Methods Article
Profilierung von Oberflächenprotein-Epitopen auf viralen Partikeln mittels Multiplex-Dual-Reporter-Strategie
In diesem Artikel
Zusammenfassung
Hier beschreiben wir einen neu entwickelten Multiplex-Fluoreszenz-Immunoassay, der ein durchflusszytometrisches System mit zwei Reportern verwendet, um gleichzeitig zwei einzigartige Spike-Protein-Epitope auf intakten Viruspartikeln des schweren akuten respiratorischen Syndroms Coronavirus 2 (SARS-CoV-2) zu detektieren, die von Angiotensin-konvertierenden Enzym-2-gekoppelten magnetischen Mikrosphären eingefangen wurden.
Zusammenfassung
Membranproteine auf behüllten Viren spielen eine wichtige Rolle bei vielen biologischen Funktionen, die die Anheftung von Viren an Zielzellrezeptoren, die Fusion von Viruspartikeln mit Wirtszellen, Wirt-Virus-Interaktionen und die Pathogenese von Krankheiten umfassen. Darüber hinaus haben sich virale Membranproteine auf Viruspartikeln und auf der Oberfläche von Wirtszellen als hervorragende Ziele für Virostatika und Impfstoffe erwiesen. Hier beschreiben wir ein Protokoll zur Untersuchung von Oberflächenproteinen auf intakten Partikeln des schweren akuten respiratorischen Syndroms Coronavirus 2 (SARS-CoV-2) mit Hilfe des Dual-Reporter-Durchflusszytometriesystems. Der Assay nutzt die Multiplex-Technologie, um einen dreifachen Nachweis von Viruspartikeln durch drei unabhängige Affinitätsreaktionen zu erzielen. Magnetische Kügelchen, die an rekombinantes humanes Angiotensin-konvertierendes Enzym-2 (ACE2) konjugiert waren, wurden verwendet, um Viruspartikel aus dem Überstand von Zellen einzufangen, die mit SARS-CoV-2 infiziert waren. Anschließend wurden zwei mit R-Phycoerythrin (PE) oder Brilliant Violet 421 (BV421) markierte Detektionsreagenzien gleichzeitig appliziert. Als Proof-of-Concept wurden Antikörperfragmente verwendet, die auf verschiedene Epitope des SARS-CoV-2-Oberflächenproteins Spike (S1) abzielen. Der Nachweis von Viruspartikeln durch drei unabhängige Affinitätsreaktionen sorgt für eine hohe Spezifität und bestätigt das Einfangen intakter Viruspartikel. Dosisabhängigkeitskurven von SARS-CoV-2-infizierten Zellüberständen wurden mit Replikatskoeffizienten-Varianzen (Mittelwert/SD) ˂14% erstellt. Die gute Assay-Leistung in beiden Kanälen bestätigte, dass zwei Zielprotein-Epitope auf der Virusoberfläche parallel nachweisbar sind. Das hier beschriebene Protokoll könnte angewendet werden für (i) High-Multiplex- und Hochdurchsatz-Profiling von Oberflächenproteinen, die auf behüllten Viren exprimiert werden; ii) Nachweis aktiver intakter Viruspartikel; und (iii) Bewertung der Spezifität und Affinität von Antikörpern und antiviralen Arzneimitteln für Oberflächenepitope viraler Antigene. Die Anwendung kann potenziell auf jede Art von extrazellulären Vesikeln und Biopartikeln ausgeweitet werden, wodurch Oberflächenantigene in Körperflüssigkeiten oder anderen flüssigen Matrizen freigelegt werden.
Einleitung
Die häufigsten pathogenen Viren wie Influenza, HIV, humane Cytomegalieviren und SARS-CoV-Stämme sind behüllte Viren. Eine Zellinfektion durch behüllte Viren erfordert die Fusion von Virus- und Wirtszellmembranen, was zur Freisetzung des viralen Genoms in das Zytoplasma führt. Die virale RNA repliziert sich dann, bevor sie in ein neues Viruspartikel gepackt wird 1,2. Während dieser Prozesse können nicht nur virale Proteine, sondern auch Proteine der Wirtsmembran in die Hülle eingebaut werden und so zu einem integralen Bestandteil des neuen Viruspartikels werden. In die Virushülle eingebaute Membranproteine der Wirtszelle können den Eintritt des Virus in eine neue Wirtszelle erleichtern und dabei die Mechanismen der Zell-Zell-Interaktionen, des Homing und der Flucht des Immunsystems ausnutzen 3,4.
Trotz der Bedeutung der Untersuchung virusassoziierter Proteine unterstützen die meisten der derzeit verfügbaren Techniken zur Virusanalyse5 keine Hochdurchsatz- und High-Multiplex-Charakterisierung des Virusoberflächenantigens. Sie sind auch nicht in der Lage, einzelne Viruspartikel nachzuweisen oder zwischen infektiösen intakten Viruspartikeln, nicht-infektiöser RNA, viralen Proteinen und Virussubpopulationen, die unterschiedliche Antigene exprimieren, zu unterscheiden. In jüngster Zeit wurde die Durchflusszytometrie modifiziert und zu einer neuartigen Methode zur Analyse von Viruspartikeln adaptiert, nämlich der Durchflussvirometrie. Die Flow-Virometrie ermöglicht die Untersuchung einzelner Viruspartikel und ihrer Oberflächenantigene. Einschränkungen wie geringer Durchsatz, geringe Multiplexfähigkeit, komplizierter Versuchsaufbau und Datenanalyse sowie eingeschränkte Nachweisbarkeit kleiner Viruspartikel bleiben jedoch bestehen 6,7.
Die mikrosphärenbasierte Multiplex-Quantifizierung von Proteinen und Nukleinsäuren ist eine etablierte Technologie mit zahlreichen Anwendungen, die von der Proteinquantifizierung in Körperflüssigkeiten über Protein-Protein-Interaktionsstudien bis hin zur Diagnose von Virusinfektionenreichen 8,9,10,11,12,13 . Ein kürzlich vorgestelltes Durchflussanalysegerät verfügt über einen Dual-Reporter-Kanal, der die Messung von zwei fluoreszierenden Reportermolekülen in derselben Reaktionsvertiefung ermöglicht. Diese neue Fähigkeit hat sich als besonders nützlich für die parallele Profilierung verschiedener Immunglobulin-Isotypenerwiesen 14. Hier wird beschrieben, wie das Dual-Reporter-System verwendet werden kann, um intakte Viruspartikel zu detektieren und dabei mehrere Oberflächenantigene parallel zu adressieren.
Als Proof of Concept beschreibt dieser Bericht die Entwicklung eines dreifachen Nachweissystems für SARS-CoV-2-Viruspartikel. SARS-CoV-2 besteht aus vier Hauptproteinen, eines davon ist das Spike-Protein (S), das aus zwei Untereinheiten besteht. Die erste Untereinheit, S1, bindet primär an das in menschlichen Zellmembranen exprimierte ACE2. Die zweite Untereinheit, S2, erleichtert den Eintritt in die Zielzelle durch ein Fusionspeptid, wodurch eine Pore in der Zielzellmembran entsteht, in die das Virion eindringen kann15. Die drei verbleibenden Bausteine von SARS-CoV-2 sind das Nukleokapsid (N), das Membranprotein (M) und das Hüllprotein (E). Das Nukleokapsid ist für die Verpackung des viralen Genoms verantwortlich, indem es mit RNA Ribonukleoproteinstrukturen bildet, während die Membran- und Hüllproteine eine zentrale Rolle bei der Virusassemblierung spielen.
Der hier beschriebene Assay zielt auf drei unabhängige Epitope der S1-Untereinheit ab, die auf der Hülloberfläche von SARS-CoV-2 exprimiert werden. Es werden serielle Verdünnungen sowohl von SARS-CoV-2-infizierten als auch von nicht infizierten Zellüberständen verwendet. Viruspartikel werden über ACE2-konjugierte Mikrosphären eingefangen, die die S1-Untereinheit an das Virus binden. Das S-Protein des Oberflächenvirus wird dann parallel zu einem kommerziell erhältlichen markierten Immunglobulin-Einzelketten-Variablen-Fragment (scFv) und einem humanen monoklonalen Anti-S1-Antikörper (Hu-anti-S1) zusammen mit einem eigens entwickelten FLAG-markierten scFv nachgewiesen. Das Hu-anti-S1 wird durch den ersten Kanal (RP1) im Dual-Reporter-System mit orangefarbenem R-Phycoerythrin (PE)-konjugiertem anti-humanem IgG-Fc-Sekundärantikörper detektiert, und das scFv wird durch den zweiten Kanal (RP2) mit einem blauen Brilliant Violet 421 (BV421)-konjugierten sekundären Anti-FLAG-Antikörper nachgewiesen. Der Viruspartikel-Assay ist in Abbildung 1 dargestellt.
Protokoll
1. Konjugation von Neutravidin und Kontrollantikörpern an magnetische Mikrosphären
HINWEIS: Fluoreszierend gefärbte magnetische Kügelchen (Polystyrol-Mikrokugeln mit einem Durchmesser von 6,5 μm und eingebettetem Magnetit) mit unterschiedlichen fluoreszierenden Markierungen, die in der Materialtabelle aufgeführt sind, werden zur Herstellung der folgenden Beadschoppenkonjugate und -kontrollen verwendet: (1) Biotinyliertes rekombinantes humanes ACE2, das an Kügelchen gebunden ist, die mit einem Neutravidin-Linker gekoppelt sind; (2) Biotin, das an Kügelchen gebunden ist, gekoppelt mit einem Neutravidin-Linker; (3) Ziegen-IgG, das direkt an die Perlen gekoppelt ist; und (4) unkonjugierte Perlen. Das Protein, das an die Kügelchen gekoppelt werden soll, sollte frei von Natriumazid, Rinderserumalbumin (BSA), Glycin, Tris(hydroxymethyl)aminomethan (Tris), Glycerin oder aminhaltigen Zusätzen sein. Der Aktivierungspuffer besteht aus 0,1 M monobasischem Natriumphosphat, wasserfrei (NaH2PO4), pH 6. 2-Morpholinethansulfonsäure (MES; 50 mM) Puffer von pH 5 wird zur Verdünnung von Konjugaten verwendet. Waschpuffer ist PBS-T (1x PBS [phosphatgepufferte Kochsalzlösung], pH 7,4 + 0,05 % (v/v) Tween-20). Der Lagerpuffer besteht aus 2,7 mg/ml Blockierungsreagenz für ELISA (BRE) + 0,1 % Antibiotika (hier ProClin 300).
- Nehmen Sie Sulfo-N-Hydroxysulfosuccinimid (NHS)-Pulver aus dem Kühlschrank und 65 mg präaliquotiertes 1-Ethyl-3-[3-dimethylaminopropyl]carbodiimidhydrochlorid (EDC) aus dem Gefrierschrank und lassen Sie es 30 Minuten lang auf Raumtemperatur (RT; 18-22 °C) kommen. Lagern Sie sowohl NHS als auch EDC in einem Umschlag mit Kieselsäurekügelchen während dieses Schritts, um eine Hydrolyse durch Luftfeuchtigkeit zu verhindern.
- Mikrosphären für die Aktivierung und Kopplung vorbereiten.
HINWEIS: Die Fluoreszenzfarbstoffe in Mikrosphären sind lichtempfindlich, und die Kügelchen sollten im Dunkeln und bei Kühlschranktemperaturen (4-8 °C) aufbewahrt werden, wenn sie nicht aktiv verwendet werden.- Resuspendieren Sie 4 verschiedene Schäfte der farbcodierten Mikrosphären (12,5 x 106/ml) (Materialtabelle) durch kurzes Vortexen, Beschallen oder Drehen (15 min bei 15-30 U/min), gemäß Produktinformationsblatt.
- Übertragen Sie 40 μl jeder Bead-Suspension (5 x 105 Mikrosphären) in zugewiesene Vertiefungen einer 96-Well-Mikrotiterplatte mit halbem Boden und flachem Boden (Materialtabelle).
- Waschen Sie die magnetischen Kügelchen.
HINWEIS: Die Waschschritte können entweder manuell oder mit einer automatischen Tellerwaschmaschine durchgeführt werden.- Geben Sie 80 μl/Well-Aktivierungspuffer zu den Beads und immobilisieren Sie die Kügelchen auf einem magnetischen Plattenseparator für 30 s. Der Überstand wird aus den Mikrosphären abgesaugt, während die Kügelchen auf dem magnetischen Plattenseparator immobilisiert werden.
- Entfernen Sie die Mikrotiterplatte vom Magnetplattenseparator und resuspendieren Sie die Kügelchen in 50 μl Aktivierungspuffer.
- Aktivieren Sie die Kügelchen mit Sulfo-NHS und EDC.
- Sulfo-NHS-Arbeitslösung mit 50 mg/ml in Aktivierungspuffer in einem 1,5-ml-Mikrofuge-Röhrchen herstellen. Stellen Sie das vorrätige NHS-Pulver wieder in den Kühlschrank (4-8 °C) und schützen Sie es vor Feuchtigkeit.
- Bereiten Sie die EDC-Arbeitslösung mit 50 mg/ml in Aktivierungspuffer im 1,5-ml-Mikrofuge-Röhrchen vor. Löse die vorgefertigten 65-mg-Aliquots EDC-Pulver in 1,3 ml Aktivierungspuffer auf.
HINWEIS: Sulfo-NHS und EDC beginnen zu hydrolysieren und verlieren ihre Aktivität, wenn sie aufgelöst werden. Vermeiden Sie es, den Kopplungsvorgang zu unterbrechen, bis NHS und EDC zu den Kügelchen hinzugefügt wurden. Bewahren Sie gelöste NHS- oder EDC-Lösungen nicht für die spätere Verwendung auf. - Bereiten Sie die Aktivierungslösung für die Bead-Aktivierung vor, indem Sie 20 % Sulfo-NHS-Stammlösung (50 mg/ml), 20 % EDC-Stammlösung (50 mg/ml) und 60 % Aktivierungspuffer volumetrisch kombinieren. Für jede Beadsaktivierungsreaktion ist eine Aktivierungslösung von 50 μl erforderlich (mit 5 × 105 Kügelchen/Reaktion) plus ausreichend zusätzliches Volumen, um Pipettierverluste auszugleichen.
- Geben Sie 50 μl vollständige Aktivierungslösung in jede Vertiefung, die gewaschene Kügelchen enthält. Mit dem bereits vorhandenen Suspensionsvolumen von 50 μl im Aktivierungspuffer pro Vertiefung beträgt die Endkonzentration von Sulfo-NHS 5 mg/ml und die Endkonzentration von EDC ebenfalls 5 mg/ml.
- Verschließen Sie die Mikrosphären-Reaktionsplatte mit einem Einweg-Klebe-, Kunststoff- oder Folienplattenversiegeler und inkubieren Sie sie 20 min lang auf einem Orbitalschüttler (650 U/min) bei Raumtemperatur (18-22 °C) im Dunkeln.
- Waschen Sie überschüssige Aktivierungslösung von den Kügelchen.
- Die Mikrotiterplatte bei 233 × g 1 min zentrifugieren.
- Immobilisieren Sie aktivierte Kügelchen auf einem magnetischen Plattenseparator für 30 s. Entfernen Sie die Plattenversiegelung und saugen Sie den Überstand aus magnetimmobilisierten Kügelchen ab, wobei die Mikrotiterplatte noch auf dem Magnetabscheider positioniert ist.
- Entfernen Sie die Mikrotiterplatte vom Magnetabscheider und geben Sie 100 μl MES-Puffer in jede Vertiefung.
- Wiederholen Sie die Schritte 1.4.2-1.4.3 ein weiteres Mal für insgesamt zwei Wäschen.
- Koppeln Sie Neutravidin und Ziegen-IgG (Kontrolle) mit geeigneten Perlensets. Bereiten Sie ausreichend Neutravidin- und Ziegen-IgG-Arbeitslösungen vor, wobei Sie 100 μl/Reaktion und ausreichend zusätzliches Material zur Aufnahme von Pipettierverlusten wie folgt planen:
HINWEIS: Neutravidin-Proteinpulver wird mit Reinstwasser rekonstituiert und dann mit PBS auf eine 1 mg/ml-Stammlösung verdünnt, bevor es für die Lagerung/Verwendung aliquotiert wird (Neutravidin-Protein ist nicht direkt in PBS löslich, aber in Wasser bis ~10 mg/ml).- Bereiten Sie die Neutravidin-Arbeitslösung in einer Konzentration von 125 μg/ml in MES-Puffer in einem 1,5-ml-Mikrofugenröhrchen mit geringer Proteinbindung vor.
- Bereiten Sie eine IgG-Kontroll-Antikörper-Arbeitslösung für Ziegen in einer Konzentration von 17,5 μg/ml in MES-Puffer vor.
- Bereiten Sie die Mikrotiterplatte mit den aktivierten Kügelchen vor. Immobilisieren Sie die Kügelchen auf einem magnetischen Plattenseparator für 30 s. Während die Mikrotiterplatte noch auf dem Magnetseparator positioniert ist, aspirieren Sie den Überstand aus magnetimmobilisierten Kügelchen.
- 100 μl Neutravidin-Arbeitslösung (125 μg/ml) in die entsprechenden Vertiefungen geben, die Kügelchen enthalten (für die Neutravidin-Biotin- und Neutravidin-ACE2-Kupplung).
- Geben Sie 100 μl Ziegen-IgG-Arbeitslösung (17,5 μg/ml) in die Vertiefung mit den Kügelchen, die als reine IgG-Kontrollen für Ziegen zugewiesen sind.
- Geben Sie 100 μl MES-Puffer in die Vertiefung, die als unkonjugierte Kontrollkügelchen zugewiesen ist.
- Die Mikrotiterplatte verschließen und 2 h auf einem Orbitalschüttler (650 U/min) bei RT (18-22 °C) im Dunkeln inkubieren. Wirbeln Sie die Platte nach 1 h Inkubation kurz vortexen, um sicherzustellen, dass die Kügelchen in Suspension bleiben.
- Waschen Sie die Perlen mit PBS-T.
- Die Mikrotiterplatte bei 233 × g 1 min zentrifugieren.
- Gekoppelte Kügelchen auf einem magnetischen Plattenseparator für 30 s immobilisieren. Während die Mikrotiterplatte noch auf dem Magnetseparator positioniert ist, aspirieren Sie den Überstand aus magnetimmobilisierten Kügelchen.
- Entfernen Sie die Mikrotiterplatte vom Magnetabscheider.
- Geben Sie 100 μl PBS-T in jede Vertiefung, die Kügelchen enthält.
- Wiederholen Sie die Waschschritte 1.6.2-1.6.4 einmal für insgesamt zwei Wäschen mit PBS-T.
- Konjugierte Kügelchen für die Lagerung vorbereiten.
- Gekoppelte Kügelchen auf einem magnetischen Plattenseparator für 30 s immobilisieren. Während die Mikrotiterplatte noch auf dem Magnetseparator positioniert ist, aspirieren Sie den Überstand aus magnetimmobilisierten Kügelchen. Entfernen Sie die Mikrotiterplatte vom Magnetabscheider.
- Geben Sie 50 μl Lagerpuffer zu jeder Mikrokugel-ID, um die verbleibende Bead-Aktivität zu löschen.
- Inkubieren Sie die Mikrotiterplatte bei Kühlschranktemperaturen (4-8 °C) im Dunkeln über Nacht (16-22 h).
- Übertragen Sie unkonjugierte und Ziegen-IgG-konjugierte Kügelchensuspensionen (50 μl) in entsprechend markierte 1,5-ml-Mikrofuge-Röhrchen mit geringer Proteinbindung, kombiniert mit zwei 100-μl-Lagerpufferspülungen der Wells, um eine maximale Bead-Rückgewinnung zu gewährleisten.
HINWEIS: Unkonjugierte und Ziegen-IgG-konjugierte Kügelchen haben beide die Nummern 5 × 105 mit einem Endvolumen von 250 μl (d. h. 2 × 103 Kügelchen/μl). Lagern Sie die Mikrofugenröhrchen bis zur Verwendung bei Kühlschranktemperaturen (4-8 °C).
- Binden Sie biotinyliertes ACE2 und Biotin an Neutravidin-konjugierte Kügelchen.
- Bereiten Sie die rekombinante humane biotinylierte ACE2-Arbeitslösung mit 18 μg/ml ACE2 in 10 mM PBS vor. Pro Reaktion werden 100 μl benötigt. Bereiten Sie die Biotin-Arbeitslösung mit 2,4 mg/ml Biotin in 10 mM PBS vor. Pro Reaktion werden 100 μl benötigt.
- Bereiten Sie die Mikrotiterplatte mit Neutravidin-konjugierten Mikrosphären vor.
- Immobilisieren Sie die Mikrokugeln auf einem magnetischen Plattenseparator für 30 s. Während die Mikrotiterplatte noch auf dem Magnetabscheider positioniert ist, entfernen Sie die Plattenversiegelung und saugen Sie den Überstand aus magnetimmobilisierten Mikrokugeln an.
- Entfernen Sie die Mikrotiterplatte vom Magnetabscheider und fügen Sie 50 μl 10 mM PBS/Vertiefung hinzu.
- Wiederholen Sie die Schritte 1.8.2.1-1.8.2.2 einmal.
- 100 μl der biotinylierten ACE2-Arbeitslösung werden in geeignete Vertiefungen gegeben, die Neutravidin-konjugierte Mikrosphären enthalten. 100 μl der Biotin-Arbeitslösung werden in geeignete Vertiefungen gegeben, die Neutravidin-konjugierte Mikrosphären enthalten.
- Die Mikrotiterplatte verschließen und 1 h auf einem Orbitalschüttler (650 U/min) bei RT (18-22 °C) im Dunkeln inkubieren.
- Waschen Sie die Mikrokügelchen wie in den Schritten 1.6.1-1.6.5 beschrieben.
- Bereiten Sie die ACE2- und Biotin-konjugierten Mikrosphären vor und lagern Sie sie, wie in den Schritten 1.7.1 bis 1.7.4 beschrieben.
HINWEIS: Biotinylierte ACE2- und Biotin-konjugierte Kügelchen haben beide die Nummern 5 × 105 in einem Endvolumen von 250 μl (d. h. 2 × 103 Kügelchen/μl).
2. Konjugationstest
- Bereiten Sie eine Bead-Mischung vor, indem Sie alle vier Arten von Mikrosphären kombinieren, die in Abschnitt 1 hergestellt wurden (d. h. Neutravidin-konjugiertes biotinyliertes ACE2, Neutravidin-konjugiertes Biotin, Ziegen-IgG-konjugiert und unkonjugiert).
HINWEIS: Die Stamm-Mikrosphären wurden bei 2 × 103 Kügelchen/μl gelagert und so kombiniert, dass die endgültige Kügelchenkonzentration in der Arbeitsperlenmischung 40 Kügelchen von jedem Set/μl beträgt.- Berechnen Sie das Volumen der Arbeitsperlenmischung, die für den Test erforderlich ist (5 μl/Reaktion), wobei das zusätzliche Volumen Pipettierverluste ausgleichen kann. Wirbeln Sie jedes Röhrchen kurz vor und kombinieren Sie gleiche berechnete Volumina jeder Bead-Suspension in einem neuen Mikrofuge-Röhrchen mit geringer Proteinbindung. Die Kügelchenkonzentration beträgt nun 400 Kügelchen jedes Sets/μL.
- Stellen Sie die Arbeitsraupenmischung her, indem Sie die kombinierte Raupensuspension zusätzlich um das 10-fache mit Lagerpuffer verdünnen (40 von jedem Satz/μl Arbeitskonzentration).
HINWEIS: Stellen Sie zuerst eine kleine Menge der Arbeitsperlenmischung her, um die Anzahl der Mikrokügelchen/μl für jede ID zu schätzen.
- Inkubieren Sie die Mikrosphären mit Ziegen-Anti-ACE2-Antikörpern.
- Pipettieren Sie 5 μl der Arbeitsperlenmischung in 3 Vertiefungen einer 96-Well-Mikrotiterplatte mit flachem Boden.
- Geben Sie jeweils 50 μl Anti-ACE2 (0,4 μg/ml verdünnt in PBS-T, Materialtabelle) in die 3 Vertiefungen mit den Kügelchen in der Mikrotiterplatte. Verschließen Sie die Mikrotiterplatte, den Vortex und inkubieren Sie auf einem Orbitalschüttler (650 U/min) bei RT (18-22 °C) für 1 h im Dunkeln.
- Die Mikrotiterplatte bei 233 × g für 1 min nach unten pulsieren und die Mikrosphären dreimal mit PBS-T waschen, wie in 1.6.2-1.6.4 beschrieben.
- Inkubieren Sie Mikrosphären mit Nachweisantikörpern.
- Pipettieren Sie 5 μl der einarbeitenden Bead-Mischung in 6 neue Vertiefungen der Mikrotiterplatte.
- Bereiten Sie jeweils 1 μg/ml der Arbeitsnachweismischungen vor: Anti-Ziegen-IgG-PE, Anti-Maus-IgG-PE und Anti-Kaninchen-IgG-PE in 3 separaten 1,5-ml-Röhrchen unter Verwendung von PBS-T als Verdünnungsmittel.
- Geben Sie 50 μl der Nachweismischungen in jeweils 3 Vertiefungen, und das Anti-Ziegen-IgG wird in die gleichen Vertiefungen wie das Anti-ACE2 aus Schritt 2.2 gegeben.
- Versiegeln, vortexen und auf einem Orbitalschüttler (650 U/min) bei RT (18-22 °C) für 30 Minuten inkubieren.
- Die Platte bei 233 × g 1 min lang nach unten pulsieren und die Mikrosphären dreimal mit PBS-T waschen, wie in 1.6.2-1.6.4 beschrieben.
- Fügen Sie 100 μl PBS-T hinzu und führen Sie es mit dem Dual-Reporter-Durchflussanalysegerät mit den folgenden Einstellungen aus:
Modus: Dualer Reporter; Auszeitüberschreitung: 45 s; DD-Anschnitt: 7500-17500; Minimale Mikrosphärenzahl: 100 Mikrosphären/Satz (niedrigster QC-Cutoff: 35 Mikrosphären/Satz).
3. Produktion von SARS-CoV-2-infizierten Zellüberständen
Das SARS-CoV-2-Virus wird in Vero E6-Zellen des Wirts (Affennieren-Epithelzelllinie; ATCC; Tabelle der Materialien). Vero E6-Zellen werden in modifiziertem Eagles-Medium (MEM) bei 37 °C in einer Atmosphäre mit 5 % CO2 und 95 % relativer Luftfeuchtigkeit kultiviert. Jeder Liter MEM wird mit 10 ml L-Glutamin (200 mM), 38 ml NaHCO3 (7,5%), 5 ml Penicillin/Streptomycin-Lösung und 50 ml fötalem Rinderserum (FCS) ergänzt; Tabelle der Materialien.
VORSICHT: Verwenden Sie beim Umgang mit SARS-CoV-2 geeignete Biosicherheitsverfahren und -ausrüstung.
- Vero E6-Zellen werden in zwei 150 cmgroßen 2-Gewebekulturflaschen bis zum Zusammenfluss gezüchtet. Infizieren Sie eine Flasche mit dem SARS-CoV-2-Virus und verwenden Sie die andere scheininfizierte Flasche als Kontrolle.
- Mischen Sie etwa 100.000 SARS-CoV-2-Infektionspartikel vom Wildtyp (WT) mit 5 mL Eagles MEM-Medium.
- Das Medium wird aus einem 150-cm-2-Kolben aspiriert und 55 ml vollständiges MEM hinzugefügt, um nicht infizierte Kontrollüberstände zu erzeugen. Das Medium wird aus dem anderen 150-cm-2-Kolben aspiriert und das Virusgemisch in die Zellen gegeben. Inkubieren Sie die Zellen 1 h lang bei 37 °C. Schütteln Sie den Kolben alle 15 Minuten leicht, um das Virus zu verteilen.
- Geben Sie 50 ml vollständiges MEM-Medium in den Kolben mit SARS-CoV-2 und inkubieren Sie die Zellen, bis zytopathische Wirkungen beobachtet werden, wobei die Kolben alle 24 Stunden visuell ausgewertet werden.
HINWEIS: Es sollte etwa 3-4 Tage nach der Infektion dauern, bis die Zytopathie auftritt. Zytopathische Effekte auf die Monoschichtstruktur der Vero E6-Zellen (z. B. Zellretraktion, Krenation, Rundung, Deadhäsion, Verlust der intrazytoplasmatischen Granularität, offene Lyse) werden qualitativ durch Beobachtung der Zellen mit einem inversen Lichtmikroskop gemäß den Richtlinien der Internationalen Normungsorganisation für In-vitro-Zytotoxizitätstests 16 bewertet. - Den Zellüberstand aus beiden Kolben auffangen und 6 Minuten lang bei 253 × g drehen, um Zelltrümmer zu sedimentieren.
- UV-inaktiviertes SARS-CoV-2-Virus im Zellüberstand
- Pipettieren Sie 0,5 ml Überstand pro Vertiefung in 12 Vertiefungen in einer 24-Well-Mikrotiterplatte. Die Mikrotiterplatte ohne Deckel 30 s lang unter einer geeigneten UV-Lampe UV-bestrahlen (Materialtabelle).
HINWEIS: Die Virusinaktivierung im Zellüberstand sollte durch Versuch der Virusvermehrung in Vero E6-Zellkulturen festgestellt werden.
- Pipettieren Sie 0,5 ml Überstand pro Vertiefung in 12 Vertiefungen in einer 24-Well-Mikrotiterplatte. Die Mikrotiterplatte ohne Deckel 30 s lang unter einer geeigneten UV-Lampe UV-bestrahlen (Materialtabelle).
- Aliquotieren Sie den Zellüberstand in 1,5-ml-Röhrchen und lagern Sie ihn bis zur weiteren Verwendung bei -20 °C.
HINWEIS: Der Küvettenüberstand kann bei -80 °C gelagert werden.
4. Assay: Nachweis von SARS-CoV-2-Viruspartikeln im Zellüberstand
- Bereiten Sie den Assay-Puffer vor, indem Sie 0,1 % Kasein, 0,5 % Polyvinylalkohol, 0,8 % Polyvinylpyrrolidon und 1 % BSA (alle w/v) (pH 7) mischen. Bereiten Sie den Probenverdünnungspuffer vor, indem Sie 10 % Kaninchen-IgG in Assay-Puffer vorbereiten.
- Berechnen und bereiten Sie das Volumen der Arbeitsperlenmischung (Schritt 2.1) vor, das für den Test erforderlich ist (5 μl/Reaktion), wobei das überschüssige Volumen zum Ausgleich von Pipettierverlusten verwendet werden kann.
- Bereiten Sie die Überstandsverdünnungsreihe vor. Berechnen Sie die erforderlichen Überstandsvolumina. Untersuchen Sie jeden Verdünnungspunkt in dreifachen Vertiefungen für jedes der fünf scFvs, was zu 15 Vertiefungen pro Verdünnungspunkt und Probentyp führt. Verwenden Sie 45 μl verdünnte Überstände in jeder Vertiefung, so dass insgesamt 675 μl benötigt werden; ein einzelnes 1,5-ml-Mikrofuruge-Röhrchen für jedes SARS-CoV-2- und Kontrollüberstand ist ausreichend.
- SARS-CoV-2-Überstände und Kontrollüberstände mindestens 1 h bei 4 °C auftauen. Halten Sie die Überstände und ihre Verdünnungen bis zur Verwendung kalt (2-8 °C). Markieren und ordnen Sie jeweils acht 1,5-ml-Mikrofuge-Röhrchen für SARS-CoV-2 und Kontrollüberstände an.
HINWEIS: Die höchste untersuchte Konzentration ist eine Überstandsverdünnung im Verhältnis 1:1 (2-fach), bei der eine Reihe von 1:2 (3-fachen) Verdünnungen unter Verwendung von Probenverdünnungspuffer durchgeführt wird, wobei die höchste Verdünnung eine Verdünnung von 1:1458 ist. Leerproben, die Probenverdünnungspuffer enthalten, dienen nur als Kontrollen ohne Überstand. Daher betragen die getesteten Verdünnungen jedes Überstandstyps (SARS-CoV-2 oder Kontrolle) das 2-, 6-, 18-, 54-, 162-, 486- und 1458-fache, mit einer reinen Pufferkontrolle. - Geben Sie Probenverdünnungspuffer in die markierten Mikrofugenröhrchen. Die 1:1 (2-fach) Verdünnungsröhrchen benötigen 600 μl Puffer und die restlichen Röhrchen 800 μl Puffer. Erzeugen Sie die höchste Verdünnung (1:1; 2-fach) jedes Überstands, indem Sie 600 μl des Überstands mit 600 μl Probenverdünnungspuffer in entsprechend gekennzeichneten Röhrchen kombinieren und das Röhrchen zum Mischen kurz vortexen.
- Die Serie wird fortgesetzt, indem 400 μl der 1:1 (2-fach) verdünnten Überstände nacheinander in das nächste Verdünnungsröhrchen überführt werden (d. h. 6-fache Verdünnung) und die 3-fache Verdünnung fortgesetzt wird, bis die niedrigste Verdünnung (1458-fach) erreicht ist. Jeden verdünnten Überstand kurz vortexen, bevor mit der nächsten Verdünnung fortgefahren wird.
- SARS-CoV-2-Überstände und Kontrollüberstände mindestens 1 h bei 4 °C auftauen. Halten Sie die Überstände und ihre Verdünnungen bis zur Verwendung kalt (2-8 °C). Markieren und ordnen Sie jeweils acht 1,5-ml-Mikrofuge-Röhrchen für SARS-CoV-2 und Kontrollüberstände an.
- Inkubieren Sie die Mikrosphären mit dem Überstand.
- Die vorbereitete Arbeitsperlenmischung wird 30 s lang vortext, um die Mikrosphären zu resuspendieren, und 5 μl der Bead-Mischung in jede zugewiesene Vertiefung einer 384-Well-Mikrotiterplatte mit flachem Boden gegeben.
HINWEIS: Es können auch 96-Well-Platten verwendet werden. - Geben Sie 45 μl der vorbereiteten Überstandsverdünnungen in die zugewiesenen Vertiefungen, die Mikrosphären in der 384-Well-Platte enthalten. Verschließen Sie die Platte und inkubieren Sie sie über Nacht (16-22 h) auf einem Orbitalschüttler (650 U/min) bei RT (18-22 °C) im Dunkeln.
- Die vorbereitete Arbeitsperlenmischung wird 30 s lang vortext, um die Mikrosphären zu resuspendieren, und 5 μl der Bead-Mischung in jede zugewiesene Vertiefung einer 384-Well-Mikrotiterplatte mit flachem Boden gegeben.
- Waschen Sie überschüssigen Überstand von den Perlen.
- Entfernen Sie die Mikrotiterplatte aus dem Orbitalschüttler und entfernen Sie die Plattenversiegelung. Die Platte bei 931 x g 1 min zentrifugieren.
- Immobilisieren Sie die Kügelchen, indem Sie die Mikrotiterplatte für 30 s auf einen Magnetplattenseparator legen. Während die Mikrotiterplatte noch auf dem Magnetabscheider positioniert ist, entfernen Sie die Plattenversiegelung und saugen Sie den Überstand aus magnetimmobilisierten Kügelchen an.
- Entfernen Sie die Mikrotiterplatte vom Magnetabscheider.
- Geben Sie 60 μl PBS-T in jede Vertiefung, die Kügelchen enthält.
- Wiederholen Sie die Waschschritte 4.5.2-4.5.4 zweimal, um insgesamt drei PBS-T-Waschgänge zu erhalten.
- Bereiten Sie die verschiedenen Detektionsmischungen in separaten 1,5-ml-Röhrchen vor. Jeder Nachweismix besteht aus einem kommerziellen humanen monoklonalen anti-S1 (Hu-anti-S1)-Antikörper (1 μg/ml) und einem von fünf verschiedenen FLAG-markierten scFvs (1 μg/ml) (Ergänzende Datei 1, Ergänzende Tabelle 1 und Ergänzende Tabelle 2), die auf das Spike-Protein auf dem SARS-CoV-2-Partikel abzielen, verdünnt in Assay-Puffer (Schritt 4.1), was zu insgesamt fünf verschiedenen Nachweismischungen führt.
- Wiederholen Sie die Schritte 4.5.2 bis 4.5.3. Resuspendieren Sie die gewaschenen Mikrosphären in 50 μl/Vertiefung des entsprechenden scFv-spezifischen Spike-Detektionsmixes. Die Mikrotiterplatte verschließen und 1 h auf einem Orbitalschüttler (650 U/min) bei RT (18-22 °C) im Dunkeln inkubieren.
- Die Mikrotiterplatte bei 931 × g 1 min zentrifugieren. Waschen Sie überschüssiges Spike-Detektionsreagenz von den Kügelchen. Führen Sie drei Waschschritte mit 60 μl PBS-T gemäß den Schritten 4.5.2-4.5.5 durch.
- Inkubieren Sie Mikrosphären mit einer fluoreszierenden Detektions-Antikörpermischung.
- Bereiten Sie eine Fluoreszenzlösungsmischung vor, die aus handelsüblichem PE-konjugiertem Anti-Human-IgG zusammen mit BV421-konjugiertem Anti-FLAG-Antikörper besteht, der im Assay-Puffer verdünnt ist, mit Arbeitskonzentrationen von 0,2 μg/ml bzw. 1 μg/ml. Pro Reaktion werden 50 μl Fluoreszenz-Detektionsreagenz benötigt.
- Wiederholen Sie die Schritte 4.5.2-4.5.3. Resuspendieren Sie die Mikrosphären in 50 μl/Vertiefung Fluoreszenzlösungsmischung. Die Mikrotiterplatte verschließen und 30 min auf einem Orbitalschüttler (650 U/min) bei RT (18-22 °C) im Dunkeln inkubieren.
- Die Mikrotiterplatte bei 931 × g 1 min eindrehen. Waschen Sie überschüssige Fluoreszenzlösungsmischung von den Mikrokügelchen. Führen Sie drei Waschschritte mit 60 μl PBS-T gemäß den Schritten 4.5.2-4.5.5 durch.
- Suspendieren Sie die Mikrosphären in 60 μl PBS-T aus dem letzten Waschschritt. Analysieren Sie die Platte auf einem Dual-Reporter-Strömungsanalysesystem mit den in Schritt 2.6 beschriebenen Einstellungen.
Ergebnisse
Konjugationstest
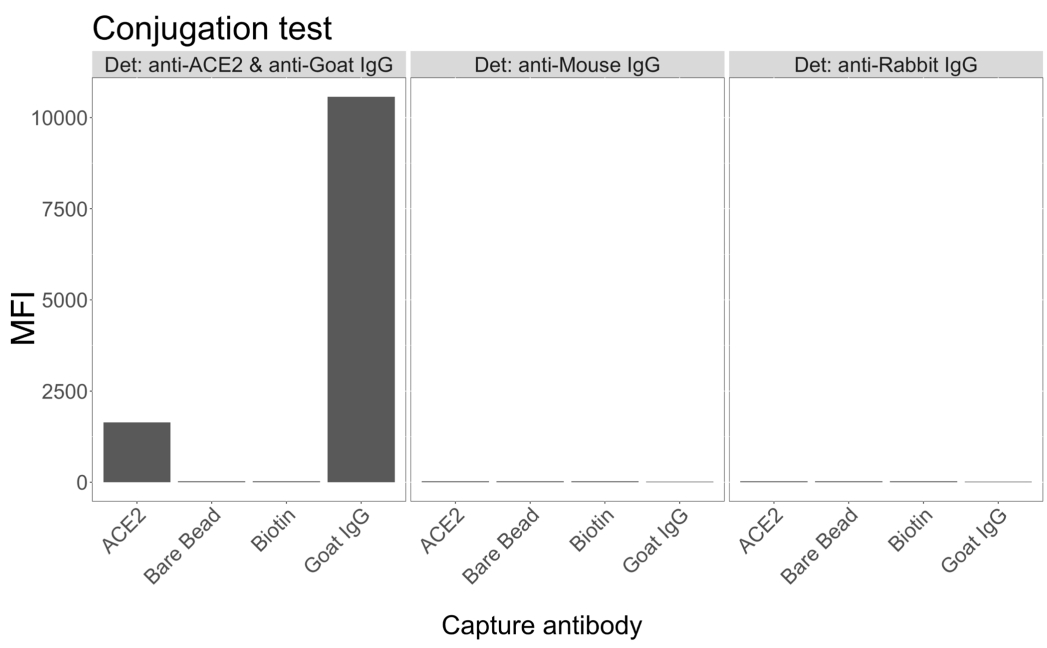
Der Konjugationstest zeigte, dass Ziegen-IgG und Neutravidin-biotinyliertes ACE2 erfolgreich an die Mikrosphären konjugiert wurden. Die Spezifität des Assay-Nachweises wurde durch die Untersuchung von ACE2-konjugierten Mikrosphären mit PE-markierten Sekundärantikörpern bestätigt, die in verschiedenen Tierarten erzeugt wurden (Abbildung 2). Es wurde keine Kreuzreaktivität zwischen den verschiedenen Nachweisantikörpern beobachtet. Bei der Untersuchung der Bead-Mischungen mit Ziegen-Anti-ACE2 + Anti-Ziegen-IgG PE wurde ein medianer Fluoreszenzintensitätswert (MFI; beliebige Einheiten) über dem Hintergrund sowohl für ACE2- als auch für Ziegen-IgG-konjugierte Mikrosphären nachgewiesen, nicht jedoch für die unkonjugierte Mikrosphäre (blank) oder für die Biotin-beschichteten Mikrosphären. Anti-Maus-IgG-PE und Anti-Kaninchen-IgG-PE wurden als Negativkontrollen verwendet, um auf falsch-positive Signale zu prüfen. Bei der Inkubation mit den Mikrosphären wurde ein vernachlässigbares Fluoreszenzsignal erzeugt, was darauf hindeutet, dass die positiven Signale für das ACE2 und das Ziegen-IgG spezifisch waren.
Nachweisbarkeit von Viruspartikeln in Zellüberständen
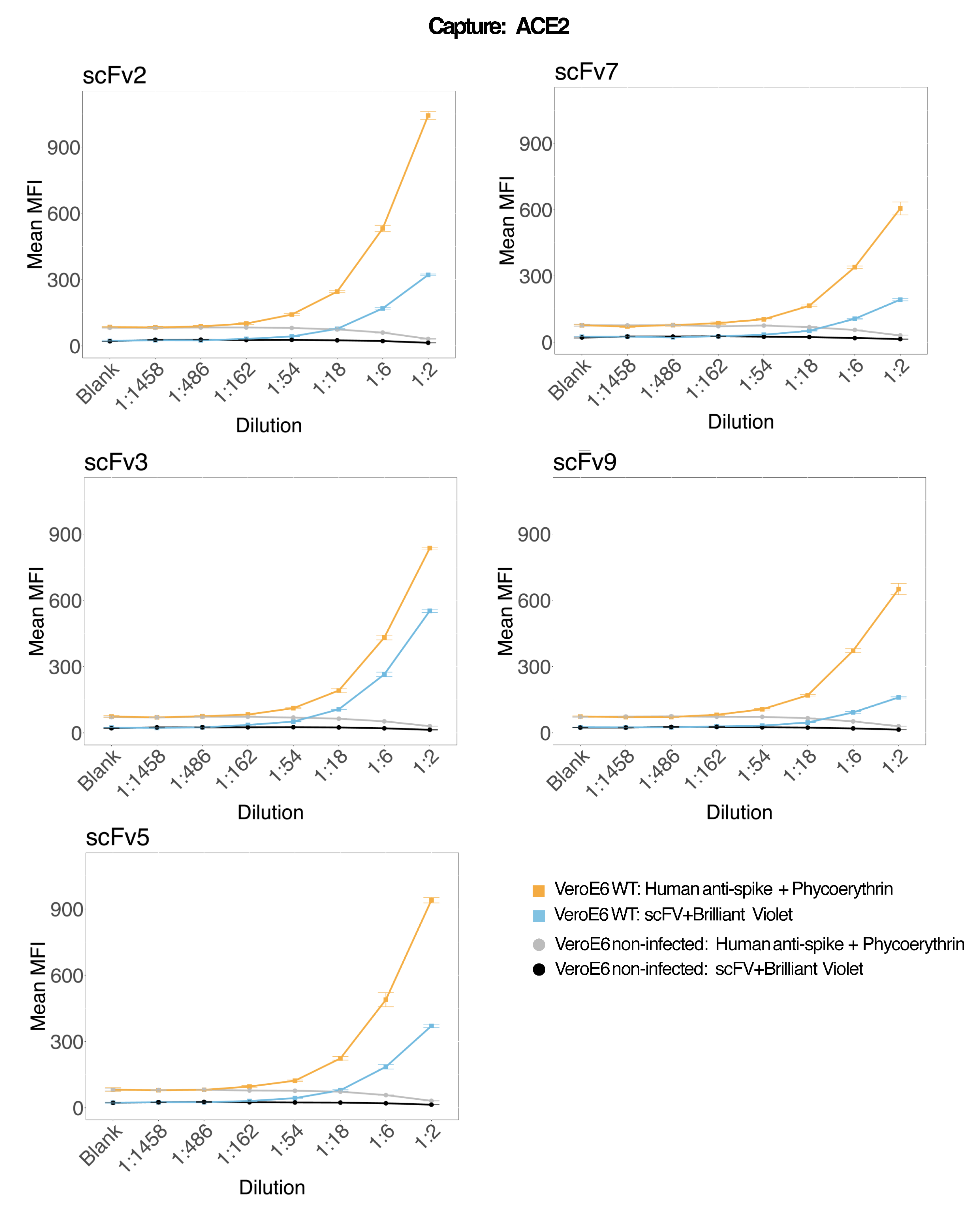
Magnetische Kügelchen, die an rekombinantes humanes ACE2 gekoppelt waren, wurden verwendet, um SARS-CoV-2-Viruspartikel von infizierten und kontrollierten (kein Virus) VeroE6-Zellkulturüberständen einzufangen, und dann gleichzeitig mit einem monoklonalen Antikörper und einem von fünf verschiedenen scFvs auf zwei verschiedene virale Spike-Regionen untersucht. In beiden Reporterkanälen (RP1 und RP2) wurde ein konzentrationsabhängiges Signal in den Verdünnungen von SARS-Cov-2-infizierten Zellüberständen beobachtet (Abbildung 3), was darauf hindeutet, dass sowohl der kommerzielle Hu-anti-S1-Antikörper als auch die verschiedenen scFvs das an die ACE2-konjugierte Mikrosphäre gebundene Viruspartikel detektierten. Bei drei von fünf scFvs ist das Virus in Verdünnungen bis hinunter zu 1:18 nachweisbar (scFv2, scFv3, scFv5); für die verbleibenden zwei scFvs (scFv7 und scFv9) ist es bis zu 1:6 Verdünnungen nachweisbar. Dies könnte auf eine andere Affinität zum Ziel zurückzuführen sein. Wie in Abbildung 3 und Tabelle 1 gezeigt, bietet scFv3 die höchste MFI-Intensität, gefolgt von scFv5, scFv2, scFv7 bzw. scFv9.
Weltweit führt die scFvs-Detektion zu einem niedrigeren MFI im Vergleich zum Hu-anti-S1. Dies könnte auf eine geringere Affinität hindeuten, aber auch um ein Artefakt aufgrund der Markierung mit unterschiedlichen Fluoreszenzfarbstoffen (PE und BV421) handeln. Ein weiterer Trend, der bei scFv7 und scFv9 zu beobachten ist, ist, dass die MFI-Werte auch für den RP1-Kanal (Anti-Spike) im Vergleich zu den anderen drei Konfigurationen etwas niedriger sind. Dies könnte darauf hindeuten, dass die scFvs entweder kreuzreagieren oder auf andere Weise mit der ACE2-Hu-anti-S1-Wechselwirkung interferieren, was auch das niedrigere Signal im RP2-Kanal erklären könnte. Im Überstand der nicht infizierten Vero E6-Zellen wurden weder im RP1- noch im RP2-Kanal Viruspartikel nachgewiesen.
Als Negativkontrollkügelchen werden die Neutravidin-Biotin-konjugierte Mikrosphäre, die Ziegen-IgG-Mikrosphäre und die unkonjugierten Mikrosphären verwendet. Die Viruspartikel wurden mit magnetischen Mikrosphären eingefangen, die an ACE2 gekoppelt waren, und mit kommerziellem humanem Anti-Spike im RP1-Reporterkanal und mit verschiedenen scFvs im RP2-Reporterkanal getestet (scFv ist oben links in jedem Panel angegeben). In keiner der infizierten und nicht infizierten Proben wurden Viruspartikel nachgewiesen.
Präzision und Robustheit des Assays
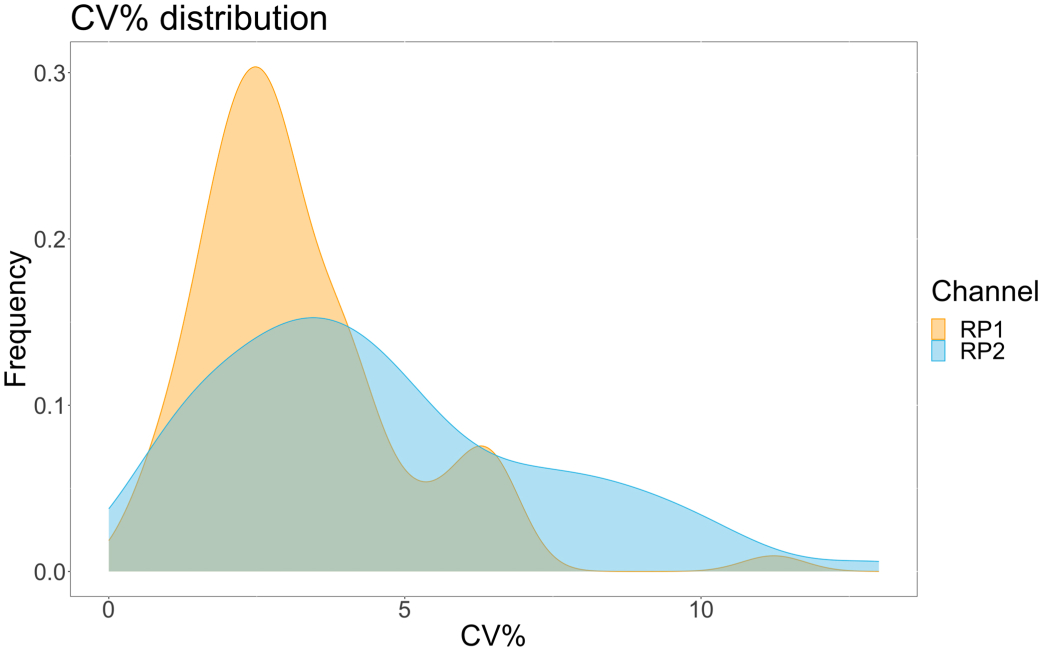
Um die Genauigkeit des Assays zu bewerten, wurden alle Bedingungen in dreifacher Ausfertigung ausgeführt. Für jeden Verdünnungspunkt wurde ein Varianzkoeffizient (CV) für die ACE2-Mikrosphäre berechnet. Alle berechneten CVs für den Assay lagen unter 15 %, wobei der höchste gemessene CV 13 % und der niedrigste CV 1 % betrug (Tabelle 2). Wie im Dichtediagramm (Abbildung 4) des RP1-Kanals zu sehen ist, zeigt die PE-Detektion des kommerziellen Hu-anti-S1 eine höhere Präzision, die sich hauptsächlich auf einen CV von 3% konzentriert. Der RP2-Kanal, BV-Detektion von scFVs, zeigt höhere CVs. Wie jedoch in Tabelle 2 zu sehen ist, wird der höhere Bereich der CVs durch die Proben mit niedrigen Konzentrationen von Viruspartikeln, wie z. B. die Blindprobe, bestimmt. Um die Robustheit des Protokolls zu testen, wurde der Assay zweimal von verschiedenen Bedienern wiederholt, wobei Bead-Mischungen verwendet wurden, die an verschiedenen Tagen erzeugt wurden, und ein geringeres Probenvolumen (72 % geringer). Eine sehr gute Pearson-Korrelation zwischen 0,98 und 1 wurde sowohl für den RP1- als auch für den RP2-Kanal beobachtet (p-Wert < 0,01), was die Robustheit des Assays und die Möglichkeit bestätigt, den Assay anzuwenden, wenn weniger Probe verfügbar ist (Abbildung 5). Diese Durchflussanalysetechnologie folgt der "Umgebungsanalyttheorie"17, wodurch der Assay empfindlich auf die Konzentration, aber nicht auf das Volumen reagiert.

Abbildung 1: Der Viruspartikel-Assay. (A) Zellüberstände sowohl von infizierten als auch von nicht infizierten Vero E6-Zellen werden in einer seriellen Verdünnung entweder auf eine 96-Well- oder 384-Well-Platte gegeben, zusammen mit magnetischen Mikrosphären, die mit Neutravidin konjugiert sind, und dann entweder an biotinyliertes humanes ACE2 oder Biotin gekoppelt. Unkonjugierte Mikrosphären, gekoppelt mit Ziegen-IgG und blanken Mikrosphären, werden zusammen mit der Neutravidin-Biotin-konjugierten Mikrosphäre als Negativkontrollen verwendet. (B) Gebildete Mikrosphären-Virus-Partikelkomplexe werden mit einem Nachweiscocktail aus Hu-anti-S1 und einem der verschiedenen scFvs mit FLAG-Tag nachgewiesen. Anschließend wird eine Fluoreszenzmischung mit Anti-Human-IgG PE, das auf Hu-anti-S1 abzielt, und Anti-FLAG Brilliant Violet 421, das auf die scFvs abzielt, zugegeben. (C) Das Dual-Detektionssystem mit drei Lasern sendet einen roten, grünen und violetten Laser aus, um den Mikropartikelkomplex zu detektieren. Der rote Laser detektiert die Farbstoffmarkierung der Mikrosphäre, während der grüne und der violette Laser das Anti-S1 bzw. das scFvs detektieren. Die generierten Daten werden anschließend analysiert. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
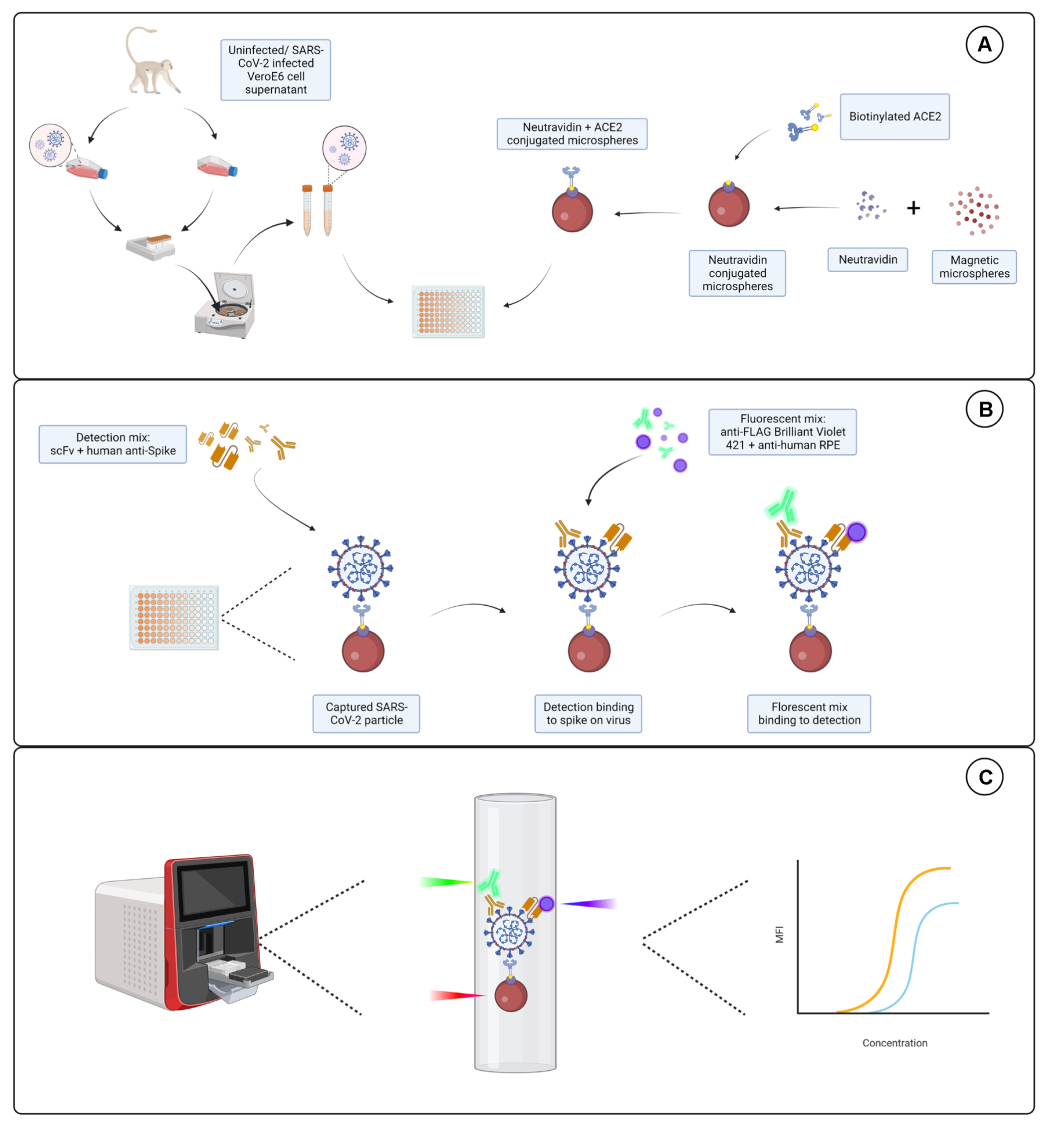{kind=link}

Abbildung 2: Diagramm zur Bestätigung der Konjugation. Die Kügelchenmischungen bestanden aus vier verschiedenen Mikrosphären-IDs, die jeweils mit einem anderen Protein konjugiert waren: Neutravidin-Biotin-ACE2 (ACE2), unkonjugierte Mikrosphäre (Bare Bead), Neutravidin-Biotin (Biotin) und Ziegen-IgG (Ziegen-IgG). Im Konjugationstest wurden drei verschiedene Konfigurationen von Detektionsfluorophoren verwendet. Nämlich Ziegen-Anti-ACE2 + Anti-Ziegen-IgG PE, Anti-Maus-IgG PE und Anti-Kaninchen-IgG PE. Die Y-Achse zeigt das durchschnittlich gemessene MFI-Signal (mediane Fluoreszenzintensität; beliebige Einheiten) von jeder Mikrosphäre unter den drei verschiedenen Bedingungen. Die X-Achse zeigt die verschiedenen angewendeten Capture-Antikörper. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 3: Multiplex-Detektion von Oberflächenproteinen. Y-Achse: Mittlere MFI (mediane Fluoreszenzintensität; beliebige Einheiten ± Standardabweichung) für jede Probe, analysiert in dreifachen Wells pro Zustand. X-Achse: Serielle Verdünnungspunkte des Zellüberstandes. Orange: Viruspartikel im Überstand von Vero E6, die mit SARS-CoV-2 WT infiziert sind, nachgewiesen mit humanem Anti-Spike + Anti-humanem PE (Phycoerythrin). Blau: Überstand von Vero E6, der mit SARS-CoV-2 WT infiziert ist und mit den verschiedenen scFvs + anti-FLAG Brilliant Violet 421 nachgewiesen wurde. Grau: Nicht infizierter Zellüberstand, der mit humanem Anti-Spike + Anti-humanem PE nachgewiesen wurde. Schwarz: Nicht infizierter Zellüberstand, der mit den fünf verschiedenen scFvs + anti-FLAG Brilliant Violet 421 nachgewiesen wurde. Die Viruspartikel wurden mit magnetischen Mikrosphären, die an ACE2 gekoppelt waren, eingefangen und mit kommerziellen humanen Anti-Spike-Antikörpern im RP1-Reporterkanal und mit verschiedenen scFvs im RP2-Reporterkanal getestet (scFv ist oben links auf jedem Panel angegeben). In keiner der nicht infizierten Proben wurden Viruspartikel nachgewiesen. Das Epitop, auf das scFv3 abzielte, hatte die höchste Affinität. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 4: Diagramm der Streuung der Variation . Die Y-Achse stellt die Häufigkeit von Ereignissen dar, und die X-Achse zeigt den Varianzkoeffizienten (CV) in Prozent für jedes Replikat der verschiedenen Stichproben. RP1 und RP2 sind der erste und zweite Reporterkanal, der die mit Phycoerythrin bzw. Brilliant Violet 421 assoziierte Fluoreszenz nachweist. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 5: Korrelationsmatrix des Laufs. (A,B) Y-Achse: Pearson-Korrelationsmatrix auf der log10-Skala zwischen drei separaten Läufen, die von drei verschiedenen Operatoren und mit unterschiedlichen Raupenmischungen ausgeführt wurden. Im dritten Durchlauf wurde ein geringeres Probenvolumen angewendet. Die Histogramme zeigen die Verteilung der verschiedenen Variablencluster basierend auf dem gemessenen MFI. (A) Korrelation für den Reporterkanal RP1 zwischen den verschiedenen Läufen. (B) Korrelation für den RP2-Reporterkanal zwischen den verschiedenen Läufen. MFI=Mediane Fluoreszenzintensität in beliebigen Einheiten. p < 0,001. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
| Erkennung | Reaktivität |
| scFv2 | ++ |
| scFv3 | +++ |
| scFv5 | ++ |
| scFv7 | + |
| scFv9 | + |
| Humanes Anti-Spike-IgG | ++++ |
Tabelle 1: Rangfolge der scFvs bei der Detektion auf der Grundlage der MFI-Intensität, die in den Standardkurven erhalten wird.
| RP1 (PE) | RP2 (BV421) | |
| Verdünnung der Probe | CV-Bereich [%] | CV-Bereich [%] |
| Leer | 3–11 | 2–13 |
| 1:1458 | 1–7 | 2–7 |
| 1:456 | 4–6 | 3–8 |
| 1:162 | 3–6 | 3–7 |
| 1:54 | 2–4 | 2–4 |
| 1:18 | 2–4 | 1–4 |
| 1:6 | 2–6 | 1–6 |
| 1:2 | 1–5 | 1–3 |
Tabelle 2: CV%-Bereich (Mittelwert/Standardabweichung × 100) jedes Verdünnungspunkts des SARS-CoV-2-infizierten Überstands sowohl für den RP1- als auch für den RP2-Reporterkanal.
Ergänzende Datei 1: Erzeugung von Immunglobulin-Einzelketten-Variablen-Fragmenten (scFv). Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Tabelle 1: Screening von scFvs in Paaren mit Fabs gegen serielle Verdünnung von rekombinantem Spike (RBD). Um die Leistung verschiedener Detektionspeptide zu bewerten, wurden 12 Kombinationen des Spike-Proteins, Fab, als Capture in Puffer verwendet, der mit rekombinanter RBD dotiert war. Zehn (10) scFvs, die auf verschiedene Epitope des Spike-Proteins abzielen, wurden als Nachweis eingesetzt. Abhängig von der Leistung des Erfassungs- und Erkennungspaars wurden sie entweder als fehlgeschlagen (-) oder als erfolgreich (+) markiert. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Ergänzende Tabelle 2: Screening von scFvs in Paaren mit Fabs gegen serielle Verdünnung von SARS-Cov-2-infizierten Calu-3-Zellüberständen. Zur Evaluierung der Leistung verschiedener Detektionspeptide wurden 12 Kombinationen des Spike-Proteins, Fab, als Capture in SARS-Cov-2-infizierten Calu-3-Zellüberständen verwendet. Zehn (10) scFvs, die auf verschiedene Epitope des Spike-Proteins abzielen, wurden als Nachweis eingesetzt. Abhängig von der Leistung des Erfassungs- und Erkennungspaars wurden sie entweder als fehlgeschlagen (-) oder als erfolgreich (+) markiert. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Diskussion
Die Bead-basierte Multiplex-Technologie hat sich in einer Reihe von klinischen Anwendungen als wertvolle Plattform für den Nachweis von Krankheitserregern mit hohem Durchsatz erwiesen. Die hohe Flexibilität der Plattform, die auf den Prinzipien der Durchflusszytometrie basiert, ermöglicht das Targeting von Antikörpern, Proteinen und Nukleinsäuren 18,19,20,21,22 und das gleichzeitige Multiplexen von Hunderten von Analyten. Unseres Wissens nach wurde diese Technologie jedoch bisher nicht zum Nachweis intakter Viruspartikel eingesetzt. In diesem Bericht wurde die Technologie für den Nachweis intakter Viruspartikel eingesetzt, indem sie auf drei unabhängige Oberflächenepitope von SARS-CoV-2 abzielte.
Behüllte RNA-Viren zeigen eine hohe strukturelle Ähnlichkeit mit extrazellulären Vesikeln (EVs), kleinen Phospholipidmembranen, die virale RNA und Proteine zusammen mit Wirtsproteinen tragen23. Sandwich-Immunoassays wurden zuvor zum Nachweis von EVs eingesetzt, wobei ein Antikörperpaar verwendet wurde, das auf zwei unterschiedliche Oberflächenproteineabzielt 24,25. Die Einschränkung von Sandwich-Assays, nur zwei Proteine gleichzeitig zu detektieren, wird durch Multiplex-Ansätze aufgehoben, die den gleichzeitigen Nachweis von mehr als zwei Proteinen pro Reaktion ermöglichen.
Das hier beschriebene Drei-Laser-Dual-Reporter-Detektionssystem ist das bisher fortschrittlichste Instrument zur Strömungsanalyse auf Bead-Basis. In Bezug auf Single-Reporter-Auslesesysteme ermöglicht der Dual-Reporter (RP1- und RP2-Kanäle) die parallele Detektion von drei Oberflächenproteinen/Epitopen. Die Ausrichtung auf mehrere virale Oberflächenproteine und Epitope ermöglicht eine genauere Darstellung der viralen Proteinlast, die nicht nur bestätigt, dass das Virus tatsächlich intakt ist, sondern auch die Möglichkeit eröffnet, virale Oberflächenantigene und die Mechanismen der Wechselwirkungen zwischen dem Virus und dem Wirtsprotein weiter zu untersuchen.
Während der COVID-19-Pandemie war es wichtig, Personen mit aktiven Viruspartikeln rechtzeitig zu identifizieren, um die Ausbreitung des Virus einzudämmen. Genomische RNA wird unabhängig von ihrer Herkunft (intakte Viruspartikel oder frei) mittels quantitativer RT-PCR nachgewiesen. Allerdings kann nur eine intakte Hülle mit zugänglichem S-Protein den Zelleintritt und die anschließende Virusvermehrung vermitteln. Frühere Studien mit mikrofluidischen Chips in Patientenproben haben gezeigt, wie der Nachweis intakter Viruspartikel in Kombination mit Point-of-Care-Tests häufige Tests und eine verbesserte Überwachung der Krankheitsausbreitung ermöglichen würde, einschließlich einer fundierteren Entscheidung über Personen, die unter Quarantäne gestellt werden sollen26. Die Anwendung eines Multiplex-Mikrosphären-basierten Assays würde die Entwicklung von Assays ermöglichen, die auf das Screening mehrerer Viren und ihrer Oberflächenantigenvarianten abzielen und so ein genaueres Bild der Virusausbreitung in der Bevölkerung erhalten.
Die Durchflussvirometrie ist eine neue Entwicklung der Durchflusszytometrie, die auf die Analyse von Viruspartikeln abzielt. Trotz der Fähigkeit, diskrete Viruspartikel zu detektieren, stellt die Analyse kleiner Viren ein aktuelles Problem für die Flussvirometriedar 27,28. Ähnlich wie bei der hier beschriebenen Methode werden bei der Strömungsvirometrie intakte Virionen durch Goldnanopartikel eingefangen, die an Antikörper gekoppelt sind. Zu den Einschränkungen beider Methoden gehören (i) die Abhängigkeit von hochaffinen Erfassungs- und Detektionsreagenzien für oberflächenexprimiertes Antigen, auf das die Mikrosphären oder Nanopartikel abzielen, (ii) die eingeschränkte Fähigkeit, zwischen Viruspartikeln und extrazellulären Vesikeln zu unterscheiden, und (iii) das Fehlen von Standards für eine ordnungsgemäße Partikelquantifizierung.
Zellen sezernieren EVs in ihre Umgebung, und wenn sie mit einem Virus infiziert sind, können sie auch Virionen sezernieren, die ähnlich groß wie die EVs sind und schließlich die gleichen Antigene exprimieren können29. Da die EVs eine ähnliche Membranzusammensetzung wie das Virus haben, könnte es schwierig sein, sie nur mit affinitätsbasierten Methoden wie dem Dual-Laser-Single-Reporter-Ansatz voneinander zu unterscheiden. Die hier beschriebenen Strategien weisen jedoch eine höhere Multiplexkapazität auf, was eine breitere und tiefere Untersuchung der Proteinzusammensetzung der Partikel ermöglicht. Strömungsbasierte Methoden ermöglichen die Verfolgung diskreter Partikel und bieten so Möglichkeiten zur digitalen Quantifizierung. Eine Strategie, um das Quantifizierungsproblem in unserer Methode zu lösen, wäre die Verwendung gut charakterisierter synthetischer Vesikel, die Antigene von Interesse als virusähnliche Partikel (VLPs) exprimieren, für die Erstellung von Standardkurven.
Ein gemeinsamer Weg des Eintritts und Austritts von SARS-CoV-2 aus den Wirtszellen ist die Interaktion zwischen dem Virus und der Wirtszellmembran 2,15. Dabei ist die Wahrscheinlichkeit, dass Wirtsmembranproteine in die Virusoberfläche eingebaut werden, hoch. Durch das Screening von eingebauten Wirtsproteinen kann man den Weg der Infektion verfolgen und möglicherweise den Krankheitsverlauf für verschiedene Risikopatienten vorhersagen, was frühere Behandlungsentscheidungen ermöglicht. Es ermöglicht auch die Charakterisierung der Viren über verschiedene Probenchargen hinweg in Forschungslabors. Dies kann weiter erforscht werden, indem getestet wird, ob unterschiedliche Merkmale mit unterschiedlichen Niveaus der viralen Infektiosität zusammenhängen, und indem Antikörper und Wirkstoffmoleküle gescreent werden, die auf virale Oberflächenproteine abzielen.
Ein wichtiger Aspekt bei der beschriebenen Methode ist, dass sie auf der Affinität der Fang- und Nachweisreagenzien zu ihren Zielproteinen auf dem Virus beruht. Die Wahl der Affinitätsreagenzien ist daher ein entscheidender Faktor für die Assay-Leistung. Möglicherweise sollten mehrere Reagenzien mit hoher Affinität gescreent und auf ihre Erfassung und Detektion getestet werden, um diejenigen mit der höchsten Affinität auszuwählen. Hier wurde die Leistung von zehn scFvs und zwölf Fab-Fragmenten vorläufig mit rekombinanter RBD und an Viruspartikeln aus dem Überstand von SARS-Cov-2-infizierten Calu-3-Lungenepithelzellen bewertet (VeroE6-Zellen wurden in allen nachfolgenden Studien zur Kultivierung/Beurteilung der Zytotoxizität verwendet). Anti-FLAG PE wurde verwendet, um die mit FLAG markierten scFvs zu detektieren (Ergänzende Tabelle 1 und Ergänzende Tabelle 2). Die fünf leistungsstärksten scFvs wurden dann ausgewählt, um im Dual-Reporter-Assay zusammen mit kommerziellem Hu-anti-S1 (Tabelle 1) auf Überstände von infizierten VeroE6-Nierenepithelzellen des afrikanischen grünen Affen angewendet zu werden.
Ein weiterer kritischer Faktor für den Erfolg des Protokolls ist das gewählte Verfahren für die Mikrosphärenkopplung. Die Kopplungsmethode sollte effizient sein und gleichzeitig Konformationsepitope oder Aminosäurereste, die an der Proteinbindung beteiligt sind, intakt und unverändert halten. Hier wurde die EDC-NHS-Reaktion angewendet, um Neutravidin direkt an Mikrosphären zu koppeln, wobei ein zuvor beschriebenes Protokoll30 und ein Neutravidin + Biotin-System zur Bindung von rekombinantem ACE2 an die gekoppelten Mikrosphären angepasst wurden. Alternative Kopplungsverfahren und deren Wirkungsgrad können getestet und verglichen werden. Schließlich wurde beobachtet, dass verschiedene fluoreszenzmarkierte Detektionsreagenzien (z. B. anti-FLAG PE (Phycoerythrin) und anti-FLAG Brilliant Violet 421) zu unterschiedlichen MFI-Werten führen können, die die Sensitivität des Assays beeinflussen können.
Zusammenfassend lässt sich sagen, dass das beschriebene Verfahren den Nachweis von intakten Viruspartikeln in Lösung unter Anwendung einer Dual-Reporter-Strategie ermöglicht. Die parallele Analyse von drei Oberflächendeterminanten bietet ein spezifischeres Werkzeug, um virale Partikel zu charakterisieren und sie schließlich von anderen EVs (z. B. die keine viralen Antigene enthalten) zu unterscheiden. Diese Strategie ist eine Alternative zur Flow-Virometrie. Obwohl der derzeitige Ansatz keine Partikelgrößen unterscheidet, bieten magnetische Bead-Strategien mit farbcodierten Mikrosphären eine breitere Möglichkeit bei der Erstellung von Oberflächenantigen-Profilen und der Versuchsplanung durch Hochmultiplex- und Hochdurchsatzanalyse. Der Assay zeigt eine hohe Präzision und Robustheit und kann auf die Analyse jeder Art von extrazellulärem Vesikel und jeder anderen Art von Biopartikeln erweitert werden, die Oberflächenantigene in Körperflüssigkeiten oder anderen flüssigen Matrices freilegen. Dabei handelte es sich um eine Proof-of-Concept-Studie, die den Nutzen der Verwendung von scFvs als ein Nachweisreagenz in einer Multiplex-Analyse mehrerer Proteinepitope auf Viruspartikeln demonstrierte. Zukünftige Studien sind notwendig, um die spezifischen Eigenschaften von scFvs (z. B. Bindungsaffinitäten, Kreuzreaktivität mit anderen Reagenzien und Zielmolekülen) zu bestimmen, wenn sie für quantitative oder klinische Zwecke verwendet werden sollen.
Offenlegungen
Die Autoren erklären, dass kein Interessenkonflikt besteht.
Danksagungen
Wir danken im SciLifeLab, Schweden, dem Team der Affinity Proteomics-Stockholm Scilifelab Unit für die Entwicklung und Anwendung der hier beschriebenen Methode, der Human Antibody Therapeutics Unit für die Bereitstellung von scFvs und Fab-Reagenzien und Jonas Klingström für die VeroE6-Zellen, die mit SARS-CoV-2-Isolaten aus klinischen Proben infiziert wurden. Die Autoren danken Sherry Dunbar, PhD, MBA der Luminex Corporation (Austin, TX), für die Unterstützung der Forschung und Matt Silverman MSci, PhD of Biomedical Publishing Solutions (Panama City, FL; mattsilver@yahoo.com) für die wissenschaftliche und redaktionelle Unterstützung. Diese Arbeit wurde durch Mittel der Knut and Alice Wallenberg Foundation und des Science for Life Laboratory (SciLifeLab) unterstützt (VC2020-0015 an Claudia Fredolini und Francesca Chiodi und VC-2022-0028 an Claudia Fredolini).
Materialien
| Name | Company | Catalog Number | Comments |
| ACE2-Biotin | Acro Biosystems (Newark, DE) | AC2-H82E6-25 ug | Conc: 340 µg/mL, LOT#BV35376-203HFI-2128 |
| Anti-Goat IgG, PE-conjugated | Jackson ImmunoResearch (West Grove, PA) | 705-116-147 | Host species: Donkey |
| Anti-Human IgG R-PE | Life Technologies/Thermo Fisher (Waltham, MA) | H10104 | Conc: 0.15 mg/mL, LOT#2079224, Host species: Goat |
| Anti-Mouse IgG, PE-conjugated | Jackson ImmunoResearch (West Grove, PA) | 115-116-146 | Host species: Goat |
| Anti-Rabbit IgG, PE-conjugated | Jackson ImmunoResearch (West Grove, PA) | 111-116-144 | Host species: Goat |
| Biotin | Thermo-Fisher Scientific (Waltham, MA) | 20RUO | 100 mM, pH 10 Conc. 1 mg/mL |
| Blocker Casein in PBS | Thermo-Fisher Scientific (Waltham, MA) | 37528 | LOT#VD301372 |
| Blocker reagent for ELISA (BRE) | Roche (Basel, Switzerland) | 11112589001 | |
| Brilliant Violet 421 anti-DYKDDDDK Tag Antibody (Anti-FLAG) 0.2 mg/ml, rat IgG2a, λ | BioLegend (Amsterdam, The Netherlands) | 637321 | |
| Bovine serum albumin (BSA) | Saveen & Werner (Limhamn, Sweden) | B2000-500 | LOT#04D5865 |
| EDC (1-Ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride) | Proteochem (Hurricane, UT) | C1100-custom (65 mg) | LOT# MK3857 |
| Fetal calf serum (FCS) | Gibco/Thermo Fisher (Waltham, MA) | 10270-106 | |
| Goat anti-ACE2 polyclonal antibody | R&D Systems/Bio-Techne (Minneapolis, MN) | AF933 | Host species: Goat |
| Goat IgG | Bethyl Labs (Montgomery, TX) | P50-200 | LOT#P50-200-6 |
| L-glutamine | Thermo-Fisher Scientific (Waltham, MA) | 25030024 | |
| Low-bind 1.5 mL microfuge tubes | VWR (Radnor, PA) | 525-0133 | |
| MagPlex-C Microspheres | Luminex Corporation (Austin, TX) | MC10XXX-01 | |
| MEM tissue cuture media | Gibco/Thermo Fisher (Waltham, MA) | 21430-020 | |
| Microplate, 96-Well, Polystyrene, Half-area, Clear | Greiner Bio-One (Kremsmünster, Austria) | 675101 | |
| NaHCO3 | Gibco/Thermo Fisher (Waltham, MA) | 25080-060 | |
| Neutravidin | Thermo-Fisher Scientific (Waltham, MA) | 31000 | LOT#UK292857 |
| PBS tablets | Medicago AB (Uppsala, Sweden) | 09-9400-100 | LOT#272320-01 |
| Penicillin/Streptomycin | Gibco/Thermo Fisher (Waltham, MA) | 15140122 | |
| Poly(vinyl alcohol) | Sigma-Aldrich (St. Louis, MO) | 360627 | |
| Polyvinylpyrrolidone | Sigma-Aldrich (St. Louis, MO) | 437190 | |
| ProClin 300 | Sigma-Aldrich (St. Louis, MO) | 48915-U | |
| Rabbit IgG | Bethyl Labs (Montgomery, TX) | P120-301 | LOT#12 |
| scFv-FAb1 | In-house production | Human Antibody Therapeutics Unit, Scilifelab, Sweden. Monoclonal scFv. Conc: 0.12 mg/mL. | |
| scFv-FAb2 | In-house production | Human Antibody Therapeutics Unit, Scilifelab, Sweden. Monoclonal scFv. Conc batch1: 0.38 mg/mL. Conc batch2: 0.45 mg/mL | |
| scFv-FAb3 | In-house production | Human Antibody Therapeutics Unit, Scilifelab, Sweden. Monoclonal scFv. Conc: 0.34 mg/mL. | |
| scFv-FAb4 | In-house production | Human Antibody Therapeutics Unit, Scilifelab, Sweden. Monoclonal scFv. Conc: 2.85 mg/mL. | |
| scFv-FAb5 | In-house production | Human Antibody Therapeutics Unit, Scilifelab, Sweden. Monoclonal scFv. Conc:2.7mg/mL. | |
| SARS-CoV-2 infectious particles, Swedish isolate | In-house production | The Public Health Agency of Sweden | |
| SARS-CoV-2 Spike Antibody (Hu-anti-S1) | Novus Biologicals (Centennial, CO) | NBP2-90980 | Monoclonal antibody. Conc: 1 mg/mL. Host: Human. Clone: CR3022. Isotype: IgG1 Kappa. LOT#T201B06 |
| Sodium phosphate monobasic, anhydrous | Sigma-Aldrich (St. Louis, MO) | S3139 | |
| Sulfo-NHS (N-hydroxysulfosuccinimide) | Thermo-Fisher Scientific (Waltham, MA) | 24510 | LOT# XH321563 |
| Tween | Thermo-Fisher Scientific (Waltham, MA) | BP337-50 | LOT#194435 |
| Ultraviolet lamp | Vilber Lourmat GmbH (Eberhardzell, Germany) | VL-215.G | Wavelength = 254 nm; 2 × 15-watt bulbs |
| Vero E6 cells | ATCC (Manassus, VA) | CRL-1586 | |
| xMAP INTELLIFLEX DR-SE (dual-reporter flow instrument) | Luminex Corporation (Austin, TX) | INTELLIFLEX-DRSE-RUO |
Referenzen
- Rey, F. A., Lok, S. M. Common features of enveloped viruses and implications for immunogen design for next-generation vaccines. Cell. 172 (6), 1319-1334 (2018).
- V'kovski, P., Kratzel, A., Steiner, S., Stalder, H., Thiel, V. Coronavirus biology and replication: implications for SARS-CoV-2. Nature Reviews Microbiology. 19 (3), 155-170 (2021).
- Burnie, J., et al. Flow virometry quantification of host proteins on the surface of HIV-1 pseudovirus particles. Viruses. 12 (11), 1296 (2020).
- Gentili, M., et al. Transmission of innate immune signaling by packaging of cGAMP in viral particles. Science. 349 (6253), 1232-1236 (2015).
- Modrow, S., Falke, D., Truyen, U., Schätzl, H. . Viruses: Definition, Structure, Classification. In Molecular Virology. , 163-181 (2013).
- Trinh, K. T. L., Do, H. D. K., Lee, N. Y. Recent advances in molecular and immunological diagnostic platform for virus detection: A review. Biosensors. 13 (4), 490 (2023).
- Zamora, J. L. R., Aguilar, H. C. Flow virometry as a tool to study viruses. Methods. 134-135, 87-97 (2018).
- Graham, H., Chandler, D. J., Dunbar, S. A. The genesis and evolution of bead-based multiplexing. Methods. 158, 2-11 (2019).
- Byström, S., et al. Affinity proteomic profiling of plasma for proteins associated to area-based mammographic breast density. Breast Cancer Research. 20 (1), 14 (2018).
- Rudberg, A. -. S., et al. SARS-CoV-2 exposure, symptoms and seroprevalence in healthcare workers in Sweden. Nature Communications. 11 (1), 5064 (2020).
- Liu, J., et al. Multiplex reverse transcription PCR Luminex assay for detection and quantitation of viral agents of gastroenteritis. Journal of Clinical Virology. 50 (4), 308-313 (2011).
- Gadsby, N. J., Hardie, A., Claas, E. C. J., Templeton, K. E. Comparison of the Luminex respiratory virus panel fast assay with in-house real-time PCR for respiratory viral infection diagnosis. Journal of Clinical Microbiology. 48 (6), 2213-2216 (2010).
- Lorenzen, E., et al. Multiplexed analysis of the secretin-like GPCR-RAMP interactome. Science Advances. 5 (9), (2019).
- Angeloni, S., Cameron, A., Pecora, N. D., Dunbar, S. A rapid, multiplex dual reporter IgG and IgM SARS-CoV-2 neutralization assay for a multiplexed bead-based flow analysis system. Journal of Visualized Experiments: JoVE. (170), e62487 (2021).
- Jackson, C. B., Farzan, M., Chen, B., Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nature Reviews Molecular Cell Biology. 23 (1), 3-20 (2022).
- ISO10993-5 Biological evaluation of medical devices - Part 5: Tests for in vitro cytotoxicity. International Standardization Organization Available from: https://nhiso.com/wp-content/uploads/2018/05/ISO-10993-5-2009.pdf (2009)
- Poetz, O., et al. Sequential multiplex analyte capturing for phosphoprotein profiling. Molecular & Cellular Proteomics. 9 (11), 2474-2481 (2010).
- Dunbar, S. A., Vander Zee, C. A., Oliver, K. G., Karem, K. L., Jacobson, J. W. Quantitative, multiplexed detection of bacterial pathogens: DNA and protein applications of the Luminex LabMAP system. Journal of Microbiological Methods. 53 (2), 245-252 (2003).
- Taniuchi, M., et al. Multiplex polymerase chain reaction method to detect Cyclospora, Cystoisospora, and Microsporidia in stool samples. Diagnostic Microbiology and Infectious Disease. 71 (4), 386-390 (2011).
- Wu, M., et al. High-throughput Luminex xMAP assay for simultaneous detection of antibodies against rabbit hemorrhagic disease virus, Sendai virus, and rabbit rotavirus. Archives of Virology. 164 (6), 1639-1646 (2019).
- Dias, D., et al. Optimization and validation of a multiplexed Luminex assay to quantify antibodies to neutralizing epitopes on human papillomaviruses 6, 11, 16, and 18. Clinical and Vaccine Immunology. 12 (8), 959-969 (2005).
- Opalka, D., et al. Simultaneous quantitation of antibodies to neutralizing epitopes on virus-like particles for human papillomavirus types 6, 11, 16, and 18 by a multiplexes lumina assay. Clinical and Diagnostic Laboratory Immunology. 10 (1), 108-115 (2003).
- Nolte-'T Hoen, E., Cremer, T., Gallo, R. C., Margolis, L. B. Extracellular vesicles and viruses: Are they close relatives. Proceedings of the National Academy of Sciences. 113 (33), 9155-9161 (2016).
- Ohmichi, T., et al. Quantification of brain-derived extracellular vesicles in plasma as a biomarker to diagnose Parkinson's and related diseases. Parkinsonism & Related Disorders. 61, 82-87 (2019).
- Ter-Ovanesyan, D., et al. Framework for rapid comparison of extracellular vesicle isolation methods. Elife. 10, e70725 (2021).
- Gamage, S. S. T., et al. Microfluidic affinity selection of active SARS-CoV-2 virus particles. Science Advances. 8 (39), (2022).
- Renner, T. M., Tang, V. A., Burger, D., Langlois, M. -. A. Intact viral particle counts measured by flow virometry provide insight into the infectivity and genome packaging efficiency of moloney murine leukemia virus. Journal of Virology. 94 (2), e01600-01619 (2020).
- Niraja, S., et al. A flow virometry process proposed for detection of SARS-CoV-2 and large-scale screening of COVID-19 cases. Future Virology. 15 (8), 525-532 (2020).
- Lippé, R. Flow virometry: A powerful tool to functionally characterize viruses. Journal of Virology. 92 (3), e01765 (2018).
- Drobin, K., Nilsson, P., Schwenk, J. M. Highly multiplexed antibody suspension bead arrays for plasma protein profiling. Methods in Molecular Biology. 1023, 137-145 (2013).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten