
Method Article
Profilazione degli epitopi proteici di superficie su particelle virali mediante la strategia multiplex dual-reporter
In questo articolo
Riepilogo
Qui, descriviamo un test immunologico fluorescente multiplex di nuova concezione che utilizza un sistema citometrico a flusso dual-reporter per rilevare contemporaneamente due epitopi unici della proteina spike su particelle virali intatte di coronavirus 2 della sindrome respiratoria acuta grave (SARS-CoV-2) che erano state catturate da microsfere magnetiche accoppiate all'enzima di conversione dell'angiotensina-2.
Abstract
Le proteine di membrana dei virus avvolti svolgono un ruolo importante in molte funzioni biologiche che coinvolgono l'attaccamento del virus ai recettori delle cellule bersaglio, la fusione di particelle virali con le cellule ospiti, le interazioni ospite-virus e la patogenesi della malattia. Inoltre, le proteine della membrana virale sulle particelle virali e presentate sulle superfici delle cellule ospiti si sono dimostrate ottimi bersagli per antivirali e vaccini. Qui, descriviamo un protocollo per studiare le proteine di superficie su particelle intatte di coronavirus 2 della sindrome respiratoria acuta grave (SARS-CoV-2) utilizzando il sistema citometrico a flusso a doppio reporter. Il test sfrutta la tecnologia multiplex per ottenere una tripla rilevazione di particelle virali mediante tre reazioni di affinità indipendenti. Le microsfere magnetiche coniugate all'enzima di conversione dell'angiotensina-2 umano ricombinante (ACE2) sono state utilizzate per catturare le particelle virali dal surnatante delle cellule infettate da SARS-CoV-2. Quindi, sono stati applicati contemporaneamente due reagenti di rilevamento marcati con R-ficoeritrina (PE) o Brilliant Violet 421 (BV421). Come prova di concetto, sono stati utilizzati frammenti di anticorpi mirati a diversi epitopi della proteina di superficie SARS-CoV-2 Spike (S1). La rilevazione di particelle virali mediante tre reazioni di affinità indipendenti fornisce una forte specificità e conferma la cattura di particelle virali intatte. Le curve dose-dipendenza del surnatante cellulare infetto da SARS-CoV-2 sono state generate con varianze del coefficiente di replicazione (media/DS) ˂14%. Le buone prestazioni del test in entrambi i canali hanno confermato che due epitopi proteici bersaglio sulla superficie del virus sono rilevabili in parallelo. Il protocollo qui descritto potrebbe essere applicato per (i) la profilazione ad alto multiplex e ad alto rendimento di proteine di superficie espresse su virus avvolti; ii) rilevamento di particelle virali attive intatte; e (iii) valutazione della specificità e dell'affinità degli anticorpi e dei farmaci antivirali per gli epitopi di superficie degli antigeni virali. L'applicazione può essere potenzialmente estesa a qualsiasi tipo di vescicole extracellulari e bioparticelle, esponendo antigeni di superficie nei fluidi corporei o in altre matrici liquide.
Introduzione
I virus patogeni più comuni, come l'influenza, l'HIV, il citomegalovirus umano e i ceppi di SARS-CoV, sono virus avvolti. L'infezione cellulare da parte di virus avvolti richiede la fusione delle membrane virali e cellulari dell'ospite, con conseguente rilascio del genoma virale nel citoplasma. L'RNA virale si replicherà quindi prima di essere impacchettato in una nuova particella virale 1,2. Durante questi processi, non solo le proteine virali, ma anche le proteine della membrana ospite possono essere incorporate nell'involucro, diventando parte integrante della nuova particella virale. Le proteine della membrana della cellula ospite incorporate nell'involucro del virus possono facilitare l'ingresso del virus in una nuova cellula ospite, sfruttando i meccanismi delle interazioni cellula-cellula, dell'homing e della fuga del sistema immunitario 3,4.
Nonostante l'importanza di studiare le proteine associate al virus, la maggior parte delle tecniche attualmente disponibili per l'analisi del virus5 non supporta la caratterizzazione ad alto rendimento e ad alto multiplex dell'antigene di superficie del virus. Né sono in grado di rilevare singole particelle virali o di discriminare tra particelle virali infettive intatte, RNA non infettivo, proteine virali e sottopopolazioni virali che esprimono diversi antigeni. Recentemente, la citometria a flusso è stata modificata e adattata in un nuovo metodo per l'analisi delle particelle virali, vale a dire la virometria a flusso. La virometria a flusso consente lo studio di singole particelle virali e dei loro antigeni di superficie. Tuttavia, rimangono limitazioni tra cui bassa produttività, bassa capacità multiplex, complicata configurazione sperimentale e analisi dei dati e limitata rilevabilità di particelle virali di piccole dimensioni 6,7.
La quantificazione multiplexata basata su microsfere di proteine e acidi nucleici è una tecnologia consolidata con numerose applicazioni che vanno dalla quantificazione delle proteine nei fluidi corporei, agli studi di interazione proteina-proteina e alla diagnosi di infezioni virali 8,9,10,11,12,13 . Uno strumento di analisi del flusso di recente introduzione è dotato di un canale a doppio reporter, che consente la misurazione di due molecole reporter fluorescenti nello stesso pozzetto di reazione. Questa nuova capacità si è dimostrata particolarmente utile per la profilazione parallela di diversi isotipi di immunoglobuline14. Qui viene descritto come il sistema a doppio reporter può essere utilizzato per rilevare particelle virali intatte, mirando a più antigeni di superficie in parallelo.
A riprova del concetto, questo rapporto descrive in dettaglio lo sviluppo di un sistema a triplo rilevamento delle particelle del virus SARS-CoV-2. SARS-CoV-2 è costituito da quattro proteine principali, una è la proteina spike (S), che consiste di due subunità. La prima subunità, S1, si lega primariamente all'ACE2 espresso nelle membrane cellulari umane. La seconda subunità, S2, facilita l'ingresso nella cellula bersaglio da parte di un peptide di fusione, creando un poro nella membrana della cellula bersaglio in cui il virione può entrare attraverso15. I tre elementi costitutivi rimanenti di SARS-CoV-2 sono il nucleocapside (N), la proteina di membrana (M) e la proteina dell'involucro (E). Il nucleocapside è responsabile dell'impacchettamento del genoma virale formando strutture ribonucleoproteiche con l'RNA, mentre le proteine di membrana e dell'involucro svolgono un ruolo centrale nell'assemblaggio del virus.
Il test qui descritto si rivolge a tre epitopi indipendenti della subunità S1 espressa sulla superficie dell'involucro di SARS-CoV-2. Vengono utilizzate diluizioni seriali di surnatanti cellulari infetti e non infetti da SARS-CoV-2. Le particelle virali vengono catturate tramite microsfere coniugate con ACE2 che legano la subunità S1 sul virus. La proteina S del virus di superficie viene quindi rilevata in parallelo con un frammento variabile a catena singola (scFv) di immunoglobulina marcato disponibile in commercio e un anticorpo monoclonale umano anti-S1 (Hu-anti-S1) insieme a un scFv marcato con FLAG sviluppato internamente. L'Hu-anti-S1 viene rilevato dal primo canale (RP1) nel sistema dual-reporter con l'anticorpo secondario IgG-Fc umano coniugato con R-ficoeritrina (PE) arancione, e l'scFv viene rilevato dal secondo canale (RP2) con un anticorpo secondario anti-FLAG coniugato blu Brilliant Violet 421 (BV421). Il test delle particelle virali è rappresentato nella Figura 1.
Protocollo
1. Coniugazione della neutravidina e anticorpi di controllo contro le microsfere magnetiche
NOTA: Le perle magnetiche colorate in fluorescenza (microsfere di polistirene di 6,5 μm di diametro con magnetite incorporata) con diverse etichette fluorescenti, elencate nella Tabella dei materiali , vengono utilizzate per generare i seguenti coniugati e controlli delle perline: (1) ACE2 umano ricombinante biotinilato legato a perle accoppiate con un linker della neutravidina; (2) Biotina legata a perle accoppiata con un linker della neutravidina; (3) IgG di capra accoppiate direttamente alle perline; e (4) perline non coniugate. La proteina da accoppiare alle perle deve essere priva di sodio azide, albumina sierica bovina (BSA), glicina, tris(idrossi-metil)amminometano (Tris), glicerolo o additivi contenenti ammine. Il tampone di attivazione è 0,1 M Fosfato di sodio monobasico, anidro (NaH2PO4), pH 6. Il tampone dell'acido 2-morfolinoetsolfonico (MES; 50 mM) di pH 5 viene utilizzato per diluire i coniugati. Il tampone di lavaggio è PBS-T (1x PBS [soluzione salina tamponata con fosfato], pH 7,4 + 0,05 % (v/v) Tween-20). Il tampone di conservazione è un reagente bloccante da 2,7 mg/mL per ELISA (BRE) + 0,1% di antibiotici (in questo caso, ProClin 300).
- Togliere la polvere di Sulfo-N-idrossisulfosuccinimide (NHS) dal frigorifero e 65 mg di 1-etil-3-[3-dimetilamminopropil]carbodiimmide cloridrato (EDC) pre-aliquotati dal congelatore e lasciare a temperatura ambiente (RT; 18-22 °C) per 30 minuti. Conservare sia l'NHS che l'EDC in una busta contenente perle di silice durante questa fase per prevenire l'idrolisi dall'umidità atmosferica.
- Preparare le microsfere per l'attivazione e l'accoppiamento.
NOTA: I coloranti fluorescenti all'interno delle microsfere sono sensibili alla luce e le perle devono essere conservate al buio e a temperature di frigorifero (4-8 °C) quando non vengono utilizzate attivamente.- Risospendere 4 diversi stock di microsfere codificate con ID colore (12,5 x 106/mL) (Tabella dei materiali) mediante breve vortice, sonicazione o rotazione (15 minuti a 15-30 giri/min), secondo la scheda informativa del prodotto.
- Trasferire 40 μl di ciascuna sospensione di perline (5 x 105 microsfere) nei pozzetti assegnati di una piastra per microtitolazione a mezzo pozzetto, a fondo piatto, da 96 pozzetti (Tabella dei materiali).
- Lavare le perline magnetiche.
NOTA: Le fasi di lavaggio possono essere eseguite manualmente o utilizzando una lavapiatti automatizzata.- Aggiungere 80 μL/pozzetto di tampone di attivazione alle perle e immobilizzare le perle su un separatore a piastre magnetiche per 30 s. Aspirare il surnatante dalle microsfere mentre le perle sono immobilizzate sul separatore a piastre magnetiche.
- Rimuovere la piastra per microtitolazione dal separatore a piastre magnetiche e risospendere le perle in 50 μL di tampone di attivazione.
- Attiva le perline con Sulfo-NHS e EDC.
- Preparare la soluzione di lavoro Sulfo-NHS a 50 mg/mL nel tampone di attivazione in una provetta microfuge da 1,5 mL. Restituire la polvere NHS in frigorifero (4-8 °C), al riparo dall'umidità.
- Preparare la soluzione di lavoro EDC a 50 mg/mL nel tampone di attivazione nella sua provetta microfuge da 1,5 mL. Sciogliere le aliquote preconfezionate da 65 mg di polvere EDC in 1,3 ml di tampone di attivazione.
NOTA: Sulfo-NHS e EDC iniziano a idrolizzarsi e a perdere attività dopo essere stati disciolti. Evitare di interrompere la procedura di accoppiamento fino a quando NHS e EDC non sono stati aggiunti ai cordoni. Non conservare le soluzioni NHS o EDC disciolte per un uso successivo. - Preparare la soluzione di attivazione per l'attivazione delle microsfere combinando volumetricamente il 20% di soluzione madre Sulfo-NHS (50 mg/mL), il 20% di soluzione madre EDC (50 mg/mL) e il 60% di tampone di attivazione. Per ogni reazione di attivazione delle microsfere è necessaria una soluzione di attivazione di 50 μl (utilizzando 5 × 105 sfere/reazione), oltre a un volume extra sufficiente per accogliere le perdite di pipettaggio.
- Aggiungere 50 μl di soluzione di attivazione completa a ciascun pozzetto contenente le perle lavate. Con il volume preesistente di sospensione di 50 μL nel tampone di attivazione, per pozzetto, la concentrazione finale di Sulfo-NHS sarà di 5 mg/mL e anche la concentrazione finale di EDC sarà di 5 mg/mL.
- Sigillare la piastra di reazione a microsfere con un sigillante adesivo monouso in plastica o in alluminio e incubare per 20 minuti su un agitatore orbitale (650 giri/min) a temperatura ambiente (18-22 °C) al buio.
- Lavare la soluzione di attivazione in eccesso dalle perline.
- Centrifugare la piastra per microtitolazione a 233 × g per 1 min.
- Immobilizzare le perle attivate su un separatore a piastre magnetiche per 30 s. Rimuovere il sigillante per piastre e aspirare il surnatante dalle perle immobilizzate con il magnete ancora posizionato sul separatore magnetico.
- Rimuovere la piastra di microtitolazione dal separatore magnetico e aggiungere 100 μl di tampone MES a ciascun pozzetto.
- Ripetere i passaggi 1.4.2-1.4.3 un'altra volta per un totale di due lavaggi.
- Accoppiare la neutravidina e le IgG di capra (controllo) ai set di perline appropriati. Preparare un numero sufficiente di soluzioni di lavoro per neutravidina e IgG di capra, pianificando 100 μl/reazione e una quantità sufficiente in più per accogliere le perdite di pipettaggio come segue:
NOTA: La polvere proteica di neutravidina viene ricostituita con acqua ultrapura e quindi diluita in una soluzione madre da 1 mg/mL con PBS prima di aliquotare per la conservazione/l'uso (la proteina di neutravidina non è direttamente solubile in PBS ma è solubile in acqua fino a ~10 mg/mL).- Preparare la soluzione di lavoro di neutravidina a una concentrazione di 125 μg/mL nel tampone MES in una provetta per microfuge a basso contenuto proteico da 1,5 mL.
- Preparare la soluzione di lavoro per gli anticorpi di controllo IgG di capra a una concentrazione di 17,5 μg/mL in tampone MES.
- Preparare la piastra per microtitolazione contenente le perle attivate. Immobilizzare le perle su un separatore a piastre magnetiche per 30 s. Con la piastra di microtitolazione ancora posizionata sul separatore magnetico, aspirare il surnatante dalle perle immobilizzate con magnete.
- Aggiungere 100 μL di soluzione di lavoro di neutravidina (125 μg/mL) nei pozzetti appropriati contenenti le perle (per l'accoppiamento neutravidina-biotina e neutravidina-ACE2).
- Aggiungere 100 μL di soluzione di lavoro IgG di capra (17,5 μg/mL) al pozzetto contenente le perle assegnate come controlli di sole IgG di capra.
- Aggiungere 100 μL di tampone MES al pozzetto assegnato come perline non coniugate di controllo.
- Sigillare la piastra per microtitolazione e incubare per 2 ore su un agitatore orbitale (650 giri/min) a RT (18-22 °C) al buio. Agitare brevemente la piastra dopo 1 ora di incubazione per assicurarsi che le perle rimangano in sospensione.
- Lavare le perline con PBS-T.
- Centrifugare la piastra per microtitolazione a 233 × g per 1 min.
- Immobilizzare le perle accoppiate su un separatore a piastre magnetiche per 30 s. Con la piastra di microtitolazione ancora posizionata sul separatore magnetico, aspirare il surnatante dalle perle immobilizzate con magnete.
- Rimuovere la piastra di microtitolazione dal separatore magnetico.
- Aggiungere 100 μl di PBS-T a ciascun pozzetto contenente le perle.
- Ripetere i passaggi di lavaggio 1.6.2-1.6.4 una volta per un totale di due lavaggi con PBS-T.
- Preparare le perline coniugate per la conservazione.
- Immobilizzare le perle accoppiate su un separatore a piastre magnetiche per 30 s. Con la piastra di microtitolazione ancora posizionata sul separatore magnetico, aspirare il surnatante dalle perle immobilizzate con magnete. Rimuovere la piastra di microtitolazione dal separatore magnetico.
- Aggiungere 50 μl di tampone di stoccaggio a ciascun ID di microsfere per estinguere l'attività residua delle microsfere.
- Incubare la piastra per microtitolazione a temperatura di frigorifero (4-8 °C) al buio per una notte (16-22 h).
- Trasferire sospensioni di perle non coniugate e coniugate con IgG di capra (50 μL) in provette per microfugi a basso contenuto proteico da 1,5 mL opportunamente marcate, combinate con due risciacqui dei pozzetti con tampone di conservazione da 100 μL per garantire il massimo recupero delle sfere.
NOTA: Le perle non coniugate e quelle coniugate con IgG di capra saranno entrambe numerate da 5 × 105 in un volume finale di 250 μL (cioè 2 × 103 perline/μL). Conservare i tubi per microfusibili a temperatura di frigorifero (4-8 °C) fino al momento dell'uso.
- Legare l'ACE2 biotinilato e la biotina alle perle coniugate con neutravidina.
- Preparare la soluzione di lavoro ACE2 biotinilata umana ricombinante a 18 μg/mL di ACE2 in 10 mM PBS. Per ogni reazione, saranno necessari 100 μl. Preparare la soluzione di lavoro della biotina a 2,4 mg/mL di biotina in 10 mM PBS. Per ogni reazione, saranno necessari 100 μl.
- Preparare la piastra per microtitolazione contenente microsfere coniugate con neutravidina.
- Immobilizzare le microsfere su un separatore a piastre magnetiche per 30 s. Con la piastra per microtitolazione ancora posizionata sul separatore magnetico, rimuovere il sigillante per piastre e aspirare il surnatante dalle microsfere immobilizzate con magnete.
- Rimuovere la piastra per microtitolazione dal separatore magnetico e aggiungere 50 μl di 10 mM PBS/pozzetto.
- Ripetere i passaggi 1.8.2.1-1.8.2.2 una volta.
- Aggiungere 100 μl della soluzione di lavoro biotinilata-ACE2 in pozzetti appropriati contenenti microsfere coniugate con neutravidina. Aggiungere 100 μl della soluzione di lavoro della biotina in pozzetti appropriati contenenti microsfere coniugate con neutravidina.
- Sigillare la piastra per microtitolazione e incubare per 1 ora su un agitatore orbitale (650 giri/min) a RT (18-22 °C) al buio.
- Lavare le microsfere come descritto nei passaggi 1.6.1-1.6.5.
- Preparare e conservare le microsfere coniugate con ACE2 e biotina come descritto nei passaggi 1.7.1-1.7.4.
NOTA: Le perle biotinilate ACE2 e le perle coniugate con biotina saranno entrambe numerate da 5 × 105 in un volume finale di 250 μl (cioè 2 × 103 perle/μl).
2. Test di coniugazione
- Preparare una miscela di perle combinando tutti e quattro i tipi di microsfere create nella sezione 1 (ad esempio, ACE2 biotinilato coniugato con neutravidina, biotina coniugata con neutravidina, IgG di capra coniugata e non coniugata).
