Method Article
Évaluation simultanée de la parenté, du numéro de division et du phénotype par cytométrie en flux pour les cellules souches hématopoïétiques et progénitrices
Dans cet article
Résumé
Présenté ici est une technique basée sur la cytométrie de flux qui permet de mesurer simultanément le nombre de divisions cellulaires, le phénotype des cellules de surface et la parenté cellulaire. Ces propriétés peuvent être testées statistiquement à l’aide d’un framework basé sur la permutation.
Résumé
Peu de techniques peuvent évaluer le phénotype et le devenir d’une même cellule simultanément. La plupart des protocoles actuels utilisés pour caractériser le phénotype, bien que capables de générer de grands ensembles de données, nécessitent la destruction de la cellule d’intérêt, rendant impossible l’évaluation de son devenir fonctionnel. Les systèmes de différenciation biologique hétérogènes comme l’hématopoïèse sont donc difficiles à décrire. En nous appuyant sur les colorants de suivi de la division cellulaire, nous avons développé un protocole pour déterminer simultanément la parenté, le numéro de division et le statut de différenciation de nombreux progéniteurs hématopoïétiques uniques. Ce protocole permet d’évaluer le potentiel de différenciation ex vivo des progéniteurs hématopoïétiques murins et humains, isolés à partir de diverses sources biologiques. De plus, comme il est basé sur la cytométrie en flux et un nombre limité de réactifs, il peut générer rapidement une grande quantité de données, au niveau d’une seule cellule, de manière relativement peu coûteuse. Nous fournissons également le pipeline analytique pour l’analyse de cellules uniques, combiné à un cadre statistique robuste. Comme ce protocole permet de lier la division cellulaire et la différenciation au niveau de la cellule unique, il peut être utilisé pour évaluer quantitativement l’engagement du destin symétrique et asymétrique, l’équilibre entre auto-renouvellement et différenciation, et le nombre de divisions pour un destin d’engagement donné. Dans l’ensemble, ce protocole peut être utilisé dans des conceptions expérimentales visant à démêler les différences biologiques entre les progéniteurs hématopoïétiques, d’un point de vue unicellulaire.
Introduction
La dernière décennie a été marquée par la diffusion mondiale des approches unicellulaires de la biologie cellulaire et moléculaire. Suivant les traces de la génomique unicellulaire1,2, il est aujourd’hui possible d’étudier de nombreux composants d’une seule cellule (par exemple, l’ADN, l’ARN, les protéines), avec de nouvelles techniques monocellulaires -omiques en plein essor chaque année. Ces techniques ont mis en lumière des questions anciennes et nouvelles pour les domaines de l’immunologie, de la neurobiologie, de l’oncologie et autres, utilisant à la fois des cellules humaines et des cellules d’organismes modèles3. En mettant en évidence les différences entre les cellules individuelles, les cellules unicellulaires -omiques ont incité à définir un nouveau modèle d’hématopoïèse, centré sur l’hétérogénéité des cellules souches et progénitrices hématopoïétiques (HSPC) et s’éloignant du modèle classique des populations homogènes discrètes 4,5.
L’un des rares inconvénients de toutes les techniques -omiques est la destruction de la cellule d’intérêt, excluant la possibilité d’évaluer sa fonctionnalité. Inversement, d’autres méthodes unicellulaires, telles que le test de transplantation de cellules uniques et les technologies de traçage de la lignée, permettent de lire la fonctionnalité de la cellule ancêtre en évaluant le devenir de cellules individuelles in vivo 6,7. Les technologies de traçage de lignée consistent à marquer la cellule d’intérêt avec un marqueur génétique héréditaire7 ou fluorescent8,9, permettant de suivre le destin de plusieurs cellules individuelles en même temps. Cependant, la caractérisation des cellules de départ est typiquement limitée à un nombre restreint de paramètres, comme l’expression de quelques protéines de surface évaluées par cytométrie de flux10. En outre, les technologies de traçage de lignées unicellulaires nécessitent une détection laborieuse du marqueur cellulaire, généralement via le séquençage ou l’imagerie de l’ADN / ARN. Ce dernier point en particulier limite le nombre de conditions pouvant être testées dans une seule expérience.
Une autre classe de méthodes utilisées pour étudier la fonctionnalité de cellules individuelles sont les systèmes de culture cellulaire ex vivo de HSPC uniques. Faciles à réaliser, ces tests de référence impliquent le tri de cellules individuelles dans des récipients de culture cellulaire à 96 puits et, après culture, caractérisant le phénotype de la descendance cellulaire, généralement par cytométrie en flux ou analyse morphologique. Ces tests ont principalement été utilisés pour caractériser la différenciation à long terme des HSPC en cellules matures, généralement après 2-3 semaines de culture11,12. Alternativement, ils ont été utilisés pour tenter de maintenir et d’étendre ex vivo les HSPC 13,14,15,16,17,18, avec la promesse d’avantages médicaux pour la greffe de cellules souches humaines 19. Enfin, ils ont été utilisés pour étudier l’engagement précoce des HSPC en utilisant la culture à court terme20, le faible nombre de cellules générées dans cette culture étant le principal facteur limitant. L’un des inconvénients de ces différents types d’essais ex vivo est qu’ils ne reflètent que partiellement la complexité in vivo; Pourtant, ils sont l’un des rares moyens d’étudier la différenciation HSPC humaine.
Une information manquante dans les méthodes monocellulaires existantes (monocellule-omic, traçage de lignée et culture ex vivo) est la détection précise des divisions cellulaires, un paramètre essentiel à prendre en compte lors de l’étude de la dynamique HSPC21. Un moyen simple d’évaluer le nombre de divisions par cytométrie en flux est l’utilisation de « colorants protéiques » solubles, comme l’ester de succinimidyl (CFSE)-diacétate de 5-(et 6)-carboxyfluorescéine (CFSE)22. Ces colorants de division diffusent à l’intérieur du cytoplasme des cellules colorées, et dilués de moitié et passent aux deux cellules filles à chaque division cellulaire, permettant de dénombrer jusqu’à 10 divisions. En combinant plusieurs colorants de division, il est possible d’ensemencer plusieurs progéniteurs individuels dans le même puits, car chaque colorant individuel permet la séparation des différents descendants. C’est le principe derrière l’utilisation de colorants cellulaires pour le clonage multiplex et le suivi de division qui a été introduit pour la première fois pour les lymphocytes murins23,24.
Nous présentons ici le développement du test MultiGen pour une utilisation avec les HSPC murins et humains. Il permet de tester simultanément de nombreuses cellules individuelles pour leurs propriétés de différenciation, de division et de parenté ex vivo. Ce test à haut débit, facile à réaliser et peu coûteux permet de mesurer le phénotype cellulaire, le nombre de divisions effectuées, la parenté cellulaire et la relation clonale avec les autres cellules du puits, le tout en même temps. Il peut être utilisé pour évaluer quantitativement l’engagement de destin symétrique et asymétrique, l’équilibre entre auto-renouvellement et différenciation, et le nombre de divisions nécessaires pour un destin d’engagement donné. Le protocole nécessite un trieur cellulaire activé par fluorescence (FACS) et un cytomètre en flux avec un lecteur de plaques, ainsi que l’équipement nécessaire pour effectuer la culture cellulaire. En plus du protocole technique pour l’exécution du test sur les HSPC humains, nous fournissons également le cadre d’analyse détaillé, y compris les tests statistiques nécessaires pour évaluer les propriétés cellulaires liées au concept de famille cellulaire25. Ce protocole a déjà été utilisé avec succès pour décrire le compartiment murinHSPC 26,27.
Le protocole suivant utilise des cellules CD34+ enrichies magnétiquement comme matériau de départ28. De cette façon, il est possible de colorer et d’isoler efficacement les HSPC humains de différentes sources sanguines (par exemple, le sang de cordon, la moelle osseuse, le sang périphérique). Il est important de ne pas jeter la fraction CD34- , car elle sera utilisée dans le cadre du protocole pour établir différents types de témoins expérimentaux. Les quantités et volumes de cellules mentionnés peuvent être augmentés ou réduits, en fonction du flux de travail expérimental et des besoins. De même, le protocole peut être adapté à l’étude de différents types de progéniteurs, simplement en modifiant les anticorps utilisés pour les étapes de tri cellulaire et de cytométrie de flux.
Protocole
Pour le protocole suivant, du sang de cordon ombilical anonymisé a été utilisé comme source HSPC et prélevé conformément aux lignes directrices définies par la biobanque de sang de cordon ombilical de l’Hôpital Saint-Louis (autorisation AC-2016-2759) et à la Déclaration d’Helsinki.
REMARQUE : Avant de commencer, assurez-vous que tous les réactifs et équipements nécessaires à ce protocole sont disponibles, tels qu’ils sont énumérés dans le tableau des matériaux et mentionnés dans le protocole. Préparez les réactifs pertinents à l’état frais et ne les stockez pas, sauf mention explicite.
1. Coloration cellulaire
REMARQUE : Cette section décrit la coloration avec quatre combinaisons de colorants de division cellulaire CFSE et de colorant violet (CTV). Traiter tous les tubes simultanément, même si aucune solution de colorant cellulaire n’est ajoutée. Toutes les étapes sont effectuées dans des conditions stériles pour permettre l’étape de culture cellulaire suivante. Temps requis : environ 100 min.
- Traiter l’unité de sang de cordon selon un protocole de tri magnétique29. Assurez-vous que deux fractions sont disponibles : une fraction CD34- grande et une fraction CD34+ plus petite. Faire tourner les deux tubes pendant 5 min à 300 x g. Aspirer le surnageant sans déranger la pastille.
- Pour la fraction CD34+ , la remettre en suspension dans 1 mL du milieu Eagle modifié (DMEM) de Dulbecco sans sérum fœtal bovin (FBS). Compter les cellules à l’aide d’un hémocytomètre; la densité cellulaire ne doit pas être supérieure à 3 x 106 cellules/mL. Si c’est le cas, adaptez le volume en conséquence. Pour la fraction CD34, remettre en suspension dans du DMEM sans FBS et régler le volume à un maximum de 6 x 106 cellules/mL.
- Aliquote 250 μL de la fraction CD34+ dans quatre tubes en polypropylène de 15 mL. Étiquetez les tubes comme suit : CD34+/CF (CFSE_only), CD34+/CV (CFSE_high CTV_low), CD34+/VC (CFSE_low CTV_high) et CD34+/VI (CTV_high). Aliquote 250 μL de la fraction CD34- dans quatre autres tubes en polypropylène de 15 mL. Étiqueter les tubes comme suit : CD34-/CF (CFSE_only), CD34-/CV (CFSE_high CTV_low), CD34-/VC (CFSE_low CTV_high) et CD34-/VI (CTV_high). Les cellules restantes de la fraction CD34- peuvent être éliminées.
- Préparez deux solutions CFSE, nommées CFSE_high et CFSE_low. Pour CFSE_high (10 μM), mélanger 1,1 mL de DMEM sans FBS avec 2,2 μL de solution mère CFSE (5 mM). Pour CFSE_low (5 μM), mélanger 550 μL de DMEM sans FBS et 0,55 μL de solution mère CFSE (5 mM).
- Ajouter 250 μL de la solution CFSE_high aux tubes CF et CV, 250 μL de la solution CFSE_low aux tubes VC et 250 μL de DMEM sans FBS au tube VI. Pour assurer un mélange efficace de suspension cellulaire et de colorant cellulaire, inclinez le tube de près de 90 degrés et déposez les solutions CFSE sur la paroi du tube. Ensuite, tenez le tube verticalement pour mélanger les deux solutions, et pipeter trois ou quatre fois pour assurer un mélange rapide des solutions CFSE avec les cellules en suspension. Incuber à 37 °C pendant 8 min précises.
- Après l’incubation, ajouter 5 mL de DMEM + 10% FBS. Maintenir les tubes à 37 °C pendant 5 min.
- Faire tourner les tubes pendant 5 min à 300 x g. Retirer le surnageant par aspiration sans déranger la pastille et laver la pastille avec 5 mL de solution saline tamponnée au phosphate 1x/acide éthylènediaminetétraacétique (PBS 1x/EDTA). Tourner à nouveau pendant 5 min à 300 x g. Jeter le surnageant sans déranger la pastille, et remettre en suspension la pastille de cellule dans 250 μL de 1x PBS / EDTA.
- Préparez deux solutions CTV, nommées CTV_high et CTV_low. Pour CTV_high (10 μM), mélanger 1,1 mL de PBS 1x/EDTA et 2,2 μL de CTV (5 mM). Pour CTV_low (5 μM), mélanger 550 μL de PBS 1x/EDTA avec 0,55 μL de CTV (5 mM).
- Ajouter 250 μL de la solution CTV_high aux tubes VC et VI, 250 μL de la solution CTV_low au tube CV et 250 μL de 1x PBS / EDTA au tube CF. Utilisez la même technique que celle décrite à l’étape 1.5. Incuber à 37 °C pendant 8 min précises.
- Après l’incubation, ajouter 5 mL de DMEM + 10% FBS. Conserver à 37 °C pendant 5 min.
- Faire tourner les tubes pendant 5 min à 300 x g, jeter le surnageant sans déranger le granulé, puis laver le granulé avec 5 mL de 1x PBS/EDTA. Tourner à nouveau pendant 5 min à 300 x g.
- Jeter le surnageant sans perturber la pastille, et remettre en suspension les fractions CD34- dans 1x PBS/EDTA pour une concentration finale de 1,5 x 106 cellules/mL. Resuspendre les fractions CD34+ dans 40 μL de tampon de coloration et transférer les cellules dans des tubes de 1,5 mL.
2. Coloration des anticorps
REMARQUE: La coloration des anticorps peut être personnalisée en fonction des besoins expérimentaux. Seules les fractions CD34+ subissent une coloration par anticorps; les fractions CD34- sont utilisées comme témoin de coloration unique pour les combinaisons de colorants de division cellulaire (fractions CV, VC, CF et VI). Le panel suivant est conçu pour la détection de quatre types de HSPC : les cellules souches hématopoïétiques (CSH), les progéniteurs multipotents (MPP), les progéniteurs multipotents à amorce lymphoïde (LMPP) et les cellules progénitrices hématopoïétiques (HPC)12. Cependant, l’identification des CSS et des MPP est présentée. Temps requis : 75 min.
- Préparez la coloration unique pour la coloration de surface, en utilisant des perles de compensation. Mélanger les billes négatives et les billes d’immunoglobuline G (IgG) dans un rapport de 1:1, pour un volume total équivalent à 20 μL x le nombre de marqueurs de surface (p. ex. 120 μL si le panneau de coloration contient six anticorps).
- Expédier 20 μL de billes dans des tubes individuels de 1,5 mL pour chaque marqueur. Ajouter le volume correspondant au facteur de dilution pour chaque anticorps dans le tube correspondant (par exemple, si le facteur de dilution est de 1:20, ajouter 1 μL).
- Pour colorer les cellules CD34+ , préparer un mélange maître d’anticorps12, basé sur le tableau 1. Mélanger les anticorps dans un seul tube de 0,5 mL. Ajouter 7 μL du mélange principal d’anticorps à chacune des quatre affections CD34+.
- Incuber les billes de compensation et les échantillons de CD34+ à 4°C pendant au moins 30 min.
NOTE: Le temps d’incubation doit être adapté aux détails techniques des anticorps utilisés pour la coloration. - Pendant l’incubation, préparer la plaque à fond rond de 96 puits à utiliser pour le tri, en ajoutant 100 μL de milieu de culture cellulaire à chaque puits à l’aide d’une pipette multicanal.
NOTE: Laissez les puits H8-H12 vides. - Étiqueter les tubes en polypropylène de 5 mL pour les témoins de coloration de surface (5, à l’aide de billes), les témoins de colorants par division cellulaire (4, à l’aide des fractions CD34) et les échantillons CD34+ (4).
- À la fin de l’incubation, laver les cellules et les perles avec 1 mL de tampon de coloration. Transférer le volume total dans les tubes en polypropylène de 5 mL. Centrifuger les tubes pendant 5 min à 300 x g, puis aspirer le surnageant sans déranger la pastille.
- Resuspendre les cellules dans un tampon de coloration, en utilisant environ 500 μL chacune pour les billes et les cellules CD34+, et 1 mL pour les tubes CD34.
Tableau 1 : Modèle pour préparer le mélange principal d’anticorps pour une expérience de tri cellulaire. Veuillez cliquer ici pour télécharger ce tableau.
3. Tri cellulaire
Remarque : Les numéros de cellules triées peuvent varier en fonction de la quantité totale de cellules disponibles. Dans le protocole, un numéro de cellule minimum pour chaque contrôle est fourni. Temps nécessaire (pour une seule plaque) : 100 min.
- Ouvrez le modèle d’expérience ou définissez un nouveau test. Créez un seul échantillon et plusieurs tubes, un pour chaque condition.
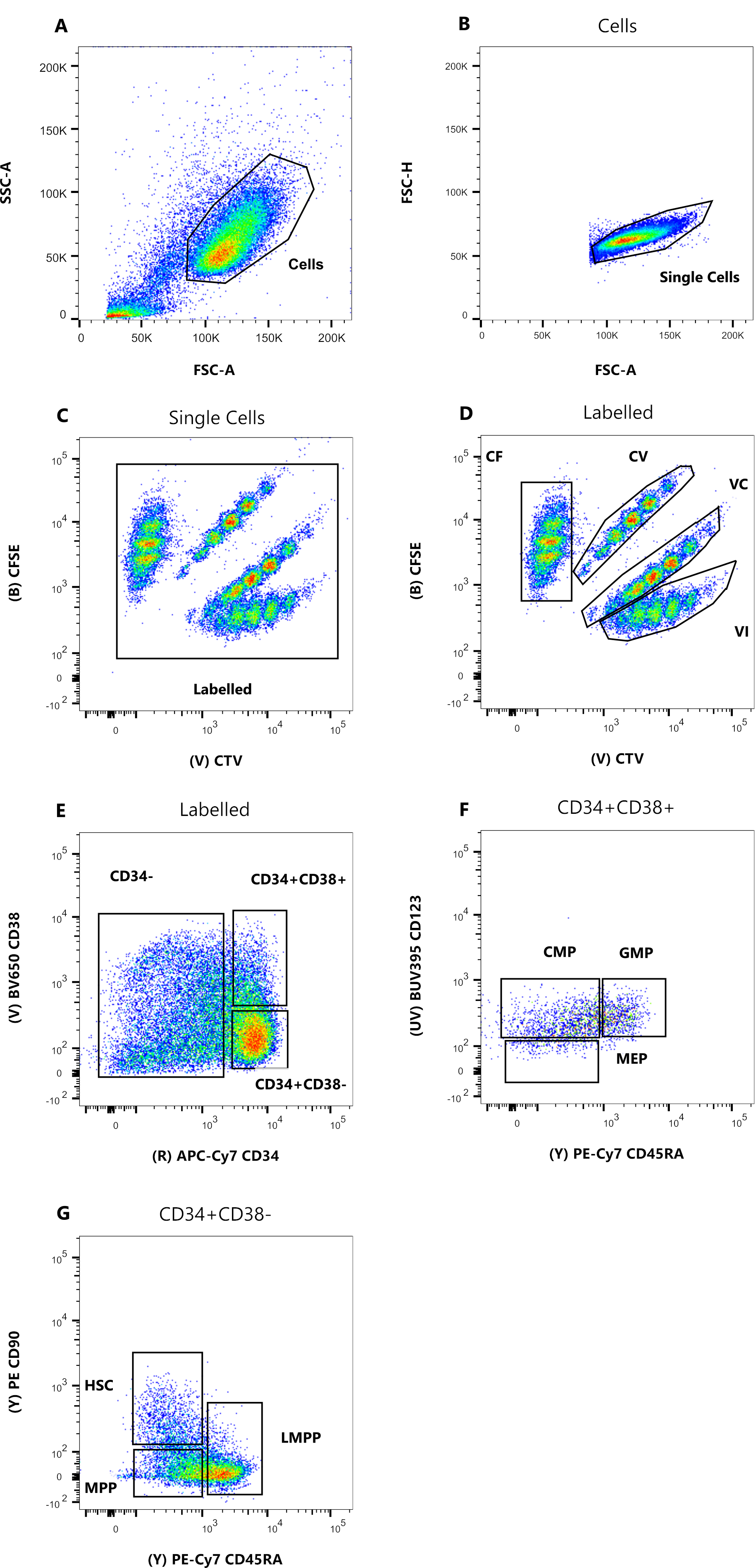
- Définissez la stratégie de contrôle détaillée à la figure 1, en créant six diagrammes de tracé à points. Tout d’abord, visualisez les cellules sur un graphique à points FSC-A/SSC-A et double-cliquez sur l’outil de contrôle polygonal pour sélectionner une population avec une faible dispersion latérale (Figure 1A). Dans le diagramme à points suivant (FSC-A/FSC-H), faites un clic droit sur le tracé et sélectionnez la porte « Cellules » dans le menu déroulant en cliquant dessus. Utilisez le même outil de contrôle pour sélectionner une population serrée sur la diagonale entre les deux axes (Figure 1B).
- Dans le troisième diagramme à points (APC vs FSH-H), affichez la population « Cellules uniques » et mettez les cellules négatives pour l’expression de la lignée APC (Lin) (Figure 1C). Dans le quatrième graphique (CFSE vs CTV), affichez la population « Lin- » et créez quatre portes séparées, une pour chaque combinaison de colorants (figure 1D).
REMARQUE: Ces portes doivent être étanches pour ne sélectionner qu’une petite fraction de cellules colorées de manière homogène. - Utilisez les cinquième et sixième graphiques (APC-Cy7 vs BV650 et PE-Cy7 vs PE) pour identifier les progéniteurs d’intérêt. Porter généreusement la population CD34+CD38- et la population CD34+CD38+ dans la cinquième parcelle (figure 1E). Ensuite, sélectionnez la population CD34+CD38- dans le sixième graphique et tracez trois portes, selon la figure 1F.
- Exécutez les tubes de coloration simples contenant les perles de compensation, en cliquant sur le bouton Acquérir . Ajustez les tensions du tube photomultiplicateur (PMT) à partir du menu déroulant Paramètres , en particulier pour les colorants de division cellulaire (entre 104 et 105 sur une échelle biexponentielle).
- Affinez la matrice de compensation en fonction du panneau utilisé pour le tri, à l’aide de l’onglet Compensation . Enregistrez au moins 5 000 événements dans la porte des perles, en cliquant sur le bouton Enregistrer .
- Exécutez les fractions CD34- et vérifiez à nouveau la matrice de compensation. Enregistrez au moins 10 000 événements dans la porte à cellule unique.
- Exécutez les fractions CD34+ en enregistrant au moins 5 000 événements dans la porte à cellule unique. Ajustez la grille pour chaque combinaison de colorants, en définissant une grille étanche pour sélectionner une population homogène (Figure 1D). De même, ajustez le point de contrôle pour sélectionner les CSS et les députés.
- Une fois l’analyse terminée et tous les tubes enregistrés, insérer la plaque dans le support approprié, après avoir effectué l’étalonnage standard Aria pour le tri sur des plaques à 96 puits. Il est recommandé de refroidir la plaque.
- Préparez le modèle de tri des plaques selon le schéma présenté dans le tableau 2, en utilisant la disposition de tri expérimental. Les puits nommés « CD34- » contiennent 5 000 à 10 000 cellules, triées sur la porte CF/CV/VC/VI. Les puits « Bulk » contiennent au moins 500 cellules, triées sur la porte CD34+CD38-. Enfin, les puits unicellulaires ne contiennent qu’un seul événement par combinaison de colorants par division cellulaire par puits, donc quatre événements par puits au total.
REMARQUE : Les populations « en vrac » peuvent être adaptées à un sous-ensemble spécifique de progéniteurs; Ne triez pas moins de 500 cellules. - Pour le tri, procédez dans l’ordre, en complétant chaque combinaison de colorant de division cellulaire avant de passer à la suivante. Par exemple, commencez par trier le CD34-CF , en mode pureté d’élasticité . Cliquez sur le bouton acquérir, puis sur le bouton de tri.
- À la fin du tri CD34, insérez le tube CF CD34+ . Acquérir, puis cliquer sur le bouton de tri, en vous assurant d’avoir coché 0/ 16/0 comme grade de pureté. Enfin, triez les cellules d’intérêt, une cellule par puits, en pureté unicellulaire , en vous assurant de cocher l’option Tri d’index .
- Passez à la combinaison de colorant de division cellulaire suivante, en répétant le même ordre. À titre de référence, le tableau 2 fournit un exemple de plaque triée.
Remarque : La fonction de tri d’index génère des fichiers individuels pour chaque condition triée. - À la fin du tri, exportez les fichiers au format .fcs 3.0. Placez les cellules dans un incubateur à 37 °C, 5% CO2 . Les cellules sont cultivées pendant plusieurs jours, selon le plan expérimental, pendant au moins 24 h26.
Tableau 2 : Modèle pour une plaque de tri cellulaire à 96 puits, en fonction des exigences spécifiques pour l’analyse par cytométrie en flux successive. Veuillez cliquer ici pour télécharger ce tableau.
4. Analyse des données de tri cellulaire
REMARQUE : Pour valider la qualité du tri cellulaire, l’analyse des données FACS est nécessaire avant d’aller plus loin. Le résultat principal de cette étape est la génération d’une feuille de calcul contenant les intensités de marqueur de chaque cellule individuelle triée.
- Téléchargez les fichiers .fcs 3.0 dans le logiciel d’analyse.
- Vérifiez le paramètre de compensation utilisé lors du tri des cellules, en utilisant les fichiers de coloration uniques enregistrés avant le tri réel.
- Définissez la stratégie de contrôle révisée à l’aide des fichiers correspondant au volume différent. Copiez et collez ces portes sur les fichiers de tri d’index.
- Vérifiez que les cellules triées d’index sont tombées dans la porte définie. Si certaines cellules triées ont été mal fermées, elles peuvent être identifiées en exportant les coordonnées de plaque enregistrées lors du tri de l’index et supprimées ultérieurement dans l’analyse.
- Exportez l’événement à partir des fichiers de tri d’index en tant que paramètres compensés. Exportez-les sous forme de fichiers .csv, en cochant les options « valeurs d’échelle » et « paramètres compensés ». Ces fichiers doivent être exportés dans un dossier nommé « Fichiers exportés ».
- Combinez tous les fichiers dans un seul fichier .csv, à l’aide du script du fichier supplémentaire 1. Définissez le bon chemin avec la fonction « setwd ». La sortie de ce script est une feuille de calcul contenant tous les événements différents et les intensités relatives pour tous les paramètres.
5. Coloration des anticorps après culture
NOTE: Exécuter cette partie du protocole dans des conditions stériles; Plusieurs réactifs sont partagés avec les étapes précédentes et doivent rester stériles. Pour l’analyse de cytométrie en flux, utilisez un cytomètre en flux avec un lecteur de plaques. Cela permet d’effectuer la coloration directement dans la plaque de culture tissulaire, réduisant la perte cellulaire au minimum en limitant la quantité de pipetage et de filature. Préparer la coloration monocolore du marqueur de surface à l’aide des billes de compensation, à l’exception des puits A1-A4, qui représentent la coloration unique pour les couleurs CF/CV/VC/VI et sont déjà présentes dans la plaque de 96 puits. Les populations en vrac triées en fonction du colorant cellulaire aident à établir la stratégie de contrôle pour le nombre de divisions et le contrôle général. Temps requis: 120 min.
- Avant de commencer le protocole, marquez les puits qui contiennent au moins une cellule en vérifiant la plaque sous un microscope inversé. Cette étape permet d’optimiser la quantité d’anticorps utilisée pour la coloration et accélère la procédure.
- Préparez le mélange d’anticorps, conformément au tableau 3. Comme il y a une quantité importante de pipetage, le tableau tient compte de l’erreur technique due au pipetage, y compris un volume supplémentaire de 5%. Les anticorps décrits dans le tableau permettent de caractériser une gamme de HSPC à partir d’échantillons de sang de cordon ombilical humain12.
- Centrifuger la plaque pendant 5 min à 300 x g. Retourner rapidement la plaque sous le capot et sur une serviette en papier, pour enlever le surnageant.
- Ajouter 8 μL de tampon de coloration aux puits A1-A4. Ajouter 8 μL du mélange aux autres puits.
- Mélanger les billes négatives et les billes de compensation IgG dans un rapport de 1:1, pour un volume total équivalent à 120 μL. Expédier 20 μL dans un tube de 1,5 mL par marqueur. Ajouter le volume d’anticorps correspondant au facteur de dilution (par exemple, si le facteur de dilution est de 1:20, ajouter 1 μL).
REMARQUE : Adapter le volume total au nombre de marqueurs utilisés pour la coloration (p. ex., 100 μL si le panneau de coloration contient cinq anticorps). - Incuber la plaque et les contrôles de compensation de tache simple à +4 °C, pendant au moins 30 min.
NOTE: Le temps d’incubation doit être adapté aux détails techniques des anticorps utilisés pour la coloration. - Lavez les billes avec 1 ml de tampon de coloration. Transférer le volume total dans les tubes de polypropylène de 5 mL précédemment étiquetés. Centrifuger les tubes pendant 5 min à 300 x g, puis retirer le surnageant par aspiration.
- Lavez les cellules de la plaque en ajoutant 100 μL de tampon de coloration par puits à l’aide d’une pipette multicanaux. Centrifuger la plaque à 300 x g pendant 5 min, puis retourner rapidement la plaque sous le capot et sur une serviette en papier, pour enlever le surnageant.
- Re-suspendre les cellules dans 85 μL de tampon de coloration, à l’aide d’une pipette multicanaux.
- Lancez l’analyse sur le cytomètre en flux (mode Acquisition), en utilisant le modèle dédié et en cliquant sur personnalisé. Ce gabarit personnalisé prend en considération les caractéristiques techniques de la plaque à fond rond de 96 puits, en particulier les dimensions de chaque puits (diamètre, profondeur et épaisseur). La sonde doit atteindre le fond du puits, alors placez-la au centre exact des puits A1 et H12.
- Après avoir sélectionné les fluorophores d’intérêt dans la liste proposée par le logiciel, paramétrer la configuration de la plaque en suivant le gabarit de plaque du tableau 2, corrigé du nombre de puits contenant au moins une cellule.
- Sélectionnez 100 μL comme limite de volume d’acquisition. Cochez l’option d’agitation . Réglez le taux d’acquisition à 1 μL/s max, car une vitesse plus faible améliore le volume total analysé par puits.
- Ajouter les solutions de nettoyage et de lavage appropriées aux puits H8-H12. Le modèle du tableau 2 laisse spécifiquement les puits H8-H12 vides, car le cytomètre en flux doit exécuter une gamme de conditions de lavage à la fin de l’analyse.
REMARQUE : Cette étape est adaptée aux spécificités du cytomètre en flux utilisé. - Dans la section des tracés et des portes, définissez d’abord la porte à cellule unique, à l’aide du nuage de points FSC-A/SSC-A, puis du nuage de points FSC-H/FSC-A. Créez un histogramme pour chaque marqueur d’intérêt.
- Une fois les paramètres confirmés, passez à la section Analyse . Analysez d’abord la coloration unique, en enregistrant pas moins de 5 000 événements (plage optimale : 5 000 à 15 000 événements), tant pour les billes de compensation que pour les fractions colorées CD34. Ajustez les tensions si nécessaire.
- Une fois que les taches simples sont toutes enregistrées, il est possible de commencer l’acquisition proprement dite, en cliquant sur la fonction Acquisition .
Tableau 3 : Mélange principal d’anticorps pour une expérience de cytométrie en flux, en particulier pour l’identification des HSPC à partir du sang de cordon ombilical humain. Veuillez cliquer ici pour télécharger ce tableau.
6. Analyse des données de cytométrie en flux post-culture
REMARQUE : L’analyse des données décrite est spécifique au logiciel mentionné dans le tableau des matériaux. Le résultat principal est la génération d’une feuille de calcul contenant des informations sur l’intensité du marqueur de surface, le nombre de divisions et la parenté pour chaque cellule analysée. Cette partie du protocole comprend un script écrit en R, nécessaire à ce flux de travail pour générer la feuille de calcul d’analyse finale.
- Exportez les fichiers du cytomètre en flux en tant que fichiers .fcs. Téléchargez-les dans le logiciel d’analyse, en les regroupant sous les formes « coloration unique », « en vrac » et « cellule unique ».
- Préparez une matrice de compensation à l’aide des fichiers de coloration uniques et appliquez-la aux deux autres groupes par glisser-déposer.
REMARQUE: Si un outil de compensation automatique est utilisé, vérifiez la qualité à la main avant de continuer. - Pour avoir un point de contrôle représentatif, concaténez les différents puits en vrac dans un seul fichier. Cette étape met rapidement en évidence si deux couleurs se chevauchent (généralement CV et VC) ou d’autres anomalies, et doivent donc être exclues. Après avoir cliqué sur l’option concaténer les populations, sélectionnez tous les paramètres non compensés dans le menu « paramètres », puis cliquez sur concaténer.
- Téléchargez le fichier concaténé dans l’espace de travail, puis appliquez la matrice de compensation par glisser-déposer.
- Préparez la stratégie de contrôle définie à la figure 2 à l’aide du fichier concaténé. Dans la porte à cellule unique, affichez les événements sur un nuage de points avec CFSE et CTV. Créez une première porte appelée Labeled, incluant les quatre couleurs et excluant une éventuelle auto-fluorescence (Figure 2C). Ensuite, fermez chaque couleur individuellement.
- Les cellules marquées avec CV et VC ont besoin d’une valeur transformée, étant donné que la couleur est le résultat des signaux CFSE et CTV. Les deux signaux coordonnés sont donc tournés sur une échelle logarithmique de 45°, pour permettre à la dilution de la division de se dérouler parallèlement à l’axe des abscisses. Cette valeur transformée est dérivée manuellement, en cliquant sur Outils puis sur Dériver Paramètre. Collez la formule suivante dans la zone de formule :

NOTE : L’équation26 suppose que CFSE et CTV sont les paramètres 03 et 17. - Pour visualiser correctement ce nouveau paramètre nommé Paramètre dérivé, définissez un axe linéaire compris entre ~3 et 7, en cliquant sur l’option Paramètre de l’axe et en sélectionnant Personnaliser l’axe.
- Appliquez le point de contrôle à chaque couleur individuellement sous forme de tracé d’histogramme : pour CF et VI, définissez CFSE-A et CTV-A sur l’axe des abscisses, respectivement. Pour CV et VC, définissez le nouveau paramètre dérivé sur l’axe des abscisses. Réglez les portes correspondant à chaque pic, comme illustré à la figure 3.
- Appliquez le point de contrôle à chaque puits individuel. Assurez-vous d’ajouter le paramètre dérivé à chaque puits analysé. Vérifiez manuellement chaque porte de couleur pour chaque puits, afin de détecter les événements qui sont incorrectement affectés à un pic donné. Des exemples de points de contrôle sont présentés à la figure 4.
- Une fois l’analyse terminée et tous les puits vérifiés, sélectionnez toutes les portes CF/CV/VC/VI qui contiennent au moins une cellule. Exportez-les sous forme de fichiers .csv, en cochant les options « valeurs d’échelle » et « paramètres compensés ». Ces fichiers sont exportés dans un dossier nommé « Fichiers exportés ».
- Combinez tous les fichiers en une seule .csv, à l’aide du script R dans le fichier supplémentaire 1. N’oubliez pas de définir le bon chemin avec la fonction « setwd ». La sortie de ce script est une feuille de calcul contenant tous les différents événements bloqués et les intensités relatives pour tous les paramètres.
- Ouvrez la feuille de calcul et renommez les colonnes pour chaque paramètre, par exemple, en utilisant les noms suivants : CFSE, CTV, CD90, CD123, CD45RA, CD34, CD38. Ces noms seront utilisés pour identifier le seuil de contrôle afin d’attribuer correctement à chaque cellule leur identité.
- Ajoutez six colonnes nommées « Well », « Condition », « Color », « Generation », « Original_cell » et « Culture_time ». Ces variables sont celles définies expérimentalement et sont déduites de chaque ligne :
export_A10 CD34 + PBS_CV_Peak 1.csv.1 = A10 (puits), CD34+ (Original_cell), PBS (état), CV (couleur) Peak_1 (génération). - Exporter les puits en vrac pour identifier les valeurs seuils pour les points de contrôle : exporter la population d’intérêt compensée (p. ex., CD34+CD38-) sous forme de fichiers .csv, en cochant les options « valeurs d’échelle » et « paramètres compensés ». Exportez ces fichiers dans un dossier nommé « Fichiers exportés ».
- Pour trouver le seuil de CD38, identifiez la plus grande valeur numérique pour ce paramètre. Inversement, pour trouver le seuil de CD34, identifiez la plus petite valeur numérique pour ce paramètre. Répétez ce processus pour tous les paramètres d’intérêt.
REMARQUE : Pour l’analyse présentée dans le protocole, le marqueur CD45RA est utilisé à la fois pour identifier les LMPP dans la porte CD34+CD38- et CMP/GMP dans la porte CD34+CD38+. Cela signifie que deux valeurs seuils différentes doivent être extraites pour ce marqueur. - Copiez et collez les valeurs de seuil dans un fichier Excel appelé « gating_matrix ». Ce fichier est organisé selon le tableau 4, et permet l’analyse de multiples expériences indépendantes. Il est très important de nommer chaque colonne exactement avec ce schéma: XXYYMMDD_xxh, où XX représente les deux initiales de l’opérateur, YY les deux derniers chiffres de l’année, MM pour le mois, DD pour le jour et xx pour le point de temps d’analyse.
Tableau 4 : Matrice de déclenchement pour l’assignation du devenir cellulaire, avant l’analyse statistique. Le CD45h fait référence à l’intensité de CD45RA pour le sous-ensemble HPC, tandis que CD45l fait référence à l’intensité de CD45RA pour les sous-ensembles CD34+CD38. Veuillez cliquer ici pour télécharger ce tableau.
7. Analyse statistique
Remarque : Le test statistique des données générées implique un pipeline d’analyse personnalisé, codé à l’aide du langage de programmation Python (fichier supplémentaire 2, fichier supplémentaire 3 et fichier supplémentaire 4). Le script est organisé en trois blocs : le premier pour le traitement de la feuille de calcul, le deuxième bloc pour générer la carte thermique pour la visualisation des données et le dernier bloc pour générer plusieurs histogrammes pour analyser et tester les propriétés de différenciation et de division.
- À partir du bloc « 0_process_data » (fichier supplémentaire 2), assurez-vous que les chemins d’gating_matrix et de feuille de calcul de données sont correctement définis dans le script.
- Définissez le dictionnaire « cell_cols », pour attribuer les destins cellulaires pertinents à chaque cellule. Dans le cas spécifique, les destins sont les CSH, les MPP, les LMPP, les progéniteurs myéloïdes communs (CMP), les progéniteurs granulo-monocytaires (GMP), les progéniteurs mégacaryocytaires-érythroïdes (MEP) et CD34-.
- À l’aide des valeurs seuils définies à partir des puits en vrac (étape 6.16), définissez la fonction « cell_class_exp_time ». Il est essentiel d’être cohérent dans la dénomination des colonnes, de définir correctement ces seuils, en utilisant le même nom que celui utilisé pour définir chaque colonne à l’étape 6.12.
- Les phénotypes cellulaires sont définis dans le script à l’aide d’une série d’instructions « if-else », basées sur les seuils détectés lors de l’analyse de cytométrie en flux.
REMARQUE : Différents phénotypes peuvent être affichés en modifiant ces énoncés pour tenir compte d’autres combinaisons de marqueurs. - Précisez les conditions expérimentales spécifiques à l’aide de la fonction « cond_rule » (p. ex., différents traitements expérimentaux). Pour le jeu de données fourni, les conditions sont nommées « GT » et « Diff ». Décrire les deux milieux de culture cellulaire différents utilisés pour cultiver les cellules. Ces informations seront utilisées par le bloc « 1_dot_plot » (Fichier supplémentaire 3) pour tracer la carte thermique.
- Dans le bloc « 2_bar_plot » (fichier supplémentaire 4), définissez le dictionnaire « class_dct », y compris les destins cellulaires discrets d’intérêt. Pour l’ensemble de données fourni, les destins cellulaires d’intérêt sont les mêmes que ceux décrits pour le dictionnaire « cell_cols ».
- Définissez « conds » (conditions), « or_cells » (cellule d’origine), « sym_labs » (étiquettes de symétrie) et « times » (le point temporel expérimental). Ce sont des filtres de réitération nécessaires au traçage. « conds » reprend les conditions définies dans « cond_rule », « or_cells » sont les CSS et les MPP, et « sym_labs » décrit le type de divisions.
- Dans le bloc « 2_bar_plot », il est possible de tracer les cellules qui ont progressé jusqu’à la division 6.
Remarque : Le jeu de données fourni inclut uniquement les cellules jusqu’à la division 4, de sorte qu’un message d’erreur apparaît, mais cela n’empêche pas le script de fonctionner. - Les figures générées par le script peuvent être récupérées dans le dossier nommé « figures » sous forme de fichiers pdf. Les fichiers nommés « Test » représentent les différents tests statistiques effectués pour l’histogramme correspondant.
Résultats
Tri FACS
Les stratégies de tri présentées dans ce protocole sont basées sur des stratégies largement acceptées 12,30,31. Pour la stratégie de déclenchement présentée à la figure 1, la matière première est constituée de progéniteurs de sang de cordon ombilical préalablement purifiés par enrichissement magnétique CD34+, ce qui explique le pourcentage négligeable de cellules positives de la lignée. Il est essentiel d’utiliser des portes serrées pour les quatre combinaisons de colorants intracellulaires (par exemple, le CTV sur la figure), d’améliorer la résolution des pics lors de l’analyse suivante et de créer la bonne population cellulaire (Figure 1D). Dans le cas illustré dans la figure, les portes sélectionnent la population la plus grande et la mieux définie. La présence de populations multiples et proches pour chaque combinaison de colorants de division cellulaire n’est, selon notre expérience, pas représentative des différences biologiques. Au lieu de cela, cela pourrait indiquer a) une procédure de coloration non optimale, ou b) une grande hétérogénéité (en particulier en taille) dans le pool de cellules de départ. Cela n’est pas surprenant lorsque l’on commence à partir de sang de cordon ombilical ou d’autres sources biologiques complexes (p. ex. aspiration de moelle osseuse, sang périphérique). Si la vanne n’est pas étroitement définie, la dilution progressive des différentes combinaisons de colorants peut conduire à la fusion des pics ultérieurs, en particulier pour les conditions CV et VC (Figure 2D). Une autre conséquence négative d’un point de déclenchement sous-optimal est l’incapacité de distinguer efficacement les différents pics après la culture cellulaire, car une population de départ hétérogène peut conduire à des pics peu profonds.

Figure 1 : Stratégie de contrôle pour le tri cellulaire. (A) FSC-A versus SSC-A, pour exclure les débris et les cellules contaminantes. (B) FSC-A versus FSC-H, pour exclure les doublets et les amas cellulaires. (C) Lin versus FSC-H, pour exclure les cellules qui sont Lin+. (D) CTV versus CFSE, pour identifier de manière univoque les cellules colorées avec les combinaisons de colorants CF, CV, VC et VI. Les portes doivent être suffisamment strictes pour inclure une population homogène. (E) CD34 versus CD38, pour séparer les progéniteurs restreints CD34+CD38+ (également appelés HPC) du compartiment multipotent CD34+CD38-. (F) CD45RA versus CD90, de la population CD34+CD38-, pour séparer les progéniteurs les plus immatures enrichis dans le CSH (CD90+CD45RA-), le LMPP (CD90midCD45RA+) et le MPP plus engagé (CD90-CD45RA-). (G) les événements triés par indice, représentés ici pour leur coloration de combinaison de colorants cellulaires et (H) l’expression des marqueurs de surface CD90 et CD45RA. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Analyse par cytométrie en flux après culture cellulaire
Les données de la figure 2 sont représentatives des CSH de sang de cordon ombilical humain, conservées en culture pendant 72 heures, en présence de multiples cytokines capables de soutenir une gamme de progéniteurs et de précurseurs myéloïdes. Les panneaux 2A à 2D représentent le point de départ nécessaire pour établir la parenté de chacune des cellules individuelles, tandis que les panneaux 2E à 2G permettent le phénotypage cellulaire. La présence réduite de MEP dans la figure est probablement la conséquence des conditions de culture utilisées pour cette expérience représentative (Figure 2F). L’utilisation de cytokines et de conditions de culture différentes modifie le pourcentage relatif de chaque sous-ensemble, de la même manière que la sélection de différentes cellules de départ pour l’expérience.

Figure 2 : Stratégie de contrôle pour l’analyse de cytométrie en flux. (A) FSC-A par rapport à SSC-A, pour exclure les débris et les cellules contaminantes. (B) FSC-A versus FSC-H, pour exclure les doublets et les amas cellulaires. (C) CTV versus CFSE, la porte étiquetée permet d’exclure tout événement auto-fluorescent qui pourrait affecter la résolution des données. (D) CTV contre CFSE. Il est extrêmement important de traiter rigoureusement les quatre populations, en fonction des dilutions de colorant de division cellulaire. (E) CD34 versus CD38, pour faire la distinction entre les précurseurs engagés (CD34-), les progéniteurs restreints (HPC) (CD34+CD38+) et les progéniteurs immatures (CD34+CD38-). (F) CD45RA par rapport à CD123, pour distinguer trois types de progéniteurs restreints : CMP (CD123+CD45RA-), MEP (CD123-CD45RA-) et GMP (CD123+CD45RA+). (G) CD45RA versus CD90, à partir du CD34+CD38-, pour identifier les CSH, les PMT et les MPP. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
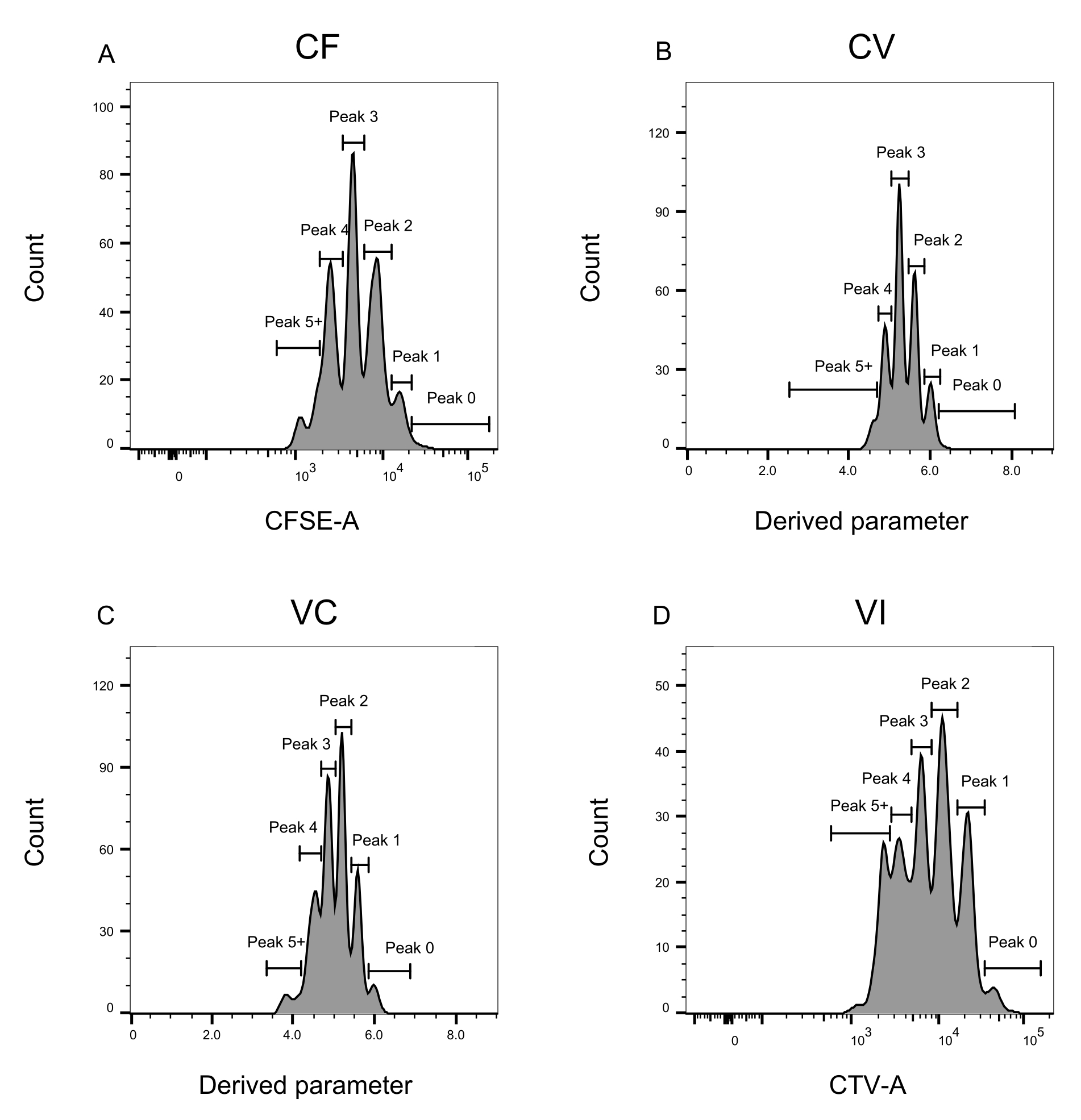
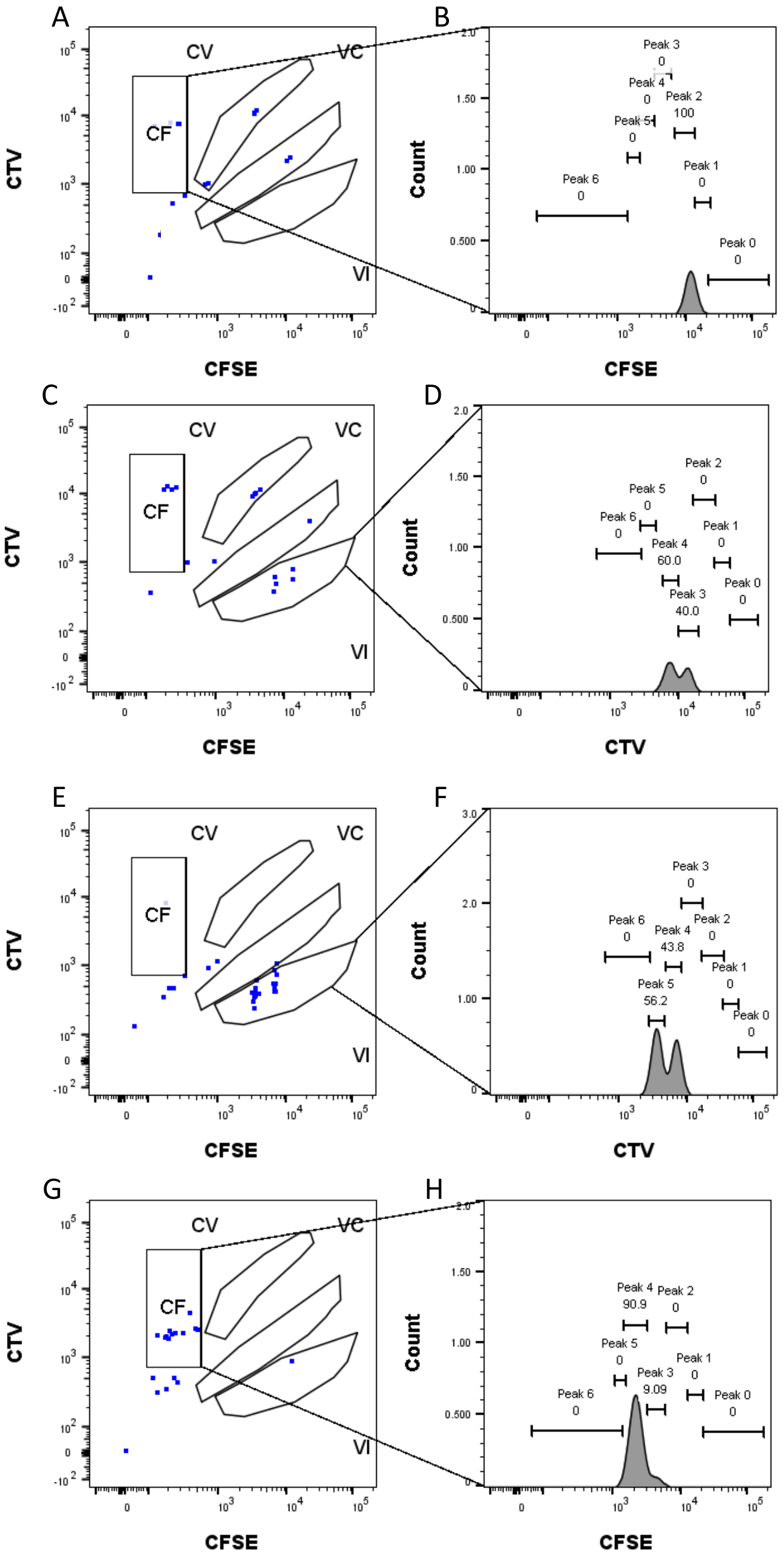
La définition des pics et les étapes d’affectation (Figure 3 et Figure 4) sont des aspects cruciaux du protocole et nécessitent la définition de portes strictes. Pour la définition du pic (Figure 3), au moins 1 000 événements sont nécessaires pour une identification fiable. En ce sens, il pourrait être bénéfique d’isoler plus de cellules lors de l’étape de tri cellulaire pour les puits « Bulk ». La figure 4 décrit quatre exemples de puits uniques contenant plusieurs familles. Cette figure clarifie l’importance des points de contrôle de la figure 2D et de la figure 3, en particulier pour l’identification de chaque famille et de chaque pic. La figure 4A illustre un exemple simple, car toutes les cellules de la grille CF sont très proches les unes des autres et peuvent être facilement affectées à un seul pic. La figure 4C montre un autre exemple de famille répartie de manière univoque sur deux pics bien séparés, comme le montre clairement l’histogramme de la figure 4D. Les figures 4E,G révèlent l’importance d’un contrôle strict fondé sur un grand nombre d’événements; Ils affichent tous deux peu d’événements qui sont proches, mais en dehors des portes de combinaison de colorants. Ces événements pourraient être inclus à tort dans les portes VI et CF, en se fondant exclusivement sur l’analyse d’un seul puits. Enfin, la figure 4F,H montre deux exemples différents de familles réparties sur plusieurs pics, avec un exemple de deux pics d’intensité similaires (Figure 4F) et un avec deux pics d’intensité inégaux (Figure 4H).

Figure 3 : Définition des pics pour l’analyse de cytométrie en flux. (A-D) Les pics doivent être définis en enregistrant au moins 500 événements, afin d’assurer une bonne représentation de chaque pic individuel. (A) Histogramme pour l’intensité CFSE-A. Plusieurs pics peuvent être identifiés, chacun correspondant à une population différente de cellules en division. (B,C) Histogrammes de l’intensité du paramètre dérivé, représentant le mélange CFSE-CTV, CV (B) et VC (C), respectivement. (D) Histogramme pour l’intensité CTV-A. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 4 : Affectation des pics. (A,B) Un seul pic peut être détecté pour ce puits, dans la vanne CF. (C, D) Deux pics d’intensité presque égale peuvent être détectés dans ce puits, dans la porte VI. Les pics sont bien résolus. (E,F) Deux pics d’intensité comparable peuvent être détectés dans ce puits, dans la porte VI. Seuls les événements dans la porte ont été pris en compte, sur la base de la stratégie définie à l’aide des puits en vrac. (G-H) Deux pics d’intensité inégale peuvent être détectés dans ce puits, dans la porte CF. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Représentation des données et tests statistiques
La figure 5 montre différents types de représentation des données de deux expériences distinctes, toutes deux réalisées après 72 h de culture cellulaire. Les CSH et les MPP ont été cultivés dans deux milieux de culture cellulaire différents, censés modifier la division cellulaire et les propriétés de différenciation. Ces médias sont nommés « Diff » (Différenciation)32 et « GT"33; le premier favorise la différenciation myéloïde et érythroïde, car il contient de l’érythropoïétine (EPO) et du facteur de stimulation des colonies granulo-monocytaires (GM-CSF), tandis que le second a été développé dans le cadre d’essais cliniques de thérapie génique, dans le but de maintenir et d’amplifier un pourcentage élevé de HSPC. La figure 5A est une carte thermique représentative de la condition « Diff », représentant une variété de familles cellulaires, à la fois dans les destins cellulaires et les divisions. Dans cette carte thermique, chaque ligne représente une famille individuelle, chaque cellule au carré une cellule individuelle et les colonnes regroupent toutes les cellules de la même génération (par exemple, les cellules de la génération 2 divisées au moins deux fois). Il est possible de distinguer des familles très homogènes, composées d’un seul type cellulaire et présentant le même nombre de divisions (ex. : famille #63), et des familles hétérogènes, incluant trois types cellulaires sur deux générations (ex. : famille #84). Étant donné que le taux de récupération cellulaire pour cette analyse est d’environ 70 %, les familles complètes, qui sont définies par le fait que toutes leurs cellules sont récupérées dans des générations éventuellement différentes (p. ex., une famille d’une cellule de génération 1 et de deux cellules de génération 2), sont rarement observées (affichage d’un hashtag à côté de leur numéro d’identification à la figure 5A). Il existe de multiples explications expliquant la détection incomplète, qui peuvent être techniques (problème de coloration, perte de cellules due au protocole) ou biologiques (mort cellulaire et/ou apoptose). Les limitations techniques peuvent être surmontées à l’aide d’un analyseur conçu pour réduire le volume mort associé à l’échantillon individuel et en effectuant la coloration cellulaire directement dans la plaque de culture cellulaire pour réduire le pipetage volumineux. Inversement, les méthodes orthogonales pour déterminer la quantité de mort cellulaire (par exemple, via des expériences d’imagerie de cellules vivantes) peuvent aider à distinguer les facteurs techniques et biologiques entraînant une détection incomplète.
La figure 5Bi montre comment visualiser l’effet de l’état de culture sur la composition du type cellulaire, comme si l’on avait effectué un essai en vrac. Ici, la condition Diff favorise un plus grand nombre de destins et un pourcentage plus élevé de cellules CD34+ (définies comme tous les types de cellules sauf CD34-). Les intervalles de confiance sont calculés dans le script via les données d’amorçage de base, avec 250 000 jeux de données amorcés34. Il convient de noter que tous les autres histogrammes de la figure 5 affichent des intervalles de confiance calculés de la même manière. Le tableau 5 récapitule toutes les informations sur le nombre de familles et le nombre de cellules dans chaque génération.
La figure 5Bii représente graphiquement le résultat des tests statistiques effectués dans le script « 2_bar_plot ». Une description formelle du cadre statistique est disponible26. En bref, ce cadre permet de tester des hypothèses statistiques tout en supposant que les cellules d’une même famille sont dépendantes (hypothèse elle-même testable), contrairement aux statistiques classiques qui nécessiteraient une indépendance entre toutes les cellules observées. Dans le cas spécifique présenté dans la figure, le test statistique remet en question l’hypothèse selon laquelle les choix de devenir cellulaire des MPP, mesurés comme les fréquences des différents types de cellules présents dans la culture, sont indépendants des conditions de culture cellulaire utilisées. Tout d’abord, la statistique du test G est utilisée pour évaluer l’écart entre les fréquences de type cellulaire de différents milieux cellulaires (pour l’exemple de Bii, cette statistique est représentée par la barre rouge). Ensuite, une randomisation des données est effectuée par permutation, échangeant des familles entières de cellules entre les deux conditions de culture cellulaire. Il s’agit de préserver la dépendance entre les cellules liées à la famille, tout en gardant le nombre de familles dans chaque ensemble cohérent avec les données originales. La statistique du test G est calculée à partir de l’ensemble de données randomisées. Les valeurs bleues représentées dans 5Bii sont la statistique du test G pour 250 000 permutations. Enfin, la valeur p est calculée pour évaluer dans quelle mesure l’ensemble de données d’origine s’écarte de la distribution des ensembles permutés. Dans l’exemple, la statistique originale s’écarte largement de la distribution, ce qui donne une petite valeur p et rejette ainsi l’hypothèse selon laquelle le devenir cellulaire des MPP est indépendant des conditions de culture.
La figure 5C représente le pourcentage de familles cellulaires par génération maximale, afin d’explorer comment différentes conditions modifient la division cellulaire par famille cellulaire. Ce diagramme de données montre qu’à 72 h, les cellules cultivées dans la condition Diff effectuent un plus grand nombre de divisions que les cellules dans la condition GT. Le nombre maximum de générations par famille est représenté, de sorte qu’une famille qui affiche des cellules dans les générations 1 et 2 est considérée comme la génération 2. Le même cadre statistique utilisé pour la figure 5B peut être utilisé pour tester statistiquement l’indépendance entre la division cellulaire et les conditions de culture.
La figure 5D explore le type de symétrie/asymétrie de la première division pour les différents types d’ancêtres (CSH ou MPP). Pour les familles cellulaires complètes de la génération 1 - la seule génération où il est possible d’établir définitivement les deux cellules filles comme cellules sœurs - quatre types différents de symétrie/asymétrie peuvent être définis: l’étiquette « Sym Undiff » décrit les familles où les deux filles conservent le phénotype de la cellule d’origine. « Sym Diff » signifie que les deux filles ont le même phénotype, et il est différent de la cellule d’origine. « Asym Undiff » signifie qu’une fille ne conserve que le phénotype de la cellule d’origine. Enfin, « Asym Diff » décrit les familles où les deux filles ont des phénotypes différents, et aucune d’entre elles n’est la même que la cellule d’origine. Pour acquérir une puissance statistique dans l’évaluation de ces destins symétriques / asymétriques, il est souhaitable d’effectuer l’analyse MultiGen aux premiers points temporels, afin d’observer plus de familles dont la progéniture se trouve dans la génération 1.
Enfin, la figure 5E représente les pourcentages de types de cellules en fonction du nombre de divisions, afin de mieux comprendre la progression du modèle de différenciation entre les divisions. Par exemple, les données affichées dans la figure suggèrent que les cellules progressent vers l’état CD34- , avec plus de 50% des cellules détectées dans cette classe après seulement trois divisions. De plus, il est possible d’en déduire que les MPP ne favorisent pas la division par auto-renouvellement, car un faible pourcentage de cellules conserve le phénotype d’origine. Certaines de ces conclusions peuvent ensuite être testées à l’aide du cadre statistique présenté dans les figures précédentes.

Figure 5 : Exemple de représentation des données pour une expérience de 72 h utilisant des HSPC de sang de cordon. (A) Cartes thermiques pour un ensemble de données sélectionné (HSC, en milieu « Diff », après 72 h de culture). Les graphiques représentent toutes les cellules individuelles (carrés) en fonction de leur parenté (lignes), du nombre de divisions effectuées (colonnes, appelées génération) et du phénotype (couleurs). (Bi) Histogramme comparant les proportions des types cellulaires des descendants cellulaires des CSH et des MPP, entre la condition GT et la condition Diff. (Bii) Le graphique représente les tests statistiques effectués dans le script « 2_bar_plot » pour les MPP à 72h de culture, en comparant les cocktails de cytokines « Diff » et « GT ». La valeur expérimentale est affichée en rouge et les valeurs générées via 250 000 permutations en bleu. La valeur p. du test G est indiquée dans le coin supérieur droit avec le nombre de familles utilisées pour le test. (C) Histogramme comparant le pourcentage de familles (314 familles au total) dans chaque génération (code couleur), pour les CSH et les MPP par condition de culture. Les intervalles de confiance sont calculés avec 250 000 jeux de données démarrés. (D) Histogramme représentant le type de symétrie/asymétrie entre le devenir des cellules filles pour les familles à deux cellules de génération 1 : Sym Undiff (les deux filles conservent le phénotype de la cellule d’origine), Sym Diff (les deux filles ont le même phénotype, et il est différent de la cellule d’origine), Asym Undiff (une seule fille conserve le phénotype de la cellule d’origine), et Asym Diff (les deux filles ont des phénotypes différents et aucune d’entre elles ne ressemble à la cellule d’origine). E) Histogrammes de la contribution des types cellulaires classés par génération pour les MPP cultivés avec le cocktail « Diff »; n = 204 cellules et 97 familles. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Tableau 5 : Description du nombre de familles et de cellules analysées pour chaque condition expérimentale (cellule d’origine et milieu de culture cellulaire). Veuillez cliquer ici pour télécharger ce tableau.
Fichier supplémentaire 1 : Veuillez cliquer ici pour télécharger ce fichier.
Fichier supplémentaire 2 : Veuillez cliquer ici pour télécharger ce fichier.
Fichier supplémentaire 3 : Veuillez cliquer ici pour télécharger ce fichier.
Fichier supplémentaire 4 : Veuillez cliquer ici pour télécharger ce fichier.
Discussion
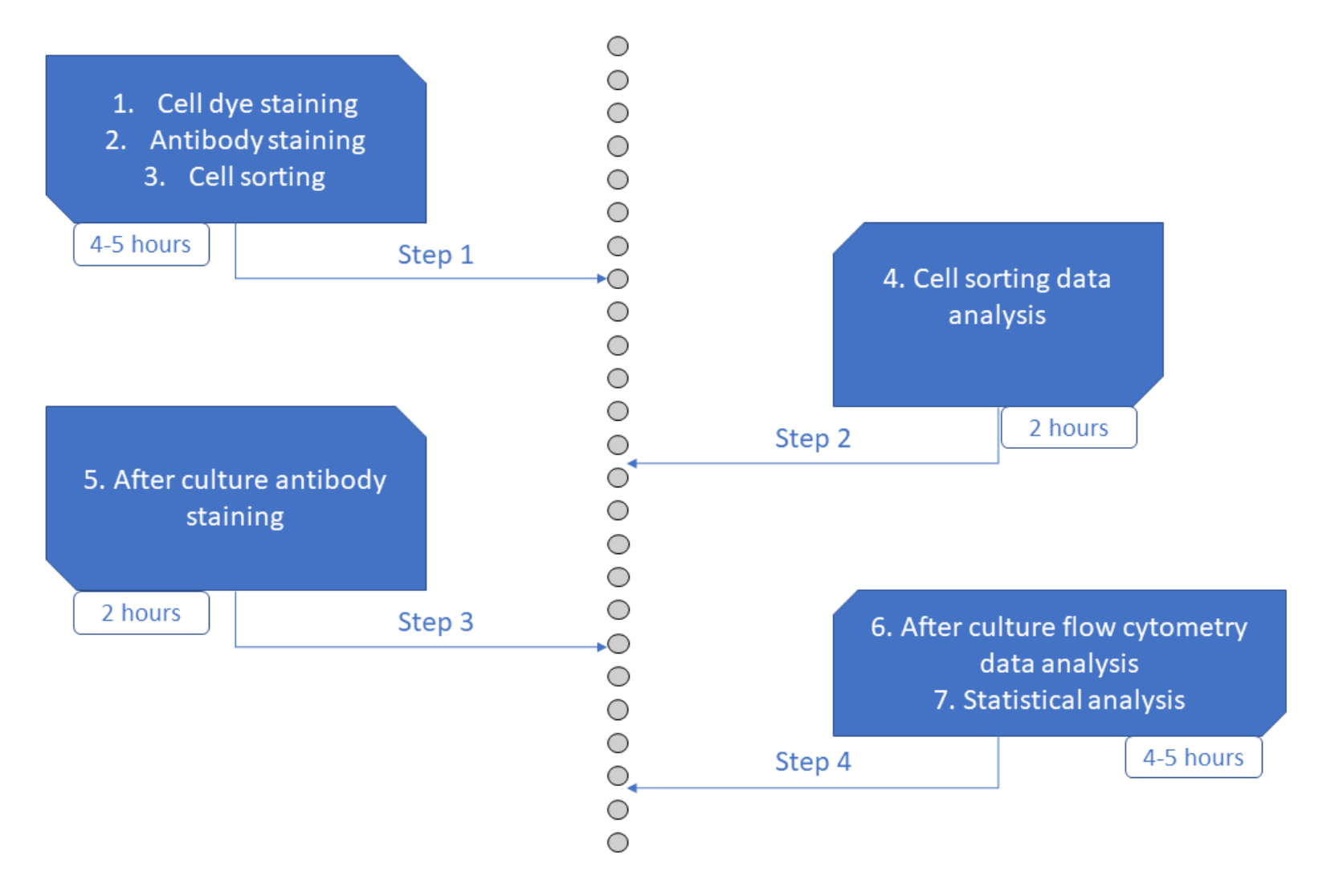
Le test MultiGen est un test à haut débit, facile à réaliser et peu coûteux, qui a joué un rôle déterminant dans l’étude des lymphocytes 23,24,35 et des cellules hématopoïétiques murines26,27. Nous présentons ici un nouveau développement de l’approche qui permet de déchiffrer ex vivo la phase précoce de l’engagement HSPC humain, au niveau de la cellule unique en utilisant une culture à court terme (Figure 6). Les systèmes de culture ex vivo unicellulaire sont généralement utilisés pour évaluer le devenir à long terme des HSPC dans les cellules matures, mais certains destins se produisent plus tôt que d’autres36, ce qui peut biaiser l’analyse en faveur de moins de destins. En outre, ces systèmes de culture manquent généralement d’informations sur les divisions lors de l’engagement du destin. Il a été démontré que les premières étapes de l’engagement se produisent dès le début de la culture, parfois sans division26,37, ce qui rend la culture à court terme et le suivi de la division essentiels pour étudier l’engagement précoce du destin. En suivant simultanément le destin, la division et la parenté, ce test permet de comprendre le rôle de la première division et de la décision du destin dans les HSPC humains. En utilisant le test, il est possible de déduire après combien de divisions le processus d’engagement se produit, l’équilibre entre l’auto-renouvellement et la différenciation pour ces premiers progéniteurs, et comment ces propriétés sont héritées à travers les générations. À notre connaissance, c’est le seul test qui permet ce type de mesures pour les HSPC humains, à une résolution unicellulaire. De plus, en utilisant différentes combinaisons de colorants de division cellulaire, nous avons augmenté le débit de l’analyse, faisant de ce test un outil précieux pour générer rapidement de grands ensembles de données. Les combinaisons de colorants permettent de suivre plusieurs familles dans les mêmes puits, augmentant ainsi le nombre de cellules disponibles pour analyse en culture à court terme. Le nombre de combinaisons pourrait être augmenté encore plus, par l’ajout d’autres colorants (p. ex., colorant jaune) ou la modification du rapport entre CFSE et CTV. Cependant, cela réduit le nombre d’autres paramètres qui peuvent être analysés.

Figure 6 : Représentation schématique du protocole. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Pour effectuer l’analyse avec succès, en raison du grand nombre de puits et du nombre réduit de cellules à analyser, il est nécessaire d’exécuter l’analyse de cytométrie en flux sur un analyseur équipé d’un lecteur de plaques. La nouvelle génération d’analyseurs de banc est particulièrement adaptée à ce test, car la plupart d’entre eux ont un volume mort plus petit pour réduire le pourcentage de perte cellulaire. Cela garantit à son tour une plus grande efficacité dans la récupération de la totalité de chaque puits, ce qui entraîne une efficacité estimée à 70%26. L’estimation de la perte cellulaire lors de l’acquisition de la cytométrie en flux est cruciale pour l’analyse de chaque famille individuelle. Par exemple, en supposant qu’il n’y ait pas de mort cellulaire et en comptant le nombre de divisions, il est possible d’estimer le nombre de cellules par famille. Néanmoins, il est souhaitable de mener quelques expériences de confirmation, notamment dans l’estimation de la mort cellulaire dans les conditions de culture testées et la mesure expérimentale du taux de récupération en utilisant un nombre défini de cellules.
L’une des étapes cruciales de ce protocole est l’affectation de pointe. Comme déjà mentionné, une distribution de pics de bonne qualité dépend fortement de l’isolement de pics très étroits lors du tri cellulaire. Néanmoins, il est encore difficile d’attribuer le nombre correct de divisions en fonction uniquement de la distribution. Comme le tri cellulaire et l’analyse par cytométrie de flux sont effectués sur deux machines différentes, il n’est pas possible de comparer directement l’intensité de chaque signal, il pourrait donc être difficile de savoir si le premier pic observé à l’extrémité droite de l’histogramme est le pic 0 ou le pic 1. À cet égard, peu de solutions sont possibles; Une façon consiste à effectuer une expérience orthogonale pour mesurer avec précision le nombre de divisions effectuées par ces cellules (par exemple, l’imagerie de cellules vivantes). Une autre possibilité consiste simplement à compter le nombre de cellules dans le puits sous un microscope à fond clair inversé, avant d’effectuer l’analyse par cytométrie en flux. Cela permettra d’en déduire un nombre moyen de divisions (en supposant qu’il n’y ait pas de mort cellulaire). Enfin, une solution post-hoc pour l’assignation de pointe est la détection d’un nombre inhabituel de « familles impossibles »; Ces familles sont composées d’un nombre de cellules plus grand que possible par génération (p. ex., cinq cellules en génération 2, ou deux cellules en génération 1 et une cellule en génération 2). La possibilité d’exclure les familles impossibles est codée dans l’étape de l’analyse statistique et signale la famille impossible. Si la fréquence de ces erreurs est trop élevée, il est raisonnable de supposer que l’affectation de pointe doit être révisée.
Dans ce protocole, nous avons inclus quelques exemples de représentation et d’analyse des données pour le test, car c’est devenu une étape essentielle dans la génération et l’interprétation de grands ensembles de données38. Le premier exemple est la carte thermique montrant la totalité de toutes les cellules analysées, organisées par famille. Il s’agit d’un outil efficace pour explorer les propriétés générales des données et les conclusions potentielles : les familles sont-elles composées de plusieurs types de cellules ou ont-elles tendance à être homogènes dans leur composition ? Les familles sont-elles réparties sur plusieurs générations ou se divisent-elles la plupart du temps le même nombre de fois? Cette analyse exploratoire doit ensuite être complétée par des placettes et des tests statistiques plus spécifiques. Il peut être utilisé pour évaluer quantitativement l’engagement de destin symétrique et asymétrique, la différenciation sans division, l’équilibre entre auto-renouvellement et différenciation, et le nombre de divisions pour un destin d’engagement donné. Il est fondamental, lors de la planification expérimentale, de définir la longueur de la culture cellulaire en fonction du type de question posée; Par exemple, pour les deux premières questions (équilibre symétrique/asymétrique et différenciation sans division), la planification d’étapes de cultures très courtes permet d’isoler un grand nombre de familles qui n’ont effectué qu’une seule division ou aucune divisiondu tout26. Inversement, des expériences plus longues permettent d’explorer le nombre de divisions requises pour un engagement cellulaire spécifique, car elles échantillonnent des familles à différents stades de différenciation. Néanmoins, cette méthode n’est pas conçue pour les cultures à long terme (2-3 semaines), car la dilution du colorant cellulaire n’est pas en mesure de suivre avec précision plus de sept ou huit divisions22. En conséquence, cet outil est principalement adapté pour étudier l’engagement précoce des progéniteurs hématopoïétiques, et n’est pas conçu pour tirer des conclusions solides sur les propriétés de différenciation à long terme de ces cellules.
Le cadre statistique a été développé spécifiquement pour l’analyse de ce type de données et basé sur le concept de permutations26. Cela était nécessaire en raison de l’observation d’une dépendance familiale à la distribution des types cellulaires et au nombre de divisions effectuées. En d’autres termes, les cellules qui font partie de la même famille sont également plus susceptibles d’afficher des phénotypes similaires et de se diviser le même nombre de fois. Bien qu’une analyse approfondie dépasse la portée de ce travail, l’ensemble de tests statistiques fourni devrait être suffisant pour évaluer différentes conditions.
En conclusion, ce protocole constitue un outil précieux pour évaluer la dynamique cellulaire des cellules souches et progénitrices hématopoïétiques ex vivo, de manière rapide et peu coûteuse. En raison de sa flexibilité et de sa polyvalence en ce qui concerne le point temporel, les conditions de culture et le type de HSPC analysés, il permet de tester une variété de conditions expérimentales. En tant que test basé sur la cytométrie en flux, il peut être mis en œuvre dans la plupart des laboratoires et ne nécessite pas de connaissances préalables approfondies, ce qui en fait un bon candidat pour les dépistages et les expériences pilotes.
Déclarations de divulgation
Les auteurs ne déclarent aucun conflit d’intérêts pertinent pour ce travail. Les bailleurs de fonds n’ont joué aucun rôle dans la conception de l’étude, la collecte et l’interprétation des données, ni dans la décision de soumettre le travail pour publication.
Remerciements
Nous tenons à remercier les membres de l’Institut Curie Flow Facility pour leur aide dans la mise en place des expériences de cytométrie en flux. Nous tenons également à souligner les contributions des autres membres de l’équipe Perié, lors de multiples discussions. Nous remercions le Dr Julia Marchingo et le professeur Phil Hodgkin (Walter end Eliza Hall Institute of Medical Research) d’avoir partagé leur protocole de multiplexage des colorants de division cellulaire sur les lymphocytes. Nous remercions la biobanque de sang de cordon de l’hôpital de Saint Louis d’avoir fourni les ressources biologiques nécessaires à l’élaboration de ce protocole. L’étude a été soutenue par une subvention ATIP-Avenir du CNRS et de la Fondation Bettencourt-Schueller (à L.P.), des subventions du Labex CelTisPhyBio (ANR-10-LBX-0038) (à L.P. et A.D.), le programme Idex Paris-Science-Lettres (ANR-10-IDEX-0001-02 PSL) (à L.P.), le Canceropole INCA Emergence (2021-1-EMERG-54b-ICR-1, à L.P.), et la subvention ITMO MIIC (21CM044, à L.P.). Outre le financement du Conseil européen de la recherche (CER) dans le cadre du programme de recherche et d’innovation Horizon 2020 de l’Union européenne ERC StG 758170-Microbar (à L.P.), A.D. a été soutenu par une bourse de la Fondation de France.
matériels
| Name | Company | Catalog Number | Comments |
| 1.5 mL polypropylene microcentrifuge tubes | vWR | 87003-294 | |
| 15-mL polypropylene tubes | vWR | 734-0451 | |
| 50-mL polypropylene tubes | vWR | 734-0448 | |
| 96-well U-bottom culture plate | Falcon | 353077 | |
| Anti-human Lin APC | Thermo Fisher | 22-7776-72 | Dilution 1/40 |
| ARIA III | BD | Can be replaced with any FACS sorter able to sort individual cells in 96-wells plate | |
| Carboxyfluorescein succinimidyl ester (CFSE) | Life Technologies | C34570 | |
| Cell Trace Violet (CTV) | Life Technologies | C34571 | |
| Compensation beads | BD | 552843 | |
| Dulbecco’s modified Eagle medium (DMEM) | Life Technologies | 11320033 | |
| Ethylenediaminetetraacetic acid (EDTA) | Thermo Scientific | J62948-36 | Prepare a solution 0.5 M, in sterilised water |
| FC block Fc1.3216 | BD | 564220 | Dilution 1/50 |
| Fetal Bovine Serum (FBS) | Dutscher | S1900-500C | Batch S00CH |
| FlowJo v10.8.1 | BD | ||
| Mouse anti-human CD10 PerCP-5.5, clone HI10a | Biolegend | 312216 | Dilution 1/20 |
| Mouse anti-human CD123 BUV395, clone 7G3 | BD | 564195 | Dilution 1/15 |
| Mouse anti-human CD34 APC-Cy7, clone 581 | Biolegend | 343513 | Dilution 1/40 |
| Mouse anti-human CD38 BV650, clone HB7 | Biolegend | 356620 | Dilution 1/40 |
| Mouse anti-human CD45RA AF700, clone HI100 | BD | 560673 | Dilution 1/20 |
| Mouse anti-human CD45RA PE-Cy7, clone HI100 | BD | 560675 | Dilution 1/20 |
| Mouse anti-human CD90 PE, clone 5E10 | Biolegend | 328110 | Dilution 1/20 |
| Phosphate Buffered Saline (PBS) 1X | Life Technologies | 10010001 | |
| Python | |||
| R | |||
| Sterile 12x75 mm conical polypropylene tubes | Falcon | ||
| ZE5 | Biorad | Can be replaced with any flow cytometry analyzer equipped with a plate reader | |
| Laboratory prepared | |||
| Cell culture media | Depends from the specific experiment. Prepare fresh daily and store at +4 °C until use | ||
| DMEM + 10% FBS | Can be stored in sterile conditions, at +4 °C up to 1 year. To prepare 500 mL, add 50 mL of FBS to 450 mL DMEM | ||
| PBS 1X + EDTA 0.1% | Can be stored in sterile conditions, at room temperature, up to 1 year. To prepare 500 mL, add 3.42 mL of EDTA 0.5 M to 500 mL PBS 1X | ||
| Staining buffer | Can be stored in sterile conditions, at +4 °C up to 1 year. To prepare 500 mL, add 2 mL of EDTA 0.5 M and 1 mL FBS to 500 mL PBS 1X |
Références
- Ginhoux, F., Yalin, A., Dutertre, C. A., Amit, I. Single-cell immunology: Past, present, and future. Immunity. 55 (3), 393-404 (2022).
- Ke, M., Elshenawy, B., Sheldon, H., Arora, A., Buffa, F. M. Single cell RNA-sequencing: A powerful yet still challenging technology to study cellular heterogeneity. Bioessays. 44 (11), 2200084 (2022).
- Regev, A., et al. The human cell atlas. Elife. 6, 27041 (2017).
- Laurenti, E., Göttgens, B. From haematopoietic stem cells to complex differentiation landscapes. Nature. 553 (7689), 418-426 (2018).
- Haas, S., Trumpp, A., Milsom, M. D. Causes and consequences of hematopoietic stem cell heterogeneity. Cell Stem Cell. 22 (5), 627-638 (2018).
- Loughran, S. J., Haas, S., Wilkinson, A. C., Klein, A. M., Brand, M. Lineage commitment of hematopoietic stem cells and progenitors: insights from recent single cell and lineage tracing technologies. Experimental Hematology. 88, 1-6 (2020).
- Perié, L., Duffy, K. R. Retracing the in vivo haematopoietic tree using single-cell methods. FEBS Letters. 590 (22), 4068-4083 (2016).
- Yu, V. W. C., et al. Epigenetic memory underlies cell-autonomous heterogeneous behavior of hematopoietic stem cells. Cell. 167 (5), 1310-1322 (2016).
- Ganuza, M., et al. Lifelong haematopoiesis is established by hundreds of precursors throughout mammalian ontogeny. Nature Cell Biology. 19 (10), 1153-1163 (2017).
- Naik, S. H., Schumacher, T. N., Perié, L. Cellular barcoding: A technical appraisal. Experimental Hematology. 42 (8), 598-608 (2014).
- Quek, L., et al. Genetically distinct leukemic stem cells in human CD34 − acute myeloid leukemia are arrested at a hemopoietic precursor-like stage. The Journal of Experimental Medicine. 213 (8), 1513-1535 (2016).
- Karamitros, D., et al. Single-cell analysis reveals the continuum of human lympho-myeloid progenitor cells. Nature Immunology. 19 (1), 85-97 (2018).
- Boitano, A. E., et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science. 329 (5997), 1345-1348 (2010).
- Delaney, C., et al. Notch-mediated expansion of human cord blood progenitor cells capable of rapid myeloid reconstitution. Nature Medicine. 16 (2), 232-236 (2010).
- Fares, I., et al. Cord blood expansion. Pyrimidoindole derivatives are agonists of human hematopoietic stem cell self-renewal. Science. 345 (6203), 1509-1512 (2014).
- Guo, B., Huang, X., Lee, M. R., Lee, S. A., Broxmeyer, H. E. Antagonism of PPAR-γ 3 signaling expands human hematopoietic stem and progenitor cells by enhancing glycolysis. Nature Medicine. 24 (3), 360-367 (2018).
- Vannini, N., et al. The NAD-booster nicotinamide riboside potently stimulates hematopoiesis through increased mitochondrial clearance. Cell Stem Cell. 24 (3), 405-418 (2019).
- Gupta, R., et al. Nov/CCN3 enhances cord blood engraftment by rapidly recruiting latent human stem cell activity. Cell Stem Cell. 26 (4), 527-541 (2020).
- Horwitz, M. E., et al. Omidubicel vs standard myeloablative umbilical cord blood transplantation: results of a phase 3 randomized study. Blood. 138 (16), 1429-1440 (2021).
- Weinreb, C., Rodriguez-Fraticelli, A., Camargo, F. D., Klein, A. M. Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science. 367 (6479), 3381 (2020).
- Loeffler, D., Schroeder, T. Understanding cell fate control by continuous single-cell quantification. Blood. 133 (13), 1406-1414 (2019).
- Tario, J. D., et al. Optimized staining and proliferation modeling methods for cell division monitoring using cell tracking dyes. Journal of Visualized Experiments. (70), e4287 (2012).
- Marchingo, J. M., et al. T-cell stimuli independently sum to regulate an inherited clonal division fate. Nature Communications. 7, 13540 (2016).
- Horton, M. B., et al. Multiplexed division tracking dyes for proliferation-based clonal lineage tracing. Journal of Immunology. 201 (3), 1097-1103 (2018).
- Lehmann, E. L., Romano, J. P., Casella, G. . Testing statistical hypotheses. , 784 (2005).
- Tak, T., et al. HSPCs display within-family homogeneity in differentiation and proliferation despite population heterogeneity. Elife. 10, 360624 (2021).
- Sommerkamp, P., et al. Mouse multipotent progenitor 5 cells are located at the interphase between hematopoietic stem and progenitor cells. Blood. 137 (23), 3218-3224 (2021).
- Kato, K., Radbruch, A. Isolation and characterization of CD34+ hematopoietic stem cells from human peripheral blood by high-gradient magnetic cell sorting. Cytometry. 14 (4), 384-392 (1993).
- Miltenyi, S., Müller, W., Weichel, W., Radbruch, A. High gradient magnetic cell separation with MACS. Cytometry. 11 (2), 231-238 (1990).
- Doulatov, S., et al. Revised map of the human progenitor hierarchy shows the origin of macrophages and dendritic cells in early lymphoid development. Nature Immunology. 11 (7), 585-593 (2010).
- Goardon, N., et al. Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer Cell. 19 (1), 138-152 (2011).
- Laurenti, E., et al. CDK6 levels regulate quiescence exit in human hematopoietic stem cells. Cell Stem Cell. 16 (3), 302-313 (2015).
- Aiuti, A., et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 341 (6148), 1233151 (2013).
- Davison, A. C., Hinkley, D. V. . Bootstrap Methods and their Application. , (1997).
- Horton, M. B., et al. Lineage tracing reveals B cell antibody class switching is stochastic, cell-autonomous, and tuneable. Immunity. 55 (10), 1843-1855 (2022).
- Notta, F., et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science. 351 (6269), 2116 (2016).
- Grinenko, T., et al. Hematopoietic stem cells can differentiate into restricted myeloid progenitors before cell division in mice. Nature Communications. 9 (1), 1898 (2018).
- Saeys, Y., Van Gassen, S., Lambrecht, B. N. Computational flow cytometry: Helping to make sense of high-dimensional immunology data. Nature Reviews Immunology. 16 (7), 449-462 (2016).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.