA) | 111-116-144 | Host species: Goat |
| Biotin | Thermo-Fisher Scientific (Waltham, MA) | 20RUO | 100 mM, pH 10 Conc. 1 mg/mL |
| Blocker Casein in PBS | Thermo-Fisher Scientific (Waltham, MA) | 37528 | LOT#VD301372 |
| Blocker reagent for ELISA (BRE) | Roche (Basel, Switzerland) | 11112589001 | |
| Brilliant Violet 421 anti-DYKDDDDK Tag Antibody (Anti-FLAG) 0.2 mg/ml, rat IgG2a, λ | BioLegend (Amsterdam, The Netherlands) | 637321 | |
| Bovine serum albumin (BSA) | Saveen & Werner (Limhamn, Sweden) | B2000-500 | LOT#04D5865 |
| EDC (1-Ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride) | Proteochem (Hurricane, UT) | C1100-custom (65 mg) | LOT# MK3857 |
| Fetal calf serum (FCS) | Gibco/Thermo Fisher (Waltham, MA) | 10270-106 | |
| Goat anti-ACE2 polyclonal antibody | R&D Systems/Bio-Techne (Minneapolis, MN) | AF933 | Host species: Goat |
| Goat IgG | Bethyl Labs (Montgomery, TX) | P50-200 | LOT#P50-200-6 |
| L-glutamine | Thermo-Fisher Scientific (Waltham, MA) | 25030024 | |
| Low-bind 1.5 mL microfuge tubes | VWR (Radnor, PA) | 525-0133 | |
| MagPlex-C Microspheres | Luminex Corporation (Austin, TX) | MC10XXX-01 | |
| MEM tissue cuture media | Gibco/Thermo Fisher (Waltham, MA) | 21430-020 | |
| Microplate, 96-Well, Polystyrene, Half-area, Clear | Greiner Bio-One (Kremsmünster, Austria) | 675101 | |
| NaHCO3 | Gibco/Thermo Fisher (Waltham, MA) | 25080-060 | |
| Neutravidin | Thermo-Fisher Scientific (Waltham, MA) | 31000 | LOT#UK292857 |
| PBS tablets | Medicago AB (Uppsala, Sweden) | 09-9400-100 | LOT#272320-01 |
| Penicillin/Streptomycin | Gibco/Thermo Fisher (Waltham, MA) | 15140122 | |
| Poly(vinyl alcohol) | Sigma-Aldrich (St. Louis, MO) | 360627 | |
| Polyvinylpyrrolidone | Sigma-Aldrich (St. Louis, MO) | 437190 | |
| ProClin 300 | Sigma-Aldrich (St. Louis, MO) | 48915-U | |
| Rabbit IgG | Bethyl Labs (Montgomery, TX) | P120-301 | LOT#12 |
| scFv-FAb1 | In-house production | Human Antibody Therapeutics Unit, Scilifelab, Sweden. Monoclonal scFv. Conc: 0.12 mg/mL. | |
| scFv-FAb2 | In-house production | Human Antibody Therapeutics Unit, Scilifelab, Sweden. Monoclonal scFv. Conc batch1: 0.38 mg/mL. Conc batch2: 0.45 mg/mL | |
| scFv-FAb3 | In-house production | Human Antibody Therapeutics Unit, Scilifelab, Sweden. Monoclonal scFv. Conc: 0.34 mg/mL. | |
| scFv-FAb4 | In-house production | Human Antibody Therapeutics Unit, Scilifelab, Sweden. Monoclonal scFv. Conc: 2.85 mg/mL. | |
| scFv-FAb5 | In-house production | Human Antibody Therapeutics Unit, Scilifelab, Sweden. Monoclonal scFv. Conc:2.7mg/mL. | |
| SARS-CoV-2 infectious particles, Swedish isolate | In-house production | The Public Health Agency of Sweden | |
| SARS-CoV-2 Spike Antibody (Hu-anti-S1) | Novus Biologicals (Centennial, CO) | NBP2-90980 | Monoclonal antibody. Conc: 1 mg/mL. Host: Human. Clone: CR3022. Isotype: IgG1 Kappa. LOT#T201B06 |
| Sodium phosphate monobasic, anhydrous | Sigma-Aldrich (St. Louis, MO) | S3139 | |
| Sulfo-NHS (N-hydroxysulfosuccinimide) | Thermo-Fisher Scientific (Waltham, MA) | 24510 | LOT# XH321563 |
| Tween | Thermo-Fisher Scientific (Waltham, MA) | BP337-50 | LOT#194435 |
| Ultraviolet lamp | Vilber Lourmat GmbH (Eberhardzell, Germany) | VL-215.G | Wavelength = 254 nm; 2 × 15-watt bulbs |
| Vero E6 cells | ATCC (Manassus, VA) | CRL-1586 | |
| xMAP INTELLIFLEX DR-SE (dual-reporter flow instrument) | Luminex Corporation (Austin, TX) | INTELLIFLEX-DRSE-RUO |
Referencias
- Rey, F. A., Lok, S. M. Common features of enveloped viruses and implications for immunogen design for next-generation vaccines. Cell. 172 (6), 1319-1334 (2018).
- V'kovski, P., Kratzel, A., Steiner, S., Stalder, H., Thiel, V. Coronavirus biology and replication: implications for SARS-CoV-2. Nature Reviews Microbiology. 19 (3), 155-170 (2021).
- Burnie, J., et al. Flow virometry quantification of host proteins on the surface of HIV-1 pseudovirus particles. Viruses. 12 (11), 1296 (2020).
- Gentili, M., et al. Transmission of innate immune signaling by packaging of cGAMP in viral particles. Science. 349 (6253), 1232-1236 (2015).
- Modrow, S., Falke, D., Truyen, U., Schätzl, H. . Viruses: Definition, Structure, Classification. In Molecular Virology. , 163-181 (2013).
- Trinh, K. T. L., Do, H. D. K., Lee, N. Y. Recent advances in molecular and immunological diagnostic platform for virus detection: A review. Biosensors. 13 (4), 490 (2023).
- Zamora, J. L. R., Aguilar, H. C. Flow virometry as a tool to study viruses. Methods. 134-135, 87-97 (2018).
- Graham, H., Chandler, D. J., Dunbar, S. A. The genesis and evolution of bead-based multiplexing. Methods. 158, 2-11 (2019).
- Byström, S., et al. Affinity proteomic profiling of plasma for proteins associated to area-based mammographic breast density. Breast Cancer Research. 20 (1), 14 (2018).
- Rudberg, A. -. S., et al. SARS-CoV-2 exposure, symptoms and seroprevalence in healthcare workers in Sweden. Nature Communications. 11 (1), 5064 (2020).
- Liu, J., et al. Multiplex reverse transcription PCR Luminex assay for detection and quantitation of viral agents of gastroenteritis. Journal of Clinical Virology. 50 (4), 308-313 (2011).
- Gadsby, N. J., Hardie, A., Claas, E. C. J., Templeton, K. E. Comparison of the Luminex respiratory virus panel fast assay with in-house real-time PCR for respiratory viral infection diagnosis. Journal of Clinical Microbiology. 48 (6), 2213-2216 (2010).
- Lorenzen, E., et al. Multiplexed analysis of the secretin-like GPCR-RAMP interactome. Science Advances. 5 (9), (2019).
- Angeloni, S., Cameron, A., Pecora, N. D., Dunbar, S. A rapid, multiplex dual reporter IgG and IgM SARS-CoV-2 neutralization assay for a multiplexed bead-based flow analysis system. Journal of Visualized Experiments: JoVE. (170), e62487 (2021).
- Jackson, C. B., Farzan, M., Chen, B., Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nature Reviews Molecular Cell Biology. 23 (1), 3-20 (2022).
- ISO10993-5 Biological evaluation of medical devices - Part 5: Tests for in vitro cytotoxicity. International Standardization Organization Available from: https://nhiso.com/wp-content/uploads/2018/05/ISO-10993-5-2009.pdf (2009)
- Poetz, O., et al. Sequential multiplex analyte capturing for phosphoprotein profiling. Molecular & Cellular Proteomics. 9 (11), 2474-2481 (2010).
- Dunbar, S. A., Vander Zee, C. A., Oliver, K. G., Karem, K. L., Jacobson, J. W. Quantitative, multiplexed detection of bacterial pathogens: DNA and protein applications of the Luminex LabMAP system. Journal of Microbiological Methods. 53 (2), 245-252 (2003).
- Taniuchi, M., et al. Multiplex polymerase chain reaction method to detect Cyclospora, Cystoisospora, and Microsporidia in stool samples. Diagnostic Microbiology and Infectious Disease. 71 (4), 386-390 (2011).
- Wu, M., et al. High-throughput Luminex xMAP assay for simultaneous detection of antibodies against rabbit hemorrhagic disease virus, Sendai virus, and rabbit rotavirus. Archives of Virology. 164 (6), 1639-1646 (2019).
- Dias, D., et al. Optimization and validation of a multiplexed Luminex assay to quantify antibodies to neutralizing epitopes on human papillomaviruses 6, 11, 16, and 18. Clinical and Vaccine Immunology. 12 (8), 959-969 (2005).
- Opalka, D., et al. Simultaneous quantitation of antibodies to neutralizing epitopes on virus-like particles for human papillomavirus types 6, 11, 16, and 18 by a multiplexes lumina assay. Clinical and Diagnostic Laboratory Immunology. 10 (1), 108-115 (2003).
- Nolte-'T Hoen, E., Cremer, T., Gallo, R. C., Margolis, L. B. Extracellular vesicles and viruses: Are they close relatives. Proceedings of the National Academy of Sciences. 113 (33), 9155-9161 (2016).
- Ohmichi, T., et al. Quantification of brain-derived extracellular vesicles in plasma as a biomarker to diagnose Parkinson's and related diseases. Parkinsonism & Related Disorders. 61, 82-87 (2019).
- Ter-Ovanesyan, D., et al. Framework for rapid comparison of extracellular vesicle isolation methods. Elife. 10, e70725 (2021).
- Gamage, S. S. T., et al. Microfluidic affinity selection of active SARS-CoV-2 virus particles. Science Advances. 8 (39), (2022).
- Renner, T. M., Tang, V. A., Burger, D., Langlois, M. -. A. Intact viral particle counts measured by flow virometry provide insight into the infectivity and genome packaging efficiency of moloney murine leukemia virus. Journal of Virology. 94 (2), e01600-01619 (2020).
- Niraja, S., et al. A flow virometry process proposed for detection of SARS-CoV-2 and large-scale screening of COVID-19 cases. Future Virology. 15 (8), 525-532 (2020).
- Lippé, R. Flow virometry: A powerful tool to functionally characterize viruses. Journal of Virology. 92 (3), e01765 (2018).
- Drobin, K., Nilsson, P., Schwenk, J. M. Highly multiplexed antibody suspension bead arrays for plasma protein profiling. Methods in Molecular Biology. 1023, 137-145 (2013).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados