
Method Article
Utilisation du pipeline Open-Source MALDI TOF-MS IDBac pour l'analyse des protéines microbiennes et des données spécialisées sur les métabolites
Dans cet article
Résumé
IDBac est un pipeline bioinformatique à base de spectrométrie de masse open source qui intègre des données provenant à la fois de protéines intactes et de spectres de métabolites spécialisés, recueillis sur des matériaux cellulaires extraits de colonies bactériennes. Le pipeline permet aux chercheurs d'organiser rapidement des centaines à des milliers de colonies bactériennes en groupes taxonomiques putatifs, et de les différencier davantage en fonction de la production spécialisée de métabolites.
Résumé
Afin de visualiser la relation entre la phylogénie bactérienne et la production spécialisée de métabolites des colonies bactériennes poussant sur l'agar nutritif, nous avons développé IDBac-une desorption/ionisation laser à faible coût et à haut débit spectrométrie de masse en temps de vol (MALDI-TOF MS) pipeline bioinformatique. Le logiciel IDBac est conçu pour les non-experts, est disponible gratuitement, et capable d'analyser quelques à des milliers de colonies bactériennes. Ici, nous présentons des procédures pour la préparation des colonies bactériennes pour l'analyse mS MALDI-TOF, l'opération des instruments de SP, et le traitement et la visualisation des données dans IDBac. En particulier, nous instruisons les utilisateurs comment regrouper les bactéries en dendrogrammes à partir d'empreintes digitales protéiques de la SP et créons de manière interactive les réseaux d'associations de métabolites (MN) à partir de données spécialisées sur les métabolites.
Introduction
Un obstacle majeur pour les chercheurs qui étudient la fonction bactérienne est la capacité d'évaluer rapidement et simultanément l'identité taxonomique d'un micro-organisme et sa capacité à produire des métabolites spécialisés. Cela a empêché des progrès significatifs dans la compréhension de la relation entre la phylogénie bactérienne et la production spécialisée de métabolites dans la majorité des bactéries isolées de l'environnement. Bien que les méthodes basées sur la SP qui utilisent des empreintes digitales protéiques pour regrouper et identifier les bactéries soient bien décrites1,2,3,4, ces études ont généralement été réalisées sur de petits groupes d'isolats, d'une manière spécifique à l'espèce. Fait important, l'information sur la production spécialisée de métabolites, un moteur majeur de la fonction microbienne dans l'environnement, n'a pas été incorporée dans ces études. Silva et coll.5 ont récemment fourni un historique complet détaillant la sous-utilisation de MALDI-TOF MS pour analyser les métabolites spécialisés et la pénurie de logiciels pour soulager les goulots d'étranglement actuels en bioinformatique. Afin de remédier à ces lacunes, nous avons créé IDBac, un pipeline bioinformatique qui intègre à la fois les modes linéaires et réfléchissants de MALDI-TOF MS6. Cela permet aux utilisateurs de visualiser et de différencier rapidement les isolats bactériens à partir de protéines et de métabolites spécialisés, respectivement, les empreintes digitales de la SP.
IDBac est rentable, à haut débit, et conçu pour l'utilisateur laïc. Il est disponible gratuitement (chasemc.github.io/IDBac), et ne nécessite l'accès qu'à un spectromètre de masse MALDI-TOF (le mode réflecteur sera nécessaire pour l'analyse spécialisée des métabolites). La préparation de l'échantillon repose sur la méthode simple de « transfert direct étendu »7,8 et les données sont recueillies avec des acquisitions linéaires et réfléchissantes consécutives sur un seul point MALDI-cible. Avec IDBac, il est possible d'analyser la phylogénie putative et la production spécialisée de métabolites de centaines de colonies en moins de quatre heures, y compris la préparation d'échantillons, l'acquisition de données et la visualisation des données. Ceci présente un avantage significatif de temps et de coût sur les méthodes traditionnelles d'identification des bactéries (telles que le séquençage de gène), et d'analyse de la sortie métabolique (spectrométrie de masse de chromatographie liquide [LCMS] et méthodes chromatographiques semblables).
À l'aide de données obtenues dans le cadre d'une analyse linéaire en mode, IDBac utilise un clusterhiérarchique pour représenter la parenté des spectres protéiques. Étant donné que les spectres représentent principalement des protéines ribosomal ionisées, ils fournissent une représentation de la diversité phylogénétique présente dans un échantillon. En outre, IDBac intègre des données en mode réflecteur pour afficher des empreintes digitales spécialisées métabolites sous forme de metabolite Association Networks (MANs). Les MAN sont des réseaux bipartites qui permettent une visualisation facile de la production de métabolites partagée et unique entre les isolats bactériens. La plate-forme IDBac permet aux chercheurs d'analyser à la fois les données protéiques et les métabolites spécialisés en tandem, mais aussi individuellement si un seul type de données est acquis. Fait important, IDBac traite les données brutes des instruments Bruker et Xiamen, ainsi que txt, onglet, csv, mzXML et mzML. Cela élimine le besoin de conversion manuelle et de mise en forme des ensembles de données, et réduit considérablement le risque d'erreur de l'utilisateur ou de mauvaise manipulation des données De SP.
Protocole
1. Préparation de la matrice MALDI
- Préparer 10 mg/mL MALDI-grade, et/ou recrystallized 'cyano-4-hydroxycinnamic acid (CHCA) dans les solvants de MS-grade : 50% acetonitrile (ACN), 47.5% eau (H2O), 2.5% acide trifluoroacetic (TFA). Exemple : 100 euros de solution l'ACN de 50 l 47,5 euros L H2O, 2,5 l TFA, 1 mg CHCA
- Préparer au moins 1 l de solution de matrice par tache de plaque MALDI et vortex ou sonicate jusqu'à ce qu'en solution (environ 5 min sonication ou pas de solides visibles).
CAUTION: TFA est un acide fort qui doit être manipulé dans une hotte de fumée chimique tout en portant un équipement de protection individuelle approprié, car il peut endommager la peau, les yeux et les voies respiratoires avec contact ou par inhalation.
REMARQUE : Le CHCA est hygroscopique et sensible à la lumière et doit être stocké dans des flacons d'ambre dans un dessiccateur. Il existe de nombreuses options de matrice MALDI disponibles. ChCA est plus commun pour le profilage des protéines des bactéries, mais travaille également pour l'analyse spécialisée des métabolites. La sélection de la matrice dépend des besoins individuels de l'utilisateur/expérience.
- Préparer au moins 1 l de solution de matrice par tache de plaque MALDI et vortex ou sonicate jusqu'à ce qu'en solution (environ 5 min sonication ou pas de solides visibles).
2. Préparation des plaques cibles MALDI
REMARQUE: Voir Sauer et al.7, pour plus de détails.
- Rincer la plaque MALDI avec du méthanol (de qualité HPLC ou plus) et essuyer à l'aulet avec des lingettes en papier mou. N'utilisez pas de brosses abrasives lorsque vous nettoyez les plaques cibles, car cela peut endommager de façon permanente la surface de la plaque cible.
- Attribuez des taches de calibrant de protéine et de métabolite spécialisés. Organiser les taches d'étalonnage uniformément dans l'échantillon, pour tenir compte des irrégularités de la plaque MALDI et de la dérive des instruments au fil du temps. Attribuer un nombre approprié de points média/matrice-blank pour l'étude; ces taches ne contiendront que des médias et une matrice, ou seulement une matrice.
- À l'aide d'un cure-dent stérile, transférer une petite partie d'une colonie bactérienne à l'endroit approprié sur la plaque MALDI. Étendre la colonie bactérienne uniformément sur place. L'endroit doit apparaître aussi plat que possible.
REMARQUE : Il sera plus facile d'aplatir les colonies bactériennes qui sont plus mucoïdes/amorphes. Pour les colonies plus rigides/solides, évitez de laisser des amas visibles de masse cellulaire sur la tache MALDI (Figure 1).

Figure 1 : plaque MALDI-cible montrant deux isolats différents avant d'ajouter l'acide formique et la matrice MALDI (top 3 taches - Bacillus sp.; bas 3 taches - Streptomyces sp.). Pour les deux, la colonne 3 représente l'échantillon excédentaire; la colonne 2 représente la quantité appropriée d'échantillon; la colonne 1 représente un échantillon insuffisant pour l'analyse MALDI. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
- Préparer une matrice/contrôle média en utilisant un cure-dent stérile pour transférer une quantité minimale d'agar/média sur l'endroit approprié sur la plaque MALDI.
- Regroupez 1 'L de 70% d'acide formique de niveau de spectrométrie de masse sur chaque tache d'échantillon, y compris les taches de contrôle de matrice. Laisser sécher complètement l'acide dans une étui à fumée chimique (environ 5 min).
CAUTION: L'acide formique est un produit chimique caustique et doit être manipulé dans des hottes de fumée chimique. Il peut endommager les voies respiratoires s'il est inhalé. - Ajoutez 1 'L de la solution de matrice MALDI préparée à chaque tache d'échantillon, ainsi qu'aux points de contrôle de matrice/médias. Laisser la solution de matrice sécher complètement à l'air (environ 5 min).
REMARQUE : Il est possible de stocker la plaque dans un dessiccateur, dans l'obscurité, jusqu'à ce qu'elle puisse être analysée sur un spectromètre de masse MALDI-TOF. Les temps de stockage autorisés peuvent varier en fonction de la stabilité de l'échantillon. - Ajouter un calibrant de 0,5 à 1,0 L aux points d'étalonnage assignés, suivi d'une solution de matrice MALDI de 1 l. Pipette la solution résultante de haut en bas pour mélanger. Laisser sécher complètement toutes les taches avant l'introduction dans le spectromètre de masse MALDI-TOF.
REMARQUE : Les calibrants de protéine et de métabolite spécialisés devraient être ajoutés dans les 30 minutes de l'analyse de MALDI, car les deux sont susceptibles à la dégradation.
3. Acquisition de données
REMARQUE : Les paramètres généraux pour l'acquisition de données sont énumérés dans le tableau 1.
| paramètre | protéine | Metabolite spécialisé |
| Mass Start (Da) | en 1920 | 60 Annonces |
| Fin de masse (Da) | en 21000 | 2700 Annonces |
| Déviation de masse (Da) | en 1900 | 50 Annonces |
| Coups | 500 Annonces | 1000 |
| Fréquence (Hz) | En 2000 | En 2000 |
| Taille laser | en liberté | taille moyenne |
| MaxStdDev (ppm) | 300 Ans et plus | 30 Ans, états-unis ( |
Tableau 1.
- En suivant les protocoles spécifiques à l'instrument utilisé, configurez à la fois des étalonnages protéiques et des métabolites spécialisés.
- Testez quelques points cibles distincts pour déterminer la puissance laser optimale et le gain de détecteur à utiliser lors de l'acquisition de spectres (cela variera au jour le jour et par instrument).
REMARQUE : La figure 2A et la figure 3A montrent des spectres optimaux, tandis que la figure 2D et la figure 3D sont des exemples de spectres de mauvaise qualité.

Figure 2 : Exemple de spectres protéiques affichant l'effet de la modification de la puissance laser et du gain du détecteur. La qualité spectrale est la meilleure dans le panneau A, et diminue jusqu'à ce que la qualité insuffisante des spectres dans les panneaux C et D. Bien que le spectre du panneau B puisse entraîner des pics utilisables, le panneau A affiche des données optimales. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 3 : Exemple de spectres de métabolites spécialisés affichant l'effet de modifier la puissance laser et le gain de détecteur. La qualité spectrale est la meilleure dans le panneau A et diminue jusqu'à ce que la qualité des spectres insuffisante s'insinvace dans les panneaux C et D. Bien que le spectre du panneau B puisse entraîner des pics utilisables, le panneau A affiche des données optimales. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
- Acquérir des spectres, enregistrer des spectres protéiques dans un dossier et des spectres de métabolites spécialisés dans un deuxième dossier séparé.
4. Nettoyage de la plaque cible MALDI (adapté de Sauer et al.7)
- Retirer la plaque cible MALDI de son support et rincer à l'acétone.
- Laver avec un savon liquide non abrasif pour enlever les protéines et les lipides traces, et les lingettes en papier mou / brosse à dents à poils doux.
- Rincer à l'eau désionisée pendant environ 2 min pour enlever complètement le savon.
- Sonicate la plaque cible dans l'eau (grade HPLC ou plus) pendant 5 min.
- Rincer la plaque cible avec de l'eau (grade HPLC ou plus).
- Rincer la plaque cible avec du méthanol (grade HPLC ou supérieur).
5. Installation du logiciel IDBac
- Téléchargez le logiciel IDBac.
REMARQUE : Des sauvegardes permanentes et versionnelles sont également disponibles en téléchargement (voir le Tableau des matériaux). - Double-cliquez sur le téléchargement "Install-IDBac.exe" pour initier l'installateur et suivre les instructions à l'écran.
6. En commençant par les données brutes
REMARQUE : Des explications détaillées et des instructions de chaque étape de traitement des données sont intégrées dans IDBac, mais les principales analyses et entrées interactives sont décrites ci-dessous.
- Double-cliquez sur le raccourci de bureau IDBac pour lancer IDBac. IDBac s'ouvre sur l'onglet Introduction par défaut.
- Utilisez le bouton Vérification des mises à jour pour vous assurer que la version la plus récente d'IDBac est utilisée (nécessite un accès à Internet). Si une nouvelle version est disponible, IDBac téléchargera et installera automatiquement la mise à jour, après quoi IDBac demandera à être redémarré.
- Cliquez sur l'onglet Démarrer avec des données brutes et choisissez parmi le menu le type de données à utiliser avec IDBac ; continuer en suivant les instructions in-app.
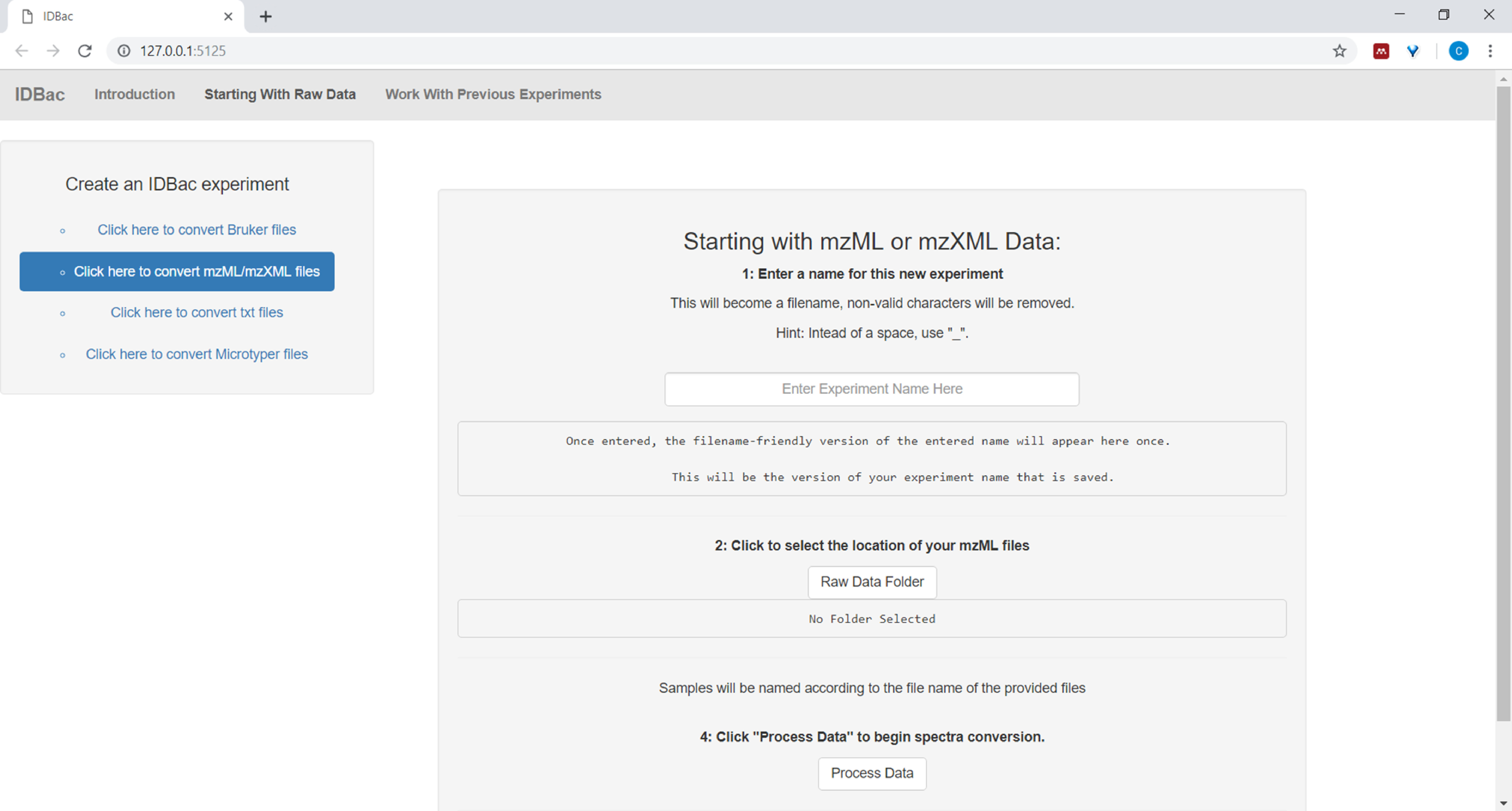
- Lors de la mise en place de la conversion et du traitement des fichiers de données, entrez un nom descriptif pour l'expérience lorsqu'il est invité (voir Figure 4). Les expériences seront ensuite affichées par ordre alphabétique, de sorte qu'une stratégie utile consiste à commencer à expérimenter des noms avec un attribut de groupe (p. ex., « bacillus-trials-experiment-1 »; "bacillus-trials-expérience-2").

Figure 4 : Étape de conversion et de prétraitement des données IDBac. IDBac convertit les spectres bruts dans le format mzML ouvert et stocke mzML, les listes de pointe et les informations d'échantillon dans une base de données pour chaque expérience. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
7. Travailler avec des expériences précédentes
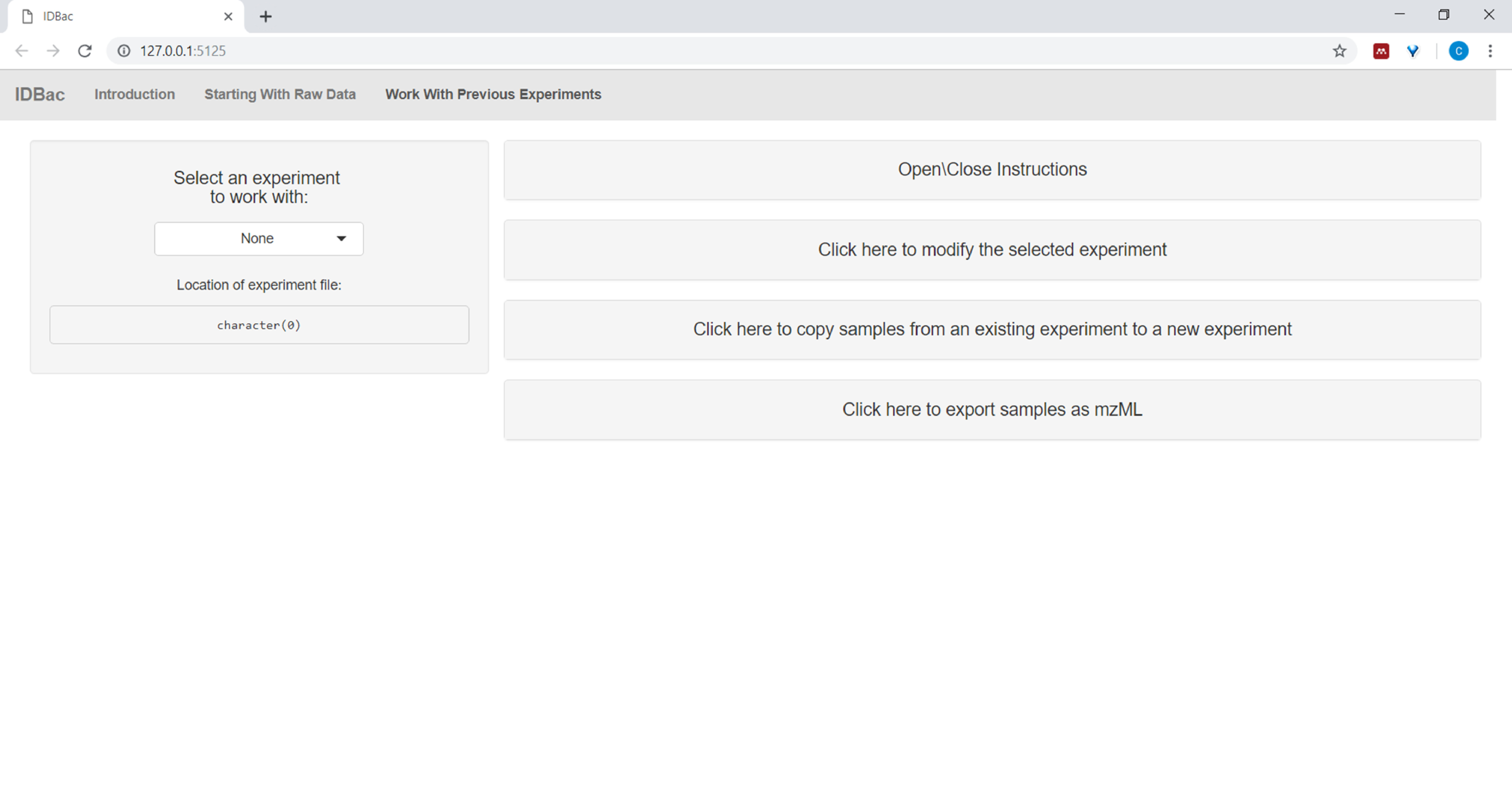
- Après avoir converti des fichiers et les avoir traités avec IDBac, ou n'importe quand on souhaite réanalyser une expérience, naviguer à la page Travail avec des expériences précédentes et Sélectionnez une expérience pour travailler avec ( Figure5).

Figure 5 : Page « Travailler avec des expériences antérieures ». Utilisez la page « Travailler avec des expériences antérieures » d'IDBac pour sélectionner une expérience à analyser ou à modifier. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
- (Facultatif) Ajouter des informations sur les échantillons à l'aide du menu Cliquez ici pour modifier l'expérience sélectionnée. Informations d'entrée dans la feuille de calcul auto-remplie et appuyez sur Enregistrer (Figure 6). Cette option permet à l'utilisateur de coder des données de couleur pendant les analyses.

Figure 6 : Informations sur l'échantillon d'entrée. Dans la page "Travailler avec des expériences antérieures" les utilisateurs peuvent entrer des informations sur des échantillons tels que l'identité taxonomique, l'emplacement de la collecte, les conditions d'isolement, etc. S'il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}
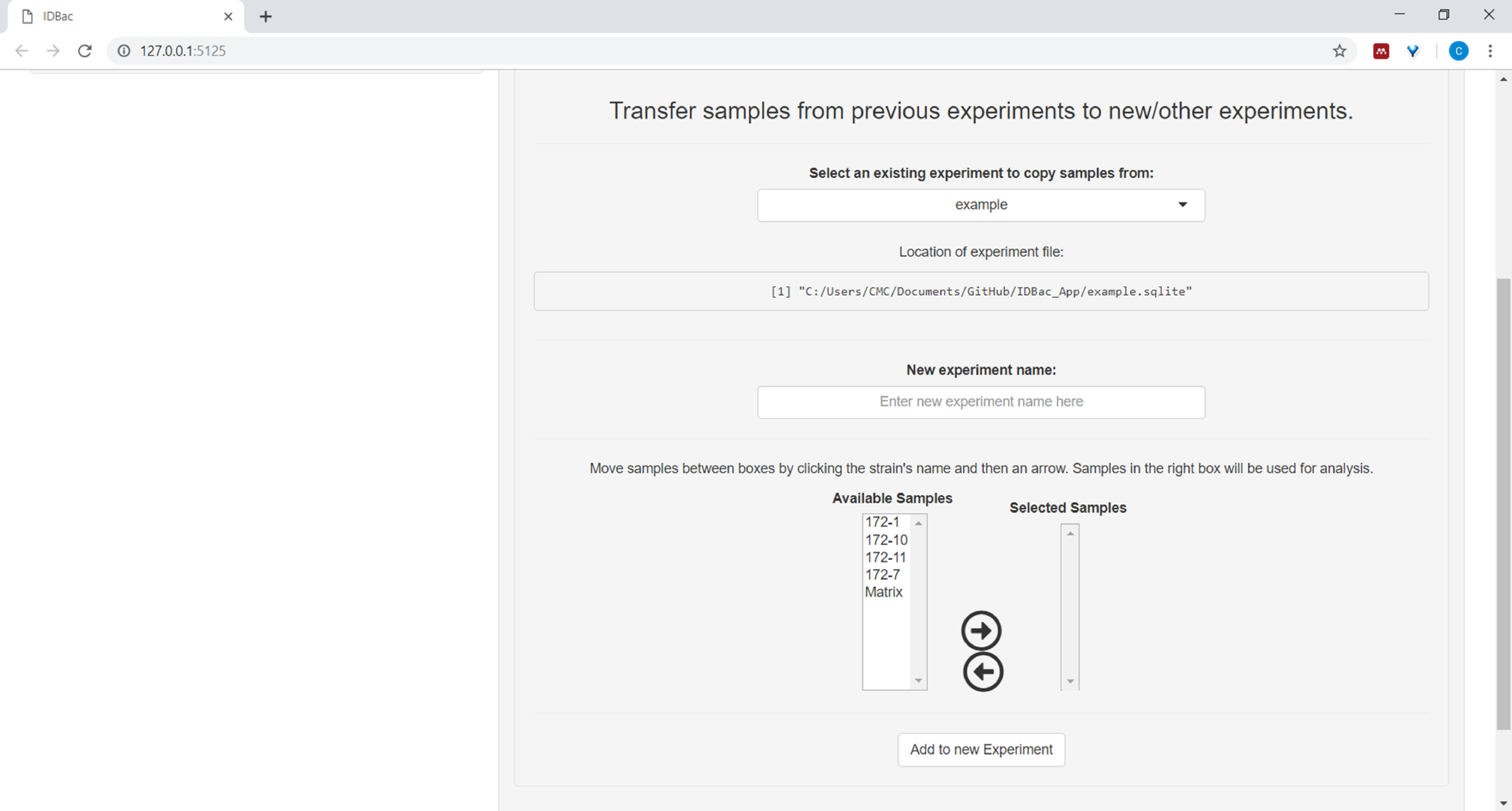
- (Facultatif) Transférer tous les échantillons, ou un sous-ensemble d'échantillons, à une nouvelle expérience ou à une autre expérience en cliquant sur les échantillons de transfert des expériences précédentes à des expériences nouvelles ou autres et en suivant les instructions fournies ( figure7).

Figure 7 : Transférer des données. La page « Travailler avec des expériences antérieures » contient la possibilité de transférer des données entre les expériences existantes et les nouvelles expériences. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
- Lorsque vous êtes prêt à commencer l'analyse, assurez-vous que l'expérience avec laquelle travailler est sélectionnée. Sélectionnez l'analyse de données protéiques ou l'analyse de données sur les petitesmolécules.
8. Mise en place de l'analyse des données protéiques et création de parcelles miroirs
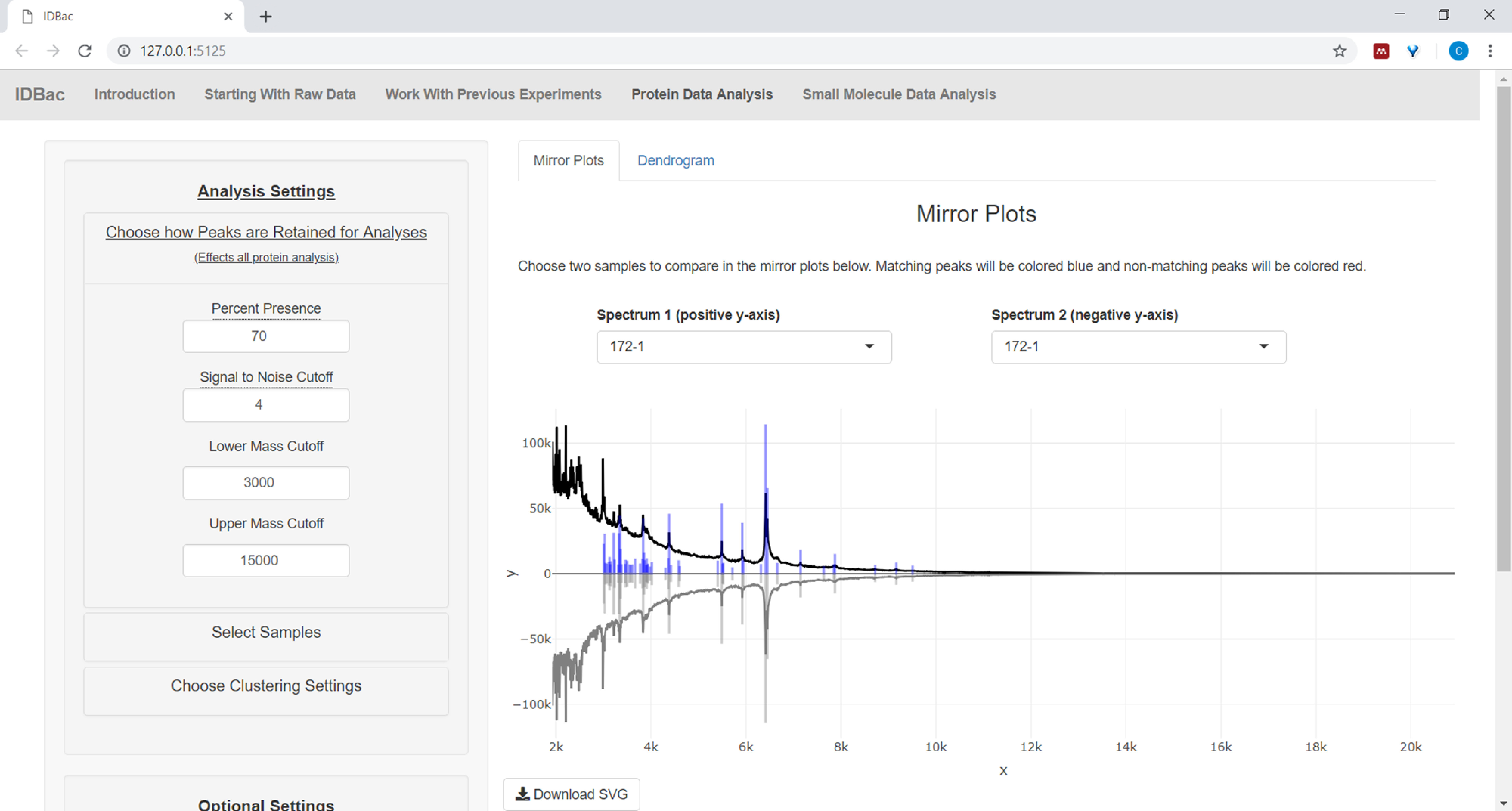
- Si vous analysez des données protéiques, naviguez d'abord vers la page d'analyse des données sur les protéines. Choisissez des réglages de pic et évaluez les spectres protéiques des échantillons par l'intermédiaire des parcelles miroir affichées (figure 8).
REMARQUE : Dans les parcelles miroirs, un pic rouge signifie la présence de ce pic uniquement dans le spectre supérieur, tandis que les pics bleus représentent ceux qui se produisent dans les deux spectres.

Figure 8 : Choisissez la façon dont les pics sont conservés pour analyse. Après avoir sélectionné une expérience à analyser, la visite de la page « Analyse des données protéinées » et l'ouverture ultérieure du menu « Choisissez comment les pics sont retenus pour l'analyse » permettent aux utilisateurs de choisir des paramètres comme le rapport signal-bruit pour la rétention des pics. L'intrigue miroir affichée (ou dendrogramme) sera automatiquement mise à jour pour refléter les paramètres choisis. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
- Ajuster le pourcentage de répliques dans lesquelles un pic doit être présent pour qu'il soit inclus pour les analyses (p. ex., si le seuil est fixé à 70 % et qu'un pic se produit dans au moins 7 répliques sur 10, il sera inclus).
- En utilisant les parcelles miroirs comme guidage visuel, ajustez le signal à la coupure de bruit qui conserve les pics les plus "véritables" et le moins de bruit, notant que plus de répliques et une plus forte "présence en pourcentage de pointe" de valeur permettra la sélection d'un signal inférieur à la coupure de bruit.
- Spécifiez les seuils m/z inférieurs et supérieurs, dictant la gamme de valeurs de masse dans chaque spectre à utiliser dans d'autres analyses par IDBac.
9. Regrouper des échantillons à l'aide de données protéiques
- Dans la page analyse des données protéiques, sélectionnez l'onglet Dendrogram. Cela permet de regrouper les échantillons en un dendrogramme selon des mesures de distance sélectionnées par l'utilisateur et des algorithmes de clustering.
- Cliquez sur Sélectionnez des échantillons sur le menu et suivez les instructions pour sélectionner les échantillons à inclure dans les analyses. Seuls les échantillons contenant des spectres protéiques seront affichés dans la case Échantillons disponibles (figure 9).

Figure 9 : Sélectionnez les échantillons de l'expérience choisie pour les inclure dans le dendrogramme affiché. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
- Utilisez les valeurs par défaut ou, sous Choisissez les paramètresde clustering, sélectionnez les algorithmes de distance et de clustering souhaités à appliquer à la génération du dendrogramme.
- Sélectionnez Présence/Absence comme entrée. Alternativement, si vous êtes sûr des hauteurs maximales des échantillons (p. ex., après avoir effectué une étude pour évaluer la variabilité de l'intensité maximale), sélectionnez Intensities comme entrée.
REMARQUE : Au moment de la publication, IDBac offre une flexibilité dans les paramètres de clustering, en s'appuyant sur les utilisateurs pour choisir les combinaisons appropriées. Si elle n'est pas familière avec ces options, il est suggéré de jumeler soit A: "cosine" distance, et "moyenne (UPGMA)" clustering; ou B: distance "Euclidean", et "Ward.D2" clustering. - Pour afficher les valeurs bootstrap sur le dendrogramme, entrez un nombre entre 2 et 1000 sous Bootstraps.
- Lorsque vous rapportez les résultats, copiez le texte dans le paragraphe Suggestions for Reporting Protein Analysis. Cela fournit les paramètres définis par l'utilisateur qui ont généré le dendrogramme spécifique.
10. Personnalisation du dendrogramme protéique
- Pour commencer à personnaliser le dendrogramme, ouvrez le menu Ajuster le dendrogramme (Figure 10).

Figure 10 : Ajustez le dendrogramme. IDBac fournit quelques options pour modifier l'apparence du dendrogramme, ceux-ci peuvent être trouvés dans le menu "Ajuster le Dendrogram". Cela comprend les branches de coloration et les étiquettes par k-moyens, ou en " coupant" le dendrogramme à une hauteur fournie par l'utilisateur. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
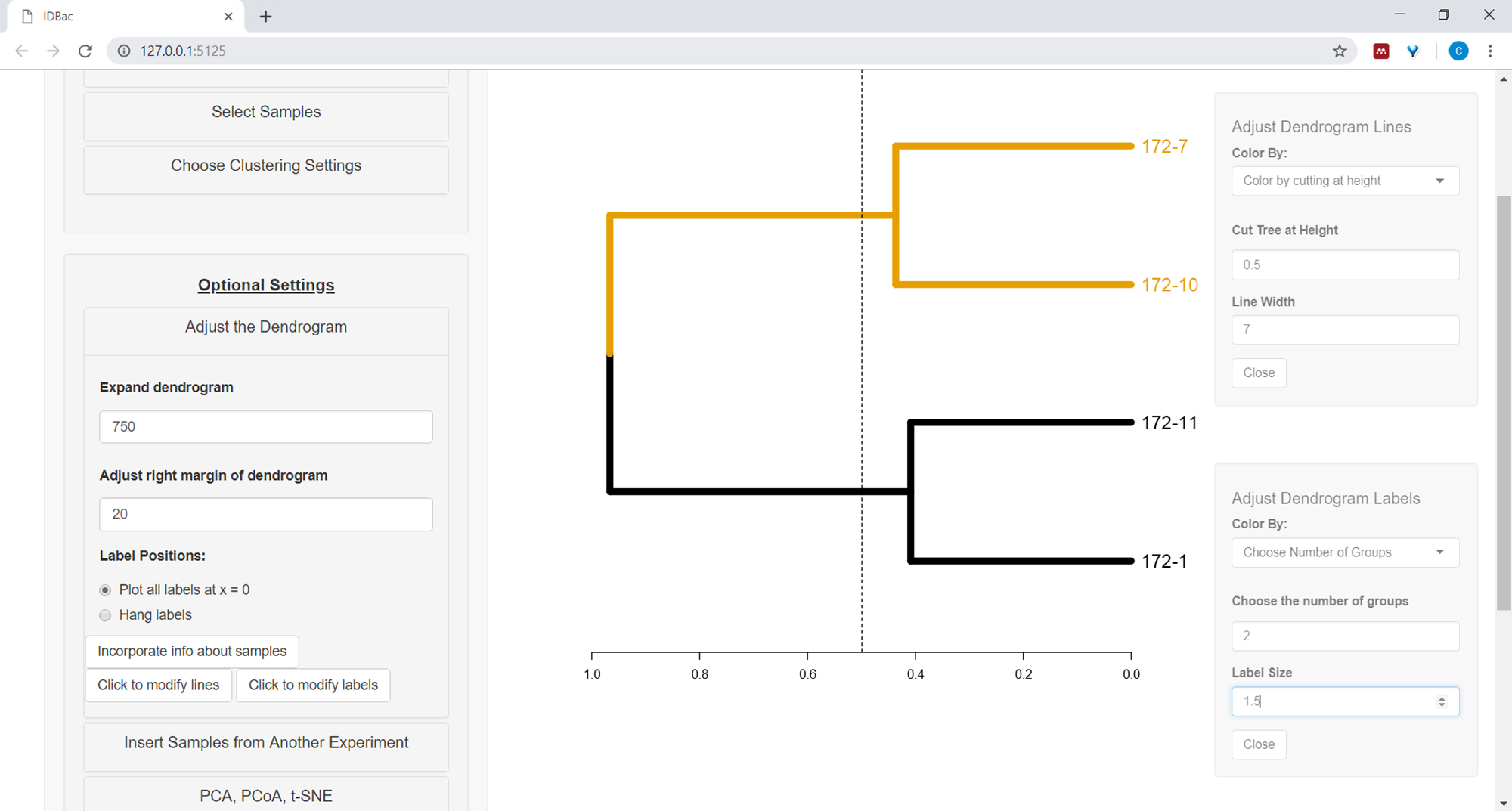{kind=link}
- Pour colorer les lignes et/ou les étiquettes du dendrogramme, sélectionnez le bouton approprié : Cliquez pour modifier les lignes ou Cliquez pour modifier les étiquettes et sélectionnez les options souhaitées.
- Pour tracer les informations de la feuille de calcul à côté du dendrogramme (voir l'étape 7.2), sélectionnez le bouton Incorporez des informations sur les échantillons. Cela ouvrira un panneau où une catégorie (colonne dans la feuille de calcul) s'auto-peuplera en fonction des valeurs saisies (figure 11).

Figure 11 : Incorporez des informations sur les échantillons. Dans le menu "Ajuster le Dendrogram" est l'option "Incorporez des informations sur les échantillons". La sélection de ce projet permettra de tracer des informations sur des échantillons à côté du dendrogramme. L'exemple d'information est entré dans la page « Travailler avec des expériences antérieures ». Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
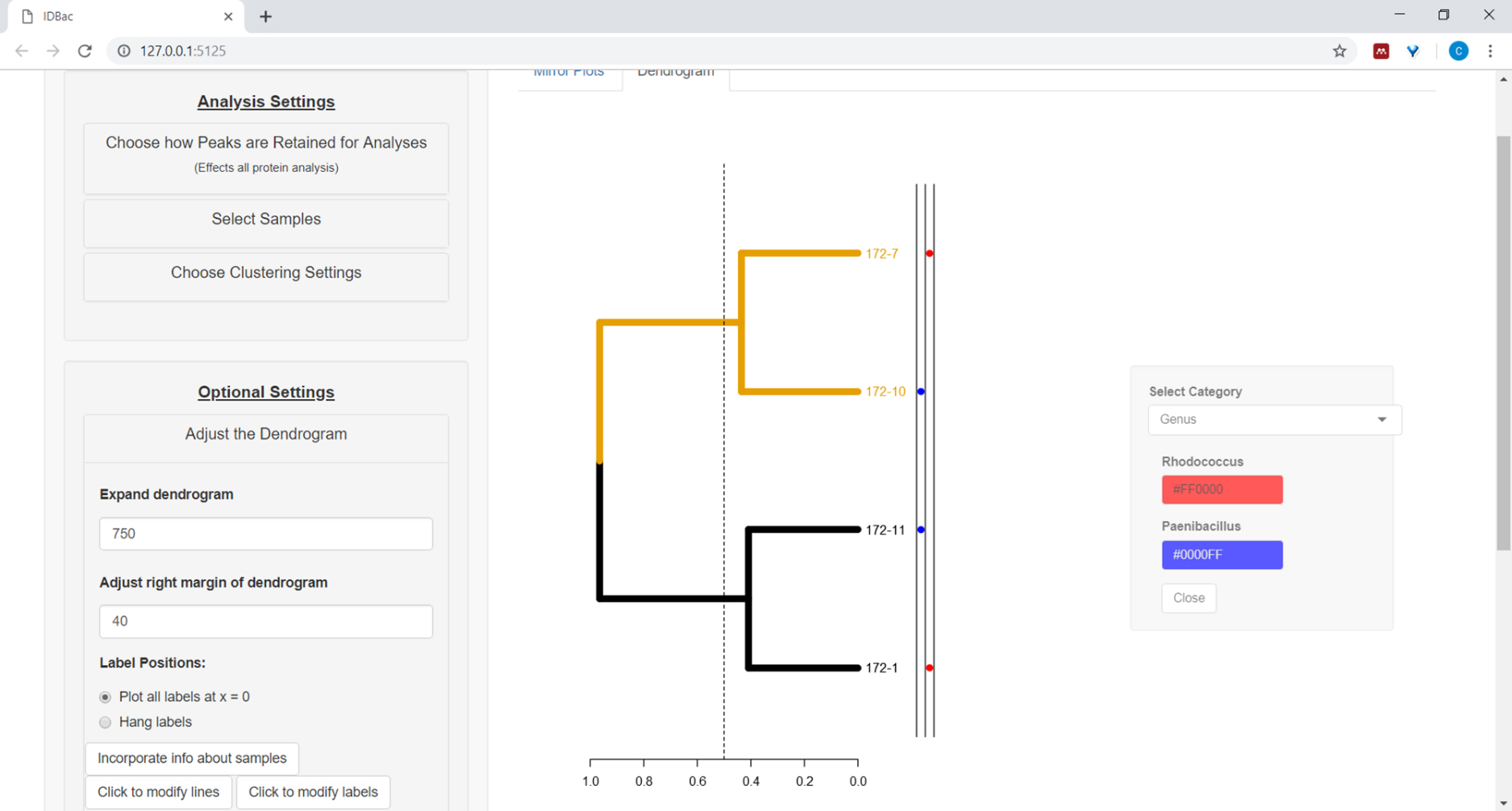{kind=link}
11. Insérer des échantillons d'une expérience distincte dans le dendrogramme
- Pour insérer des échantillons d'une autre expérience, sélectionnez le bouton menu Insérer des échantillons d'une autre expérience. Suivez les instructions dans le panneau nouvellement ouvert (figure 12).

Figure 12 : Insérez des échantillons d'un autre menu d'expérience. Parfois, il est utile de comparer des échantillons d'une autre expérience. Utilisez le menu « Insérer des échantillons d'une autre expérience » pour choisir des échantillons à inclure dans le dendrogramme actuellement affiché. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
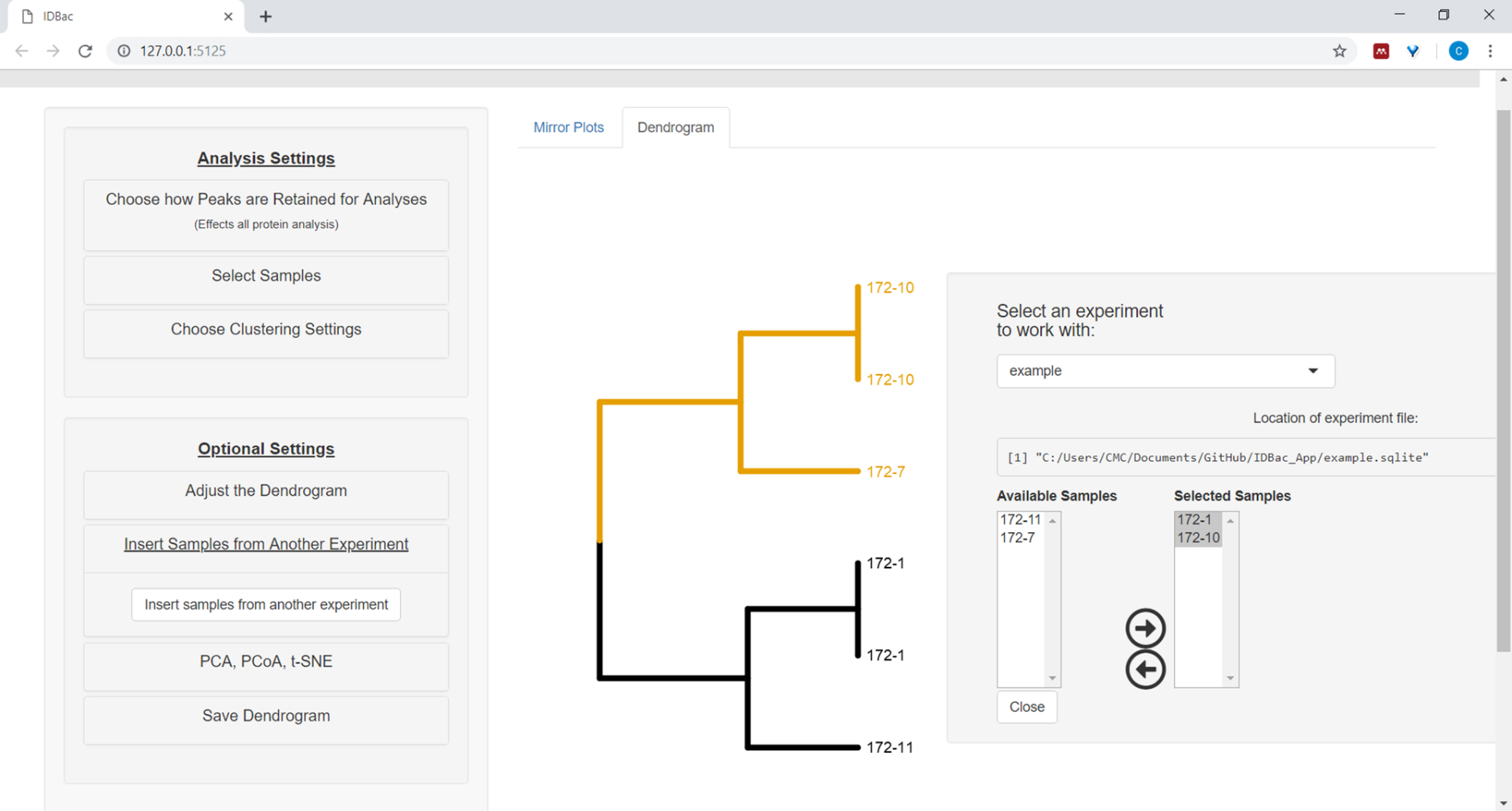{kind=link}
12. Analyse des données spécialisées sur les métabolites et les réseaux d'associations de métabolites (NAM)
- Passez à la page Metabolite Association Network (Small-Molecule). Cette page permet la visualisation des données par l'analyse des composants principaux (PCA) et les MAN, qui utilisent des réseaux bipartites pour afficher la corrélation des petites molécules de valeurs m/z avec les échantillons.
- Si un dendrogramme protéique a été créé (section 9), il sera également affiché sur cette page. Cliquez et faites glisser sur le dendrogramme pour mettre en évidence certains échantillons d'intérêt à analyser. Si aucun échantillon n'est mis en évidence ou qu'aucun dendrogramme protéique n'a été créé, un HOMME d'un sous-ensemble aléatoire ou de tous les échantillons apparaîtra, respectueusement (figure 13).

Figure 13 : Page d'analyse des données sur les petites molécules. Si un dendrogramme a été créé à partir de spectres protéiques, il sera affiché dans la page « Analyse des données des petites molécules ». Cette page affichera également les réseaux associés de métabolites (AMN) et l'analyse des composants de principe (PCA) pour les données de petites molécules. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
- Pour soustraire une matrice/media blank dans le MAN, ouvrez le menu Sélectionnez un échantillon à soustraire et choisissez l'échantillon approprié à utiliser comme un blanc.
- Ouvrez le menu Afficher/Masquer les paramètres de MAN pour sélectionner les valeurs souhaitées pour le pourcentage de présence maximale dans les répliques, le signal au bruit et les coupures de masse supérieures et inférieures, comme cela a été fait pour les spectres protéiques dans la section 9. Utilisez les parcelles miroir petite molécule pour guider la sélection de ces paramètres.
- Sélectionnez "Télécharger les données réseau actuelles" pour enregistrer les données de l'homme qui est actuellement affichée. Ces données peuvent être utilisées dans des logiciels d'analyse de réseau autres que IDBac.
- Pour les résultats de déclaration, copiez le texte dans le paragraphe Suggestions for Reporting MAN Analysis. Cela fournit les paramètres définis par l'utilisateur utilisés pour générer l'HOMME créé.
13. Partage de données
- Chaque « expérience » IDBac est enregistrée sous la forme d'une base de données SQLite unique. Il contient les spectres bruts mzML convertis, les pics détectés et toutes les informations utilisateur-entrée sur les échantillons. Par conséquent, pour partager une expérience IDBac il suffit de partager le fichier SQlite qui a le même nom que l'expérience (l'emplacement du fichier est affiché sur la page Travailler avec des expériences précédentes).
Résultats
Nous avons analysé six souches de Micromonospora chokoriensis et deux souches de Bacillus subtilis, qui étaient auparavant caractérisées6, en utilisant des données disponibles à DOI: 10.5281/zenodo.2574096. Suivant les instructions dans l'onglet Démarrage avec données brutes, nous avons sélectionné l'option Cliquez ici pour convertir les fichiers Bruker et suivi les instructions fournies par IDBac pour chaque jeu de données (Figure 14).
Une fois les étapes automatisées de conversion et de prétraitement/pic, nous avons procédé à la création d'une nouvelle expérience combinée IDBac en transférant des échantillons des deux expériences dans une seule expérience contenant à la fois Bacillus et Échantillons de micromonosopora (figure 15). L'analyse qui en a résulté a consisté à comparer les spectres protéiques à l'aide de parcelles miroirs, telles qu'illustrées à la figure 16, ce qui a été utile pour évaluer la qualité des spectres et ajuster les paramètres de pic-picking. Figure 17 affiche une capture d'écran des résultats de regroupement de protéines avec les paramètres par défaut sélectionnés. Le dendrogramme a été coloré en ajustant le seuil sur l'intrigue (apparaît comme une ligne pointillée). Il convient de noter la séparation claire entre les genres, avec à la fois M. chokoriensis et B. subtilis isole groupage séparément.
Figure 18, Figure 19, et Figure 20 mettent en évidence la capacité de générer des NAM de régions sélectionnées par l'utilisateur en cliquant et en faisant glisser à travers le dendrogramme de protéines. Avec cela, nous avons été en mesure de créer rapidement DES NMA pour comparer seulement les souches B. subtilis (Figure 18), seulement les souches de M. chokoriensis (Figure 19), et toutes les souches simultanément (Figure 20). La fonction principale de ces réseaux est de fournir aux chercheurs une vue d'ensemble du degré de chevauchement des métabolites spécialisés entre les bactéries. Avec ces données en main, les chercheurs ont maintenant la capacité de prendre des décisions éclairées à partir seulement d'une petite quantité de matériel gratté à partir d'une colonie bactérienne.

Figure 14 : Traitement spectral. Les spectres autoFlex Bruker téléchargés ont été convertis et traités à l'aide d'IDBac. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 15 : Expérience combinée IDBac. Étant donné que les spectres Micromonospora et Bacillus ont été recueillis sur différentes plaques cibles MALDI, les deux expériences ont ensuite été combinées en une seule expérience - "Bacillus-Micromonsopora". Cela a été fait dans l'onglet "Travailler avec des expériences antérieures", en suivant les instructions dans le menu "Transférer des échantillons d'expériences précédentes à de nouvelles /autres expériences". Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
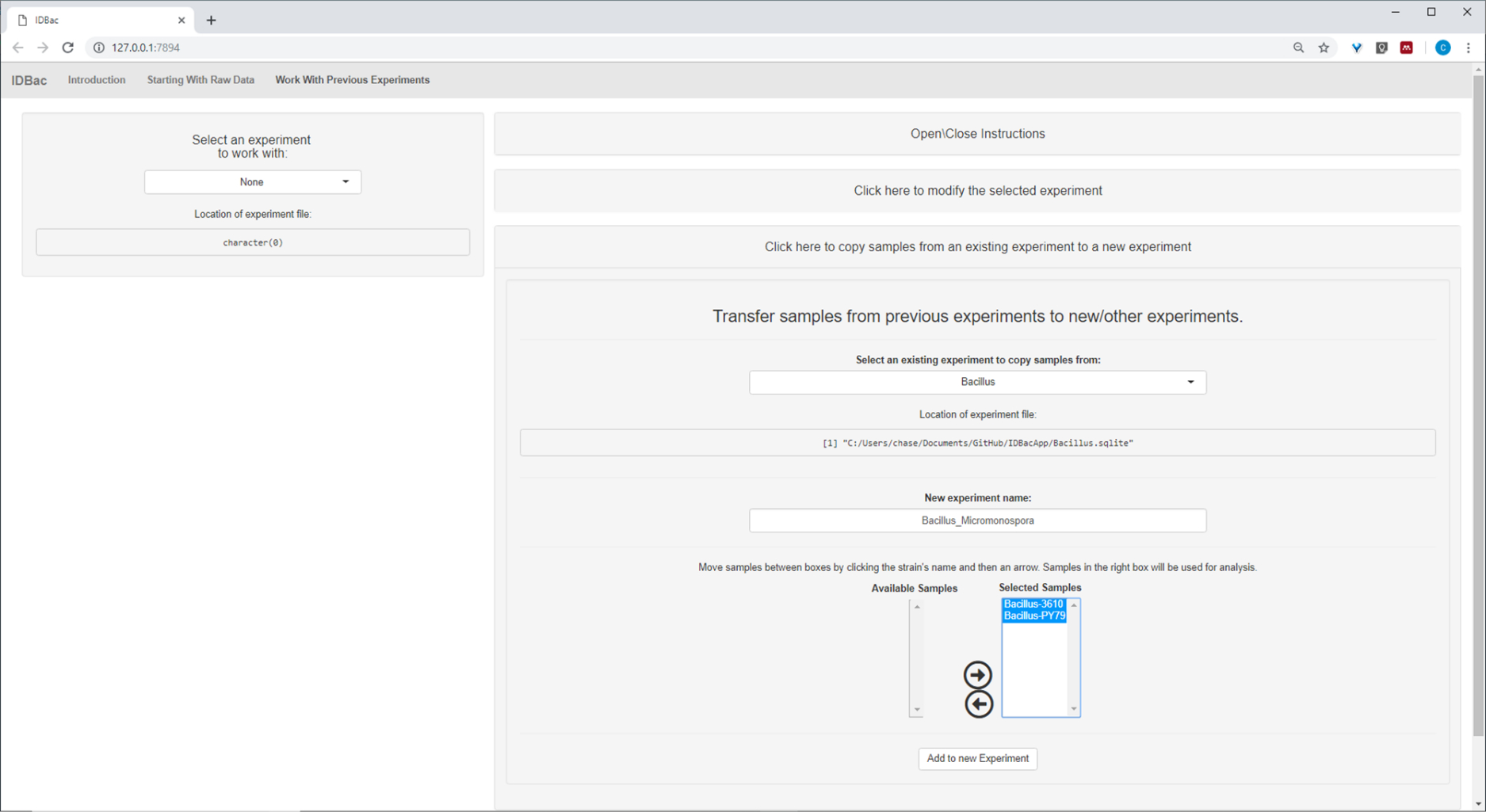{kind=link}

Figure 16 : Comparaison. Les spectres De Micromonspora et Bacillus ont été comparés à l'aide des parcelles miroirs de la page « Analyse des données protéiques ». En fin de compte, les paramètres de pointe par défaut ont été choisis. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

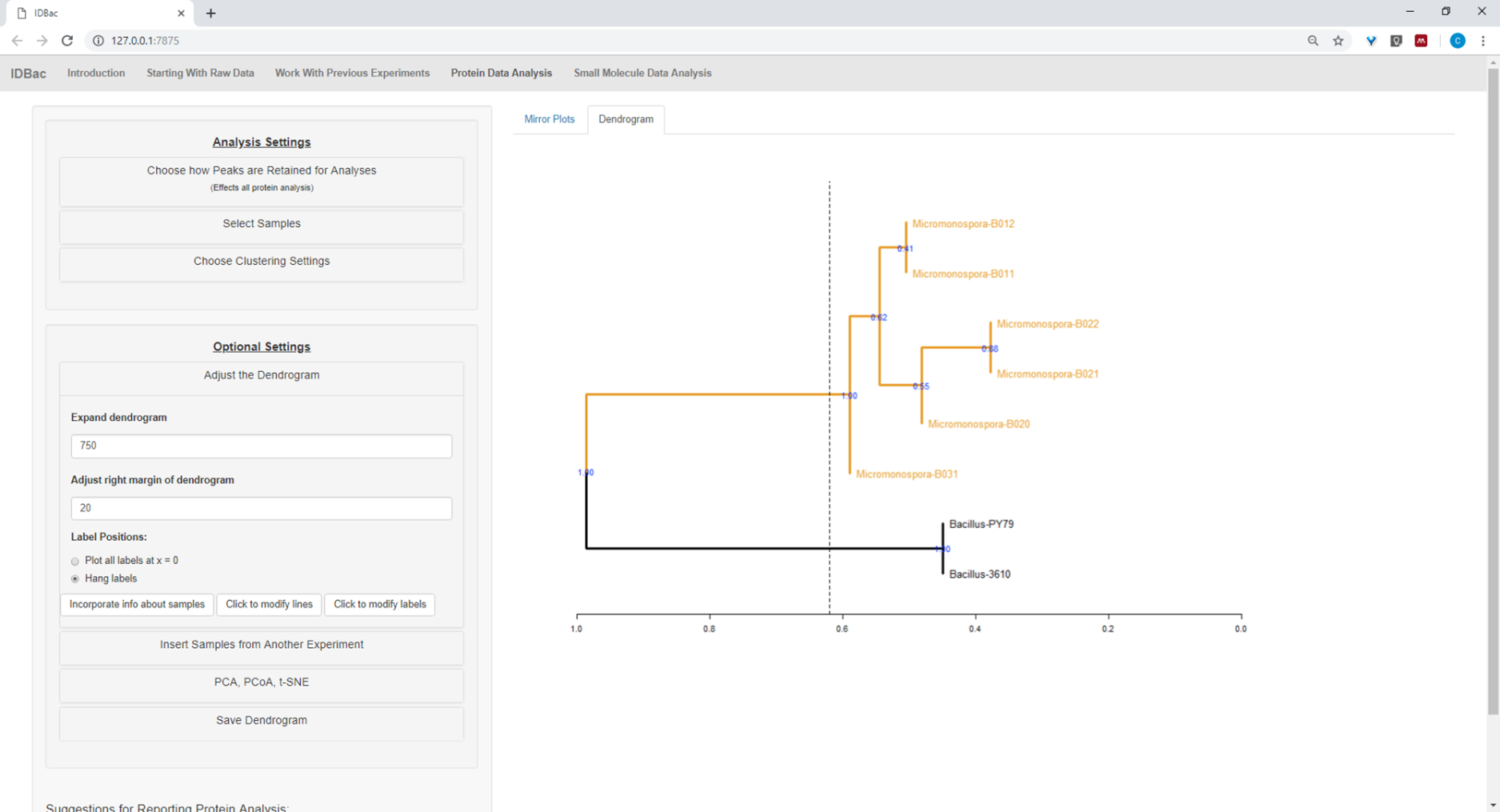
Figure 17 : Regroupement hiérarchique. Le clustering hiérarchique, à l'aide de paramètres par défaut, a correctement regroupé les isolats Bacillus et Micromonospora. Le dendrogramme a été coloré en « coupant » le dendrogramme à une hauteur arbitraire (affichée comme une ligne pointillée) et 100 bootstraps utilisés pour montrer la confiance dans la ramification. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 18 : L'homme créé en sélectionnant les souches de Bacillus sp. du dendrogramme protéique a montré une production différentielle de métabolitesspécialisés. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

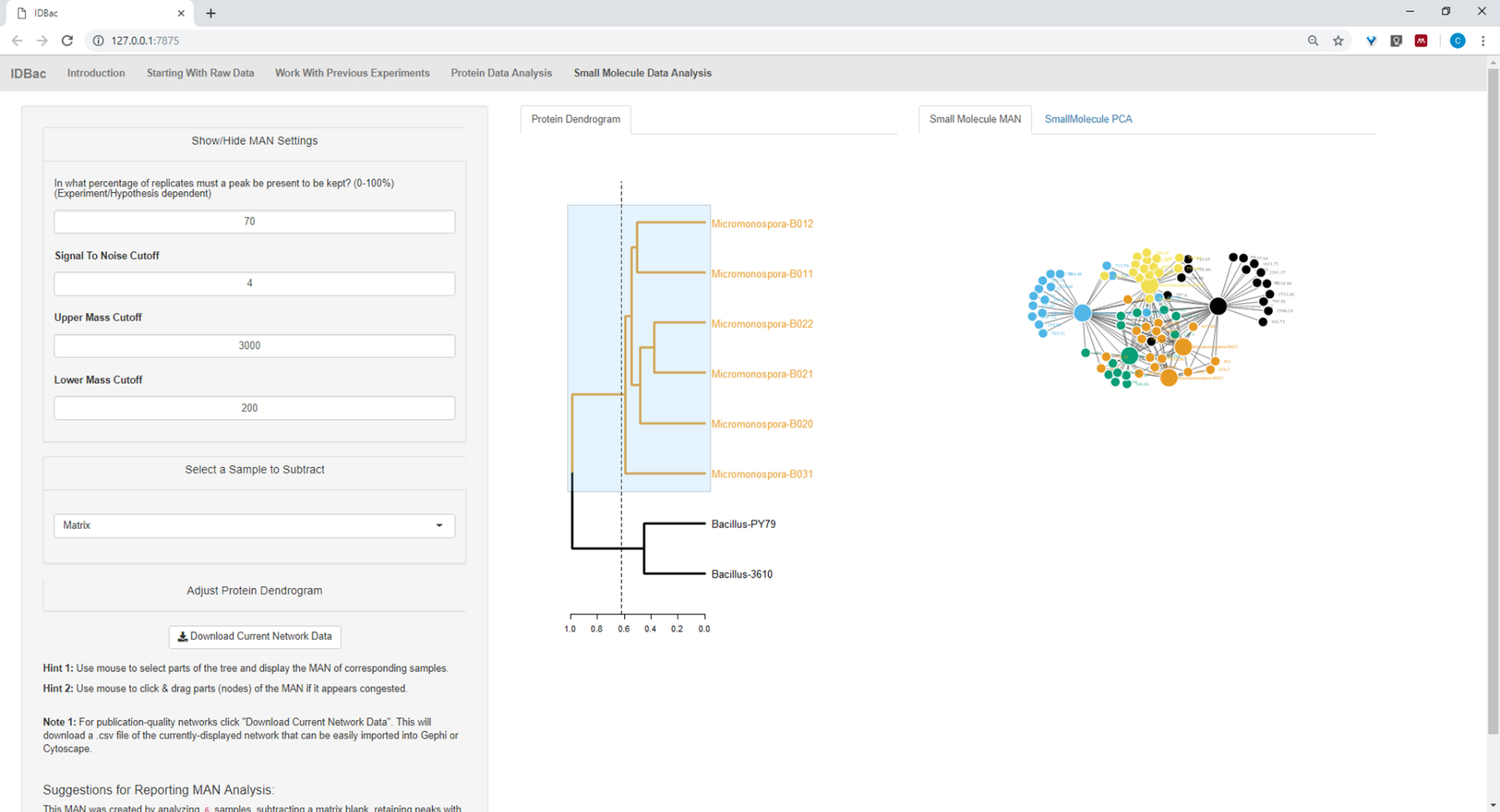
Figure 19 : L'homme créé en sélectionnant les six souches de Micromonospora sp. du dendrogramme protéique a montré la production différentielle de métabolites spécialisés. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

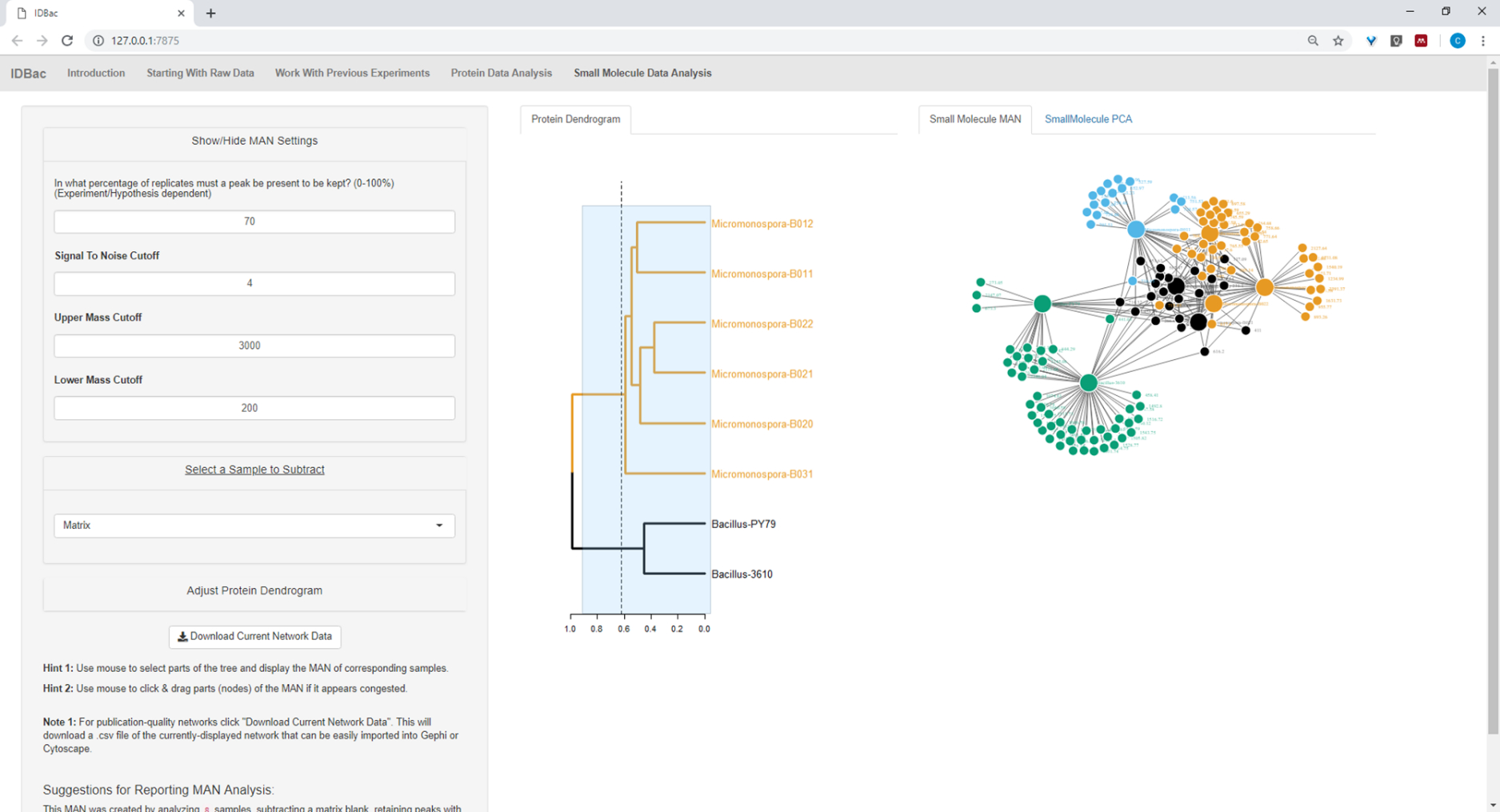
Figure 20 : MAN de Bacillus sp. et Micromonospora sp. souches montrant une production différentielle de métabolites spécialisés. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
Discussion
Le protocole IDBac détaille l'acquisition et l'analyse de données de métabolites spécialisés de jusqu'à 384 isolats bactériens en 4 h par un seul chercheur. Avec IDBac il n'est pas nécessaire d'extraire l'ADN des isolats bactériens ou de générer des extraits spécialisés de métabolites à partir de bouillons de fermentation liquide et de les analyser à l'aide de méthodes chromatographiques. Au lieu de cela, les données de protéine et de métabolite spécialisés sont rassemblées en répandant simplement le matériel des colonies bactériennes directement sur une plaque cible de MALDI. Cela réduit considérablement le temps et le coût associés à des techniques alternatives telles que le séquençage du gène 16S rRNA et LCMS9.
Il est important d'ajouter une matrice vierge et des taches d'étalonnage à la plaque MALDI, et nous recommandons d'utiliser un nombre approprié de répliques pour assurer la reproductibilité et la confiance statistique. Le nombre de répliques dépendra de l'expérience. Par exemple, si un utilisateur a l'intention de différencier des milliers de colonies d'une collection de plaques de diversité environnementale, moins de répliques peuvent être nécessaires (notre laboratoire recueille trois répliques techniques par colonie). Alternativement, si un utilisateur souhaite créer une base de données personnalisée de souches de taxons bactériens spécifiques pour déterminer rapidement les classifications sous-espèces d'isolats inconnus, alors d'autres répliques sont appropriées (notre laboratoire recueille huit répliques biologiques par souche).
IDBac est un outil permettant de différencier rapidement les isolats bactériens hautement liés à partir d'informations taxonomiques présumées et de la production spécialisée de métabolites. Il peut compléter ou servir de précurseur à des méthodes orthogonales telles que des analyses génétiques approfondies, des études impliquant la production et la fonction des métabolites, ou la caractérisation de la structure spécialisée des métabolites par spectroscopie par résonance magnétique nucléaire et/ou LC-MS/MS.
La production spécialisée de métabolites (IDBac MANs) est très sensible aux conditions de croissance bactérienne, en particulier en utilisant différents médias, ce qui est une limitation potentielle de la méthode. Toutefois, ces traits peuvent être exploités par l'utilisateur, car IDBac peut facilement générer des NMA montrant les différences dans la production spécialisée de métabolites dans une variété de conditions de croissance. Il est important de noter que même si les empreintes digitales spécialisées des métabolites peuvent varier selon l'état de croissance, nous avons déjà montré que les empreintes digitales protéiques demeurent relativement stables entre ces variables (voir Clark et coll.6). Lorsqu'il s'agit de plaques de diversité environnementale, nous recommandons de purifier les isolats bactériens avant l'analyse afin de réduire les contributions possibles des cross-talk bactériens voisins.
Enfin, l'absence d'une base de données publique consultable sur les empreintes digitales protéiques de la SP est une lacune majeure dans l'utilisation de cette méthode pour classer les bactéries environnementales inconnues. Nous avons créé IDBac dans cet esprit, et inclus la conversion automatisée des données dans un format open-source accepté par la communauté (mzML)10,11,12 et conçu le logiciel pour permettre la recherche, le partage et la création de bases de données personnalisées. Nous sommes en train de créer une grande base de données publique (10 000 souches entièrement caractérisées), qui permettra la classification de certains isolats au niveau de l'espèce, y compris des liens vers les numéros d'adhésion de GenBank lorsqu'ils seront disponibles.
IDBac est open source et le code est disponible pour tout le monde pour personnaliser leurs besoins d'analyse de données et de visualisation. Nous recommandons aux utilisateurs de consulter un vaste corpus de littérature (Sauer et al.7, Silva et coll.5) pour les aider à soutenir et à concevoir leurs objectifs expérimentaux. Nous organisons un forum de discussion à: https://groups.google.com/forum/#!forum/idbac et un moyen de signaler les problèmes avec le logiciel à: https://github.com/chasemc/IDBacApp/issues.
Déclarations de divulgation
Les auteurs n'ont rien à révéler.
Remerciements
Ce travail a été soutenu par national Institute of General Medical Sciences Grant R01 GM125943, National Geographic Grant CP-044R-17; Subvention du Fonds islandais de recherche 152336-051; et l'Université de l'Illinois à Chicago fonds de démarrage. De plus, nous remercions les contributeurs suivants : Dr Amanda Bulman pour son aide aux paramètres d'acquisition de protéines MALDI-TOF MS; Dr Terry Moore et Dr Atul Jain pour recrystallizing alpha-cyano-4-hydroxycinnamic acid matrix (CHCA).
matériels
| Name | Company | Catalog Number | Comments |
| Acetonitrile | Fisher | 60-002-65 | LC-MS Ultra CHROMASOLV |
| Autoflex Speed LEF MALDI-TOF instrument | Bruker Daltonics | ||
| Bruker Daltonics Bacterial test standard | Fisher | NC0884024 | Bruker Daltonics 8604530 |
| Bruker Peptide Calibration standard | Fisher | NC9846988 | Bruker Daltonics 8206195 |
| Formic Acid | Fisher Chemical | A117-50 | 99.5+%, Optima LC/MS Grade |
| MALDI-TOF target Plate | Bruker Daltonics | ||
| Methanol | Fisher Chemical | A456-500 | Optima LC/MS Grade |
| Toothpicks | any is ok | ||
| Trifluoroacetic acid | Fisher | AC293810010 | 99.5%, for biochemistry, ACROS Organics |
| Water | VWR | 7732-18-5 | LC-MS |
| α-Cyano-4-hydroxycinnamic acid | Sigma | 28166-41-8 | (C2020-25G) ≥98% (TLC), powder |
Références
- Sandrin, T. R., Goldstein, J. E., Schumaker, S. MALDI TOF MS profiling of bacteria at the strain level: A review. Mass Spectrometry Reviews. 32 (3), 188-217 (2013).
- Cain, T. C., Lubman, D. M., Weber, W. J., Vertes, A. Differentiation of bacteria using protein profiles from matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Communications in Mass Spectrometry. 8 (12), 1026-1030 (1994).
- Holland, R. D., Wilkes, J. G., et al. Rapid identification of intact whole bacteria based on spectral patterns using matrix-assisted laser desorption/ionization with time-of-flight mass spectrometry. Rapid Communications in Mass Spectrometry. 10 (10), 1227-1232 (1996).
- Rahi, P., Prakash, O., Shouche, Y. S. Matrix-assisted laser desorption/ionization time-of-flight mass-spectrometry (MALDI-TOF MS) based microbial identifications: challenges and scopes for microbial ecologists. Frontiers in Microbiology. 7, 1359 (2016).
- Silva, R., Lopes, N. P., Silva, D. B. Application of MALDI mass spectrometry in natural products analysis. Planta Medica. 82, 671-689 (2016).
- Clark, C. M., Costa, M. S., Sanchez, L. M., Murphy, B. T. Coupling MALDI-TOF mass spectrometry protein and specialized metabolite analyses to rapidly discriminate bacterial function. Proceedings of the National Academy of Sciences of the United States of America. 115 (19), 4981-4986 (2018).
- Freiwald, A., Sauer, S. Phylogenetic classification and identification of bacteria by mass spectrometry. Nature Protocols. 4 (5), 732-742 (2009).
- Schulthess, B., Bloemberg, G. V., Zbinden, R., Böttger, E. C., Hombach, M. Evaluation of the Bruker MALDI Biotyper for identification of Gram-positive rods: development of a diagnostic algorithm for the clinical laboratory. Journal of Clinical Microbiology. 52 (4), 1089-1097 (2014).
- Schumann, P., Maier, T. MALDI-TOF mass spectrometry applied to classification and identification of bacteria. Methods in Microbiology. 41, 275-306 (2014).
- Chambers, M. C., Maclean, B., et al. A cross-platform toolkit for mass spectrometry and proteomics. Nature Biotechnology. 30 (10), 918-920 (2012).
- Kessner, D., Chambers, M., Burke, R., Agus, D., Mallick, P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics. 24 (21), 2534 (2008).
- Martens, L., Chambers, M., et al. mzML-a community standard for mass spectrometry data. Molecular & Cellular Proteomics. 10 (1), (2011).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.