
Method Article
Profilage d’épitopes de protéines de surface sur des particules virales par stratégie de double rapporteur multiplex
Dans cet article
Résumé
Ici, nous décrivons un immunodosage fluorescent multiplex nouvellement développé qui utilise un système de cytométrie en flux à double rapporteur pour détecter simultanément deux épitopes uniques de protéine de pointe sur des particules virales intactes du coronavirus 2 du syndrome respiratoire aigu sévère sévère (SRAS-CoV-2) qui avaient été capturées par des microsphères magnétiques couplées à l’enzyme de conversion de l’angiotensine 2.
Résumé
Les protéines membranaires des virus enveloppés jouent un rôle important dans de nombreuses fonctions biologiques impliquant l’attachement du virus aux récepteurs cellulaires cibles, la fusion de particules virales aux cellules hôtes, les interactions hôte-virus et la pathogenèse de la maladie. De plus, les protéines membranaires virales présentes sur les particules virales et présentées à la surface des cellules hôtes se sont avérées être d’excellentes cibles pour les antiviraux et les vaccins. Nous décrivons ici un protocole permettant d’étudier les protéines de surface sur des particules intactes du coronavirus 2 du syndrome respiratoire aigu sévère (SRAS-CoV-2) à l’aide du système de cytométrie en flux à double rapporteur. Le test exploite la technologie multiplex pour obtenir une triple détection des particules virales par trois réactions d’affinité indépendantes. Des billes magnétiques conjuguées à l’enzyme de conversion de l’angiotensine-2 humaine recombinante (ACE2) ont été utilisées pour capturer les particules virales du surnageant des cellules infectées par le SRAS-CoV-2. Ensuite, deux réactifs de détection marqués à la R-phycoérythrine (PE) ou au Brilliant Violet 421 (BV421) ont été appliqués simultanément. En tant que preuve de concept, des fragments d’anticorps ciblant différents épitopes de la protéine de surface Spike (S1) du SRAS-CoV-2 ont été utilisés. La détection de particules virales par trois réactions d’affinité indépendantes apporte une forte spécificité et confirme la capture de particules virales intactes. Des courbes dose-dépendance du surnageant cellulaire infecté par le SRAS-CoV-2 ont été générées avec des variances de coefficient de réplication (moyenne/ET) ˂14%. Les bonnes performances de dosage dans les deux canaux ont confirmé que deux épitopes protéiques cibles à la surface du virus sont détectables en parallèle. Le protocole décrit ici pourrait être appliqué pour (i) le profilage à haut multiplex et à haut débit des protéines de surface exprimées sur des virus enveloppés ; ii) détection de particules virales intactes actives ; et (iii) l’évaluation de la spécificité et de l’affinité des anticorps et des médicaments antiviraux pour les épitopes de surface des antigènes viraux. L’application peut être potentiellement étendue à tout type de vésicules extracellulaires et de bioparticules, exposant des antigènes de surface dans des fluides corporels ou d’autres matrices liquides.
Introduction
Les virus pathogènes les plus courants, tels que la grippe, le VIH, le cytomégalovirus humain et les souches du SRAS-CoV, sont des virus enveloppés. L’infection cellulaire par des virus enveloppés nécessite la fusion des membranes virales et des cellules hôtes, ce qui entraîne la libération du génome viral dans le cytoplasme. L’ARN viral se répliquera ensuite avant d’être emballé dans une nouvelle particule virale 1,2. Au cours de ces processus, non seulement les protéines virales, mais aussi les protéines membranaires de l’hôte peuvent être incorporées dans l’enveloppe, devenant ainsi une partie intégrante de la nouvelle particule virale. Les protéines membranaires de la cellule hôte incorporées dans l’enveloppe du virus peuvent faciliter l’entrée du virus dans une nouvelle cellule hôte, en exploitant les mécanismes des interactions cellule-cellule, du guidage et de l’échappement du système immunitaire 3,4.
Malgré l’importance de l’étude des protéines associées aux virus, la plupart des techniques actuellement disponibles pour l’analyse des virus5 ne permettent pas une caractérisation à haut débit et à haut multiplex de l’antigène de surface du virus. Ils ne sont pas non plus capables de détecter des particules virales individuelles ou de faire la distinction entre les particules virales infectieuses intactes, l’ARN non infectieux, les protéines virales et les sous-populations virales exprimant des antigènes différents. Récemment, la cytométrie en flux a été modifiée et adaptée en une nouvelle méthode d’analyse des particules virales, à savoir la virométrie en flux. La virométrie en flux permet d’étudier des particules virales uniques et leurs antigènes de surface. Cependant, des limites telles qu’un faible débit, une faible capacité de multiplexage, une configuration expérimentale et une analyse de données compliquées et une détectabilité limitée des particules virales de petite taille demeurent 6,7.
La quantification multiplexée des protéines et des acides nucléiques basée sur les microsphères est une technologie bien établie avec de nombreuses applications allant de la quantification des protéines dans les fluides corporels, aux études d’interaction protéine-protéine et au diagnostic des infections virales 8,9,10,11,12,13 . Un instrument d’analyse de flux récemment introduit est doté d’un double canal rapporteur, permettant de mesurer deux molécules rapporteures fluorescentes dans le même puits de réaction. Cette nouvelle capacité s’est avérée particulièrement utile pour le profilage parallèle de différents isotypes14 de l’immunoglobuline. Ici, il est décrit comment le système à double rapporteur peut être utilisé pour détecter des particules virales intactes, en ciblant plusieurs antigènes de surface en parallèle.
En tant que preuve de concept, ce rapport détaille le développement d’un système de triple détection des particules du virus SARS-CoV-2. Le SRAS-CoV-2 se compose de quatre protéines principales, l’une est la protéine de pointe (S), qui se compose de deux sous-unités. La première sous-unité, S1, effectue la liaison primaire à l’ACE2 exprimé dans les membranes cellulaires humaines. La deuxième sous-unité, S2, facilite l’entrée dans la cellule cible par un peptide de fusion, créant un pore dans la membrane cellulaire cible dans lequel le virion peut pénétrer par15. Les trois éléments constitutifs restants du SRAS-CoV-2 sont la nucléocapside (N), la protéine membranaire (M) et la protéine d’enveloppe (E). La nucléocapside est responsable de l’emballage du génome viral en formant des structures ribonucléoprotéiques avec l’ARN, tandis que les protéines de membrane et d’enveloppe jouent un rôle central dans l’assemblage du virus.
Le test décrit ici cible trois épitopes indépendants de la sous-unité S1 exprimés à la surface de l’enveloppe du SARS-CoV-2. Des dilutions en série de surnageants cellulaires infectés et non infectés par le SRAS-CoV-2 sont utilisées. Les particules virales sont capturées via des microsphères conjuguées à l’ACE2 qui se lient à la sous-unité S1 du virus. La protéine S du virus de surface est ensuite détectée en parallèle avec un fragment variable à chaîne unique (scFv) d’immunoglobuline marqué dans le commerce et un anticorps monoclonal anti-S1 humain (Hu-anti-S1) ainsi qu’un scFv marqué FLAG développé en interne. Le Hu-anti-S1 est détecté par le premier canal (RP1) dans le système à double rapporteur avec l’anticorps secondaire IgG-Fc humain conjugué à la R-phycoérythrine (PE) orange, et le scFv est détecté par le deuxième canal (RP2) avec un anticorps secondaire anti-FLAG conjugué au bleu Brilliant Violet 421 (BV421). Le dosage des particules virales est représenté à la figure 1.
Protocole
1. Conjugaison de la neutravidine et des anticorps de contrôle aux microsphères magnétiques
REMARQUE : Des billes magnétiques teintées par fluorescence (microsphères de polystyrène de 6,5 m de diamètre avec magnétite intégrée) avec différentes étiquettes fluorescentes, énumérées dans le Tableau des matériaux , sont utilisées pour générer les conjugués et les témoins de billes suivants : (1) ACE2 humain recombinant biotinylé lié à des billes couplé à un agent de liaison de neutravidines ; (2) la biotine liée aux billes couplée à un agent de liaison de neutravidine ; (3) IgG de chèvre couplées directement aux billes ; et (4) les perles non conjuguées. La protéine à coupler aux billes doit être exempte d’azoture de sodium, d’albumine sérique bovine (BSA), de glycine, de tris(hydroxy-méthyl)aminométhane (Tris), de glycérol ou d’additifs contenant des amines. Le tampon d’activation est de 0,1 M de phosphate de sodium monobasique, anhydre (NaH2PO4), pH 6. L’acide 2-morpholinoéthanesulfonique (MES ; 50 mM) Un tampon de pH 5 est utilisé pour diluer les conjugués. Le tampon de lavage est PBS-T (1x PBS [solution saline tamponnée au phosphate], pH 7,4 + 0,05 % (v/v) Tween-20). Le tampon de stockage est un réactif bloquant pour ELISA (BRE) de 2,7 mg/mL + 0,1 % d’antibiotiques (ici, ProClin 300).
- Retirer la poudre de sulfo-N-hydroxysulfosuccinimide (NHS) du réfrigérateur et 65 mg de chlorhydrate de 1-éthyl-3-[3-diméthylaminopropyl]carbodiimide (EDC) pré-aliquote du congélateur et laisser revenir à température ambiante (RT ; 18-22 °C) pendant 30 min. Conservez le NHS et l’EDC dans une enveloppe contenant des billes de silice au cours de cette étape pour prévenir l’hydrolyse due à l’humidité atmosphérique.
- Préparez les microsphères pour l’activation et le couplage.
REMARQUE : Les colorants fluorescents à l’intérieur des microsphères sont sensibles à la lumière, et les billes doivent être conservées dans l’obscurité et à la température du réfrigérateur (4-8 °C) lorsqu’elles ne sont pas utilisées activement.- Remettez en suspension 4 stocks différents de microsphères codées par couleur (12,5 x 106/mL) (Table des matériaux) en vortexant, soniquant ou tournant brièvement (15 min à 15-30 tr/min), selon la fiche d’information du produit.
- Transférez 40 μL de chaque suspension de billes (5 x 105 microsphères) dans les puits assignés d’une plaque de microtitration à demi-puits, à fond plat et à 96 puits (Tableau des matériaux).
- Lavez les perles magnétiques.
REMARQUE : Les étapes de lavage peuvent être effectuées manuellement ou à l’aide d’un laveur de plaques automatisé.- Ajouter un tampon d’activation de 80 μL/puits aux billes et immobiliser les billes sur un séparateur à plaques magnétiques pendant 30 s. Aspirez le surnageant des microsphères pendant que les billes sont immobilisées sur le séparateur de plaques magnétiques.
- Retirez la plaque de microtitration du séparateur de plaques magnétiques et remettez les billes en suspension dans 50 μL de tampon d’activation.
- Activez les perles avec Sulfo-NHS et EDC.
- Préparez la solution de travail Sulfo-NHS à 50 mg/mL dans un tampon d’activation dans un tube de microfuge de 1,5 mL. Remettre la poudre NHS au réfrigérateur (4-8 °C), à l’abri de l’humidité.
- Préparez une solution de travail EDC à 50 mg/mL dans le tampon d’activation dans son tube de microfuge de 1,5 mL. Dissoudre les aliquotes préfabriquées de 65 mg de poudre EDC dans 1,3 mL de tampon d’activation.
REMARQUE : Sulfo-NHS et EDC commencent à s’hydrolyser et à perdre de leur activité lorsqu’ils sont dissous. Évitez d’interrompre la procédure d’accouplement jusqu’à ce que le NHS et l’EDC aient été ajoutés aux billes. Ne conservez pas les solutions NHS ou EDC dissoutes pour une utilisation ultérieure. - Préparez la solution d’activation pour l’activation des billes en combinant volumétriquement la solution mère Sulfo-NHS à 20 % (50 mg/mL), la solution mère EDC à 20 % (50 mg/mL) et le tampon d’activation à 60 %. Une solution d’activation de 50 μL est nécessaire pour chaque réaction d’activation de bille (en utilisant 5 × 105 billes/réaction), ainsi qu’un volume supplémentaire suffisant pour compenser les pertes de pipetage.
- Ajouter 50 μL de solution d’activation complète dans chaque puits contenant des billes lavées. Avec le volume de suspension de billes préexistant de 50 μL dans le tampon d’activation, par puits, la concentration finale de Sulfo-NHS sera de 5 mg/mL, et la concentration finale d’EDC sera également de 5 mg/mL.
- Scellez la plaque de réaction des microsphères à l’aide d’un scelleur jetable en plastique adhésif ou en feuille d’aluminium et incubez pendant 20 min sur un agitateur orbital (650 tr/min) à température ambiante (18-22 °C) dans l’obscurité.
- Lavez l’excès de solution d’activation des billes.
- Centrifuger la plaque de microtitration à 233 × g pendant 1 min.
- Immobiliser les billes activées sur un séparateur à plaques magnétiques pendant 30 s. Retirez la scelleuse de plaques et aspirez le surnageant des billes immobilisées par aimant avec la plaque de microtitration toujours positionnée sur le séparateur magnétique.
- Retirez la plaque de microtitration du séparateur magnétique et ajoutez 100 μL de tampon MES dans chaque puits.
- Répétez les étapes 1.4.2 à 1.4.3 une fois de plus pour un total de deux lavages.
- Coupler la neutravidine et les IgG de chèvre (témoin) aux ensembles de billes appropriés. Préparez suffisamment de solutions de travail pour les neutravidines et les IgG de chèvre, en prévoyant 100 μL/réaction et suffisamment de solution supplémentaire pour compenser les pertes par pipetage, comme suit :
REMARQUE : La poudre de protéine de neutravidine est reconstituée avec de l’eau ultrapure, puis diluée dans une solution mère de 1 mg/mL avec du PBS avant d’être aliquote pour le stockage/l’utilisation (la protéine de neutravidine n’est pas directement soluble dans le PBS mais est soluble dans l’eau à ~10 mg/mL).- Préparer une solution de travail de neutravidine à une concentration de 125 μg/mL dans un tampon MES dans un tube microfuge à faible liaison protéique de 1,5 mL.
- Préparer une solution de travail pour les anticorps de contrôle IgG de chèvre à une concentration de 17,5 μg/mL dans le tampon MES.
- Préparez la plaque de microtitration contenant les billes activées. Perles d’immobilisation sur un séparateur à plaques magnétiques pendant 30 s. Avec la plaque de microtitration toujours positionnée sur le séparateur magnétique, aspirez le surnageant des billes immobilisées par aimant.
- Ajouter 100 μL de solution de travail de neutravidine (125 μg/mL) dans les puits appropriés contenant des billes (pour le couplage neutravidine-biotine et neutravidine-ACE2).
- Ajouter 100 μL de solution de travail des IgG de chèvre (17,5 μg/mL) dans le puits contenant les billes désignées comme témoins IgG de chèvre seulement.
- Ajouter 100 μL de tampon MES dans le puits désigné comme billes non conjuguées de contrôle.
- Scellez la plaque de microtitration et incubez pendant 2 h sur un agitateur orbital (650 tr/min) à RT (18-22 °C) dans l’obscurité. Agitez brièvement la plaque après 1 h d’incubation pour vous assurer que les billes restent en suspension.
- Laver les perles avec PBS-T.
- Centrifuger la plaque de microtitration à 233 × g pendant 1 min.
- Immobiliser les billes couplées sur un séparateur à plaques magnétiques pendant 30 s. Avec la plaque de microtitration toujours positionnée sur le séparateur magnétique, aspirez le surnageant des billes immobilisées par aimant.
- Retirez la plaque de microtitration du séparateur magnétique.
- Ajouter 100 μL de PBS-T dans chaque puits contenant des billes.
- Répétez les étapes de lavage 1.6.2-1.6.4 une fois pour un total de deux lavages avec PBS-T.
- Préparez des perles conjuguées pour le stockage.
- Immobiliser les billes couplées sur un séparateur à plaques magnétiques pendant 30 s. Avec la plaque de microtitration toujours positionnée sur le séparateur magnétique, aspirez le surnageant des billes immobilisées par aimant. Retirez la plaque de microtitration du séparateur magnétique.
- Ajoutez 50 μL de tampon de stockage à chaque ID de microsphère pour éteindre l’activité restante des billes.
- Incuber la plaque de microtitration à la température du réfrigérateur (4-8 °C) dans l’obscurité pendant la nuit (16-22 h).
- Transférez les suspensions de billes non conjuguées et conjuguées aux IgG de chèvre (50 μL) dans des tubes de microfuge à faible teneur en protéines de 1,5 mL correctement étiquetés, combinés à deux rinçages de tampon de stockage de 100 μL des puits pour assurer une récupération maximale des billes.
REMARQUE : Les billes non conjuguées et les billes conjuguées aux IgG de chèvre seront toutes deux au nombre de 5 × 105 dans un volume final de 250 μL (c’est-à-dire 2 × 103 billes/μL). Conservez les tubes microfuge à la température du réfrigérateur (4-8 °C) jusqu’à utilisation.
- Lient l’ACE2 biotinylée et la biotine aux billes conjuguées aux neutravidines.
- Préparer la solution de travail d’ACE2 biotinylée humaine recombinante à 18 μg/mL d’ACE2 dans 10 mM de PBS. Par réaction, 100 μL seront nécessaires. Préparez la solution de travail de biotine à 2,4 mg/mL de biotine dans 10 mM de PBS. Par réaction, 100 μL seront nécessaires.
- Préparez la plaque de microtitration contenant les microsphères conjuguées aux neutravidines.
- Immobiliser les microsphères sur un séparateur à plaques magnétiques pendant 30 s. Avec la plaque de microtitration toujours positionnée sur le séparateur magnétique, retirez la scelleuse de plaques et aspirez le surnageant des microsphères immobilisées par aimant.
- Retirez la plaque de microtitration du séparateur magnétique et ajoutez 50 μL de 10 mM de PBS/puits.
- Répétez les étapes 1.8.2.1 à 1.8.2.2 une fois.
- Ajouter 100 μL de la solution de travail biotinylée-ACE2 dans les puits appropriés contenant des microsphères conjuguées aux neutravidines. Ajouter 100 μL de la solution de travail de biotine dans les puits appropriés contenant des microsphères conjuguées aux neutravidines.
- Scellez la plaque de microtitration et incubez pendant 1 h sur un agitateur orbital (650 tr/min) à RT (18-22 °C) dans l’obscurité.
- Lavez les microsphères comme décrit aux étapes 1.6.1 à 1.6.5.
- Préparez et stockez les microsphères conjuguées à l’ACE2 et à la biotine comme décrit aux étapes 1.7.1 à 1.7.4.
REMARQUE : Les billes biotinylées ACE2 et conjuguées à la biotine seront toutes deux au nombre de 5 × 105 dans un volume final de 250 μL (c’est-à-dire 2 × 103 billes/μL).
2. Test de conjugaison
- Préparez un mélange de billes en combinant les quatre types de microsphères créées dans la section 1 (c’est-à-dire l’ACE2 biotinylée conjuguée aux neutravidines, la biotine conjuguée aux neutravidines, la biotine conjuguée aux IgG de chèvre et l’ACE2 non conjuguée).
REMARQUE : Les microsphères mères ont été stockées à 2 × 103 billes/μL et combinées de sorte que la concentration finale de billes dans le mélange de billes de travail est de 40 billes de chaque ensemble/μL.- Calculez le volume du mélange de billes de travail nécessaire pour l’essai (5 μL/réaction), en laissant le volume supplémentaire pour compenser les pertes de pipetage. Vortex brièvement chaque tube et combinez des volumes calculés égaux de chaque suspension de bille dans un nouveau tube de microfuge à faible liaison protéique. La concentration des billes est maintenant de 400 billes de chaque ensemble/μL.
- Créez le mélange de billes de travail en diluant la suspension de billes combinée 10 fois plus avec le tampon de stockage (40 de chaque ensemble/μL de concentration de travail).
REMARQUE : Préparez d’abord une petite quantité de mélange de billes de travail pour estimer le nombre de microsphères/μL pour chaque ID.
- Incuber les microsphères avec l’anticorps anti-ACE2 de chèvre.
- Pipeter 5 μL du mélange de billes de travail dans 3 puits d’une plaque de microtitration à fond plat, demi-puits et 96 puits.
- Ajouter 50 μL d’anti-ACE2 de chèvre (0,4 μg/mL dilué dans du PBS-T, Table des matériaux) chacun dans les 3 puits contenant des billes dans la plaque de microtitration. Scellez la plaque de microtitration, le vortex et incubez sur un agitateur orbital (650 tr/min) à RT (18-22 °C) pendant 1 h dans l’obscurité.
- Administrer des impulsions sur la plaque de microtitration à 233 × g pendant 1 min et laver les microsphères trois fois avec du PBS-T comme décrit aux points 1.6.2 à 1.6.4.
- Incuber des microsphères avec des anticorps de détection.
- Pipeter 5 μL du mélange de billes de travail dans 6 nouveaux puits de la plaque de microtitration.
- Préparez 1 μg/mL de chacun des mélanges de détection fonctionnels : IgG PE anti-chèvre, PE IgG anti-souris et IgG PE anti-lapin dans 3 tubes séparés de 1,5 mL, en utilisant du PBS-T comme diluant.
- Ajoutez 50 μL des mélanges de détection dans 3 puits chacun, et l’IgG anti-chèvre est ajouté dans les mêmes puits que l’anti-ACE2 à partir de l’étape 2.2.
- Scellez, vortex et incubez sur un agitateur orbital (650 tr/min) à RT (18-22 °C) pendant 30 min.
- Pulser la plaque à 233 × g pendant 1 min et laver les microsphères trois fois avec du PBS-T comme décrit aux points 1.6.2 à 1.6.4.
- Ajoutez 100 μL de PBS-T et exécutez l’appareil sur l’instrument d’analyse de flux à double rapporteur avec les paramètres suivants :
Mode : Double rapporteur ; Temps mort : 45 s ; DD-gating : 7500-17500 ; Nombre minimum de microsphères : 100 microsphères/ensemble (seuil de CQ le plus bas : 35 microsphères/ensemble).
3. Production de surnageant cellulaire infecté par le SRAS-CoV-2
Le virus SARS-CoV-2 se propage dans les cellules hôtes Vero E6 (lignée cellulaire épithéliale du rein de singe ; ATCC ; Table des matériaux). Les cellules Vero E6 sont cultivées dans un milieu Modified Eagles (MEM) à 37°C dans une atmosphère à 5% de CO2 et 95% d’humidité relative. Chaque litre de MEM est complété par 10 mL de L-glutamine (200 mM), 38 mL de NaHCO3 (7,5 %), 5 mL de solution de pénicilline/streptomycine et 50 mL de sérum fœtal bovin (SCA) ; Table des matériaux.
ATTENTION : Utilisez des procédures et des équipements de biosécurité appropriés lors de la manipulation du SRAS-CoV-2.
- Les cellules Vero E6 sont cultivées jusqu’à la confluence dans deux flacons de culture tissulaire de150 cm 2 . Infectez une fiole avec le virus SARS-CoV-2 et utilisez l’autre fictive infectée comme contrôle.
- Mélangez environ 100 000 particules infectieuses de SARS-CoV-2 de type sauvage (WT) avec 5 ml de milieu MEM Eagles.
- Aspirer le milieu d’une fiole de 2 cm de150 cm et ajouter 55 mL de MEM complet pour générer des surnageants témoins non infectés. Aspirez le milieu de l’autre fiole de150 cm et ajoutez le mélange de virus dans les cellules. Incuber les cellules pendant 1 h à 37 °C. Agitez légèrement la fiole toutes les 15 minutes pour répartir le virus.
- Ajouter 50 ml de milieu MEM complet dans le flacon auquel le SARS-CoV-2 a été ajouté et incuber les cellules jusqu’à ce que des effets cytopathiques soient observés, en évaluant visuellement les flacons toutes les 24 h.
REMARQUE : Il devrait falloir environ 3-4 jours après l’infection pour que la cytopathie se produise. Les effets cytopathiques sur la structure monocouche des cellules Vero E6 (p. ex., rétraction cellulaire, crénation, arrondissement, désadhésion, perte de granularité intracytoplasmique, lyse manifeste) sont évalués qualitativement en observant les cellules à l’aide d’un microscope à lumière inversée, conformément aux lignes directrices de l’Organisation internationale de normalisation pour les essais de cytotoxicité in vitro 16. - Prélever le surnageant cellulaire dans les deux flacons et faire tourner pendant 6 min à 253 × g pour sédimenter les débris cellulaires.
- Virus SARS-CoV-2 inactivé par les UV dans le surnageant cellulaire
- Pipeter 0,5 mL de surnageant par puits dans 12 puits dans une plaque de microtitration de 24 puits. Irradier la plaque de microtitration, sans couvercle, pendant 30 s sous une lampe ultraviolette appropriée (Tableau des matériaux).
REMARQUE : L’inactivation virale dans le surnageant cellulaire doit être vérifiée en tentant de propager le virus dans des cultures de cellules Vero E6.
- Pipeter 0,5 mL de surnageant par puits dans 12 puits dans une plaque de microtitration de 24 puits. Irradier la plaque de microtitration, sans couvercle, pendant 30 s sous une lampe ultraviolette appropriée (Tableau des matériaux).
- Aliquote le surnageant cellulaire dans des tubes de 1,5 mL et conserver à -20 °C jusqu’à nouvel ordre.
REMARQUE : Le surnageant cellulaire peut être stocké à -80 °C.
4. Essai : Détection des particules virales du SRAS-CoV-2 dans le surnageant cellulaire
- Préparez le tampon de dosage en mélangeant 0,1 % de caséine, 0,5 % d’alcool polyvinylique, 0,8 % de polyvinylpyrrolidone et 1 % de BSA (tout p/v) (pH 7). Préparez le tampon de dilution de l’échantillon en préparant 10 % d’IgG de lapin dans le tampon de dosage.
- Calculer et préparer le volume du mélange de billes de travail (étape 2.1) nécessaire pour l’essai (5 μL/réaction), en laissant le volume excédentaire pour compenser les pertes de pipetage.
- Préparer la série de dilution surnageante. Calculez les volumes surnageants nécessaires. Analysez chaque point de dilution dans des puits en trois exemplaires pour chacun des cinq scFv, ce qui donne 15 puits par point de dilution et par type d’échantillon. Utiliser 45 μL de surnageants dilués dans chaque puits, pour un total de 675 μL requis ; un seul tube de microfuge de 1,5 mL pour chacun des surnageants du SRAS-CoV-2 et des surnageants de contrôle est suffisant.
- Décongeler les surnageants du SRAS-CoV-2 et les surnageants témoins à 4 °C pendant au moins 1 h. Conservez les surnageants et leurs dilutions au froid (2-8 °C) jusqu’à leur utilisation. Étiquetez et disposez huit tubes de microfuge de 1,5 ml chacun pour le SRAS-CoV-2 et les surnageants de contrôle.
REMARQUE : La concentration la plus élevée mesurée sera une dilution surnageante de 1:1 (2x), à partir de laquelle une série de dilutions de 1:2 (3x) sera effectuée à l’aide d’un tampon de dilution d’échantillon, la dilution la plus élevée étant une dilution de 1:1458. Les blancs contenant un tampon de dilution de l’échantillon ne servent que de contrôles sans surnageant. Ainsi, les dilutions testées de chaque type de surnageant (SARS-CoV-2 ou contrôle) seront de 2, 6, 18, 54, 162, 486 et 1458 fois, avec un contrôle tampon uniquement. - Ajoutez un tampon de dilution d’échantillon dans les tubes de microfuge étiquetés. Les tubes de dilution 1:1 (2 fois) nécessitent 600 μL de tampon, et les autres tubes nécessitent 800 μL de tampon. Créez la dilution la plus élevée (1:1 ; 2 fois) de chaque surnageant en combinant 600 μL du surnageant avec 600 μL de tampon de dilution d’échantillon dans des tubes étiquetés de manière appropriée, puis en faisant brièvement tourbillonner le tube pour le mélanger.
- Poursuivre la série en transférant séquentiellement 400 μL des surnageants dilués 1:1 (2 fois) dans le tube de dilution suivant (c.-à-d. dilution 6 fois), et continuer les dilutions 3 fois jusqu’à ce que la dilution la plus faible (1458 fois) ait été créée. Vortex brièvement chaque surnageant dilué avant de procéder à la dilution suivante.
- Décongeler les surnageants du SRAS-CoV-2 et les surnageants témoins à 4 °C pendant au moins 1 h. Conservez les surnageants et leurs dilutions au froid (2-8 °C) jusqu’à leur utilisation. Étiquetez et disposez huit tubes de microfuge de 1,5 ml chacun pour le SRAS-CoV-2 et les surnageants de contrôle.
- Incuber les microsphères avec le surnageant.
- Vortex le mélange de billes de travail pré-préparé pendant 30 s pour remettre les microsphères en suspension et ajouter 5 μL du mélange de billes dans chaque puits assigné d’une plaque de microtitration à fond plat de 384 puits.
REMARQUE : Des plaques à 96 puits peuvent également être utilisées. - Ajouter 45 μL des dilutions surnageantes préparées aux puits désignés contenant des microsphères dans la plaque à 384 puits. Scellez la plaque et incubez pendant une nuit (16-22 h) sur un agitateur orbital (650 tr/min) à RT (18-22°C) dans l’obscurité.
- Vortex le mélange de billes de travail pré-préparé pendant 30 s pour remettre les microsphères en suspension et ajouter 5 μL du mélange de billes dans chaque puits assigné d’une plaque de microtitration à fond plat de 384 puits.
- Laver l’excès de surnageant des billes.
- Retirez la plaque de microtitration de l’agitateur orbital et retirez la scelleuse de plaques. Centrifuger la plaque à 931 x g pendant 1 min.
- Immobiliser les billes en plaçant la plaque de microtitration sur un séparateur de plaques magnétiques pendant 30 s. Avec la plaque de microtitration toujours positionnée sur le séparateur magnétique, retirez la scelleuse de plaques et aspirez le surnageant des billes immobilisées par aimant.
- Retirez la plaque de microtitration du séparateur magnétique.
- Ajouter 60 μL de PBS-T dans chaque puits contenant des billes.
- Répétez les étapes de lavage 4.5.2-4.5.4 deux fois pour un total de trois lavages PBS-T.
- Préparez les différents mélanges de détection dans des tubes séparés de 1,5 ml. Chaque mélange de détection se compose d’un anticorps monoclonal anti-S1 (Hu-anti-S1) commercial (1 μg/mL) et de l’un des cinq scFv différents marqués FLAG (1 μg/mL) (fichier supplémentaire 1, tableau supplémentaire 1 et tableau supplémentaire 2) ciblant la protéine de spicule sur la particule du SRAS-CoV-2, dilué dans un tampon de dosage (étape 4.1), ce qui donne un total de cinq mélanges de détection différents.
- Répétez les étapes 4.5.2 à 4.5.3. Remettre en suspension les microsphères lavées dans 50 μL/puits du mélange de détection de pointes spécifique au scFv approprié. Scellez la plaque de microtitration et incubez pendant 1 h sur un agitateur orbital (650 tr/min) à RT (18-22 °C) dans l’obscurité.
- Centrifuger la plaque de microtitration à 931 × g pendant 1 min. Laver l’excédent de réactif de détection de pointes des billes. Effectuez trois étapes de lavage avec 60 μL de PBS-T conformément aux étapes 4.5.2-4.5.5.
- Incuber des microsphères avec un mélange d’anticorps de détection fluorescente.
- Préparez un mélange de solution fluorescente composé d’IgG anti-humaines conjuguées au PE disponibles dans le commerce et d’anticorps anti-FLAG conjugués au BV421 dilués dans le tampon de dosage, avec des concentrations de travail de 0,2 μg/mL et 1 μg/mL, respectivement. Par réaction, 50 μL de réactif de détection fluorescente sont nécessaires.
- Répétez les étapes 4.5.2 à 4.5.3. Remettre les microsphères en suspension dans 50 μL/puits de mélange de solution fluorescente. Scellez la plaque de microtitration et incubez pendant 30 min sur un agitateur orbital (650 tr/min) à RT (18-22 °C) dans l’obscurité.
- Tourner la plaque de microtitration à 931 × g pendant 1 min. Lavez l’excès de mélange de solution fluorescente des microsphères. Effectuez trois étapes de lavage avec 60 μL de PBS-T conformément aux étapes 4.5.2-4.5.5.
- Suspendre les microsphères dans 60 μL de PBS-T à partir de la dernière étape de lavage. Analysez la plaque sur un système d’analyse de flux à double rapporteur avec les paramètres décrits à l’étape 2.6.
Résultats
Test de conjugaison
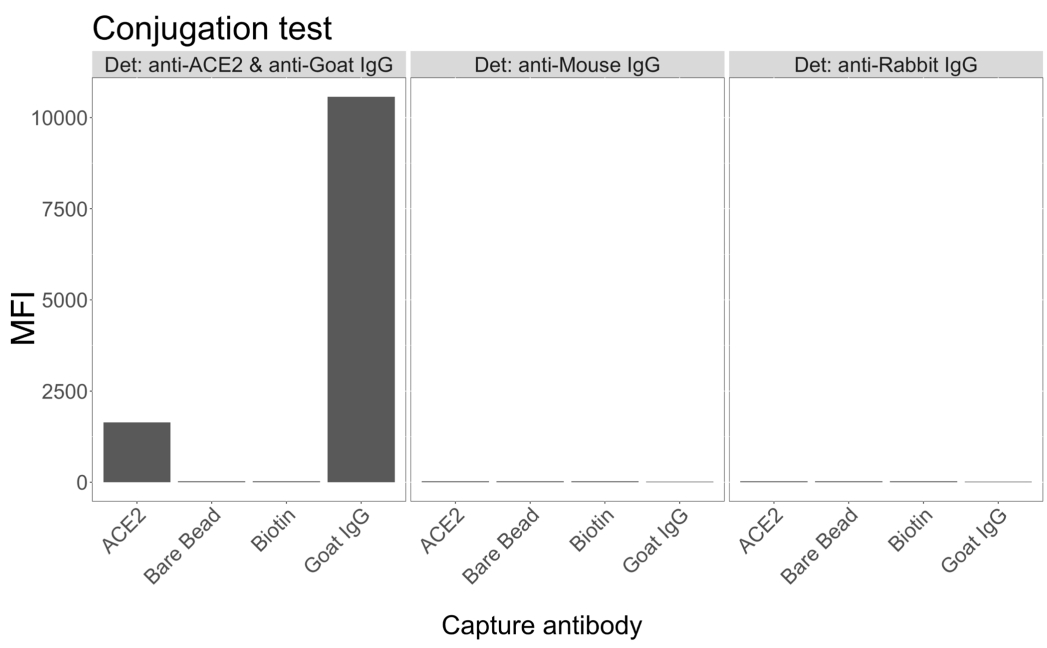
Le test de conjugaison a montré que les IgG de chèvre et l’ACE2 biotinylée par les neutravidines ont été conjuguées avec succès aux microsphères. La spécificité de détection du test a été confirmée en sondant des microsphères conjuguées à l’ACE2 avec des anticorps secondaires marqués au PE générés chez différentes espèces animales (Figure 2). Aucune réactivité croisée entre les différents anticorps de détection n’a été observée. Lorsque les mélanges de billes ont été sondés avec des IgG PE anti-ACE2 + anti-chèvre, une valeur médiane d’intensité de fluorescence (MFI ; unités arbitraires) supérieure au bruit de fond a été détectée pour les microsphères conjuguées aux IgG ACE2 et caprins, mais pas pour la microsphère non conjuguée (nue) ou pour les microsphères enrobées de biotine. L’IgG PE anti-souris et l’IgG PE anti-lapin ont été utilisés comme contrôles négatifs pour vérifier les signaux faussement positifs. Un signal de fluorescence négligeable a été généré lors de l’incubation avec les microsphères, indiquant que les signaux positifs pour l’ACE2 et les IgG de chèvre étaient spécifiques.
Détectabilité des particules virales dans les surnageants cellulaires
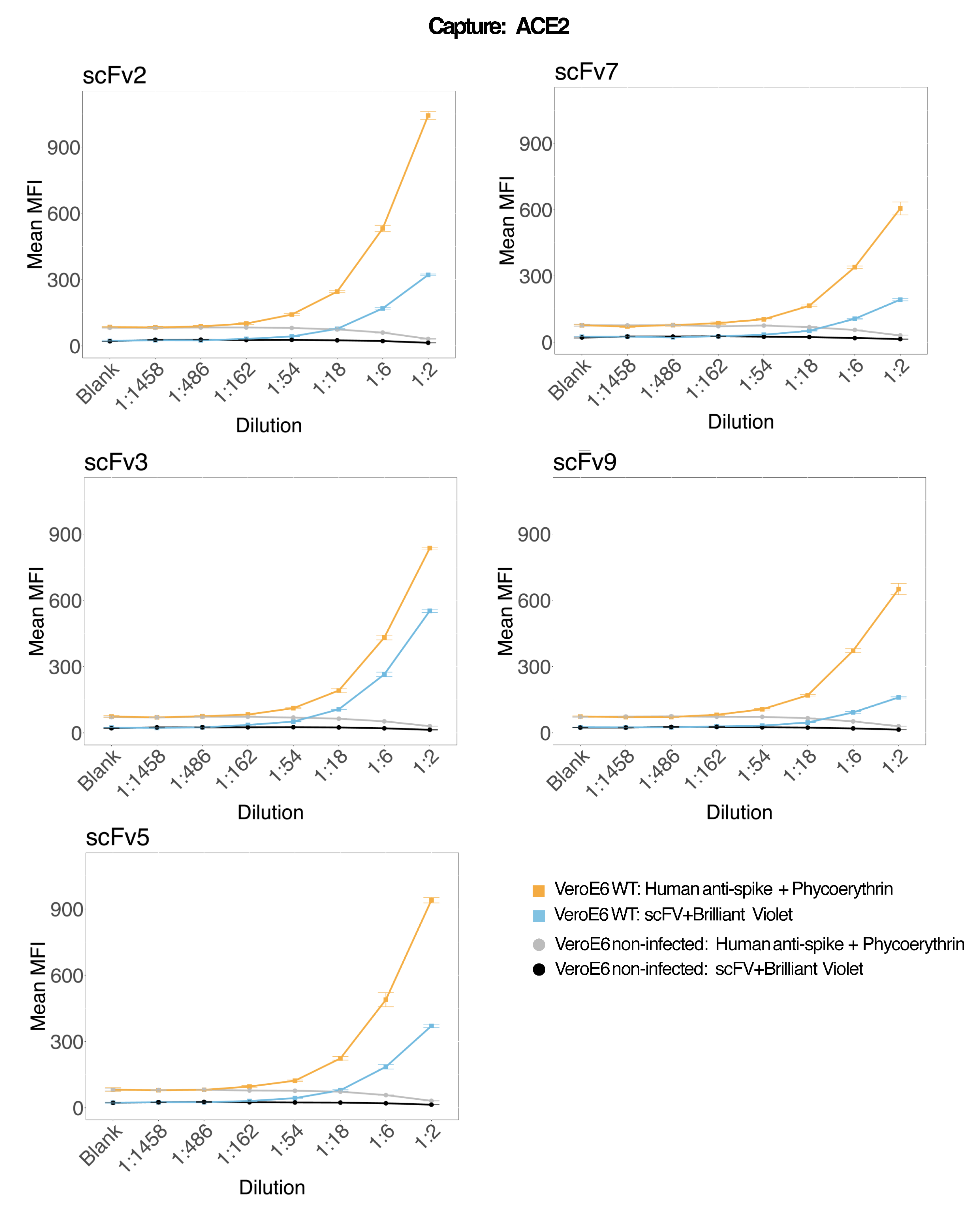
Des billes magnétiques couplées à de l’ACE2 humain recombinant ont été utilisées pour capturer des particules virales du SRAS-CoV-2 à partir de surnageants de culture cellulaire VeroE6 infectés et témoins (sans virus), puis ont été sondées simultanément pour deux régions de spicule virales distinctes à l’aide d’un anticorps monoclonal et de l’un des cinq scFv distincts. Un signal dépendant de la concentration dans les dilutions des surnageants cellulaires infectés par le SRAS-Cov-2 a été observé dans les deux canaux rapporteurs (RP1 et RP2) (Figure 3), indiquant que l’anticorps commercial Hu-anti-S1 et les différents scFvs ont détecté la particule virale liée à la microsphère conjuguée à l’ACE2. Avec trois scFv sur cinq, le virus est détectable dans des dilutions allant jusqu’à 1:18 (scFv2, scFv3, scFv5) ; pour les deux scFv restants (scFv7 et scFv9), il est détectable jusqu’à des dilutions de 1:6. Cela pourrait être attribuable à une affinité différente pour la cible. Comme le montrent la figure 3 et le tableau 1, scFv3 fournit l’intensité MFI la plus élevée, suivi respectivement par scFv5, scFv2, scFv7 et scFv9.
À l’échelle mondiale, la détection des scFvs se traduit par un MFI inférieur à celui du Hu-anti-S1. Cela pourrait indiquer une affinité plus faible, mais il pourrait également s’agir d’un artefact dû au marquage avec différents colorants fluorescents (PE et BV421). Une autre tendance que l’on peut observer pour scFv7 et scFv9 est que les valeurs MFI sont légèrement inférieures pour le canal RP1 (anti-pointe) par rapport aux trois autres configurations. Cela pourrait indiquer que les scFv réagissent de manière croisée ou interfèrent d’une autre manière avec l’interaction ACE2-Hu-anti-S1, ce qui pourrait également expliquer le signal plus faible dans le canal RP2. Aucune particule virale n’a été détectée dans le surnageant des cellules Vero E6 non infectées, que ce soit dans le canal RP1 ou RP2.
La microsphère conjuguée neutravidine-biotine, la microsphère chèvre-IgG et les microsphères non conjuguées sont utilisées comme billes de contrôle négatives. Les particules virales ont été capturées avec des microsphères magnétiques couplées à l’ACE2 et testées avec un anti-spicule humain commercial dans le canal rapporteur RP1 et avec différents scFv dans le canal rapporteur RP2 (scFv est indiqué en haut à gauche de chaque panneau). Aucune particule virale n’a été détectée dans les échantillons infectés et non infectés.
Précision et robustesse du dosage
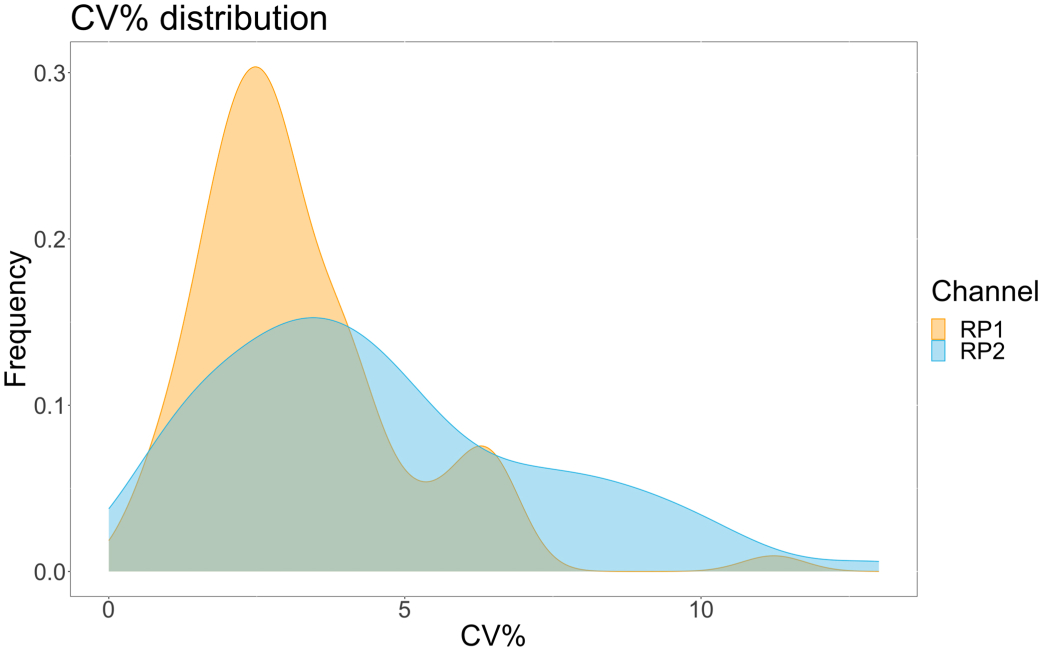
Pour évaluer la précision du dosage, toutes les conditions ont été exécutées en trois exemplaires. Un coefficient de variance (CV) pour la microsphère ACE2 a été calculé pour chaque point de dilution. Tous les CV calculés pour l’essai étaient inférieurs à 15 %, le CV mesuré le plus élevé étant de 13 % et le CV le plus bas étant de 1 % (tableau 2). Comme on peut le voir sur le graphique de densité (Figure 4) du canal RP1, la détection PE du commercial Hu-anti-S1 montre une précision plus élevée, principalement concentrée autour d’un CV de 3%. Le canal RP2, détection BV des scFV, présente des CV plus élevés. Cependant, comme le montre le tableau 2, la fourchette supérieure de CV est déterminée par les échantillons contenant de faibles concentrations de particules virales, comme le blanc. Pour tester la robustesse du protocole, le test a été répété deux fois par différents opérateurs, en utilisant des mélanges de billes générés à des jours différents et un volume d’échantillon plus faible (72 % plus faible). Une très bonne corrélation de Pearson, variant entre 0,98 et 1, a été observée pour les canaux RP1 et RP2 (valeur p < 0,01), confirmant la robustesse du test et la possibilité d’appliquer le test lorsque moins d’échantillon est disponible (Figure 5). Cette technologie d’analyse de flux suit la « théorie de l’analyte ambiant »17, ce qui rend le test sensible à la concentration mais pas au volume.

Figure 1 : Le dosage des particules virales. (A) Des cellules surnageantes de cellules Vero E6 infectées et non infectées sont ajoutées en dilution en série à une plaque de 96 puits ou de 384 puits, avec des microsphères magnétiques conjuguées à la neutravidine, puis couplées à de l’ACE2 humain biotinylé ou à de la biotine. Les microsphères non conjuguées couplées aux IgG de chèvre et aux microsphères nues sont utilisées comme témoins négatifs avec la microsphère conjuguée neutravidine-biotine. (B) Les complexes de particules microsphère-virus qui se sont formés sont détectés à l’aide d’un cocktail de détection composé de Hu-anti-S1 et de l’un des différents scFv avec FLAG-tag. Un mélange fluorescent avec des IgG PE anti-humaines ciblant le Hu-anti-S1 et un anti-FLAG Brilliant Violet 421 ciblant les scFvs est ensuite ajouté. (C) Le système à trois lasers à double détection émet un laser rouge, vert et violet pour détecter le complexe de microparticules. Le laser rouge détecte l’étiquette du colorant à microsphères, tandis que les lasers vert et violet détectent respectivement l’anti-S1 et le scFvs. Les données générées sont ensuite analysées. Veuillez cliquer ici pour voir une version agrandie de cette figure.
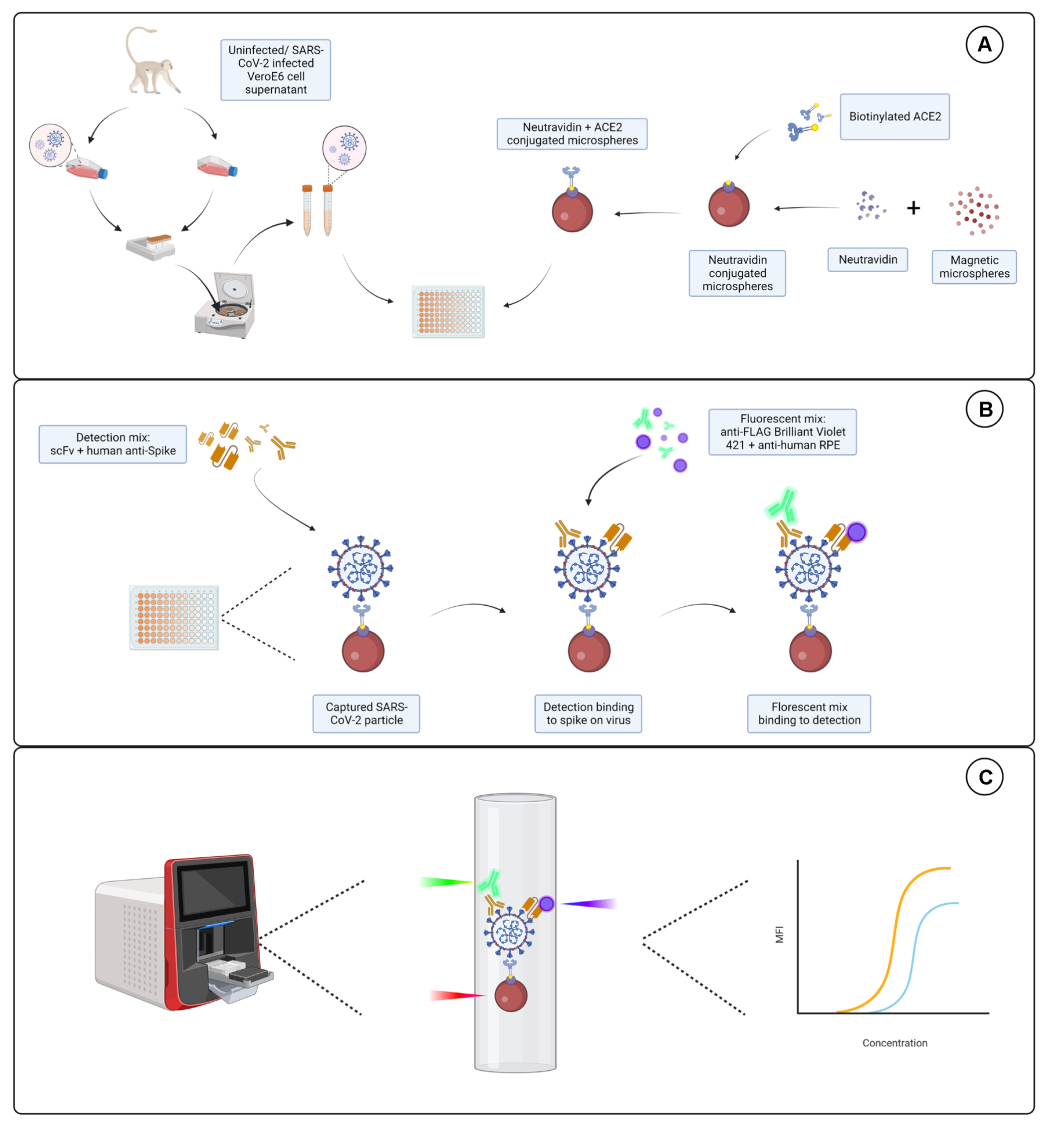{kind=link}

Figure 2 : Graphique de confirmation de la conjugaison. Les mélanges de billes étaient constitués de quatre ID de microsphères différents, chacun conjugué à une protéine différente : neutravidine-biotine-ACE2 (ACE2), microsphère non conjuguée (Bare Bead), neutravidine-biotine (biotine) et IgG de chèvre (IgG de chèvre). Dans le test de conjugaison, trois configurations différentes de fluorophores de détection ont été utilisées. À savoir, l’IgG PE anti-ACE2 + anti-chèvre, l’IgG PE anti-souris et l’IgG PE anti-lapin. L’axe Y montre le signal MFI (intensité de fluorescence médiane ; unités arbitraires) moyen mesuré de chaque microsphère dans trois conditions différentes. L’axe des X montre les différents anticorps de capture appliqués. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 3 : Détection multiplexée de protéines de surface. Axe Y : MFI moyen (intensité médiane de fluorescence ; unités arbitraires ± l’écart-type) pour chaque échantillon, analysé en trois puits par condition. Axe X : Points de dilution en série du surnageant cellulaire. Orange : Particules virales dans le surnageant de Vero E6 infecté par le SARS-CoV-2 WT détectées avec un anti-spike humain + PE (phycoérythrine) anti-humain. Bleu : Surnageant de Vero E6 infecté par le SARS-CoV-2 WT détecté avec les différents scFvs + anti-FLAG Brilliant Violet 421. Gris : Surnageant cellulaire non infecté détecté par l’anti-spike humain + le PE anti-humain. Noir : Surnageant cellulaire non infecté détecté avec les cinq différents scFvs + anti-FLAG Brilliant Violet 421. Les particules virales ont été capturées avec des microsphères magnétiques couplées à l’ACE2 et testées avec des anticorps anti-spicule humains commerciaux dans le canal rapporteur RP1 et avec différents scFv dans le canal rapporteur RP2 (scFv est indiqué en haut à gauche de chaque panneau). Aucune particule virale n’a été détectée dans les échantillons non infectés. L’épitope ciblé par scFv3 avait la plus grande affinité. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 4 : Graphique de dispersion des variations. L’axe des Y représente la fréquence des événements, et l’axe des X montre le coefficient de variance (CV) en pourcentage pour chaque répétition des différents échantillons. RP1 et RP2 sont les premier et deuxième canaux rapporteurs qui détectent la fluorescence associée à la phycoérythrine et au Brilliant Violet 421, respectivement. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 5 : Matrice de corrélation des exécutions. (A,B) Axe Y : Matrice de corrélation de Pearson à l’échelle log10 entre trois exécutions distinctes, exécutées par trois opérateurs différents et avec des mélanges de billes différents. Un volume d’échantillon plus faible a été appliqué lors de la troisième analyse. Les histogrammes montrent la distribution des différents groupes de variables en fonction de l’IMF mesurée. (A) Corrélation pour le canal rapporteur RP1 entre les différents cycles. (B) Corrélation pour le canal rapporteur RP2 entre les différents cycles. MFI = intensité médiane de fluorescence en unités arbitraires. p < 0,001. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
| Détection | Réactivité |
| scFv2 | ++ |
| scFv3 | +++ |
| scFv5 | ++ |
| scFv7 | + |
| scFv9 | + |
| IgG anti-Spike humain | ++++ |
Tableau 1 : Classement des scFvs en détection en fonction de l’intensité MFI obtenue dans les courbes standard.
| RP1 (PE) | RP2 (BV421) | |
| Dilution de l’échantillon | Portée CV [%] | Portée CV [%] |
| Blanc | 3–11 | 2–13 |
| 1:1458 | 1–7 | 2–7 |
| 1:456 | 4–6 | 3–8 |
| 1:162 | 3–6 | 3–7 |
| 1:54 | 2–4 | 2–4 |
| 1:18 | 2–4 | 1–4 |
| 1:6 | 2–6 | 1–6 |
| 1:2 | 1–5 | 1–3 |
Tableau 2 : Plage en % CV/écart type (moyenne/écart-type × 100) de chaque point de dilution du surnageant infecté par le SRAS-CoV-2 pour les canaux rapporteurs RP1 et RP2.
Fichier supplémentaire 1 : Génération de fragments variables à chaîne unique (scFv) d’immunoglobulines. Veuillez cliquer ici pour télécharger ce fichier.
Tableau supplémentaire 1 : Criblage des scFvs par paires avec des Fabs contre la dilution en série du Spike recombinant (RBD). Pour évaluer la performance de différents peptides de détection, 12 combinaisons de la protéine de pointe, Fab, ont été utilisées comme capture dans un tampon enrichi avec un RBD recombinant. Dix (10) scFv ciblant différents épitopes de la protéine de pointe ont été appliqués comme détection. En fonction des performances de la paire capture-détection, elles ont été marquées comme ayant échoué (-) ou réussi (+). Veuillez cliquer ici pour télécharger ce fichier.
Tableau supplémentaire 2 : Dépistage des scFvs en paires avec des Fabs contre la dilution en série du surnageant de cellules Calu-3 infecté par le SRAS-Cov-2. Pour l’évaluation des performances de différents peptides de détection, 12 combinaisons de la protéine de pointe, Fab, ont été utilisées comme capture dans le surnageant de cellules Calu-3 infectées par le SRAS-Cov-2. Dix (10) scFv ciblant différents épitopes de la protéine de pointe ont été appliqués comme détection. En fonction des performances de la paire capture-détection, elles ont été marquées comme ayant échoué (-) ou réussi (+). Veuillez cliquer ici pour télécharger ce fichier.
Discussion
La technologie multiplex à base de billes s’est avérée être une plate-forme précieuse pour la détection d’agents pathogènes à haut débit dans un certain nombre d’applications cliniques. La grande flexibilité de la plateforme, basée sur les principes de la cytométrie en flux, permet de cibler les anticorps, les protéines et les acides nucléiques 18,19,20,21,22, en multiplexant des centaines d’analytes simultanément. Cependant, à notre connaissance, cette technologie n’a pas encore été appliquée pour détecter des particules virales intactes. Dans ce rapport, la technologie a été appliquée pour la détection de particules virales intactes en ciblant trois épitopes de surface indépendants du SRAS-CoV-2.
Les virus à ARN enveloppé présentent une grande similitude structurelle avec les vésicules extracellulaires (VE), de petites membranes phospholipidiques transportant l’ARN viral et les protéines ainsi que les protéines de l’hôte23. Des immunoessais sandwich ont déjà été appliqués à la détection des VE, en utilisant une paire d’anticorps ciblant deux protéines de surface distinctes24,25. La limitation des tests sandwich à ne détecter simultanément que deux protéines est éliminée par des approches multiplex qui permettent la détection simultanée de plus de deux protéines par réaction.
Le système de détection à trois lasers à double rapporteur décrit ici est l’instrument d’analyse d’écoulement à base de billes le plus avancé à ce jour. En ce qui concerne les systèmes de lecture à rapporteur unique, le double rapporteur (canaux RP1 et RP2) permet la détection de trois protéines/épitopes de surface en parallèle. Le ciblage de plusieurs protéines de surface et épitopes viraux fournit une représentation plus précise de la charge protéique virale, ce qui, au-delà de la confirmation que le virus est bien intact, ouvre également la possibilité d’étudier davantage les antigènes de surface viraux et les mécanismes des interactions entre les protéines virales et l’hôte.
Pendant la pandémie de COVID-19, il était important d’identifier rapidement les personnes porteuses de particules virales actives dans les efforts visant à contenir la propagation du virus. L’ARN génomique est détecté par RT-PCR quantitative quelle que soit son origine (particules virales intactes ou libres). Cependant, seule une enveloppe intacte avec la protéine S accessible peut servir de médiateur à l’entrée cellulaire et à la réplication ultérieure du virus. Des études antérieures avec des puces microfluidiques dans des échantillons de patients ont montré comment la détection de particules virales intactes combinée à des tests au point de service permettrait des tests fréquents et une surveillance accrue de la propagation de la maladie, y compris un choix plus éclairé des personnes à mettre en quarantaine26. L’application d’un test multiplexé basé sur des microsphères permettrait de concevoir des tests visant à cribler plusieurs virus et leurs variantes d’antigènes de surface, obtenant ainsi une image plus précise de la propagation du virus dans la population.
La virométrie en flux est un développement récent de la cytométrie en flux visant à l’analyse des particules virales. Bien qu’elle soit capable de détecter des particules virales discrètes, l’analyse de petits virus pose un problème actuel pour la virométrie en flux27,28. À l’instar de la méthode décrite ici, la virométrie en flux consiste à capturer des virions intacts par des nanoparticules d’or couplées à des anticorps. Les limites des deux méthodes comprennent (i) la dépendance à l’égard de réactifs de capture et de détection de haute affinité pour l’antigène exprimé en surface ciblé par les microsphères ou les nanoparticules, (ii) la capacité limitée de distinguer les particules virales des vésicules extracellulaires, et (iii) l’absence de normes pour une quantification appropriée des particules.
Les cellules sécrètent des VE dans leur environnement et, lorsqu’elles sont infectées par un virus, elles peuvent également sécréter des virions de taille similaire à celle des VE et peuvent éventuellement exprimer les mêmes antigènes29. Étant donné que les VE auront des compositions membranaires similaires à celles du virus, il pourrait être difficile de les distinguer les uns des autres en utilisant uniquement des méthodes basées sur l’affinité telles que l’approche à double laser à rapporteur unique. Cependant, les stratégies décrites ici présentent une capacité multiplex plus élevée, ce qui permet une étude plus large et plus approfondie de la composition protéique des particules. Les méthodes basées sur l’écoulement permettent de suivre des particules discrètes, offrant ainsi des possibilités de quantification numérique. Une stratégie pour résoudre le problème de la quantification dans notre méthode serait d’utiliser des vésicules synthétiques bien caractérisées exprimant des antigènes d’intérêt sous forme de particules pseudo-virales (VLP) pour préparer des courbes standard.
L’une des voies courantes d’entrée et de sortie du SRAS-CoV-2 à partir des cellules hôtes est l’interaction du virus et de la membrane de la cellule hôte 2,15. Dans ce processus, la probabilité que les protéines de la membrane de l’hôte soient incorporées à la surface du virus est élevée. En criblant les protéines de l’hôte incorporées, on peut suivre la voie de l’infection et potentiellement prédire l’évolution de la maladie pour différents patients à risque, ce qui permet de prendre des décisions de traitement plus précoces. Il permet également de caractériser les virus sur différents lots d’échantillons dans les laboratoires de recherche. Cela peut être exploré plus en profondeur en testant si différentes caractéristiques sont liées à différents niveaux d’infectiosité virale et pour le criblage d’anticorps et de molécules médicamenteuses qui ciblent les protéines de surface virales.
Un aspect important de la méthode décrite est qu’elle repose sur l’affinité des réactifs de capture et de détection contre leurs protéines cibles sur le virus. Le choix des réactifs d’affinité est donc un facteur déterminant dans la performance du dosage. Il est possible que plusieurs réactifs d’affinité doivent être examinés et testés pour la capture et la détection afin de sélectionner ceux qui ont la plus grande affinité. Ici, la performance de dix scFvs et de douze fragments de Fab a été évaluée de manière préliminaire à l’aide de RBD recombinante et sur des particules virales provenant du surnageant de cellules épithéliales pulmonaires Calu-3 infectées par le SRAS-Cov-2 (les cellules VeroE6 ont été utilisées pour cultiver/évaluer la cytotoxicité dans toutes les études ultérieures). L’anti-FLAG PE a été utilisé pour détecter les scFv marqués FLAG (tableau supplémentaire 1 et tableau supplémentaire 2). Les cinq scFv les plus performants ont ensuite été sélectionnés pour être appliqués dans le test à double rapporteur, en même temps que le Hu-anti-S1 commercial (tableau 1), sur des surnageants provenant de cellules épithéliales rénales de singe vert africain infectées par VeroE6.
Un autre facteur critique du succès du protocole est la procédure choisie pour le couplage des microsphères. La méthode de couplage doit être efficace et, en même temps, maintenir intacts et non modifiés les épitopes conformationnels ou les résidus d’acides aminés impliqués dans la liaison aux protéines. Ici, la réaction EDC-NHS a été appliquée pour coupler la neutravidine directement aux microsphères, en adaptant un protocole précédemment décrit30 et un système neutravidine + biotine pour lier l’ACE2 recombinant aux microsphères couplées. D’autres méthodes de couplage et leur efficacité peuvent être testées et comparées. Enfin, il a été observé que différents réactifs de détection marqués par fluorescence (p. ex., anti-FLAG PE (phycoérythrine) et anti-FLAG Brilliant Violet 421) peuvent entraîner des niveaux de MFI différents qui peuvent affecter la sensibilité du dosage.
En conclusion, la méthode décrite permet de détecter des particules virales intactes en solution, en appliquant une stratégie à double rapporteur. L’analyse de trois déterminants de surface en parallèle fournit un outil plus spécifique pour caractériser les particules virales et éventuellement les distinguer des autres VE (par exemple, ne contenant pas d’antigènes viraux). Cette stratégie est une alternative à la virométrie en flux. Bien que l’approche actuelle ne discrimine pas la taille des particules, les stratégies de billes magnétiques utilisant des microsphères codées par couleur offrent une capacité plus large de profilage d’antigènes de surface et de conception expérimentale par analyse à haut multiplex et à haut débit. Le test fait preuve d’une grande précision et d’une grande robustesse et peut être étendu à l’analyse de tout type de vésicule extracellulaire et de tout autre type de bioparticule exposant des antigènes de surface dans des fluides corporels ou d’autres matrices liquides. Il s’agissait d’une étude de preuve de concept qui a démontré l’utilité d’utiliser les scFvs comme réactif de détection dans une analyse multiplex de plusieurs épitopes protéiques sur des particules virales. Des études futures sont nécessaires pour déterminer les caractéristiques spécifiques des scFv (par exemple, les affinités de liaison, la réactivité croisée avec d’autres réactifs et cibles) s’ils doivent être utilisés à des fins quantitatives ou cliniques.
Déclarations de divulgation
Les auteurs ne déclarent aucun conflit d’intérêts.
Remerciements
Nous reconnaissons à SciLifeLab, en Suède, l’équipe de l’unité Scilifelab d’Affinity Proteomics-Stockholm pour le développement et l’application de la méthode décrite ici, l’unité Human Antibody Therapeutics pour la fourniture de réactifs scFvs et Fab, et Jonas Klingström pour les cellules VeroE6 infectées par des isolats de SARS-CoV-2 provenant d’échantillons cliniques. Les auteurs remercient Sherry Dunbar, PhD, MBA de Luminex Corporation (Austin, TX), pour son soutien à la recherche, et Matt Silverman MSci, PhD of Biomedical Publishing Solutions (Panama City, FL ; mattsilver@yahoo.com) pour son aide scientifique et rédactionnelle. Ce travail a été soutenu par des fonds de la Fondation Knut et Alice Wallenberg et du Laboratoire Science for Life (SciLifeLab) (VC2020-0015 à Claudia Fredolini et Francesca Chiodi et VC-2022-0028 à Claudia Fredolini).
matériels
| Name | Company | Catalog Number | Comments |
| ACE2-Biotin | Acro Biosystems (Newark, DE) | AC2-H82E6-25 ug | Conc: 340 µg/mL, LOT#BV35376-203HFI-2128 |
| Anti-Goat IgG, PE-conjugated | Jackson ImmunoResearch (West Grove, PA) | 705-116-147 | Host species: Donkey |
| Anti-Human IgG R-PE | Life Technologies/Thermo Fisher (Waltham, MA) | H10104 | Conc: 0.15 mg/mL, LOT#2079224, Host species: Goat |
| Anti-Mouse IgG, PE-conjugated | Jackson ImmunoResearch (West Grove, PA) | 115-116-146 | Host species: Goat |
| Anti-Rabbit IgG, PE-conjugated | Jackson ImmunoResearch (West Grove, PA) | 111-116-144 | Host species: Goat |
| Biotin | Thermo-Fisher Scientific (Waltham, MA) | 20RUO | 100 mM, pH 10 Conc. 1 mg/mL |
| Blocker Casein in PBS | Thermo-Fisher Scientific (Waltham, MA) | 37528 | LOT#VD301372 |
| Blocker reagent for ELISA (BRE) | Roche (Basel, Switzerland) | 11112589001 | |
| Brilliant Violet 421 anti-DYKDDDDK Tag Antibody (Anti-FLAG) 0.2 mg/ml, rat IgG2a, λ | BioLegend (Amsterdam, The Netherlands) | 637321 | |
| Bovine serum albumin (BSA) | Saveen & Werner (Limhamn, Sweden) | B2000-500 | LOT#04D5865 |
| EDC (1-Ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride) | Proteochem (Hurricane, UT) | C1100-custom (65 mg) | LOT# MK3857 |
| Fetal calf serum (FCS) | Gibco/Thermo Fisher (Waltham, MA) | 10270-106 | |
| Goat anti-ACE2 polyclonal antibody | R&D Systems/Bio-Techne (Minneapolis, MN) | AF933 | Host species: Goat |
| Goat IgG | Bethyl Labs (Montgomery, TX) | P50-200 | LOT#P50-200-6 |
| L-glutamine | Thermo-Fisher Scientific (Waltham, MA) | 25030024 | |
| Low-bind 1.5 mL microfuge tubes | VWR (Radnor, PA) | 525-0133 | |
| MagPlex-C Microspheres | Luminex Corporation (Austin, TX) | MC10XXX-01 | |
| MEM tissue cuture media | Gibco/Thermo Fisher (Waltham, MA) | 21430-020 | |
| Microplate, 96-Well, Polystyrene, Half-area, Clear | Greiner Bio-One (Kremsmünster, Austria) | 675101 | |
| NaHCO3 | Gibco/Thermo Fisher (Waltham, MA) | 25080-060 | |
| Neutravidin | Thermo-Fisher Scientific (Waltham, MA) | 31000 | LOT#UK292857 |
| PBS tablets | Medicago AB (Uppsala, Sweden) | 09-9400-100 | LOT#272320-01 |
| Penicillin/Streptomycin | Gibco/Thermo Fisher (Waltham, MA) | 15140122 | |
| Poly(vinyl alcohol) | Sigma-Aldrich (St. Louis, MO) | 360627 | |
| Polyvinylpyrrolidone | Sigma-Aldrich (St. Louis, MO) | 437190 | |
| ProClin 300 | Sigma-Aldrich (St. Louis, MO) | 48915-U | |
| Rabbit IgG | Bethyl Labs (Montgomery, TX) | P120-301 | LOT#12 |
| scFv-FAb1 | In-house production | Human Antibody Therapeutics Unit, Scilifelab, Sweden. Monoclonal scFv. Conc: 0.12 mg/mL. | |
| scFv-FAb2 | In-house production | Human Antibody Therapeutics Unit, Scilifelab, Sweden. Monoclonal scFv. Conc batch1: 0.38 mg/mL. Conc batch2: 0.45 mg/mL | |
| scFv-FAb3 | In-house production | Human Antibody Therapeutics Unit, Scilifelab, Sweden. Monoclonal scFv. Conc: 0.34 mg/mL. | |
| scFv-FAb4 | In-house production | Human Antibody Therapeutics Unit, Scilifelab, Sweden. Monoclonal scFv. Conc: 2.85 mg/mL. | |
| scFv-FAb5 | In-house production | Human Antibody Therapeutics Unit, Scilifelab, Sweden. Monoclonal scFv. Conc:2.7mg/mL. | |
| SARS-CoV-2 infectious particles, Swedish isolate | In-house production | The Public Health Agency of Sweden | |
| SARS-CoV-2 Spike Antibody (Hu-anti-S1) | Novus Biologicals (Centennial, CO) | NBP2-90980 | Monoclonal antibody. Conc: 1 mg/mL. Host: Human. Clone: CR3022. Isotype: IgG1 Kappa. LOT#T201B06 |
| Sodium phosphate monobasic, anhydrous | Sigma-Aldrich (St. Louis, MO) | S3139 | |
| Sulfo-NHS (N-hydroxysulfosuccinimide) | Thermo-Fisher Scientific (Waltham, MA) | 24510 | LOT# XH321563 |
| Tween | Thermo-Fisher Scientific (Waltham, MA) | BP337-50 | LOT#194435 |
| Ultraviolet lamp | Vilber Lourmat GmbH (Eberhardzell, Germany) | VL-215.G | Wavelength = 254 nm; 2 × 15-watt bulbs |
| Vero E6 cells | ATCC (Manassus, VA) | CRL-1586 | |
| xMAP INTELLIFLEX DR-SE (dual-reporter flow instrument) | Luminex Corporation (Austin, TX) | INTELLIFLEX-DRSE-RUO |
Références
- Rey, F. A., Lok, S. M. Common features of enveloped viruses and implications for immunogen design for next-generation vaccines. Cell. 172 (6), 1319-1334 (2018).
- V'kovski, P., Kratzel, A., Steiner, S., Stalder, H., Thiel, V. Coronavirus biology and replication: implications for SARS-CoV-2. Nature Reviews Microbiology. 19 (3), 155-170 (2021).
- Burnie, J., et al. Flow virometry quantification of host proteins on the surface of HIV-1 pseudovirus particles. Viruses. 12 (11), 1296 (2020).
- Gentili, M., et al. Transmission of innate immune signaling by packaging of cGAMP in viral particles. Science. 349 (6253), 1232-1236 (2015).
- Modrow, S., Falke, D., Truyen, U., Schätzl, H. . Viruses: Definition, Structure, Classification. In Molecular Virology. , 163-181 (2013).
- Trinh, K. T. L., Do, H. D. K., Lee, N. Y. Recent advances in molecular and immunological diagnostic platform for virus detection: A review. Biosensors. 13 (4), 490 (2023).
- Zamora, J. L. R., Aguilar, H. C. Flow virometry as a tool to study viruses. Methods. 134-135, 87-97 (2018).
- Graham, H., Chandler, D. J., Dunbar, S. A. The genesis and evolution of bead-based multiplexing. Methods. 158, 2-11 (2019).
- Byström, S., et al. Affinity proteomic profiling of plasma for proteins associated to area-based mammographic breast density. Breast Cancer Research. 20 (1), 14 (2018).
- Rudberg, A. -. S., et al. SARS-CoV-2 exposure, symptoms and seroprevalence in healthcare workers in Sweden. Nature Communications. 11 (1), 5064 (2020).
- Liu, J., et al. Multiplex reverse transcription PCR Luminex assay for detection and quantitation of viral agents of gastroenteritis. Journal of Clinical Virology. 50 (4), 308-313 (2011).
- Gadsby, N. J., Hardie, A., Claas, E. C. J., Templeton, K. E. Comparison of the Luminex respiratory virus panel fast assay with in-house real-time PCR for respiratory viral infection diagnosis. Journal of Clinical Microbiology. 48 (6), 2213-2216 (2010).
- Lorenzen, E., et al. Multiplexed analysis of the secretin-like GPCR-RAMP interactome. Science Advances. 5 (9), (2019).
- Angeloni, S., Cameron, A., Pecora, N. D., Dunbar, S. A rapid, multiplex dual reporter IgG and IgM SARS-CoV-2 neutralization assay for a multiplexed bead-based flow analysis system. Journal of Visualized Experiments: JoVE. (170), e62487 (2021).
- Jackson, C. B., Farzan, M., Chen, B., Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nature Reviews Molecular Cell Biology. 23 (1), 3-20 (2022).
- ISO10993-5 Biological evaluation of medical devices - Part 5: Tests for in vitro cytotoxicity. International Standardization Organization Available from: https://nhiso.com/wp-content/uploads/2018/05/ISO-10993-5-2009.pdf (2009)
- Poetz, O., et al. Sequential multiplex analyte capturing for phosphoprotein profiling. Molecular & Cellular Proteomics. 9 (11), 2474-2481 (2010).
- Dunbar, S. A., Vander Zee, C. A., Oliver, K. G., Karem, K. L., Jacobson, J. W. Quantitative, multiplexed detection of bacterial pathogens: DNA and protein applications of the Luminex LabMAP system. Journal of Microbiological Methods. 53 (2), 245-252 (2003).
- Taniuchi, M., et al. Multiplex polymerase chain reaction method to detect Cyclospora, Cystoisospora, and Microsporidia in stool samples. Diagnostic Microbiology and Infectious Disease. 71 (4), 386-390 (2011).
- Wu, M., et al. High-throughput Luminex xMAP assay for simultaneous detection of antibodies against rabbit hemorrhagic disease virus, Sendai virus, and rabbit rotavirus. Archives of Virology. 164 (6), 1639-1646 (2019).
- Dias, D., et al. Optimization and validation of a multiplexed Luminex assay to quantify antibodies to neutralizing epitopes on human papillomaviruses 6, 11, 16, and 18. Clinical and Vaccine Immunology. 12 (8), 959-969 (2005).
- Opalka, D., et al. Simultaneous quantitation of antibodies to neutralizing epitopes on virus-like particles for human papillomavirus types 6, 11, 16, and 18 by a multiplexes lumina assay. Clinical and Diagnostic Laboratory Immunology. 10 (1), 108-115 (2003).
- Nolte-'T Hoen, E., Cremer, T., Gallo, R. C., Margolis, L. B. Extracellular vesicles and viruses: Are they close relatives. Proceedings of the National Academy of Sciences. 113 (33), 9155-9161 (2016).
- Ohmichi, T., et al. Quantification of brain-derived extracellular vesicles in plasma as a biomarker to diagnose Parkinson's and related diseases. Parkinsonism & Related Disorders. 61, 82-87 (2019).
- Ter-Ovanesyan, D., et al. Framework for rapid comparison of extracellular vesicle isolation methods. Elife. 10, e70725 (2021).
- Gamage, S. S. T., et al. Microfluidic affinity selection of active SARS-CoV-2 virus particles. Science Advances. 8 (39), (2022).
- Renner, T. M., Tang, V. A., Burger, D., Langlois, M. -. A. Intact viral particle counts measured by flow virometry provide insight into the infectivity and genome packaging efficiency of moloney murine leukemia virus. Journal of Virology. 94 (2), e01600-01619 (2020).
- Niraja, S., et al. A flow virometry process proposed for detection of SARS-CoV-2 and large-scale screening of COVID-19 cases. Future Virology. 15 (8), 525-532 (2020).
- Lippé, R. Flow virometry: A powerful tool to functionally characterize viruses. Journal of Virology. 92 (3), e01765 (2018).
- Drobin, K., Nilsson, P., Schwenk, J. M. Highly multiplexed antibody suspension bead arrays for plasma protein profiling. Methods in Molecular Biology. 1023, 137-145 (2013).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.