NOTA: Le microsfere stock sono state conservate a 2 × 103 perle/μl e combinate in modo tale che la concentrazione finale delle microsfere nella miscela di perle di lavoro sia di 40 perle di ciascun set/μl.- Calcolare il volume della miscela di microsfere di lavoro necessaria per il test (5 μL/reazione), consentendo al volume extra di adattarsi alle perdite di pipettaggio. Agitare brevemente ogni tubo e combinare volumi calcolati uguali di ciascuna sospensione di perline in un nuovo tubo microfuge a basso legame proteico. La concentrazione delle microsfere è ora di 400 perle di ciascun set/μL.
- Creare la miscela di perle di lavoro diluendo la sospensione di perle combinata di altre 10 volte con il tampone di stoccaggio (40 di ogni set/μl di concentrazione di lavoro).
NOTA: Preparare prima una piccola quantità di miscela di perline di lavoro per stimare il numero di microsfere/μl per ogni ID.
- Incubare le microsfere con l'anticorpo anti-ACE2 di capra.
- Pipettare 5 μl di miscela di perline di lavoro in 3 pozzetti di una piastra per microtitolazione a fondo piatto, a mezzo pozzetto, da 96 pozzetti.
- Aggiungere 50 μL di anti-ACE2 di capra (0,4 μg/mL diluito in PBS-T, Tabella dei materiali) ciascuno ai 3 pozzetti contenenti le perle nella piastra per microtitolazione. Sigillare la piastra per microtitolazione, il vortice e incubare su un agitatore orbitale (650 giri/min) a RT (18-22 °C) per 1 ora al buio.
- Pulsare la piastra per microtitolazione a 233 × g per 1 minuto e lavare le microsfere tre volte con PBS-T come descritto nei punti 1.6.2-1.6.4.
- Incubare le microsfere con gli anticorpi di rilevamento.
- Pipettare 5 μL di miscela di perline di lavoro in 6 nuovi pozzetti della piastra per microtitolazione.
- Preparare 1 μg/mL di ciascuna delle miscele di rilevazione di lavoro: IgG PE anti-capra, IgG PE anti-topo e IgG PE anti-coniglio in 3 provette separate da 1,5 mL, utilizzando PBS-T come diluente.
- Aggiungere 50 μl delle miscele di rilevamento a 3 pozzetti ciascuna e l'IgG anti-capra viene aggiunto agli stessi pozzetti dell'anti-ACE2 dal passaggio 2.2.
- Sigillare, agitare e incubare su un agitatore orbitale (650 giri/min) a RT (18-22 °C) per 30 minuti.
- Frullare la piastra a 233 × g per 1 minuto e lavare le microsfere tre volte con PBS-T come descritto nei punti 1.6.2-1.6.4.
- Aggiungere 100 μl di PBS-T ed eseguire l'esecuzione sullo strumento di analisi del flusso a doppio reporter con le seguenti impostazioni:
Modalità: doppio reporter; Time-out: 45 s; DD-gating: 7500-17500; Numero minimo di microsfere: 100 microsfere/set (cut-off QC minimo: 35 microsfere/set).
3. Produzione di surnatante cellulare infetto da SARS-CoV-2
Il virus SARS-CoV-2 si propaga nelle cellule Vero E6 dell'ospite (linea cellulare epiteliale del rene di scimmia; ATCC; Tabella dei materiali). Le cellule Vero E6 sono coltivate in terreno Modified Eagles (MEM) a 37°C in un'atmosfera del 5% di CO2 e del 95% di umidità relativa. Ogni litro di MEM è integrato con 10 ml di L-glutammina (200 mM), 38 ml di NaHCO3 (7,5%), 5 ml di soluzione di penicillina/streptomicina e 50 ml di siero fetale bovino (FCS); Tabella dei materiali.
ATTENZIONE: Utilizzare procedure e attrezzature di biosicurezza appropriate durante la manipolazione di SARS-CoV-2.
- Le cellule Vero E6 vengono coltivate fino alla confluenza in due fiasche per colture tissutali da 150 cm2 . Infettare una fiaschetta con il virus SARS-CoV-2 e utilizzare l'altra infetta da finzione come controllo.
- Miscelare circa 100.000 particelle infettive SARS-CoV-2 wild-type (WT) con 5 ml di terreno MEM Eagles.
- Aspirare il terreno da un pallone da 150 cm2 e aggiungere 55 ml di MEM completo per generare surnatanti di controllo non infetti. Aspirare il terreno dall'altro pallone da 150 cm2 e aggiungere la miscela virale alle cellule. Incubare le cellule per 1 ora a 37 °C. Agitare leggermente il pallone ogni 15 minuti per distribuire il virus.
- Aggiungere 50 mL di terreno MEM completo al pallone con l'aggiunta di SARS-CoV-2 e incubare le cellule fino a quando non si osservano effetti citopatici, valutando visivamente i palloni ogni 24 ore.
NOTA: Dovrebbero essere necessari circa 3-4 giorni dopo l'infezione perché si verifichi la citopatia. Gli effetti citopatici sulla struttura del monostrato cellulare Vero E6 (ad esempio, retrazione cellulare, crenazione, arrotondamento, deadesione, perdita di granularità intracitoplasmatica, lisi conclamata) sono valutati qualitativamente osservando le cellule utilizzando un microscopio ottico invertito, secondo le linee guida dell'Organizzazione internazionale di standardizzazione per i test di citotossicità in vitro 16. - Raccogliere il surnatante cellulare da entrambi i palloni e centrifugare per 6 minuti a 253 × g per sedimentare i detriti cellulari.
- Virus SARS-CoV-2 inattivato dai raggi UV nel surnatante cellulare
- Pipettare 0,5 mL di surnatante per pozzetto in 12 pozzetti in una piastra per microtitolazione a 24 pozzetti. Irradiare con raggi UV la piastra per microtitolazione, senza coperchio, per 30 s sotto una lampada ultravioletta adatta (Tabella dei materiali).
NOTA: L'inattivazione virale nel surnatante cellulare deve essere accertata tentando la propagazione del virus in colture cellulari Vero E6.
- Pipettare 0,5 mL di surnatante per pozzetto in 12 pozzetti in una piastra per microtitolazione a 24 pozzetti. Irradiare con raggi UV la piastra per microtitolazione, senza coperchio, per 30 s sotto una lampada ultravioletta adatta (Tabella dei materiali).
- Aliquotare il surnatante cellulare in provette da 1,5 mL e conservare a -20 °C fino a nuovo utilizzo.
NOTA: Il surnatante cellulare può essere conservato a -80 °C.
4. Saggio: Rilevamento di particelle virali SARS-CoV-2 nel surnatante cellulare
- Preparare il tampone dei saggi mescolando lo 0,1% di caseina, lo 0,5% di alcol polivinilico, lo 0,8% di polivinilpirrolidone e l'1% di BSA (tutti p/v) (pH 7). Preparare il tampone di diluizione del campione preparando il 10% di IgG di coniglio nel tampone di saggio.
- Calcolare e preparare il volume della miscela di microsfere di lavoro (fase 2.1) necessario per l'analisi (5 μl/reazione), in modo che il volume in eccesso possa compensare le perdite di pipettaggio.
- Preparare una serie di diluizioni del surnatante. Calcolare i volumi di surnatante necessari. Analizzare ogni punto di diluizione in pozzetti triplicati per ciascuno dei cinque scFvs, ottenendo 15 pozzetti per punto di diluizione e tipo di campione. Utilizzare 45 μl di surnatanti diluiti in ciascun pozzetto, per un totale di 675 μl richiesti; è sufficiente una singola provetta per microfugo da 1,5 mL per ciascuno dei surnatanti SARS-CoV-2 e di controllo.
- Scongelare i surnatanti di SARS-CoV-2 e controllarli a 4 °C per almeno 1 ora. Conservare i surnatanti e le loro diluizioni a freddo (2-8 °C) fino al momento dell'uso. Etichettare e sistemare otto provette per microfugi da 1,5 ml ciascuna per SARS-CoV-2 e surnatanti di controllo.
NOTA: La concentrazione più alta analizzata sarà una diluizione surnatante 1:1 (2 volte), da cui verrà effettuata una serie di diluizioni 1:2 (3 volte) utilizzando il tampone di diluizione del campione, con la diluizione più alta di una diluizione 1:1458. I bianchi contenenti il tampone di diluizione del campione servono solo come controlli senza surnatante. Pertanto, le diluizioni testate di ciascun tipo di surnatante (SARS-CoV-2 o controllo) saranno 2, 6, 18, 54, 162, 486 e 1458 volte, con un controllo solo tampone. - Aggiungere il tampone di diluizione del campione alle provette per microfuge etichettate. Le provette di diluizione 1:1 (2 volte) richiedono 600 μL di tampone, mentre le restanti provette richiedono 800 μL di tampone. Creare la massima diluizione (1:1; 2 volte) di ciascun surnatante combinando 600 μl di surnatante con 600 μl di tampone di diluizione del campione in provette opportunamente etichettate, quindi agitando brevemente la provetta per mescolarla.
- Continuare la serie trasferendo in sequenza 400 μl di surnatanti diluiti 1:1 (2 volte) alla provetta di diluizione successiva (cioè diluizione 6 volte) e continuare le diluizioni 3 volte fino a quando non è stata creata la diluizione più bassa (1458 volte). Agitare brevemente ogni surnatante diluito prima di procedere con la diluizione successiva.
- Scongelare i surnatanti di SARS-CoV-2 e controllarli a 4 °C per almeno 1 ora. Conservare i surnatanti e le loro diluizioni a freddo (2-8 °C) fino al momento dell'uso. Etichettare e sistemare otto provette per microfugi da 1,5 ml ciascuna per SARS-CoV-2 e surnatanti di controllo.
- Incubare le microsfere con il surnatante.
- Agitare la miscela di perle di lavoro pre-preparata per 30 s per risospendere le microsfere e aggiungere 5 μl di miscela di perline a ciascun pozzetto assegnato di una piastra per microtitolazione a fondo piatto da 384 pozzetti.
NOTA: È possibile utilizzare anche piastre a 96 pozzetti. - Aggiungere 45 μl delle diluizioni del surnatante preparate ai pozzetti assegnati contenenti microsfere nella piastra a 384 pozzetti. Sigillare la piastra e incubare per una notte (16-22 h) su un agitatore orbitale (650 giri/min) a RT (18-22°C) al buio.
- Agitare la miscela di perle di lavoro pre-preparata per 30 s per risospendere le microsfere e aggiungere 5 μl di miscela di perline a ciascun pozzetto assegnato di una piastra per microtitolazione a fondo piatto da 384 pozzetti.
- Lavare il surnatante in eccesso dalle perline.
- Rimuovere la piastra per microtitolazione dall'agitatore orbitale e rimuovere il sigillante per piastre. Centrifugare la piastra a 931 x g per 1 minuto.
- Immobilizzare le perle posizionando la piastra di microtitolazione su un separatore di piastre magnetiche per 30 s. Con la piastra per microtitolazione ancora posizionata sul separatore magnetico, rimuovere il sigillante per piastre e aspirare il surnatante dalle perle immobilizzate con magnete.
- Rimuovere la piastra di microtitolazione dal separatore magnetico.
- Aggiungere 60 μl di PBS-T a ciascun pozzetto contenente le microsfere.
- Ripetere i passaggi di lavaggio 4.5.2-4.5.4 due volte per un totale di tre lavaggi PBS-T.
- Preparare le diverse miscele di rilevamento in provette separate da 1,5 mL. Ogni miscela di rilevazione è costituita da un anticorpo monoclonale umano commerciale anti-S1 (Hu-anti-S1) (1 μg/mL) e da una delle cinque diverse scFvs marcate con FLAG (1 μg/mL) (File supplementare 1, Tabella supplementare 1 e Tabella supplementare 2) che hanno come bersaglio la proteina spike sulla particella SARS-CoV-2, diluita in Assay Buffer (fase 4.1), per un totale di cinque diverse miscele di rilevamento.
- Ripetere i passaggi 4.5.2-4.5.3. Risospendere le microsfere lavate in 50 μL/pozzetto della miscela di rilevamento spike specifica per scFv appropriata. Sigillare la piastra per microtitolazione e incubare per 1 ora su un agitatore orbitale (650 giri/min) a RT (18-22 °C) al buio.
- Centrifugare la piastra per microtitolazione a 931 × g per 1 min. Lavare il reagente di rilevamento delle punte in eccesso dalle perline. Eseguire tre fasi di lavaggio con 60 μL di PBS-T secondo i passaggi 4.5.2-4.5.5.
- Incubare le microsfere con una miscela di anticorpi fluorescenti.
- Preparare una miscela di soluzioni fluorescenti composta da IgG anti-umane coniugate con PE disponibili in commercio insieme all'anticorpo anti-FLAG coniugato con BV421 diluito nel tampone del saggio, con concentrazioni di lavoro rispettivamente di 0,2 μg/mL e 1 μg/mL. Per ogni reazione, sono necessari 50 μl di reagente di rivelazione fluorescente.
- Ripetere i passaggi 4.5.2-4.5.3. Risospendere le microsfere in 50 μL/pozzetto di miscela di soluzione fluorescente. Sigillare la piastra per microtitolazione e incubare per 30 minuti su un agitatore orbitale (650 giri/min) a RT (18-22 °C) al buio.
- Centrifugare la piastra per microtitolazione a 931 × g per 1 min. Lavare la miscela di soluzione fluorescente in eccesso dalle microsfere. Eseguire tre fasi di lavaggio con 60 μL di PBS-T secondo i passaggi 4.5.2-4.5.5.
- Sospendi le microsfere in 60 μL di PBS-T dall'ultima fase di lavaggio. Analizzare la piastra su un sistema di analisi del flusso a doppio reporter con le impostazioni descritte al punto 2.6.
Risultati
Test di coniugazione
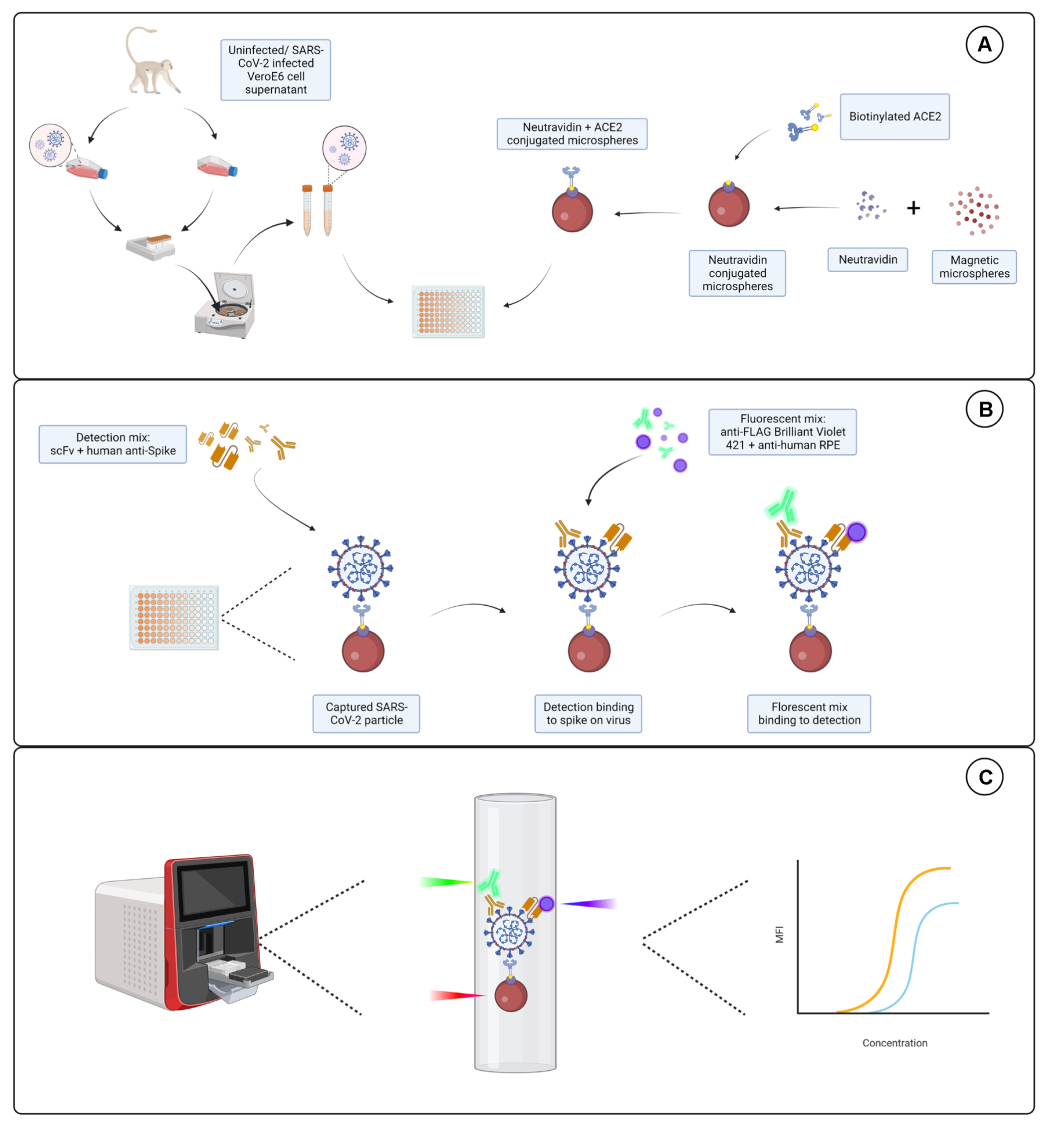
Il test di coniugazione ha mostrato che le IgG di capra e l'ACE2 biotinilato con neutravidina sono state coniugate con successo alle microsfere. La specificità di rilevamento del test è stata confermata sondando microsfere coniugate con ACE2 con anticorpi secondari marcati con PE generati in diverse specie animali (Figura 2). Non è stata osservata alcuna cross-reattività tra i diversi anticorpi di rilevamento. Quando le miscele di perle sono state sondate con PE anti-ACE2 di capra + IgG PE anti-capra, è stato rilevato un valore mediano di intensità di fluorescenza (MFI; unità arbitrarie) al di sopra del fondo sia per le microsfere coniugate con ACE2 che per quelle coniugate con IgG di capra, ma non per la microsfera non coniugata (nuda) o per le microsfere rivestite di biotina. Le IgG PE anti-topo e le IgG PE anti-coniglio sono state utilizzate come controlli negativi per verificare la presenza di segnali falsi positivi. Un segnale di fluorescenza trascurabile è stato generato dopo l'incubazione con le microsfere, indicando che i segnali positivi per l'ACE2 e le IgG di capra erano specifici.
Rilevabilità delle particelle virali nei surnatanti cellulari
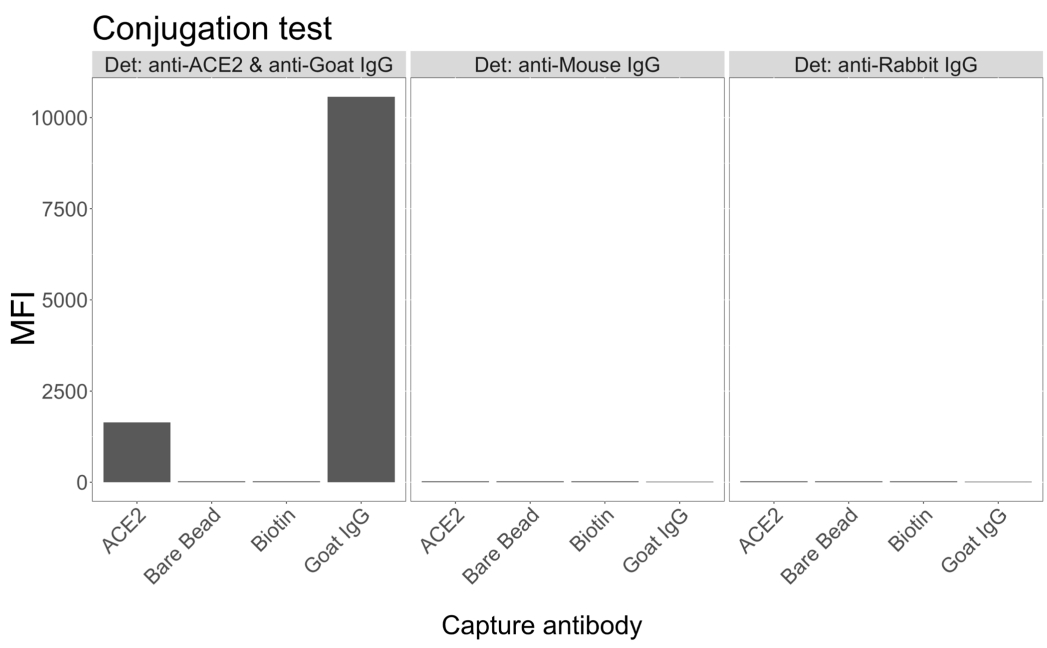
Le microsfere magnetiche accoppiate ad ACE2 umano ricombinante sono state utilizzate per catturare le particelle virali di SARS-CoV-2 da surnatanti di colture cellulari VeroE6 infettate e di controllo (senza virus) e sono state quindi sondate simultaneamente per due distinte regioni di spike virale utilizzando un anticorpo monoclonale e uno dei cinque scFvs distinti. Un segnale concentrazione-dipendente nelle diluizioni dei surnatanti cellulari infettati da SARS-Cov-2 è stato osservato in entrambi i canali reporter (RP1 e RP2) (Figura 3), indicando che sia l'anticorpo commerciale Hu-anti-S1 che i diversi scFvs hanno rilevato la particella virale legata alla microsfera coniugata con ACE2. Con tre scFv su cinque, il virus è rilevabile in diluizioni fino a 1:18 (scFv2, scFv3, scFv5); per le restanti due scFv (scFv7 e scFv9), è rilevabile fino a diluizioni 1:6. Ciò potrebbe essere attribuibile a una diversa affinità per il target. Come mostrato nella Figura 3 e nella Tabella 1, scFv3 fornisce la massima intensità di MFI, seguita rispettivamente da scFv5, scFv2, scFv7 e scFv9.
A livello globale, il rilevamento di scFvs si traduce in un MFI inferiore rispetto a Hu-anti-S1. Questo potrebbe indicare una minore affinità, ma potrebbe anche essere un artefatto dovuto all'etichettatura con diversi coloranti fluorescenti (PE e BV421). Un'altra tendenza che si può osservare per scFv7 e scFv9 è che i valori MFI sono leggermente inferiori per il canale RP1 (anti-spike) rispetto alle altre tre configurazioni. Ciò potrebbe indicare che gli scFvs stanno reagendo in modo incrociato o interferendo in altro modo con l'interazione ACE2-Hu-anti-S1, il che potrebbe anche spiegare il segnale più basso nel canale RP2. Nessuna particella virale è stata rilevata nel surnatante delle cellule Vero E6 non infette né nel canale RP1 né in quello RP2.
La microsfera coniugata neutravidina-biotina, la microsfera capra-IgG e le microsfere non coniugate sono utilizzate come sfere di controllo negativo. Le particelle virali sono state catturate con microsfere magnetiche accoppiate ad ACE2 e testate con anti-spike umano commerciale nel canale reporter RP1 e con diversi scFvs nel canale reporter RP2 (scFv è indicato in alto a sinistra di ogni pannello). Nessuna particella virale è stata rilevata in nessuno dei campioni infetti e non infetti.
Precisione e robustezza del test
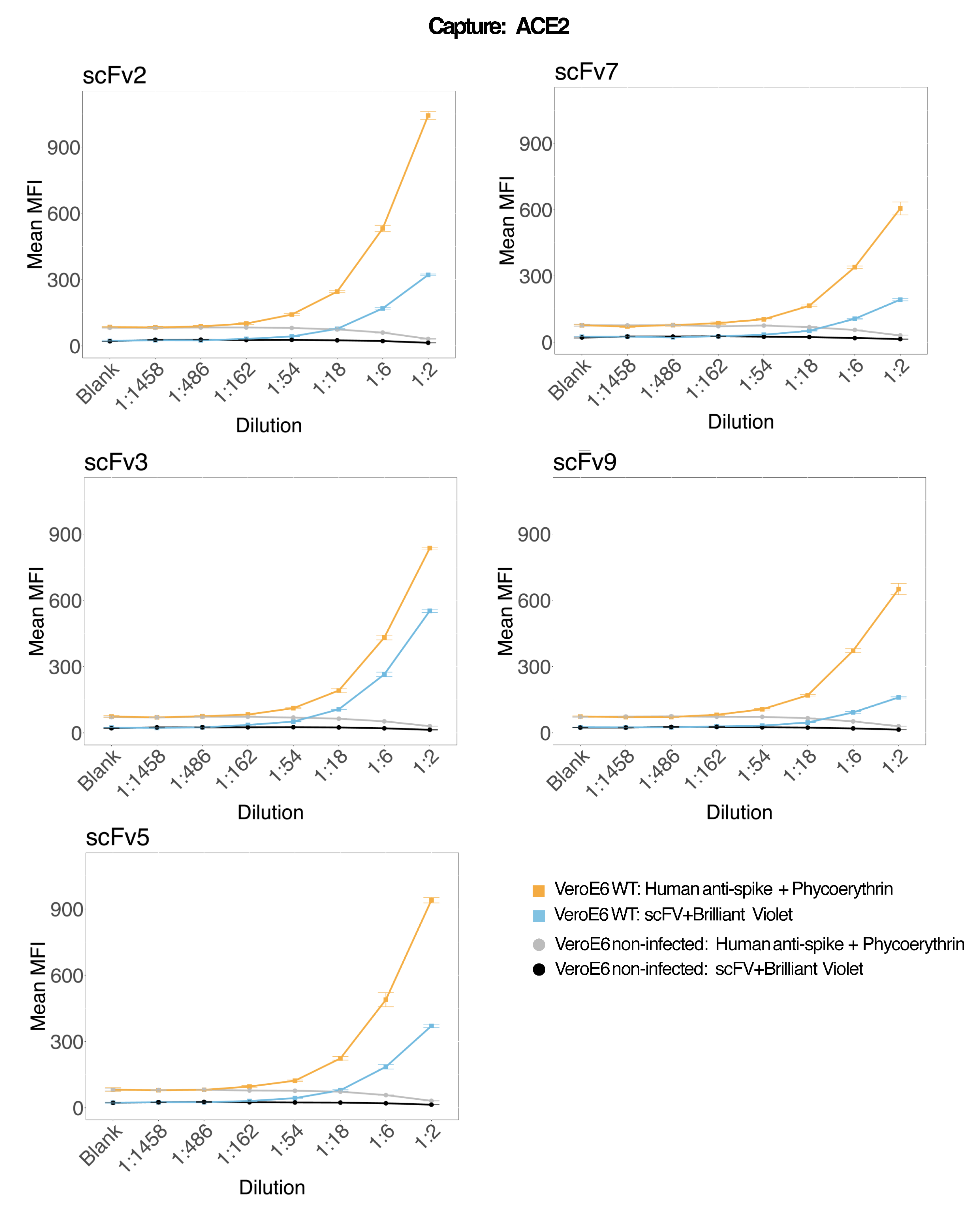
Per valutare la precisione del saggio, tutte le condizioni sono state eseguite in triplice copia. Per ogni punto di diluizione è stato calcolato un coefficiente di varianza (CV) per la microsfera ACE2. Tutti i CV calcolati per il test erano inferiori al 15%, dove il CV più alto misurato era del 13% e il CV più basso era dell'1% (Tabella 2). Come si può vedere nel grafico della densità (Figura 4) del canale RP1, la rilevazione PE dell'Hu-anti-S1 commerciale mostra una precisione più elevata, concentrata principalmente intorno a un CV del 3%. Il canale RP2, BV detection of scFV, mostra CV più elevati. Tuttavia, come si può vedere nella Tabella 2, l'intervallo più elevato di CV è guidato dai campioni con basse concentrazioni di particelle virali, come il bianco. Per testare la robustezza del protocollo, il test è stato ripetuto due volte da operatori diversi, utilizzando miscele di perline generate in giorni diversi e un volume di campione inferiore (72% inferiore). Un'ottima correlazione di Pearson, compresa tra 0,98 e 1, è stata osservata sia per i canali RP1 che per quelli RP2 (valore p < 0,01), confermando la robustezza del test e la possibilità di applicare il test quando è disponibile meno campione (Figura 5). Questa tecnologia di analisi del flusso segue la "teoria dell'analita ambientale"17, rendendo il saggio sensibile alla concentrazione ma non al volume.

Figura 1: Il saggio delle particelle virali. (A) Il surnatante cellulare proveniente da cellule Vero E6 infette e non infette viene aggiunto in una diluizione seriale a una piastra da 96 pozzetti o da 384 pozzetti, insieme a microsfere magnetiche coniugate con neutravidina, e quindi accoppiato a ACE2 umano biotinilato o biotina. Le microsfere non coniugate accoppiate con IgG di capra e microsfere nude sono utilizzate come controlli negativi insieme alla microsfera coniugata neutravidina-biotina. (B) I complessi di particelle di microsfera-virus che si sono formati vengono rilevati con un cocktail di rilevamento costituito da Hu-anti-S1 e uno dei diversi scFvs con FLAG-tag. Viene quindi aggiunta una miscela fluorescente con IgG PE anti-umane che ha come bersaglio l'Hu-anti-S1 e l'anti-FLAG Brilliant Violet 421 che ha come bersaglio gli scFvs. (C) Il sistema a tre laser a doppio rilevamento emette un laser rosso, verde e viola per rilevare il complesso di microparticelle. Il laser rosso rileva l'etichetta del colorante a microsfera, mentre i laser verde e viola rilevano rispettivamente l'anti-S1 e l'scFvs. I dati generati vengono quindi analizzati. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2: Grafico di conferma della coniugazione. Le miscele di perle consistevano in quattro diversi ID di microsfere, ciascuna coniugata con una proteina diversa: neutravidina-biotina-ACE2 (ACE2), microsfera non coniugata (Bare Bead), neutravidina-biotina (Biotina) e IgG di capra (IgG di capra). Nel test di coniugazione sono state utilizzate tre diverse configurazioni di fluorofori di rilevamento. Vale a dire, capra anti-ACE2 + anti-capra IgG PE, anti-topo IgG PE e anti-coniglio IgG PE. L'asse Y mostra il segnale MFI (intensità di fluorescenza mediana; unità arbitrarie) medio misurato da ciascuna microsfera con le tre diverse condizioni. L'asse X mostra i diversi anticorpi di cattura applicati. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3: Rilevamento multiplexato di proteine di superficie. Asse Y: MFI medio (intensità di fluorescenza mediana; unità arbitrarie ± deviazione standard) per ciascun campione, analizzato in pozzetti triplicati per condizione. Asse X: punti di diluizione seriali del surnatante cellulare. Arancione: particelle virali nel surnatante di Vero E6 infettate da SARS-CoV-2 WT rilevate con EP anti-spike + anti-PE umano (ficoeritrina). Blu: surnatante di Vero E6 infettato da SARS-CoV-2 WT rilevato con i diversi scFvs + anti-FLAG Brilliant Violet 421. Grigio: surnatante cellulare non infetto rilevato con EP anti-spike + anti-EP umana. Nero: surnatante di cellule non infette rilevato con i cinque diversi scFvs + anti-FLAG Brilliant Violet 421. Le particelle virali sono state catturate con microsfere magnetiche accoppiate ad ACE2 e testate con anticorpi anti-spike umani commerciali nel canale reporter RP1 e con diversi scFvs nel canale reporter RP2 (scFv è indicato in alto a sinistra di ciascun pannello). Nessuna particella virale è stata rilevata in nessuno dei campioni non infetti. L'epitopo bersaglio di scFv3 aveva la più alta affinità. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 4: Grafico della dispersione delle variazioni. L'asse Y è la frequenza degli eventi e l'asse X mostra il coefficiente di varianza (CV) in percentuale per ogni replica dei diversi campioni. RP1 e RP2 sono il primo e il secondo canale reporter che rilevano la fluorescenza associata rispettivamente alla ficoeritrina e al Brilliant Violet 421. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 5: Esecuzione della matrice di correlazione. (A,B) Asse Y: Matrice di correlazione di Pearson in scala log10 tra tre esecuzioni separate, eseguite da tre operatori diversi e con diverse miscele di perline. Nella terza corsa è stato applicato un volume di campione inferiore. Gli istogrammi mostrano la distribuzione dei diversi cluster di variabili in base all'MFI misurato. (A) Correlazione per il canale reporter RP1 tra le diverse esecuzioni. (B) Correlazione per il canale reporter RP2 tra le diverse esecuzioni. MFI=intensità mediana di fluorescenza in unità arbitrarie. p < 0,001. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
| Scoperta | Reattività |
| scFv2 | ++ |
| scFv3 | +++ |
| scFv5 | ++ |
| scFv7 | + |
| scFv9 | + |
| IgG umane anti-spike | ++++ |
Tabella 1: Classificazione delle scFvs nel rilevamento in base all'intensità dell'MFI ottenuta nelle curve standard.
| RP1 (PE) | RP2 (BV421) | |
| Diluizione del campione | Gamma CV [%] | Gamma CV [%] |
| Vuoto | 3–11 | 2–13 |
| 1:1458 | 1–7 | 2–7 |
| 1:456 | 4–6 | 3–8 |
| 1:162 | 3–6 | 3–7 |
| 1:54 | 2–4 | 2–4 |
| 1:18 | 2–4 | 1–4 |
| 1:6 | 2–6 | 1–6 |
| 1:2 | 1–5 | 1–3 |
Tabella 2: Intervallo CV% (media/deviazione standard × 100) di ciascun punto di diluizione del surnatante infetto da SARS-CoV-2 per entrambi i canali reporter RP1 e RP2.
File supplementare 1: Generazione di frammenti variabili a catena singola di immunoglobuline (scFv). Clicca qui per scaricare questo file.
Tabella supplementare 1: Screening di scFvs in coppia con Fabs contro la diluizione seriale di Spike ricombinante (RBD). Per valutare le prestazioni di diversi peptidi di rilevazione, sono state utilizzate 12 combinazioni di proteina spike, Fab, come cattura in tampone addizionato con RBD ricombinante. Dieci (10) scFv mirati a diversi epitopi della proteina spike sono stati applicati come rilevamento. A seconda delle prestazioni della coppia acquisizione-rilevamento, sono stati contrassegnati come non riusciti (-) o come riusciti (+). Clicca qui per scaricare questo file.
Tabella supplementare 2: Screening di scFvs in coppia con Fabs contro la diluizione seriale del surnatante cellulare Calu-3 infettato da SARS-Cov-2. Per la valutazione delle prestazioni dei diversi peptidi di rilevamento, 12 combinazioni di proteina spike, Fab, sono state utilizzate come cattura nel surnatante di cellule Calu-3 infettate da SARS-Cov-2. Dieci (10) scFv mirati a diversi epitopi della proteina spike sono stati applicati come rilevamento. A seconda delle prestazioni della coppia acquisizione-rilevamento, sono stati contrassegnati come non riusciti (-) o come riusciti (+). Clicca qui per scaricare questo file.
Discussione
La tecnologia multiplex basata su microsfere ha dimostrato di essere una piattaforma preziosa per il rilevamento di agenti patogeni ad alto rendimento in una serie di applicazioni cliniche. L'elevata flessibilità della piattaforma, basata sui principi della citometria a flusso, consente di mirare ad anticorpi, proteine e acidi nucleici 18,19,20,21,22, moltiplicando centinaia di analiti contemporaneamente. Tuttavia, per quanto ne sappiamo, questa tecnologia non è stata precedentemente applicata per rilevare particelle virali intatte. In questo rapporto, la tecnologia è stata applicata per il rilevamento di particelle virali intatte prendendo di mira tre epitopi superficiali indipendenti di SARS-CoV-2.
I virus a RNA avvolti mostrano un'elevata somiglianza strutturale con le vescicole extracellulari (EV), piccole membrane fosfolipidiche che trasportano RNA e proteine virali insieme alle proteine dell'ospite23. I saggi immunologici a sandwich sono stati precedentemente applicati alla rilevazione delle vescicole extracellulari, utilizzando una coppia di anticorpi mirati a due distinte proteine di superficie24,25. La limitazione dei saggi a sandwich di rilevare contemporaneamente solo due proteine viene rimossa con approcci multiplex che consentono la rilevazione simultanea di più di due proteine per reazione.
Il sistema di rilevamento a doppio reporter a tre laser qui descritto è lo strumento di analisi del flusso basato su microsfere più avanzato fino ad oggi. Per quanto riguarda i sistemi di lettura single-reporter, il dual-reporter (canali RP1 e RP2) consente la rilevazione di tre proteine/epitopi di superficie in parallelo. Il targeting di più proteine ed epitopi di superficie virale fornisce una rappresentazione più accurata della carica proteica virale che, oltre a confermare che il virus è effettivamente intatto, apre anche l'opportunità di studiare ulteriormente gli antigeni di superficie virale e i meccanismi delle interazioni tra le proteine virali e quelle dell'ospite.
Durante la pandemia di COVID-19, l'importanza di identificare tempestivamente gli individui portatori di particelle virali attive è stata importante negli sforzi per contenere la diffusione del virus. L'RNA genomico viene rilevato mediante RT-PCR quantitativa indipendentemente dalla sua origine (particelle virali intatte o libere). Tuttavia, solo un involucro intatto con proteina S accessibile può mediare l'ingresso nelle cellule e la successiva replicazione del virus. Precedenti studi con chip microfluidici in campioni di pazienti hanno dimostrato come il rilevamento di particelle virali intatte combinato con test point-of-care consentirebbe test frequenti e una maggiore sorveglianza della diffusione della malattia, compresa una scelta più informata degli individui da mettere in quarantena26. L'applicazione di un test basato su microsfere multiplexate consentirebbe la progettazione di test finalizzati allo screening di più virus e delle loro varianti di antigene di superficie, ottenendo un quadro più accurato della diffusione del virus nella popolazione.
La virometria a flusso è un recente sviluppo della citometria a flusso che mira all'analisi delle particelle virali. Nonostante sia in grado di rilevare particelle virali discrete, l'analisi di piccoli virus pone un problema attuale per la virometria a flusso27,28. Analogamente al metodo qui descritto, la virometria a flusso prevede la cattura di virioni intatti da parte di nanoparticelle d'oro accoppiate ad anticorpi. Le limitazioni per entrambi i metodi includono (i) la dipendenza da reagenti di cattura e rilevamento ad alta affinità per l'antigene espresso in superficie bersaglio dalle microsfere o dalle nanoparticelle, (ii) la limitata capacità di discriminare tra particelle virali e vescicole extracellulari e (iii) la mancanza di standard per una corretta quantificazione delle particelle.
Le cellule secernono vescicole extracellulari nell'ambiente circostante e, quando infettate da un virus, possono anche secernere virioni di dimensioni simili a quelle delle vescicole extracellulari e possono eventualmente esprimere gli stessi antigeni29. Poiché le vescicole extracellulari avranno composizioni di membrana simili a quelle del virus, potrebbe essere difficile distinguerle l'una dall'altra utilizzando solo metodi basati sull'affinità, come l'approccio a doppio laser single-reporter. Tuttavia, le strategie qui descritte presentano una maggiore capacità multiplex, che consente un'indagine più ampia e approfondita della composizione proteica delle particelle. I metodi basati sul flusso consentono il tracciamento di particelle discrete, offrendo opportunità per la quantificazione digitale. Una strategia per affrontare il problema della quantificazione nel nostro metodo sarebbe quella di utilizzare vescicole sintetiche ben caratterizzate che esprimono antigeni di interesse come particelle simili a virus (VLP) per preparare curve standard.
Un percorso comune di ingresso e uscita di SARS-CoV-2 dalle cellule ospiti è attraverso l'interazione del virus e della membrana della cellula ospite 2,15. In questo processo, la probabilità che le proteine della membrana dell'ospite vengano incorporate nella superficie del virus è elevata. Mediante lo screening delle proteine dell'ospite incorporate, è possibile tracciare il percorso dell'infezione e potenzialmente prevedere il decorso della malattia per i diversi pazienti a rischio, consentendo decisioni terapeutiche più precoci. Consente inoltre la caratterizzazione dei virus in diversi lotti di campioni nei laboratori di ricerca. Questo può essere ulteriormente esplorato testando se caratteristiche diverse sono correlate a diversi livelli di infettività virale e per lo screening di anticorpi e molecole di farmaci che hanno come bersaglio le proteine di superficie virali.
Un aspetto importante del metodo descritto è che si basa sull'affinità dei reagenti di cattura e rilevamento contro le loro proteine bersaglio sul virus. La scelta dei reagenti di affinità è, quindi, un fattore determinante per le prestazioni del saggio. È possibile eseguire lo screening e il test di più reagenti di affinità per la cattura e il rilevamento per selezionare quelli con la più alta affinità. Qui, le prestazioni di dieci scFvs e dodici frammenti Fab sono state valutate preliminarmente utilizzando RBD ricombinante e su particelle virali del surnatante di cellule epiteliali polmonari Calu-3 infettate da SARS-Cov-2 (le cellule VeroE6 sono state utilizzate per coltivare/valutare la citotossicità in tutti gli studi successivi). Anti-FLAG PE è stato utilizzato per rilevare gli scFvs contrassegnati con FLAG (Tabella supplementare 1 e Tabella supplementare 2). I cinque scFvs con le migliori prestazioni sono stati quindi selezionati per essere applicati nel saggio dual-reporter, insieme a Hu-anti-S1 commerciale (Tabella 1), su surnatanti provenienti da cellule epiteliali renali di scimmia verde africana VeroE6 infette.
Un altro fattore critico per il successo del protocollo è la procedura selezionata per l'accoppiamento delle microsfere. Il metodo di accoppiamento dovrebbe essere efficiente e, allo stesso tempo, mantenere intatti e non modificati gli epitopi conformazionali o i residui amminoacidici coinvolti nel legame proteico. Qui, la reazione EDC-NHS è stata applicata per accoppiare la neutravidina direttamente alle microsfere, adattando un protocollo precedentemente descritto30 e un sistema neutravidina + biotina per legare l'ACE2 ricombinante alle microsfere accoppiate. I metodi di accoppiamento alternativi e la loro efficienza possono essere testati e confrontati. Infine, è stato osservato che diversi reagenti di rivelazione marcati in fluorescenza (ad esempio, anti-FLAG PE (ficoeritrina) e anti-FLAG Brilliant Violet 421) possono determinare diversi livelli di MFI che possono influenzare la sensibilità del saggio.
In conclusione, il metodo descritto consente la rilevazione di particelle virali intatte in soluzione, applicando una strategia dual-reporter. L'analisi di tre determinanti di superficie in parallelo fornisce uno strumento più specifico per caratterizzare le particelle virali ed eventualmente discriminarle da altre vescicole extracellulari (ad esempio, non contenenti antigeni virali). Questa strategia è un'alternativa alla virometria a flusso. Sebbene l'approccio attuale non discrimini le dimensioni delle particelle, le strategie di biglie magnetiche che utilizzano microsfere codificate a colori offrono una capacità più ampia nella profilazione dell'antigene di superficie e nella progettazione sperimentale mediante analisi ad alto multiplex e ad alto rendimento. Il test mostra un'elevata precisione e robustezza e può essere esteso all'analisi di qualsiasi tipo di vescicola extracellulare e di qualsiasi altro tipo di bioparticella che espone antigeni superficiali nei fluidi corporei o in altre matrici liquide. Si trattava di uno studio proof-of-concept che ha dimostrato l'utilità dell'utilizzo di scFvs come reagente di rilevamento in un'analisi multiplex di più epitopi proteici su particelle virali. Sono necessari studi futuri per determinare le caratteristiche specifiche degli scFvs (ad esempio, affinità di legame, cross-reattività con altri reagenti e bersagli) se devono essere utilizzati per scopi quantitativi o clinici.
Divulgazioni
Gli autori dichiarano di non avere conflitti di interesse.
Riconoscimenti
Ringraziamo SciLifeLab, Svezia, il team dell'Unità ScilifeLab di Affinity Proteomics-Stoccolma per lo sviluppo e l'applicazione del metodo qui descritto, l'Unità di Terapia con Anticorpi Umani per la fornitura di scFvs e reagenti Fab e Jonas Klingström per le cellule VeroE6 infettate con isolati di SARS-CoV-2 derivanti da campioni clinici. Gli autori ringraziano Sherry Dunbar, PhD, MBA di Luminex Corporation (Austin, TX), per il supporto alla ricerca, e Matt Silverman MSci, PhD of Biomedical Publishing Solutions (Panama City, FL; mattsilver@yahoo.com) per l'assistenza scientifica e di scrittura. Questo lavoro è stato sostenuto dai fondi della Knut and Alice Wallenberg Foundation and Science for Life Laboratory (SciLifeLab) (VC2020-0015 a Claudia Fredolini e Francesca Chiodi e VC-2022-0028 a Claudia Fredolini).
Materiali
| Name | Company | Catalog Number | Comments |
| ACE2-Biotin | Acro Biosystems (Newark, DE) | AC2-H82E6-25 ug | Conc: 340 µg/mL, LOT#BV35376-203HFI-2128 |
| Anti-Goat IgG, PE-conjugated | Jackson ImmunoResearch (West Grove, PA) | 705-116-147 | Host species: Donkey |
| Anti-Human IgG R-PE | Life Technologies/Thermo Fisher (Waltham, MA) | H10104 | Conc: 0.15 mg/mL, LOT#2079224, Host species: Goat |
| Anti-Mouse IgG, PE-conjugated | Jackson ImmunoResearch (West Grove, PA) | 115-116-146 | Host species: Goat |
| Anti-Rabbit IgG, PE-conjugated | Jackson ImmunoResearch (West Grove, PA) | 111-116-144 | Host species: Goat |
| Biotin | Thermo-Fisher Scientific (Waltham, MA) | 20RUO | 100 mM, pH 10 Conc. 1 mg/mL |
| Blocker Casein in PBS | Thermo-Fisher Scientific (Waltham, MA) | 37528 | LOT#VD301372 |
| Blocker reagent for ELISA (BRE) | Roche (Basel, Switzerland) | 11112589001 | |
| Brilliant Violet 421 anti-DYKDDDDK Tag Antibody (Anti-FLAG) 0.2 mg/ml, rat IgG2a, λ | BioLegend (Amsterdam, The Netherlands) | 637321 | |
| Bovine serum albumin (BSA) | Saveen & Werner (Limhamn, Sweden) | B2000-500 | LOT#04D5865 |
| EDC (1-Ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride) | Proteochem (Hurricane, UT) | C1100-custom (65 mg) | LOT# MK3857 |
| Fetal calf serum (FCS) | Gibco/Thermo Fisher (Waltham, MA) | 10270-106 | |
| Goat anti-ACE2 polyclonal antibody | R&D Systems/Bio-Techne (Minneapolis, MN) | AF933 | Host species: Goat |
| Goat IgG | Bethyl Labs (Montgomery, TX) | P50-200 | LOT#P50-200-6 |
| L-glutamine | Thermo-Fisher Scientific (Waltham, MA) | 25030024 | |
| Low-bind 1.5 mL microfuge tubes | VWR (Radnor, PA) | 525-0133 | |
| MagPlex-C Microspheres | Luminex Corporation (Austin, TX) | MC10XXX-01 | |
| MEM tissue cuture media | Gibco/Thermo Fisher (Waltham, MA) | 21430-020 | |
| Microplate, 96-Well, Polystyrene, Half-area, Clear | Greiner Bio-One (Kremsmünster, Austria) | 675101 | |
| NaHCO3 | Gibco/Thermo Fisher (Waltham, MA) | 25080-060 | |
| Neutravidin | Thermo-Fisher Scientific (Waltham, MA) | 31000 | LOT#UK292857 |
| PBS tablets | Medicago AB (Uppsala, Sweden) | 09-9400-100 | LOT#272320-01 |
| Penicillin/Streptomycin | Gibco/Thermo Fisher (Waltham, MA) | 15140122 | |
| Poly(vinyl alcohol) | Sigma-Aldrich (St. Louis, MO) | 360627 | |
| Polyvinylpyrrolidone | Sigma-Aldrich (St. Louis, MO) | 437190 | |
| ProClin 300 | Sigma-Aldrich (St. Louis, MO) | 48915-U | |
| Rabbit IgG | Bethyl Labs (Montgomery, TX) | P120-301 | LOT#12 |
| scFv-FAb1 | In-house production | Human Antibody Therapeutics Unit, Scilifelab, Sweden. Monoclonal scFv. Conc: 0.12 mg/mL. | |
| scFv-FAb2 | In-house production | Human Antibody Therapeutics Unit, Scilifelab, Sweden. Monoclonal scFv. Conc batch1: 0.38 mg/mL. Conc batch2: 0.45 mg/mL | |
| scFv-FAb3 | In-house production | Human Antibody Therapeutics Unit, Scilifelab, Sweden. Monoclonal scFv. Conc: 0.34 mg/mL. | |
| scFv-FAb4 | In-house production | Human Antibody Therapeutics Unit, Scilifelab, Sweden. Monoclonal scFv. Conc: 2.85 mg/mL. | |
| scFv-FAb5 | In-house production | Human Antibody Therapeutics Unit, Scilifelab, Sweden. Monoclonal scFv. Conc:2.7mg/mL. | |
| SARS-CoV-2 infectious particles, Swedish isolate | In-house production | The Public Health Agency of Sweden | |
| SARS-CoV-2 Spike Antibody (Hu-anti-S1) | Novus Biologicals (Centennial, CO) | NBP2-90980 | Monoclonal antibody. Conc: 1 mg/mL. Host: Human. Clone: CR3022. Isotype: IgG1 Kappa. LOT#T201B06 |
| Sodium phosphate monobasic, anhydrous | Sigma-Aldrich (St. Louis, MO) | S3139 | |
| Sulfo-NHS (N-hydroxysulfosuccinimide) | Thermo-Fisher Scientific (Waltham, MA) | 24510 | LOT# XH321563 |
| Tween | Thermo-Fisher Scientific (Waltham, MA) | BP337-50 | LOT#194435 |
| Ultraviolet lamp | Vilber Lourmat GmbH (Eberhardzell, Germany) | VL-215.G | Wavelength = 254 nm; 2 × 15-watt bulbs |
| Vero E6 cells | ATCC (Manassus, VA) | CRL-1586 | |
| xMAP INTELLIFLEX DR-SE (dual-reporter flow instrument) | Luminex Corporation (Austin, TX) | INTELLIFLEX-DRSE-RUO |
Riferimenti
- Rey, F. A., Lok, S. M. Common features of enveloped viruses and implications for immunogen design for next-generation vaccines. Cell. 172 (6), 1319-1334 (2018).
- V'kovski, P., Kratzel, A., Steiner, S., Stalder, H., Thiel, V. Coronavirus biology and replication: implications for SARS-CoV-2. Nature Reviews Microbiology. 19 (3), 155-170 (2021).
- Burnie, J., et al. Flow virometry quantification of host proteins on the surface of HIV-1 pseudovirus particles. Viruses. 12 (11), 1296 (2020).
- Gentili, M., et al. Transmission of innate immune signaling by packaging of cGAMP in viral particles. Science. 349 (6253), 1232-1236 (2015).
- Modrow, S., Falke, D., Truyen, U., Schätzl, H. . Viruses: Definition, Structure, Classification. In Molecular Virology. , 163-181 (2013).
- Trinh, K. T. L., Do, H. D. K., Lee, N. Y. Recent advances in molecular and immunological diagnostic platform for virus detection: A review. Biosensors. 13 (4), 490 (2023).
- Zamora, J. L. R., Aguilar, H. C. Flow virometry as a tool to study viruses. Methods. 134-135, 87-97 (2018).
- Graham, H., Chandler, D. J., Dunbar, S. A. The genesis and evolution of bead-based multiplexing. Methods. 158, 2-11 (2019).
- Byström, S., et al. Affinity proteomic profiling of plasma for proteins associated to area-based mammographic breast density. Breast Cancer Research. 20 (1), 14 (2018).
- Rudberg, A. -. S., et al. SARS-CoV-2 exposure, symptoms and seroprevalence in healthcare workers in Sweden. Nature Communications. 11 (1), 5064 (2020).
- Liu, J., et al. Multiplex reverse transcription PCR Luminex assay for detection and quantitation of viral agents of gastroenteritis. Journal of Clinical Virology. 50 (4), 308-313 (2011).
- Gadsby, N. J., Hardie, A., Claas, E. C. J., Templeton, K. E. Comparison of the Luminex respiratory virus panel fast assay with in-house real-time PCR for respiratory viral infection diagnosis. Journal of Clinical Microbiology. 48 (6), 2213-2216 (2010).
- Lorenzen, E., et al. Multiplexed analysis of the secretin-like GPCR-RAMP interactome. Science Advances. 5 (9), (2019).
- Angeloni, S., Cameron, A., Pecora, N. D., Dunbar, S. A rapid, multiplex dual reporter IgG and IgM SARS-CoV-2 neutralization assay for a multiplexed bead-based flow analysis system. Journal of Visualized Experiments: JoVE. (170), e62487 (2021).
- Jackson, C. B., Farzan, M., Chen, B., Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nature Reviews Molecular Cell Biology. 23 (1), 3-20 (2022).
- ISO10993-5 Biological evaluation of medical devices - Part 5: Tests for in vitro cytotoxicity. International Standardization Organization Available from: https://nhiso.com/wp-content/uploads/2018/05/ISO-10993-5-2009.pdf (2009)
- Poetz, O., et al. Sequential multiplex analyte capturing for phosphoprotein profiling. Molecular & Cellular Proteomics. 9 (11), 2474-2481 (2010).
- Dunbar, S. A., Vander Zee, C. A., Oliver, K. G., Karem, K. L., Jacobson, J. W. Quantitative, multiplexed detection of bacterial pathogens: DNA and protein applications of the Luminex LabMAP system. Journal of Microbiological Methods. 53 (2), 245-252 (2003).
- Taniuchi, M., et al. Multiplex polymerase chain reaction method to detect Cyclospora, Cystoisospora, and Microsporidia in stool samples. Diagnostic Microbiology and Infectious Disease. 71 (4), 386-390 (2011).
- Wu, M., et al. High-throughput Luminex xMAP assay for simultaneous detection of antibodies against rabbit hemorrhagic disease virus, Sendai virus, and rabbit rotavirus. Archives of Virology. 164 (6), 1639-1646 (2019).
- Dias, D., et al. Optimization and validation of a multiplexed Luminex assay to quantify antibodies to neutralizing epitopes on human papillomaviruses 6, 11, 16, and 18. Clinical and Vaccine Immunology. 12 (8), 959-969 (2005).
- Opalka, D., et al. Simultaneous quantitation of antibodies to neutralizing epitopes on virus-like particles for human papillomavirus types 6, 11, 16, and 18 by a multiplexes lumina assay. Clinical and Diagnostic Laboratory Immunology. 10 (1), 108-115 (2003).
- Nolte-'T Hoen, E., Cremer, T., Gallo, R. C., Margolis, L. B. Extracellular vesicles and viruses: Are they close relatives. Proceedings of the National Academy of Sciences. 113 (33), 9155-9161 (2016).
- Ohmichi, T., et al. Quantification of brain-derived extracellular vesicles in plasma as a biomarker to diagnose Parkinson's and related diseases. Parkinsonism & Related Disorders. 61, 82-87 (2019).
- Ter-Ovanesyan, D., et al. Framework for rapid comparison of extracellular vesicle isolation methods. Elife. 10, e70725 (2021).
- Gamage, S. S. T., et al. Microfluidic affinity selection of active SARS-CoV-2 virus particles. Science Advances. 8 (39), (2022).
- Renner, T. M., Tang, V. A., Burger, D., Langlois, M. -. A. Intact viral particle counts measured by flow virometry provide insight into the infectivity and genome packaging efficiency of moloney murine leukemia virus. Journal of Virology. 94 (2), e01600-01619 (2020).
- Niraja, S., et al. A flow virometry process proposed for detection of SARS-CoV-2 and large-scale screening of COVID-19 cases. Future Virology. 15 (8), 525-532 (2020).
- Lippé, R. Flow virometry: A powerful tool to functionally characterize viruses. Journal of Virology. 92 (3), e01765 (2018).
- Drobin, K., Nilsson, P., Schwenk, J. M. Highly multiplexed antibody suspension bead arrays for plasma protein profiling. Methods in Molecular Biology. 1023, 137-145 (2013).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati