
Method Article
マルチプレックスデュアルレポーター法によるウイルス粒子上の表面タンパク質エピトープのプロファイリング
要約
ここでは、デュアルレポーターフローサイトメトリーシステムを使用して、アンジオテンシン変換酵素-2結合磁気マイクロスフェアによって捕捉された無傷の重症急性呼吸器症候群コロナウイルス2(SARS-CoV-2)ウイルス粒子上の2つのユニークなスパイクタンパク質エピトープを同時に検出する、新たに開発されたマルチプレックス蛍光免疫測定法について説明します。
要約
エンベロープウイルス上の膜タンパク質は、標的細胞受容体へのウイルス付着、ウイルス粒子の宿主細胞への融合、宿主とウイルスの相互作用、および疾患の病因を含む多くの生物学的機能において重要な役割を果たしています。さらに、ウイルス粒子上のウイルス膜タンパク質や宿主細胞表面に提示されるタンパク質は、抗ウイルス薬やワクチンの優れた標的であることが証明されています。ここでは、デュアルレポーターフローサイトメトリーシステムを使用して、無傷の重症急性呼吸器症候群コロナウイルス2(SARS-CoV-2)粒子上の表面タンパク質を調査するプロトコルについて説明します。このアッセイは、マルチプレックス技術を利用して、3つの独立したアフィニティー反応によるウイルス粒子のトリプル検出を実現します。組換えヒトアンジオテンシン変換酵素-2(ACE2)に結合した磁気ビーズを使用して、SARS-CoV-2に感染した細胞の上清からウイルス粒子を捕捉しました。次に、R-フィコエリトリン(PE)またはブリリアントバイオレット421(BV421)で標識された2つの検出試薬を同時に適用しました。概念実証として、SARS-CoV-2表面タンパク質Spike(S1)の異なるエピトープを標的とする抗体フラグメントを使用しました。3つの独立したアフィニティー反応によるウイルス粒子の検出は、強力な特異性を提供し、無傷のウイルス粒子の捕捉を確認します。SARS-CoV-2 感染細胞上清の用量依存性曲線は、反復係数分散 (平均/SD) ˂14% で生成されました。両方のチャネルで良好なアッセイ性能を発揮した結果、2つのウイルス表面標的タンパク質エピトープが並行して検出できることが確認されました。ここで説明するプロトコルは、(i)エンベロープウイルス上に発現する表面タンパク質の高マルチプレックス、ハイスループットプロファイリングに適用できます。ii)活性な無傷のウイルス粒子の検出。(iii)ウイルス抗原の表面エピトープに対する抗体および抗ウイルス薬の特異性と親和性の評価。このアプリケーションは、あらゆるタイプの細胞外小胞や生体粒子に拡張できる可能性があり、体液やその他の液体マトリックス中の表面抗原を露出させることができます。
概要
インフルエンザ、HIV、ヒトサイトメガロウイルス、SARS-CoV株などの最も一般的な病原性ウイルスは、エンベロープウイルスです。エンベロープウイルスによる細胞感染には、ウイルスと宿主の細胞膜の融合が必要であり、その結果、ウイルスゲノムが細胞質に放出されます。その後、ウイルスRNAは複製されてから、新しいウイルス粒子1,2に詰められます。これらの過程では、ウイルスタンパク質だけでなく、宿主膜タンパク質もエンベロープに取り込まれ、新しいウイルス粒子の不可欠な部分となることがあります。ウイルスエンベロープに組み込まれた宿主細胞膜タンパク質は、細胞間相互作用、ホーミング、および免疫系の脱出のメカニズムを利用して、新しい宿主細胞へのウイルスの侵入を促進する可能性があります3,4。
ウイルス関連タンパク質の研究は重要であるにもかかわらず、現在利用可能なウイルス解析5の手法のほとんどは、ウイルス表面抗原のハイスループットおよびハイマルチプレックス特性評価をサポートしていません。また、個々のウイルス粒子を検出することも、感染性の無傷のウイルス粒子、非感染性RNA、ウイルスタンパク質、および異なる抗原を発現するウイルス亜集団を区別することもできません。最近、フローサイトメトリーは、ウイルス粒子の分析のための新しい方法、すなわちフロービロメトリーに改良され、適応されました。フロービロメトリーは、単一のウイルス粒子とその表面抗原の調査を可能にします。しかし、低スループット、低マルチプレックス機能、複雑な実験セットアップとデータ解析、小型ウイルス粒子の検出可能性の制限などの制限は依然として存在します6,7。
タンパク質と核酸のミクロスフェアベースのマルチプレックス定量は、体液中のタンパク質定量、タンパク質間相互作用研究、ウイルス感染の診断など、数多くのアプリケーションを持つ確立された技術です8,9,10,11,12,13 .最近導入されたフロー分析装置は、デュアルレポーターチャンネルを備えており、同じ反応ウェル内の2つの蛍光レポーター分子の測定が可能です。この新しい機能は、異なる免疫グロブリンアイソタイプの並行プロファイリングに特に有用であることが示されています14。ここでは、デュアルレポーターシステムを使用して、複数の表面抗原を並行して標的とする無傷のウイルス粒子を検出する方法について説明します。
概念実証として、このレポートでは、SARS-CoV-2ウイルス粒子のトリプル検出システムの開発について詳しく説明します。SARS-CoV-2は4つの主要なタンパク質で構成されており、1つはスパイクタンパク質(S)で、2つのサブユニットで構成されています。最初のサブユニットであるS1は、ヒト細胞膜で発現するACE2への一次結合を行います。第2のサブユニットであるS2は、融合ペプチドによる標的細胞への侵入を促進し、ビリオンが15を介して侵入できる細孔を標的細胞膜に形成する。SARS-CoV-2の残りの3つの構成要素は、ヌクレオカプシド(N)、膜タンパク質(M)、およびエンベロープタンパク質(E)です。ヌクレオカプシドは、RNAとリボ核タンパク質構造を形成することによりウイルスゲノムのパッケージングに関与し、膜タンパク質とエンベロープタンパク質はウイルスの集合において中心的な役割を果たします。
ここで説明するアッセイは、SARS-CoV-2のエンベロープ表面に発現するS1サブユニットの3つの独立したエピトープを標的としています。SARS-CoV-2に感染した細胞上清と非感染した細胞上清の両方の段階希釈が使用されます。ウイルス粒子は、ウイルス上のS1サブユニットに結合するACE2標識マイクロスフェアを介して捕捉されます。次に、表面ウイルスSタンパク質を、市販のタグ付き免疫グロブリン一本鎖可変フラグメント(scFv)およびヒトモノクローナル抗S1抗体(Hu-anti-S1)と並行して検出します。Hu-抗S1は、オレンジ色のR-フィコエリトリン(PE)標識抗ヒトIgG-Fc二次抗体を用いたデュアルレポーター系の第1チャネル(RP1)によって検出され、scFvは、青色のブリリアントバイオレット421(BV421)結合二次抗FLAG抗体を用いた第2チャネル(RP2)によって検出されます。ウイルス粒子アッセイを 図1に示します。
プロトコル
1. ニュートラアビジンと制御抗体の磁気ミクロスフェアへの結合
注: 材料表 に記載されている、異なる蛍光標識を持つ蛍光染色磁気ビーズ(マグネタイトが埋め込まれた直径6.5μmのポリスチレン微小球)は、次のビーズコンジュゲートおよびコントロールを生成するために使用されます:(1)ニュートラアビジンリンカーと結合したビーズに結合したビオチン化組換えヒトACE2;(2)ニュートラアビジンリンカーと結合したビーズに結合したビオチン。(3)ビーズに直接結合したヤギIgG。(4)非共役ビーズ。ビーズに結合するタンパク質は、アジ化ナトリウム、ウシ血清アルブミン(BSA)、グリシン、トリス(ヒドロキシメチル)アミノメタン(トリス)、グリセロール、またはアミン含有添加物を含まないものでなければなりません。活性化バッファーは、0.1 M リン酸ナトリウム一塩基性、無水 (NaH2PO4)、pH 6 です。pH 5の2-モルフォリノエタンスルホン酸(MES;50 mM)バッファーは、コンジュゲートの希釈に使用されます。洗浄バッファーはPBS-T(1x PBS[リン酸緩衝生理食塩水]、pH 7.4 + 0.05 % (v/v) Tween-20) です。保存バッファーは、ELISA(BRE)+ 0.1%抗生物質(ここではProClin 300)用の2.7 mg/mLブロッキング試薬です。
- スルホ-N-ヒドロキシスルホスクシンイミド(NHS)粉末を冷蔵庫から取り出し、65 mgの1-エチル-3-[3-ジメチルアミノプロピル]カルボジイミド塩酸塩(EDC)を冷凍庫から取り出し、室温(RT;18-22°C)で30分間放置します。このステップでは、NHSとEDCの両方をシリカビーズの入った封筒に保管して、大気中の水分による加水分解を防ぎます。
- 活性化と結合のためのミクロスフェアを準備します。
注:マイクロスフェア内の蛍光色素は光に敏感であり、ビーズは、アクティブに使用していないときは、暗所および冷蔵庫の温度(4〜8°C)に保管する必要があります。- 製品情報シートに従って、色IDコード化されたマイクロスフェア(12.5 x 106 / mL)(材料表)の4つの異なるストックを再懸濁します。
- 各ビーズ懸濁液(5 x 105 マイクロスフィア)40 μLを、ハーフウェル、平底、96ウェルマイクロタイタープレートの割り当てられたウェルに移します(材料表)。
- 磁気ビーズを洗います。
注:洗浄手順は、手動または自動プレートウォッシャーを使用して実行できます。- ビーズに80 μL/wellの活性化バッファーを加え、ビーズを磁気プレートセパレーターに30秒間固定します。ビーズを磁気プレートセパレーターに固定している間に、ミクロスフェアから上清を吸引します。
- マイクロタイタープレートをマグネティックプレートセパレーターから取り外し、ビーズを50 μLの活性化バッファーに再懸濁します。
- Sulfo-NHSおよびEDCでビーズを活性化します。
- 1.5 mLマイクロフュージチューブ内の活性化バッファー中で、50 mg/mLのSulfo-NHSワーキング溶液を調製します。在庫のNHSパウダーを湿気から保護された冷蔵庫(4〜8°C)に戻します。
- 1.5 mLマイクロフュージチューブ内の活性化バッファーに50 mg/mLのEDCワーキング溶液を調製します。既製の65 mgアリコートのEDC粉末を1.3 mLの活性化バッファーに溶解します。
注:Sulfo-NHSとEDCは、溶解すると加水分解を開始し、活性を失います。NHSとEDCがビーズに追加されるまで、カップリング手順を中断しないでください。溶解したNHSまたはEDC溶液を後で使用するために保存しないでください。 - ビーズ活性化用の活性化溶液を調製するには、20% Sulfo-NHSストック溶液(50 mg/mL)、20% EDCストック溶液(50 mg/mL)、および60%活性化バッファーを容量的に組み合わせます。各ビーズ活性化反応(5 × 105 ビーズ/反応を使用)には50 μLの活性化溶液が必要であり、さらにピペッティング損失に対応するのに十分な追加容量が必要です。
- 洗浄したビーズを含む各ウェルに50 μLの完全活性化溶液を加えます。活性化バッファー中の既存の50 μLビーズ懸濁液容量をウェルあたりにすると、Sulfo-NHSの最終濃度は5 mg/mL、EDCの最終濃度も5 mg/mLになります。
- マイクロスフィア反応プレートを使い捨ての接着性プラスチックまたはホイルプレートシーラーで密封し、室温(18-22°C)の暗所でオービタルシェーカー(650rpm)で20分間インキュベートします。
- ビーズから余分な活性化溶液を洗い流します。
- マイクロタイタープレートを233 × g で1分間遠心分離します。
- 活性化ビーズを磁気プレートセパレーターに30秒間固定します。プレートシーラーを取り外し、マイクロタイタープレートを磁気セパレーターに置いたまま、磁石固定ビーズから上清を吸引します。
- マイクロタイタープレートをマグネティックセパレーターから取り外し、各ウェルに100 μLのMES Bufferを加えます。
- 手順1.4.2〜1.4.3をさらに1回繰り返し、合計2回の洗浄を行います。
- ニュートラアビジンとヤギIgG(コントロール)を適切なビーズセットに結合します。十分なニュートラアビジンとヤギIgGのワーキング溶液を準備し、100 μL/反応を計画し、次のようにピペッティング損失に対応するのに十分な追加量を計画します。
注:ニュートラアビジンタンパク質粉末を超純水で再構成し、PBSを含む1 mg / mLストック溶液に希釈してから、保存/使用のために分注します(ニュートラビジンタンパク質はPBSに直接溶解しませんが、水には~10 mg / mLに溶解します)。- 1.5 mLの低タンパク質吸着マイクロチューブ内のMES Bufferに125 μg/mLの濃度のニュートラアビジンワーキング溶液を調製します。
- MES Buffer中に17.5 μg/mLの濃度でヤギIgGコントロール抗体ワーキング溶液を調製します。
- 活性化ビーズを含むマイクロタイタープレートを準備します。ビーズを磁気プレートセパレーターに30秒間固定します。マイクロタイタープレートを磁気セパレーターに置いたまま、磁石で固定化されたビーズから上清を吸引します。
- ビーズを含む適切なウェル(ニュートラアビジン-ビオチンおよびニュートラアビジン-ACE2カップリング用)に100 μLのニュートラアビジンワーキング溶液(125 μg/mL)を加えます。
- 100 μLのヤギIgGワーキング溶液(17.5 μg/mL)を、ヤギIgGのみのコントロールとして割り当てたウェル含有ビーズに加えます。
- 100 μLのMESバッファーを、コントロールの非標識ビーズとしてウェルに添加します。
- マイクロタイタープレートをシールし、オービタルシェーカー(650rpm)で、暗闇の中でRT(18-22°C)で2時間インキュベートします。インキュベーションの1時間後にプレートを短時間ボルテックスして、ビーズが懸濁液のままであることを確認します。
- ビーズをPBS-Tで洗います。
- マイクロタイタープレートを233 × g で1分間遠心分離します。
- 結合ビーズを磁気プレートセパレーターに固定し、30秒間固定します。マイクロタイタープレートを磁気セパレーターに置いたまま、磁石で固定化されたビーズから上清を吸引します。
- マイクロタイタープレートをマグネティックセパレーターから取り外します。
- ビーズを含む各ウェルに100 μLのPBS-Tを添加します。
- 洗濯手順1.6.2〜1.6.4を1回繰り返し、PBS-Tで合計2回洗います。
- 保管用のコンジュゲートビーズを準備します。
- 結合ビーズを磁気プレートセパレーターに固定し、30秒間固定します。マイクロタイタープレートを磁気セパレーターに置いたまま、磁石で固定化されたビーズから上清を吸引します。マイクロタイタープレートをマグネティックセパレーターから取り外します。
- 各マイクロスフィアIDに50 μLのStorage Bufferを添加して、残りのビーズ活性をクエンチします。
- マイクロタイタープレートを冷蔵庫の温度(4〜8°C)で暗所で一晩(16〜22時間)インキュベートします。
- 非標識ビーズおよびヤギ-IgG標識ビーズ懸濁液(50 μL)を、適切に標識された1.5 mLの低タンパク質結合性マイクロフュージチューブに移し、ウェルの2つの100 μL保存バッファーリンスと組み合わせて、ビーズの回収率を最大化します。
注:非標識ビーズとヤギIgG標識ビーズは、最終容量250μL(すなわち、2 × 103ビーズ/μL)で5×105番になります。マイクロフュージチューブは、使用するまで冷蔵庫の温度(4〜8°C)で保管してください。
- ビオチン化ACE2とビオチンをニュートラアビジン結合ビーズに結合します。
- 組換えヒトビオチン化ACE2ワーキング溶液を18 μg/mL ACE2で調製し、10 mM PBS溶液溶液を調製します。1反応につき、100 μLが必要です。10 mM PBS中の2.4 mg/mLビオチンでビオチンワーキング溶液を調製します。1反応につき、100 μLが必要です。
- ニュートラアビジン結合ミクロスフェアを含むマイクロタイタープレートを準備します。
- マイクロスフィアを磁気プレートセパレーターに30秒間固定します。マイクロタイタープレートを磁気セパレーター上に置いたまま、プレートシーラーを取り外し、磁石で固定化されたマイクロスフェアから上清を吸引します。
- マイクロタイタープレートをマグネティックセパレーターから取り外し、10 mM PBS/ウェルを50 μL加えます。
- 手順 1.8.2.1 から 1.8.2.2 を 1 回繰り返します。
- 100 μLのビオチン化ACE2ワーキング溶液を、ニュートラアビジン結合マイクロスフェアを含む適切なウェルに加えます。100 μLのビオチンワーキング溶液を、ニュートラアビジン結合マイクロスフェアを含む適切なウェルに加えます。
- マイクロタイタープレートをシールし、暗闇の中でRT(18-22°C)でオービタルシェーカー(650rpm)で1時間インキュベートします。
- 手順1.6.1〜1.6.5の説明に従って、マイクロスフィアを洗浄します。
- ACE2およびビオチン標識マイクロスフェアを、ステップ1.7.1-1.7.4で説明したように調製し、保存します。
注:ビオチン化ACE2ビーズおよびビオチン標識ビーズは、最終容量250μL(すなわち、2 ×10 3ビーズ/μL)で5×105番になります。
2. コンジュゲーション試験
- セクション1で作成した4種類のマイクロスフェア(ニュートラアビジン結合ビオチン化ACE2、ニュートラアビジン結合ビオチン化ACE2、ニュートラアビジン結合ビオチン、ヤギIgG結合、非結合)をすべて組み合わせて、ビーズ混合物を調製します。
注:ストックマイクロスフェアは、2 × 103 ビーズ/μLで保存し、ワーキングビーズ混合物の最終ビーズ濃度が各セットの40ビーズ/μLになるように組み合わせました。- 試験に必要な作業ビーズ混合物の容量(5 μL/反応)を計算し、ピペッティング損失に対応するための追加容量を確保します。各チューブを短時間ボルテックスし、計算した各ビーズ懸濁液の同量を新しい低タンパク質結合マイクロチューブに組み合わせます。ビーズ濃度は、各セットのビーズ400個/μLになりました。
- 結合したビーズ懸濁液を保存バッファー(各セット40個/μLの作業濃度)でさらに10倍に希釈することにより、ワーキングビーズ混合物を作成します。
注:最初に少量の作業ビーズ混合物を作成して、各IDのマイクロスフェア/μLの数を推定します。
- ミクロスフェアをヤギ抗ACE2抗体とインキュベートします。
- 5 μLの作業用ビーズ混合物を平底、ハーフウェル、96ウェルのマイクロタイタープレートの3ウェルにピペットで移します。
- ヤギ抗ACE2 (0.4 μg/mL をPBS-Tで希釈、 材料表) をマイクロタイタープレート内のビーズを含む3ウェルにそれぞれ50 μL加えます。マイクロタイタープレートを密封し、ボルテックスし、オービタルシェーカー(650rpm)でRT(18-22°C)で暗所で1時間インキュベートします。
- マイクロタイタープレートを233 × g で1分間パルスダウンし、1.6.2-1.6.4で説明されているようにPBS-Tでマイクロスフェアを3回洗浄します。
- 検出抗体とミクロスフェアをインキュベートします。
- 5 μLの作業用ビーズ混合物をマイクロタイタープレートの6つの新しいウェルにピペットで移します。
- PBS-Tを希釈剤として使用して、3本の別々の1.5 mLチューブに、抗ヤギIgG PE、抗マウスIgG PE、および抗ウサギIgG PEの各1 μg/mLを調製します。
- 検出混合物50μLをそれぞれ3つのウェルに加え、ステップ2.2の抗ACE2と同じウェルに抗ヤギIgGを添加します。
- オービタルシェーカー(650 rpm)でRT(18-22°C)で30分間、シールし、ボルテックスし、インキュベートします。
- プレートを233 × g で1分間パルスダウンし、1.6.2-1.6.4で説明されているように、PBS-Tでマイクロスフェアを3回洗浄します。
- 100 μL の PBS-T を添加し、以下の設定でデュアルレポーターフロー分析装置で実行します。
モード:デュアルレポーター。タイムアウト: 45 秒;DDゲーティング:7500-17500;最小マイクロスフィア数:100マイクロスフィア/セット(最小QCカットオフ:35マイクロスフェア/セット)。
3. SARS-CoV-2感染細胞上清の産生
SARS-CoV-2ウイルスは、宿主のVero E6細胞(サル腎臓上皮細胞株;ATCC; 資料の表)。Vero E6細胞は、5%のCO2 および95%の相対湿度雰囲気で、37°CのModified Eagles培地(MEM)で培養されます。MEMの各リットルには、10 mLのL-グルタミン(200 mM)、38 mLのNaHCO3 (7.5%)、5 mLのペニシリン/ストレプトマイシン溶液、および50 mLのウシ胎児血清(FCS)が補充されます。 資料の表。
注意: SARS-CoV-2を取り扱うときは、適切なバイオセーフティ手順と機器を使用してください。
- Vero E6細胞は、2つの150 cm2 組織培養フラスコでコンフルエントに増殖します。1つのフラスコをSARS-CoV-2ウイルスに感染させ、もう1つの偽感染物をコントロールとして使用します。
- 約100,000個の野生型(WT)SARS-CoV-2感染性粒子を5 mLのEagles MEM培地と混合します。
- 150 cm2 フラスコ 1 本から培地を吸引し、55 mL の完全 MEM を添加して、感染していないコントロール上清を生成します。他の150 cm2 フラスコから培地を吸引し、ウイルス混合物を細胞に加えます。細胞を37°Cで1時間インキュベートします。 15分ごとにフラスコを軽く振ってウイルスを配布します。
- SARS-CoV-2を添加したフラスコに50 mLの完全MEM培地を加え、細胞変性効果が観察されるまで細胞をインキュベートし、24時間ごとにフラスコを視覚的に評価します。
注:細胞障害が発生するには、感染後約3〜4日かかります。.Vero E6細胞単層構造に対する細胞変性の影響(例:細胞収縮、クリネーション、丸め、脱接着、細胞質内粒度の喪失、明白な溶解)は、 in vitro 細胞毒性試験に関する国際標準化機構のガイドラインに従って、倒立光学顕微鏡を使用して細胞を観察することにより定性的に評価されます16。 - 両方のフラスコから細胞上清を採取し、253 × g で6分間回転して細胞破片を沈殿させます。
- 細胞上清中のSARS-CoV-2ウイルスをUV不活化
- 24ウェルマイクロタイタープレートで12ウェルにウェルあたり0.5mLの上清をピペットで移します。蓋をせずに、適切な紫外線ランプの下で30秒間マイクロタイタープレートにUV照射します(材料表)。
注:細胞上清中のウイルス不活性化は、Vero E6細胞培養物でウイルス増殖を試みることによって確認する必要があります。
- 24ウェルマイクロタイタープレートで12ウェルにウェルあたり0.5mLの上清をピペットで移します。蓋をせずに、適切な紫外線ランプの下で30秒間マイクロタイタープレートにUV照射します(材料表)。
- 細胞上清を1.5 mLチューブに分注し、さらに使用するまで-20°Cで保存します。
注:細胞上清は-80°Cで保存できます。
4. アッセイ:細胞上清中のSARS-CoV-2ウイルス粒子の検出
- 0.1%カゼイン、0.5%ポリビニルアルコール、0.8%ポリビニルピロリドン、および1%BSA(すべてw/v)(pH 7)を混合して、アッセイバッファーを調製します。アッセイバッファーに10%ウサギIgGを調製して、サンプル希釈バッファーを調製します。
- 試験に必要な作業用ビーズ混合物の容量(ステップ2.1)を計算して調製し(5 μL/反応)、ピペッティング損失に対応するために余分な容量を確保します。
- 上清希釈シリーズを準備します。必要な上清の量を計算します。5 つの scFv のそれぞれについて、トリプリケートウェルの各希釈点をアッセイし、希釈点およびサンプルタイプごとに 15 ウェルになります。各ウェルに希釈した上清45μLを使用し、合計675μLが必要です。SARS-CoV-2およびコントロール上清のそれぞれに1.5 mLのマイクロチューブ1本で十分です。
- SARS-CoV-2上清を解凍し、上清を4°Cで少なくとも1時間制御します。上清とその希釈液は、使用するまで冷たく(2〜8°C)保管してください。SARS-CoV-2および制御上清のために、それぞれ8本の1.5 mLマイクロフュージチューブを標識して配置します。
注:アッセイされる最高濃度は1:1(2倍)上清希釈であり、そこからサンプル希釈バッファーを使用して一連の1:2(3倍)希釈が行われ、最高希釈は1:1458希釈です。Sample Dilution Bufferを含むブランクは、上清なしのコントロールとしてのみ機能します。したがって、各上清タイプ(SARS-CoV-2またはコントロール)の試験希釈は、バッファーのみのコントロールで2倍、6倍、18倍、54倍、162倍、486倍、1458倍になります。 - サンプル希釈バッファーを標識されたマイクロフュージチューブに加えます。1:1(2倍)希釈チューブには600μLのバッファーが必要で、残りのチューブには800μLのバッファーが必要です。上清600 μLとサンプル希釈バッファー600 μLを適切な標識チューブに組み合わせ、各上清の最高希釈率(1:1、2倍)を作成し、その後、チューブを短時間ボルテックスして混合します。
- 1:1 (2倍) に希釈した上清 400 μL を次の希釈チューブに順次移し (6倍希釈)、最低希釈 (1458倍) が作成されるまで 3 倍希釈を続けます。希釈した各上清を短時間ボルテックスしてから、次の希釈に進みます。
- SARS-CoV-2上清を解凍し、上清を4°Cで少なくとも1時間制御します。上清とその希釈液は、使用するまで冷たく(2〜8°C)保管してください。SARS-CoV-2および制御上清のために、それぞれ8本の1.5 mLマイクロフュージチューブを標識して配置します。
- ミクロスフェアを上清とインキュベートします。
- 事前に調製した作業用ビーズ混合物を30秒間ボルテックスしてミクロスフェアを再懸濁し、平底の384ウェルマイクロタイタープレートの各割り当てられたウェルに5μLのビーズ混合物を追加します。
注:96ウェルプレートも使用できます。 - 調製した上清希釈液45 μLを、384ウェルプレートのマイクロスフェアを含むウェルに添加します。プレートをシールし、オービタルシェーカー(650rpm)で暗闇の中でRT(18-22°C)で一晩(16-22時間)インキュベートします。
- 事前に調製した作業用ビーズ混合物を30秒間ボルテックスしてミクロスフェアを再懸濁し、平底の384ウェルマイクロタイタープレートの各割り当てられたウェルに5μLのビーズ混合物を追加します。
- ビーズから余分な上清を洗い流します。
- オービタルシェーカーからマイクロタイタープレートを取り外し、プレートシーラーを取り外します。プレートを931 x g で1分間遠心分離します。
- マイクロタイタープレートを磁気プレートセパレーターに30秒間置いて、ビーズを固定します。マイクロタイタープレートを磁気セパレーターに置いたまま、プレートシーラーを取り外し、磁石固定ビーズから上清を吸引します。
- マイクロタイタープレートをマグネティックセパレーターから取り外します。
- ビーズを含む各ウェルに60 μLのPBS-Tを添加します。
- 洗浄手順4.5.2〜4.5.4を2回繰り返し、合計3回のPBS-T洗浄を行います。
- 異なる検出ミックスを別々の1.5 mLチューブで調製します。各検出ミックスは、市販のヒトモノクローナル抗S1(Hu-anti-S1)抗体(1 μg/mL)と、SARS-CoV-2粒子上のスパイクタンパク質を標的とする5種類のFLAGタグ付きscFvs抗体(1 μg/mL)(補足ファイル1、補足表1、 補足表2)のうちの1つで構成され、アッセイバッファーで希釈(ステップ4.1)することで、合計5種類の検出ミックスが得られます。
- 手順4.5.2〜4.5.3を繰り返します。洗浄したマイクロスフィアを、適切なscFv特異的スパイク検出ミックスの50 μL/ウェルに再懸濁します。マイクロタイタープレートをシールし、暗闇の中でRT(18-22°C)でオービタルシェーカー(650rpm)で1時間インキュベートします。
- マイクロタイタープレートを931 × g で1分間遠心分離します。ビーズから余分なスパイク検出試薬を洗い流します。60μLのPBS-Tで、手順4.5.2-4.5.5に従って3回の洗浄を行います。
- ミクロスフェアを蛍光検出抗体混合物とインキュベートします。
- 市販のPE標識抗ヒトIgGとBV421標識抗FLAG抗体をアッセイバッファーで希釈した蛍光溶液ミックスを調製し、使用濃度はそれぞれ0.2 μg/mL、使用濃度は1 μg/mLです。1反応につき、50 μLの蛍光検出試薬が必要です。
- 手順4.5.2〜4.5.3を繰り返します。ミクロスフェアを50 μL/ウェルの蛍光溶液ミックスに再懸濁します。マイクロタイタープレートを密封し、暗闇の中で軌道シェーカー(650 rpm)でRT(18-22°C)で30分間インキュベートします。
- マイクロタイタープレートを931 × g で1分間スピンダウンします。ミクロスフェアから余分な蛍光溶液ミックスを洗い流します。60μLのPBS-Tで、手順4.5.2-4.5.5に従って3回の洗浄を行います。
- 最後の洗浄ステップから60μLのPBS-Tにミクロスフェアを懸濁します。デュアルレポーター流量分析システムで、ステップ2.6で説明した設定でプレートを分析します。
結果
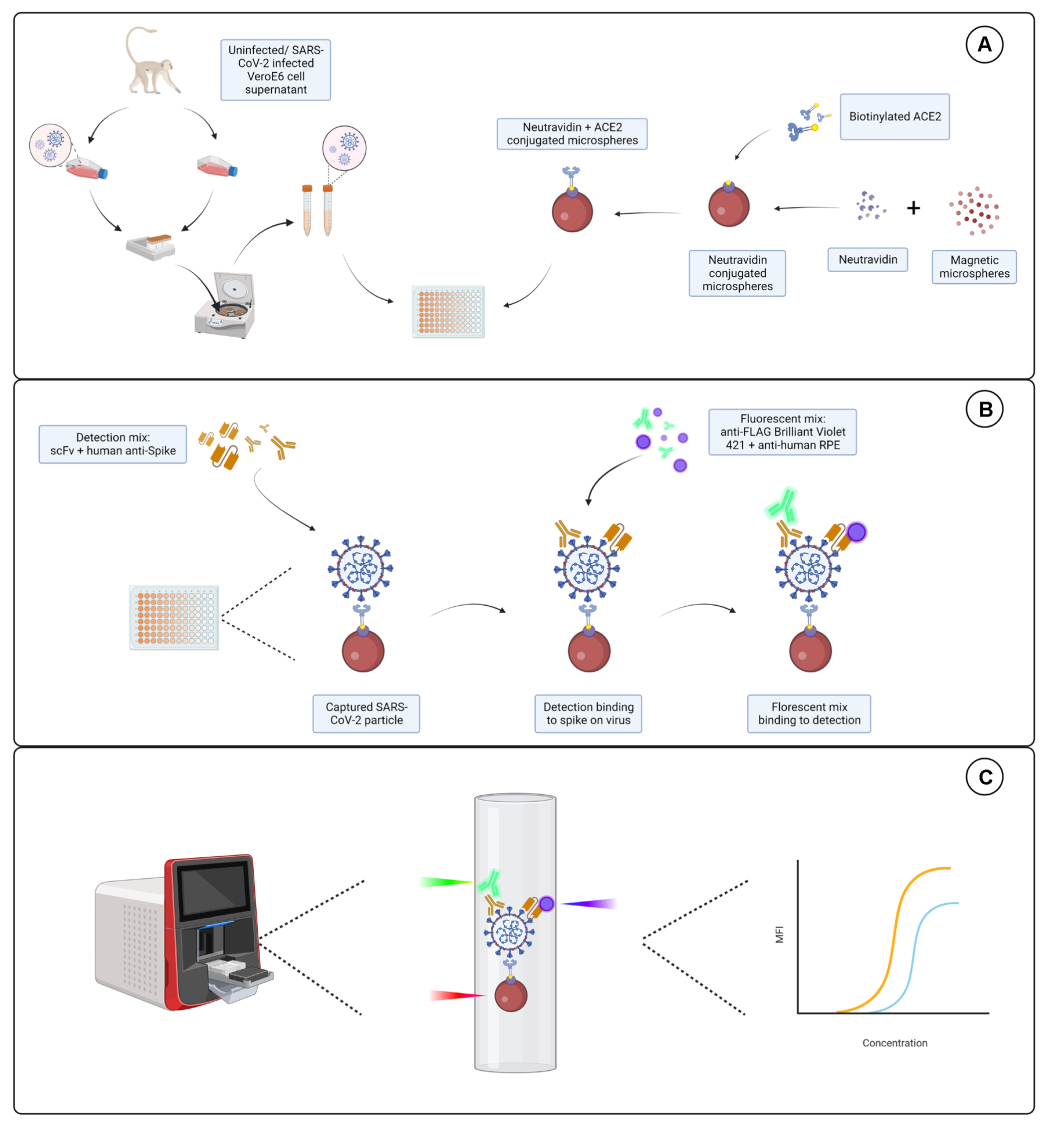
コンジュゲーションテスト
結合試験では、ヤギ-IgGおよびニュートラアビジン-ビオチン化ACE2がマイクロスフェアにうまく結合されることが示されました。アッセイ検出特異性は、ACE2標識マイクロスフェアを、異なる動物種で生成されたPE標識二次抗体でプローブすることにより確認しました(図2)。異なる検出抗体間の交差反応性は観察されませんでした。ビーズ混合物をヤギ抗ACE2 + 抗ヤギIgG PEでプローブしたところ、ACE2とヤギIgG標識マイクロスフェアの両方でバックグラウンドより上の蛍光強度中央値(MFI;任意の単位)が検出されましたが、非標識マイクロスフェア(ベア)またはビオチン被覆マイクロスフェアでは検出されませんでした。抗マウスIgG PEおよび抗ウサギIgG PEをネガティブコントロールとして使用して、偽陽性シグナルをチェックしました。マイクロスフェアとのインキュベーション時に生成された蛍光シグナルはごくわずかであり、ACE2とヤギIgGの正のシグナルが特異的であることを示しました。
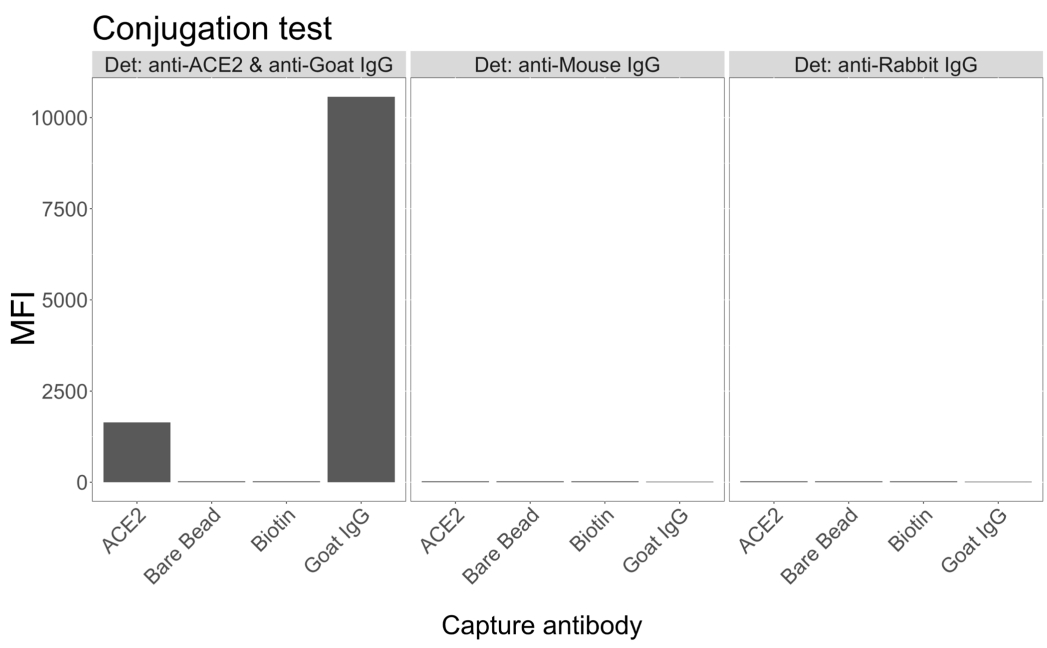
細胞上清中のウイルス粒子検出能
組換えヒトACE2に結合した磁気ビーズを使用して、感染した(ウイルスなしの)VeroE6細胞培養上清からSARS-CoV-2ウイルス粒子を捕捉し、次にモノクローナル抗体と5つの異なるscFvのうちの1つを使用して、2つの異なるウイルススパイク領域を同時にプローブしました。SARS-Cov-2に感染した細胞上清の希釈液中の濃度依存性シグナルが両方のレポーターチャネル(RP1およびRP2)で観察され(図3)、市販のHu-anti-S1抗体と異なるscFvの両方がACE2標識マイクロスフェアに結合したウイルス粒子を検出したことを示しています。5 つの scFv のうち 3 つで、ウイルスは 1:18 までの希釈で検出できます (scFv2、scFv3、scFv5)。残りの2つのscFv(scFv7およびscFv9)については、1:6希釈まで検出可能です。これは、ターゲットに対する親和性が異なることに起因している可能性があります。 図3 と 表1に示すように、scFv3が最もMFI強度が高く、次いでscFv5、scFv2、scFv7、scFv9の順となっています。
世界的に見ると、scFvs 検出は Hu-anti-S1 と比較して MFI が低くなります。これは親和性が低いことを示している可能性がありますが、異なる蛍光色素(PEおよびBV421)による標識によるアーティファクトである可能性もあります。scFv7 と scFv9 に見られる別の傾向は、RP1 チャネル (アンチスパイク) の MFI 値も他の 3 つのコンフィギュレーションと比較してわずかに低いことです。これは、scFvがACE2-Hu-anti-S1相互作用と交差反応または別の方法で干渉していることを示している可能性があり、RP2チャネルの信号が低いことも説明できます。RP1チャネルまたはRP2チャネルのいずれにおいても、非感染Vero E6細胞の上清からウイルス粒子は検出されませんでした。
ニュートラアビジン-ビオチン結合ミクロスフェア、ヤギ-IgGマイクロスフェア、および非結合マイクロスフェアは、ネガティブコントロールビーズとして使用されます。ウイルス粒子は、ACE2に結合した磁気マイクロスフェアで捕捉し、RP1レポーターチャネルで市販のヒト抗スパイクで、RP2レポーターチャネルで異なるscFvでテストしました(scFvは各パネルの左上に示されています)。感染したサンプルと感染していないサンプルのいずれにもウイルス粒子は検出されませんでした。
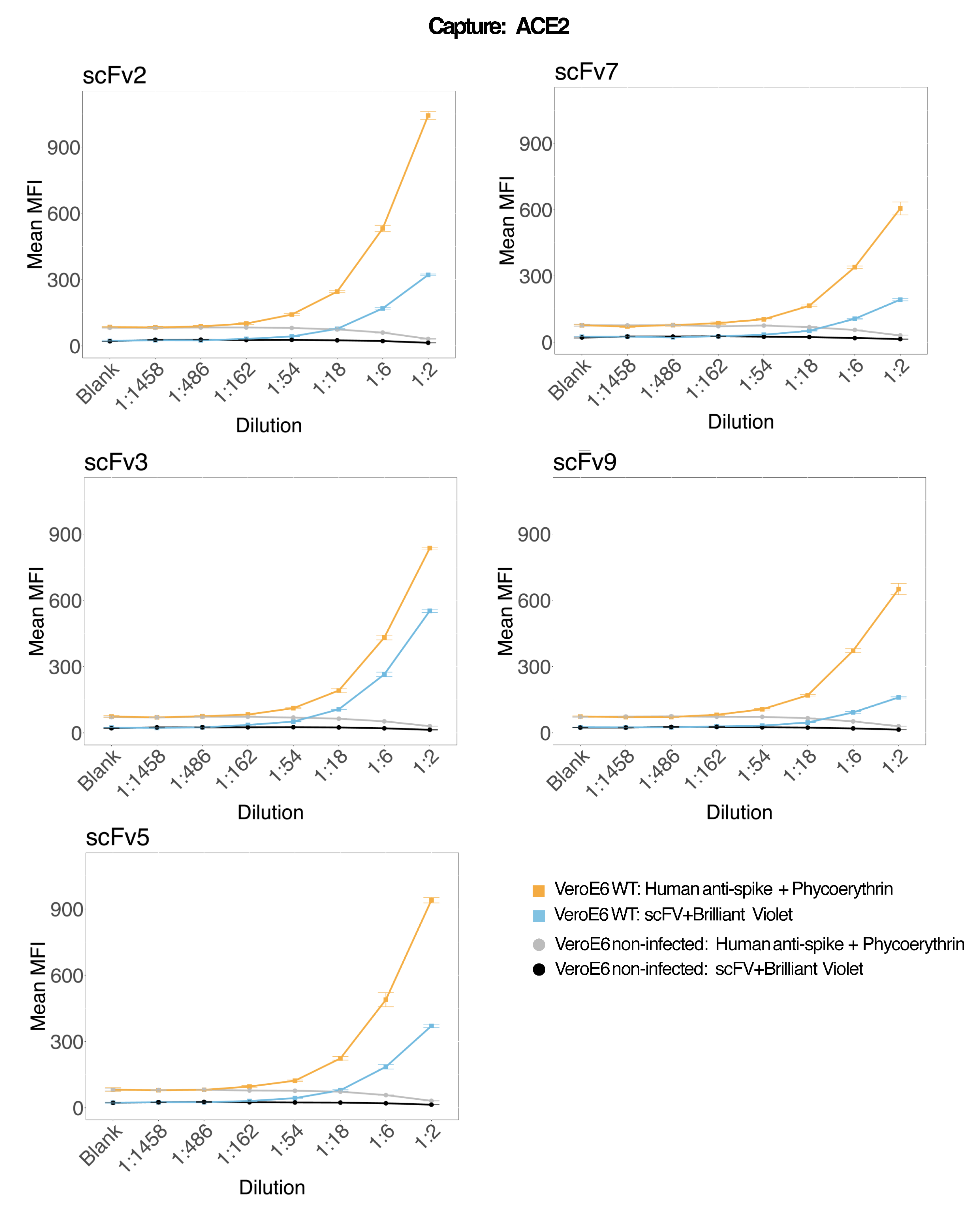
アッセイの精度と堅牢性
アッセイの精度を評価するために、すべての条件をトリプリケートで実行しました。ACE2 マイクロスフェアの分散係数 (CV) は、希釈点ごとに計算されました。アッセイで計算されたCVはすべて15%未満であり、測定された最高CVは13%、最低CVは1%でした(表2)。RP1チャネルの密度プロット(図4)からわかるように、市販のHu-anti-S1のPE検出は、主にCVが3%程度に集中し、より高い精度を示しています。RP2チャネルであるscFVのBV検出は、より高いCVを示しています。ただし、 表2 に見られるように、CVの範囲が広いのは、ブランクなどのウイルス粒子の濃度が低いサンプルによって駆動されます。プロトコールの頑健性をテストするために、アッセイは、異なる日に生成されたビーズ混合物とより少ないサンプル量(72%低い)を使用して、異なるオペレーターによって2回繰り返されました。RP1 チャネルと RP2 チャネルの両方で 0.98 から 1 の範囲の非常に良好なピアソン相関が観察され(p 値 < 0.01)、アッセイの堅牢性と、利用可能なサンプルが少ない場合にアッセイを適用できる可能性が確認されました(図 5)。このフロー分析技術は「アンビエント分析種理論」17に従っており、アッセイは濃度に敏感ですが、体積には敏感ではありません。

図1:ウイルス粒子アッセイ。 (A)感染したVero E6細胞と非感染したVero E6細胞の両方からの細胞上清を、ニュートラアビジンと結合した磁気マイクロスフェアとともに、96ウェルまたは384ウェルプレートのいずれかに段階希釈液で添加し、次いでビオチン化ヒトACE2またはビオチンのいずれかに結合させる。ヤギ-IgGおよび裸のマイクロスフェアと結合した非結合マイクロスフェアは、ニュートラアビジン-ビオチン結合マイクロスフェアとともにネガティブコントロールとして使用されます。(B)形成されたミクロスフェア-ウイルス粒子複合体は、Hu-anti-S1とFLAG-tagを持つ異なるscFvの1つからなる検出カクテルで検出される。次に、Hu-anti-S1を標的とする抗ヒトIgG PEと、scFvsを標的とする抗FLAGブリリアントバイオレット421を添加する蛍光混合物を添加します。(C)3レーザー、デュアル検出システムは、赤、緑、紫のレーザーを放射して微粒子複合体を検出します。赤色レーザーはマイクロスフィア色素ラベルを検出し、緑色レーザーと紫レーザーはそれぞれ抗S1とscFvsを検出します。その後、生成されたデータが分析されます。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図2:コンジュゲーション確認プロット。 ビーズ混合物は、ニュートラアビジン-ビオチン-ACE2(ACE2)、非結合ミクロスフェア(Bare Bead)、ニュートラアビジン-ビオチン(ビオチン)、およびヤギ-IgG(ヤギIgG)の4つの異なるマイクロスフェアIDで構成され、それぞれが異なるタンパク質と結合していました。標識試験では、3つの異なる形状の検出蛍光色素を使用しました。すなわち、ヤギ抗ACE2 +抗ヤギIgG PE、抗マウスIgG PE、および抗ウサギIgG PE。Y軸は、3つの異なる条件で各マイクロスフェアから測定された平均MFI(中央蛍光強度、任意の単位)信号を示しています。X軸は、適用されたさまざまな捕捉抗体を示しています。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図3:表面タンパク質のマルチプレックス検出。 Y軸:各サンプルの平均MFI(蛍光強度の中央値、標準偏差±任意の単位)で、条件ごとに3重のウェルで分析されます。X軸:細胞上清の段階希釈点。オレンジ:SARS-CoV-2 WTに感染したVero E6の上清中のウイルス粒子で、ヒト抗スパイク+抗ヒトPE(フィコエリスリン)で検出されました。青:SARS-CoV-2 WTに感染したVero E6の上清は、異なるscFvs + anti-FLAG Brilliant Violet 421で検出されました。灰色:ヒト抗スパイク+抗ヒトPEで検出された非感染細胞上清。黒:5種類のscFvs + 抗FLAG Brilliant Violet 421で検出された非感染細胞上清。ウイルス粒子は、ACE2に結合した磁気マイクロスフェアで捕捉され、RP1レポーターチャネルで市販のヒト抗スパイク抗体で、RP2レポーターチャネルで異なるscFvで試験されました(scFvは各パネルの左上に示されています)。感染していないサンプルのいずれにもウイルス粒子は検出されませんでした。scFv3が標的とするエピトープは、最も高い親和性を持っていました。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図4:ばらつき分散プロット。 Y 軸はイベントの頻度で、X 軸は異なるサンプルの各反復の分散係数 (CV) をパーセンテージで示します。RP1およびRP2は、それぞれフィコエリトリンおよびブリリアントバイオレット421に関連する蛍光を検出する第1および第2のレポーターチャネルです。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図5:相関行列の実行 (A,B) Y軸:3つの異なるオペレーターによって異なるビーズ混合物を使用して実行された、3つの別々の実行間のPearson相関行列(log10スケール)。3 回目の実行では、より少ないサンプル量を適用しました。ヒストグラムは、測定された MFI に基づくさまざまな変数クラスターの分布を示しています。(A)異なる実行間のRP1レポーターチャネルの相関関係。(B)異なる実行間のRP2レポーターチャネルの相関。MFI=任意の単位での蛍光強度の中央値。p < 0.001 です。この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
| 検出 | 反応 |
| scFv2の | ++ |
| scFv3の | +++ |
| scFv5の | ++ |
| scFv7 | + |
| scFv9 | + |
| ヒト抗スパイクIgG | ++++ |
表1:標準曲線で得られたMFI強度に基づく検出におけるscFvのランキング。
| RP1 (PE) | RP2 (BV421) | |
| サンプル希釈 | CV範囲 [%] | CV範囲 [%] |
| 空砲 | 3–11 | 2–13 |
| 1:1458 | 1–7 | 2–7 |
| 1:456 | 4–6 | 3–8 |
| 1:162 | 3–6 | 3–7 |
| 1:54 | 2–4 | 2–4 |
| 1:18 | 2–4 | 1–4 |
| 1:6 | 2–6 | 1–6 |
| 1:2 | 1–5 | 1–3 |
表2:RP1およびRP2レポーターチャネルの両方について、SARS-CoV-2感染上清の各希釈点のCV%(平均/標準偏差×100)範囲。
補足ファイル1:免疫グロブリン一本鎖可変フラグメント(scFv)の生成。このファイルをダウンロードするには、ここをクリックしてください。
補足表1:FabsとペアでscFvをリコンビナントスパイク(RBD)の段階希釈に対してスクリーニングします。 さまざまな検出ペプチドの性能を評価するために、スパイクタンパク質であるFabの12の組み合わせを、組換えRBDをスパイクしたバッファーの捕捉として使用しました。スパイクタンパク質の異なるエピトープを標的とする10のscFvを検出として適用しました。キャプチャと検出のペアのパフォーマンスに応じて、失敗 (-) または成功 (+) としてマークされました。 このファイルをダウンロードするには、ここをクリックしてください。
補足表2:SARS-Cov-2に感染したCalu-3細胞上清の段階希釈に対するFabsとペアのscFvのスクリーニング。 さまざまな検出ペプチドの性能を評価するために、SARS-Cov-2に感染したCalu-3細胞上清の捕捉として、スパイクタンパク質であるFabの12の組み合わせを使用しました。スパイクタンパク質の異なるエピトープを標的とする10のscFvを検出として適用しました。キャプチャと検出のペアのパフォーマンスに応じて、失敗 (-) または成功 (+) としてマークされました。 このファイルをダウンロードするには、ここをクリックしてください。
ディスカッション
ビーズベースのマルチプレックス技術は、多くの臨床アプリケーションにおいて、ハイスループットの病原体検出のための貴重なプラットフォームであることが示されています。フローサイトメトリーの原理に基づくこのプラットフォームの高い柔軟性により、抗体、タンパク質、核酸18、19、20、21、22を標的とし、数百の分析物を同時にマルチプレックス化することができます。しかし、私たちの知る限り、この技術はこれまで無傷のウイルス粒子を検出するために適用されていませんでした。このレポートでは、SARS-CoV-2の3つの独立した表面エピトープを標的とすることにより、無傷のウイルス粒子の検出にこの技術を適用しました。
エンベロープRNAウイルスは、宿主タンパク質23とともにウイルスのRNAとタンパク質を運ぶ小さなリン脂質膜である細胞外小胞(EV)と高い構造的類似性を示す。サンドイッチイムノアッセイは、2つの異なる表面タンパク質を標的とする抗体ペアを用いて、以前にEVの検出に適用されてきた24,25。サンドイッチアッセイでは、2つのタンパク質のみを同時に検出するという制限が取り除かれ、1回の反応で2つ以上のタンパク質を同時に検出できるマルチプレックスアプローチが可能になります。
ここで説明する3レーザーデュアルレポーター検出システムは、これまでで最も先進的なビーズベースのフロー分析装置です。シングルレポーター読み出しシステムに関しては、デュアルレポーター(RP1およびRP2チャンネル)により、3つの表面タンパク質/エピトープを並行して検出できます。複数のウイルス表面タンパク質とエピトープを標的とすることで、ウイルスタンパク質の負荷をより正確に表現することができ、ウイルスが実際に無傷であることを確認するだけでなく、ウイルス表面抗原やウイルスと宿主タンパク質の相互作用のメカニズムをさらに研究する機会も開きます。
COVID-19のパンデミック時には、ウイルスの拡散を封じ込める取り組みにおいて、活性ウイルス粒子を保有する個人を迅速に特定することの重要性が重要でした。ゲノムRNAは、その起源(無傷のウイルス粒子または遊離)に関係なく、定量的RT-PCRによって検出されます。しかし、アクセス可能なSタンパク質を持つ無傷のエンベロープのみが、細胞の侵入とその後のウイルス複製を媒介することができます。患者サンプルにマイクロ流体チップを使用した以前の研究では、無傷のウイルス粒子の検出とポイントオブケア検査を組み合わせることで、頻繁な検査が可能になり、病気の広がりの監視が強化されることが示されています。これには、隔離される個人のより多くの情報に基づいた選択が含まれます26。マルチプレックスマイクロスフェアベースのアッセイを適用することで、複数のウイルスとその表面抗原変異体のスクリーニングを目的としたアッセイの設計が可能になり、集団内でのウイルスの広がりをより正確に把握することができます。
フロービロメトリーは、ウイルス粒子の分析を目的としたフローサイトメトリーの最近の開発です。離散的なウイルス粒子を検出できるにもかかわらず、小さなウイルスの分析は、フローウイルスメトリー27,28の現在の問題を引き起こしています。ここで説明する方法と同様に、フロービロメトリーでは、抗体に結合した金ナノ粒子による無傷のビリオンの捕捉を行います。どちらの方法にも、(i)マイクロスフェアまたはナノ粒子を標的とする表面発現抗原の高親和性捕捉および検出試薬への依存、(ii)ウイルス粒子と細胞外小胞を区別する能力が限られていること、(iii)適切な粒子定量のための基準がないこと、などの制限があります。
細胞はEVを周囲に分泌し、ウイルスに感染すると、EVと同程度の大きさのビリオンも分泌し、最終的には同じ抗原を発現する可能性がある29。EVはウイルスと同様の膜組成を持つため、デュアルレーザーシングルレポーターアプローチなどの親和性ベースの方法だけでは、それらを互いに区別するのは難しいかもしれません。しかし、ここで説明する戦略は、より高いマルチプレックス容量を特徴としており、粒子のタンパク質組成をより広く、より深く研究することができます。フローベースの方法により、離散粒子の追跡が可能になり、デジタル定量化の機会が得られます。私たちの分析法で定量の問題に対処するための 1 つの戦略は、目的の抗原をウイルス様粒子(VLP)として発現する十分に特性評価された合成小胞を使用して標準曲線を調製することです。
SARS-CoV-2の宿主細胞への出入りの一般的な経路は、ウイルスと宿主細胞膜の相互作用を経由する2,15。このプロセスでは、宿主膜タンパク質がウイルス表面に取り込まれる可能性が高くなります。組み込まれた宿主タンパク質をスクリーニングすることで、感染経路を追跡し、さまざまなリスク患者の疾患経過を予測できる可能性があり、早期の治療決定が可能になります。また、研究室の異なるサンプルバッチ間でウイルスの特性評価を行うこともできます。これは、さまざまな特性がウイルス感染力の異なるレベルに関連しているかどうかをテストし、ウイルス表面タンパク質を標的とする抗体や薬物分子をスクリーニングすることで、さらに調査できます。
記載されている方法に関する重要な側面は、ウイルス上の標的タンパク質に対する捕捉試薬および検出試薬の親和性に依存していることです。したがって、アフィニティー試薬の選択は、アッセイ性能の決定要因となります。おそらく、複数のアフィニティー試薬をスクリーニングし、捕捉と検出のためにテストして、最も親和性の高い試薬を選択する必要があります。ここでは、10個のscFvおよび12個のFabフラグメントの性能を、組換えRBDおよびSARS-Cov-2に感染したCalu-3肺上皮細胞の上清からのウイルス粒子を用いて予備的に評価しました(VeroE6細胞は、その後のすべての研究で細胞毒性の培養/評価に使用されました)。FLAGタグ付きscFvs(補足表1 および 補足表2)を検出するために、Anti-FLAG PEを使用しました。次に、最も優れた性能を発揮する5つのscFvを選択し、市販のHu-anti-S1(表1)とともに、感染したVeroE6アフリカミドリザル腎臓上皮細胞からの上清に対するデュアルレポーターアッセイに適用しました。
このプロトコルの成功のもう一つの重要な要素は、マイクロスフェア結合のために選択された手順です。カップリング法は効率的であると同時に、タンパク質結合に関与するコンフォメーションエピトープまたはアミノ酸残基を無傷で修飾されていない状態に保つ必要があります。ここでは、EDC-NHS反応をニュートラアビジンを直接結合させるために適用し、前述のプロトコル30 とニュートラアビジン+ビオチン系を適応させて、組換えACE2を結合したマイクロスフェアに結合させた。代替のカップリング方法とその効率をテストおよび比較できます。最後に、異なる蛍光標識検出試薬(抗FLAG PE(フィコエリトリン)と抗FLAGブリリアントバイオレット421など)は、アッセイ感度に影響を与える可能性のある異なるMFIレベルをもたらす可能性があることが観察されました。
結論として、記載されている方法では、デュアルレポーター戦略を適用して、溶液中の無傷のウイルス粒子を検出できます。3つの表面決定要因を並行して分析することで、ウイルス粒子を特徴付け、最終的に他のEV(ウイルス抗原を含まないなど)と区別するためのより具体的なツールが提供されます。この戦略は、フロービロメトリーの代替手段です。現在のアプローチでは粒子サイズを区別しませんが、色分けされたマイクロスフェアを使用した磁気ビーズ戦略は、高マルチプレックスおよびハイスループット分析による表面抗原プロファイリングと実験デザインにおいてより広範な機能を提供します。このアッセイは高い精度と堅牢性を示しており、体液やその他の液体マトリックス中の表面抗原を露出するあらゆるタイプの細胞外小胞やその他のタイプの生体粒子の分析に拡張できます。これは、ウイルス粒子上の複数のタンパク質エピトープのマルチプレックス解析において、scFvsを1つの検出試薬として使用することの有用性を実証した概念実証研究でした。scFvを定量的または臨床目的で使用する場合、scFvの特定の特性(結合親和性、他の試薬や標的との交差反応性など)を決定するには、今後の研究が必要です。
開示事項
著者は、利益相反を宣言しません。
謝辞
スウェーデンのSciLifeLabでは、ここで説明した方法の開発と適用を行ったAffinity Proteomics-Stockholm Scilifelab Unitチーム、scFvsおよびFab試薬を提供したHuman Antibody Therapeutics Unit、臨床サンプル由来のSARS-CoV-2分離株に感染したVeroE6細胞を担当したJonas Klingströmに感謝します。著者らは、研究支援を提供してくださったLuminex Corporation(テキサス州オースティン)のSherry Dunbar博士(MBA)と、科学および執筆の支援を提供してくださったBiomedical Publishing Solutions(フロリダ州パナマシティ)のMatt Silverman MSci博士(mattsilver@yahoo.com)に感謝します。この研究は、クヌート・アンド・アリス・ウォレンバーグ財団とScience for Life Laboratory(SciLifeLab)からの資金提供を受けました(VC2020-0015はClaudia FredoliniとFrancesca Chiodiに、VC-2022-0028はClaudia Fredoliniに)。
資料
| Name | Company | Catalog Number | Comments |
| ACE2-Biotin | Acro Biosystems (Newark, DE) | AC2-H82E6-25 ug | Conc: 340 µg/mL, LOT#BV35376-203HFI-2128 |
| Anti-Goat IgG, PE-conjugated | Jackson ImmunoResearch (West Grove, PA) | 705-116-147 | Host species: Donkey |
| Anti-Human IgG R-PE | Life Technologies/Thermo Fisher (Waltham, MA) | H10104 | Conc: 0.15 mg/mL, LOT#2079224, Host species: Goat |
| Anti-Mouse IgG, PE-conjugated | Jackson ImmunoResearch (West Grove, PA) | 115-116-146 | Host species: Goat |
| Anti-Rabbit IgG, PE-conjugated | Jackson ImmunoResearch (West Grove, PA) | 111-116-144 | Host species: Goat |
| Biotin | Thermo-Fisher Scientific (Waltham, MA) | 20RUO | 100 mM, pH 10 Conc. 1 mg/mL |
| Blocker Casein in PBS | Thermo-Fisher Scientific (Waltham, MA) | 37528 | LOT#VD301372 |
| Blocker reagent for ELISA (BRE) | Roche (Basel, Switzerland) | 11112589001 | |
| Brilliant Violet 421 anti-DYKDDDDK Tag Antibody (Anti-FLAG) 0.2 mg/ml, rat IgG2a, λ | BioLegend (Amsterdam, The Netherlands) | 637321 | |
| Bovine serum albumin (BSA) | Saveen & Werner (Limhamn, Sweden) | B2000-500 | LOT#04D5865 |
| EDC (1-Ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride) | Proteochem (Hurricane, UT) | C1100-custom (65 mg) | LOT# MK3857 |
| Fetal calf serum (FCS) | Gibco/Thermo Fisher (Waltham, MA) | 10270-106 | |
| Goat anti-ACE2 polyclonal antibody | R&D Systems/Bio-Techne (Minneapolis, MN) | AF933 | Host species: Goat |
| Goat IgG | Bethyl Labs (Montgomery, TX) | P50-200 | LOT#P50-200-6 |
| L-glutamine | Thermo-Fisher Scientific (Waltham, MA) | 25030024 | |
| Low-bind 1.5 mL microfuge tubes | VWR (Radnor, PA) | 525-0133 | |
| MagPlex-C Microspheres | Luminex Corporation (Austin, TX) | MC10XXX-01 | |
| MEM tissue cuture media | Gibco/Thermo Fisher (Waltham, MA) | 21430-020 | |
| Microplate, 96-Well, Polystyrene, Half-area, Clear | Greiner Bio-One (Kremsmünster, Austria) | 675101 | |
| NaHCO3 | Gibco/Thermo Fisher (Waltham, MA) | 25080-060 | |
| Neutravidin | Thermo-Fisher Scientific (Waltham, MA) | 31000 | LOT#UK292857 |
| PBS tablets | Medicago AB (Uppsala, Sweden) | 09-9400-100 | LOT#272320-01 |
| Penicillin/Streptomycin | Gibco/Thermo Fisher (Waltham, MA) | 15140122 | |
| Poly(vinyl alcohol) | Sigma-Aldrich (St. Louis, MO) | 360627 | |
| Polyvinylpyrrolidone | Sigma-Aldrich (St. Louis, MO) | 437190 | |
| ProClin 300 | Sigma-Aldrich (St. Louis, MO) | 48915-U | |
| Rabbit IgG | Bethyl Labs (Montgomery, TX) | P120-301 | LOT#12 |
| scFv-FAb1 | In-house production | Human Antibody Therapeutics Unit, Scilifelab, Sweden. Monoclonal scFv. Conc: 0.12 mg/mL. | |
| scFv-FAb2 | In-house production | Human Antibody Therapeutics Unit, Scilifelab, Sweden. Monoclonal scFv. Conc batch1: 0.38 mg/mL. Conc batch2: 0.45 mg/mL | |
| scFv-FAb3 | In-house production | Human Antibody Therapeutics Unit, Scilifelab, Sweden. Monoclonal scFv. Conc: 0.34 mg/mL. | |
| scFv-FAb4 | In-house production | Human Antibody Therapeutics Unit, Scilifelab, Sweden. Monoclonal scFv. Conc: 2.85 mg/mL. | |
| scFv-FAb5 | In-house production | Human Antibody Therapeutics Unit, Scilifelab, Sweden. Monoclonal scFv. Conc:2.7mg/mL. | |
| SARS-CoV-2 infectious particles, Swedish isolate | In-house production | The Public Health Agency of Sweden | |
| SARS-CoV-2 Spike Antibody (Hu-anti-S1) | Novus Biologicals (Centennial, CO) | NBP2-90980 | Monoclonal antibody. Conc: 1 mg/mL. Host: Human. Clone: CR3022. Isotype: IgG1 Kappa. LOT#T201B06 |
| Sodium phosphate monobasic, anhydrous | Sigma-Aldrich (St. Louis, MO) | S3139 | |
| Sulfo-NHS (N-hydroxysulfosuccinimide) | Thermo-Fisher Scientific (Waltham, MA) | 24510 | LOT# XH321563 |
| Tween | Thermo-Fisher Scientific (Waltham, MA) | BP337-50 | LOT#194435 |
| Ultraviolet lamp | Vilber Lourmat GmbH (Eberhardzell, Germany) | VL-215.G | Wavelength = 254 nm; 2 × 15-watt bulbs |
| Vero E6 cells | ATCC (Manassus, VA) | CRL-1586 | |
| xMAP INTELLIFLEX DR-SE (dual-reporter flow instrument) | Luminex Corporation (Austin, TX) | INTELLIFLEX-DRSE-RUO |
参考文献
- Rey, F. A., Lok, S. M. Common features of enveloped viruses and implications for immunogen design for next-generation vaccines. Cell. 172 (6), 1319-1334 (2018).
- V'kovski, P., Kratzel, A., Steiner, S., Stalder, H., Thiel, V. Coronavirus biology and replication: implications for SARS-CoV-2. Nature Reviews Microbiology. 19 (3), 155-170 (2021).
- Burnie, J., et al. Flow virometry quantification of host proteins on the surface of HIV-1 pseudovirus particles. Viruses. 12 (11), 1296 (2020).
- Gentili, M., et al. Transmission of innate immune signaling by packaging of cGAMP in viral particles. Science. 349 (6253), 1232-1236 (2015).
- Modrow, S., Falke, D., Truyen, U., Schätzl, H. . Viruses: Definition, Structure, Classification. In Molecular Virology. , 163-181 (2013).
- Trinh, K. T. L., Do, H. D. K., Lee, N. Y. Recent advances in molecular and immunological diagnostic platform for virus detection: A review. Biosensors. 13 (4), 490 (2023).
- Zamora, J. L. R., Aguilar, H. C. Flow virometry as a tool to study viruses. Methods. 134-135, 87-97 (2018).
- Graham, H., Chandler, D. J., Dunbar, S. A. The genesis and evolution of bead-based multiplexing. Methods. 158, 2-11 (2019).
- Byström, S., et al. Affinity proteomic profiling of plasma for proteins associated to area-based mammographic breast density. Breast Cancer Research. 20 (1), 14 (2018).
- Rudberg, A. -. S., et al. SARS-CoV-2 exposure, symptoms and seroprevalence in healthcare workers in Sweden. Nature Communications. 11 (1), 5064 (2020).
- Liu, J., et al. Multiplex reverse transcription PCR Luminex assay for detection and quantitation of viral agents of gastroenteritis. Journal of Clinical Virology. 50 (4), 308-313 (2011).
- Gadsby, N. J., Hardie, A., Claas, E. C. J., Templeton, K. E. Comparison of the Luminex respiratory virus panel fast assay with in-house real-time PCR for respiratory viral infection diagnosis. Journal of Clinical Microbiology. 48 (6), 2213-2216 (2010).
- Lorenzen, E., et al. Multiplexed analysis of the secretin-like GPCR-RAMP interactome. Science Advances. 5 (9), (2019).
- Angeloni, S., Cameron, A., Pecora, N. D., Dunbar, S. A rapid, multiplex dual reporter IgG and IgM SARS-CoV-2 neutralization assay for a multiplexed bead-based flow analysis system. Journal of Visualized Experiments: JoVE. (170), e62487 (2021).
- Jackson, C. B., Farzan, M., Chen, B., Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nature Reviews Molecular Cell Biology. 23 (1), 3-20 (2022).
- ISO10993-5 Biological evaluation of medical devices - Part 5: Tests for in vitro cytotoxicity. International Standardization Organization Available from: https://nhiso.com/wp-content/uploads/2018/05/ISO-10993-5-2009.pdf (2009)
- Poetz, O., et al. Sequential multiplex analyte capturing for phosphoprotein profiling. Molecular & Cellular Proteomics. 9 (11), 2474-2481 (2010).
- Dunbar, S. A., Vander Zee, C. A., Oliver, K. G., Karem, K. L., Jacobson, J. W. Quantitative, multiplexed detection of bacterial pathogens: DNA and protein applications of the Luminex LabMAP system. Journal of Microbiological Methods. 53 (2), 245-252 (2003).
- Taniuchi, M., et al. Multiplex polymerase chain reaction method to detect Cyclospora, Cystoisospora, and Microsporidia in stool samples. Diagnostic Microbiology and Infectious Disease. 71 (4), 386-390 (2011).
- Wu, M., et al. High-throughput Luminex xMAP assay for simultaneous detection of antibodies against rabbit hemorrhagic disease virus, Sendai virus, and rabbit rotavirus. Archives of Virology. 164 (6), 1639-1646 (2019).
- Dias, D., et al. Optimization and validation of a multiplexed Luminex assay to quantify antibodies to neutralizing epitopes on human papillomaviruses 6, 11, 16, and 18. Clinical and Vaccine Immunology. 12 (8), 959-969 (2005).
- Opalka, D., et al. Simultaneous quantitation of antibodies to neutralizing epitopes on virus-like particles for human papillomavirus types 6, 11, 16, and 18 by a multiplexes lumina assay. Clinical and Diagnostic Laboratory Immunology. 10 (1), 108-115 (2003).
- Nolte-'T Hoen, E., Cremer, T., Gallo, R. C., Margolis, L. B. Extracellular vesicles and viruses: Are they close relatives. Proceedings of the National Academy of Sciences. 113 (33), 9155-9161 (2016).
- Ohmichi, T., et al. Quantification of brain-derived extracellular vesicles in plasma as a biomarker to diagnose Parkinson's and related diseases. Parkinsonism & Related Disorders. 61, 82-87 (2019).
- Ter-Ovanesyan, D., et al. Framework for rapid comparison of extracellular vesicle isolation methods. Elife. 10, e70725 (2021).
- Gamage, S. S. T., et al. Microfluidic affinity selection of active SARS-CoV-2 virus particles. Science Advances. 8 (39), (2022).
- Renner, T. M., Tang, V. A., Burger, D., Langlois, M. -. A. Intact viral particle counts measured by flow virometry provide insight into the infectivity and genome packaging efficiency of moloney murine leukemia virus. Journal of Virology. 94 (2), e01600-01619 (2020).
- Niraja, S., et al. A flow virometry process proposed for detection of SARS-CoV-2 and large-scale screening of COVID-19 cases. Future Virology. 15 (8), 525-532 (2020).
- Lippé, R. Flow virometry: A powerful tool to functionally characterize viruses. Journal of Virology. 92 (3), e01765 (2018).
- Drobin, K., Nilsson, P., Schwenk, J. M. Highly multiplexed antibody suspension bead arrays for plasma protein profiling. Methods in Molecular Biology. 1023, 137-145 (2013).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved