Method Article
Flujo de trabajo completo para el análisis de las histonas modificaciones post-traduccionales El uso de espectrometría de masas de abajo hacia arriba: A partir de la histona Extracción de Análisis de Datos
En este artículo
Resumen
Este protocolo describe un flujo de trabajo totalmente integrado para la caracterización de las histonas modificaciones post-traduccionales utilizando espectrometría de masas (MS). El flujo de trabajo incluye la purificación de la histona de cultivos de células o tejidos, la derivación de histonas y la digestión, el análisis de MS utilizando cromatografía líquida de flujo nano e instrucciones para el análisis de datos. El protocolo está diseñado para dentro de 2 - 3 días.
Resumen
Nucleosomas son la unidad estructural más pequeña de la cromatina, compuesta de 147 pares de bases de ADN envueltos alrededor de un octámero de proteínas histonas. la función de la histona está mediada por una amplia modificación posterior a la traducción por una miríada de proteínas nucleares. Estas modificaciones son críticas para la integridad nuclear ya que regulan la estructura de la cromatina y reclutar enzimas implicadas en la regulación génica, la reparación del ADN y la condensación de los cromosomas. A pesar de que una gran parte de la comunidad científica adopta técnicas basadas en anticuerpos para caracterizar la histona abundancia PTM, estos enfoques son el bajo rendimiento y sesgada contra proteínas hypermodified, ya que el epítopo puede ser obstruida por modificaciones cercanas. Este protocolo describe el uso de cromatografía líquida de nano (NLC) y espectrometría de masas (MS) para la cuantificación precisa de modificaciones de las histonas. Este método está diseñado para caracterizar una gran variedad de PTM histonas y la abundancia relativa de varios de histonas variantes dentro de sanaliza la ingle. En este protocolo, las histonas se derivatizan con anhídrido propiónico seguido por la digestión con tripsina para generar péptidos de 5-20 aa de longitud. Después de la digestión, la recién expuesta N-terminales de los péptidos de histonas se derivatizan para mejorar la retención cromatográfica durante nLC-MS. Este método permite la cuantificación relativa de PTM histonas que abarcan cuatro órdenes de magnitud.
Introducción
La epigenética se define como el estudio de los cambios heredables en la expresión de genes que se presentan por mecanismos distintos de la alteración de la secuencia de ADN subyacente 1. Regulación epigenética es crítica durante el desarrollo como el organismo se somete a fenotípica dramático cambia a pesar de que su contenido de ADN no cambia. Hay varios componentes críticos necesarios para el mantenimiento epigenética adecuada, incluyendo histonas modificaciones post-traduccionales (PTMs), variantes de las histonas, ARN no codificantes, la metilación del ADN y factores de unión de ADN, cada uno de los cuales afectan a la expresión génica a través de diferentes mecanismos 2. Por ejemplo, mientras que la metilación del ADN es una modificación muy estable que reprime la traducción del gen 3, las variantes de las histonas y PTM histonas son mucho más dinámica y puede influir en la cromatina en una variedad de maneras 4.
PTM histonas se localizan principalmente en las colas N-terminal, ya que son la región más expuesta y flexiblede la proteína. Sin embargo, el núcleo del nucleosoma está también muy modificada en comparación con un promedio de 5 proteínas. A pesar de que las marcas de las histonas se han caracterizado ampliamente en la última década, muchos enlaces entre histonas marcas conocidas y su función aún no están claros. Esto se debe en gran parte al hecho de que la mayoría de los PTM histonas no trabajan solos, sino más bien la función en conjunto con otra PTM ( "cross-talk") para alterar un proceso específico, como la transcripción de 6,7. Por ejemplo, la marca H3S10K14ac combinatoria en el gen p21 activa su transcripción, lo que no ocurriría con sólo uno de los dos PTM 8. Los compactos HP1 proteínas cromatina mediante el reconocimiento de H3K9me2 / ME3 y la difusión de la modificación de los nucleosomas cercanas. Sin embargo, no se puede enlazar HP1 H3K9me2 / 3 cuando el S10 adyacente es fosforilada 9. La acetilación de H3K4 inhibe la unión de la proteína a la spChp1 H3K9me2 / ME3 en Schizosaccharomyces pombe 10. Además, la histona lisina demethylase PHF8 tiene la mayor eficiencia de unión cuando tres nucleosoma PTM H3K4me3, K9ac, y K14ac están presentes 11. Estos ejemplos ponen de relieve la importancia de lograr una visión global de los cambios PTM histonas en lugar de centrarse en las modificaciones individuales.
La presencia de variantes de secuencia también aumenta la complejidad del análisis de la histona, como isotipos histonas generalmente tienen secuencias muy similares, pero a menudo tienen diferentes funciones en la cromatina. Por ejemplo, H2A.X tiene una secuencia C-terminal que está más fácilmente fosforilados sobre el daño del ADN en comparación con H2A canónica 12, y se requiere para la inactivación de los cromosomas sexuales en la meiosis masculina ratón 13; Del mismo modo, CENP-A sustituye a la histona H3 canónica en centrómeros 14. A pesar de sus diferentes funciones, estas variantes comparten una gran parte de su secuencia de aminoácidos con el respectivo histona canónica, lo que hace difícil identificar y cuantificar por separado.
técnicas basadas en anticuerpos, tales como transferencia Western han sido ampliamente adoptado para caracterizar las histonas. Sin embargo, los enfoques basados en anticuerpos son limitadas por las siguientes razones: (i) que sólo se puede confirmar la presencia de una modificación y no pueden identificar PTM desconocidos; (Ii) que tienen un sesgo debido a la presencia de las marcas de co-existentes, lo que puede influir en la afinidad de unión; (iii) que no pueden identificar marcas combinatorias, ya que sólo muy pocos anticuerpos están disponibles para tal propósito y (iv) que una reacción cruzada entre las variantes de las histonas altamente similares o PTMs similares (por ejemplo, di- y trimethylation de residuos de lisina). Egelhofer et al. describe que más del 25% de los anticuerpos comerciales no superen las pruebas de especificidad por transferencia de puntos o transferencia de Western, y entre los anticuerpos específicos de más de 20% de fracaso en experimentos de inmunoprecipitación de la cromatina 15. Espectrometría de masas (MS) es actualmente la herramienta de análisis más adecuado para estudiar nuevos y / o combinatorios PTM,y se ha aplicado ampliamente para las proteínas histonas (revisado en 16). Esto se debe principalmente a la alta sensibilidad y exactitud de la masa de la EM, y la posibilidad de realizar análisis a gran escala.
La estrategia de abajo hacia arriba es la estrategia proteómica basadas en MS más comúnmente utilizado para la caracterización de histonas y sus PTM, en el que la proteína intacta es digerido enzimáticamente en péptidos cortos (de 5 - 20 aa). Esta digestión facilita tanto la separación y detección LC MS. Masas en el rango de 600 - 2.000 Da son comúnmente más fácilmente ionizadas y se identificaron con una mayor exactitud de la masa y de la resolución de masas mayores. MS / MS de fragmentación también se mejora, ya que los péptidos cortos son generalmente muy adecuados para disociación inducida por colisión (CID). Sin embargo, las histonas presentan un desafío para abajo hacia arriba MS, ya que son altamente enriquecido en residuos de aminoácidos básicos, a saber, la lisina y la arginina. Por lo tanto, la digestión con tripsina da lugar a la generación de péptidos que son demasiado smtodo para la retención de LC y la localización no ambigua de la PTM. Para sortear este problema, nuestro protocolo incluye la lisina y la derivación química péptido N-terminal de 17. El uso de anhídrido propiónico se recomienda para la derivatización química eficiente en comparación con otros reactivos 18. Tales bloques de derivatización de los grupos ɛ-amino de residuos de lisina no modificados y monometil, permitiendo que la tripsina para llevar a cabo la proteolisis sólo en el C-terminal de los residuos de arginina. aminas derivatizados no pueden intercambiar protones con la solución y por lo tanto los péptidos generalmente sólo se doble o triplemente cargado, facilitando MS y detección MS / MS. Por otra parte, la derivatización N-terminal aumenta la hidrofobicidad del péptido y la retención cromatográfica de fase inversa de este modo. A continuación, se describe el flujo de trabajo para purificar las histonas y prepararlos para el análisis de proteómica PTM a través de abajo hacia arriba (Figura 1). Esta estrategia logra cuantificación de marcas de histona individuales y marcas combinatorias fo PTM histonas que están relativamente cerca en la secuencia de aminoácidos.
Protocolo
1. Recolección de células de Cultura
- Si las células se hicieron crecer en suspensión, recoger las células por centrifugación a 300 rcf durante 5 min. Si adherente, aspirado y desechar medio celular. Enjuagar las células unidas con PBS sin Ca2 + y Mg2 + (denominado de ahora en adelante PBS). Se incuban las células en tripsina o tripsina-EDTA (0,025% - 0,5%, dependiendo de la línea celular) con un volumen suficiente para cubrir la superficie de las placas a 37 ° C hasta que las células se separan (el tiempo varía para diferentes líneas de células).
- Recoger las células por centrifugación a 300 rcf durante 5 min. Lavar las células dos veces más en PBS y recoger mediante centrifugación.
- Estimar el volumen de células empaquetadas aproximadamente desde las graduaciones marcadas en los tubos de 1,8 ml o 15 ml tubos cónicos.
Nota: Las células de cultivo se pueden snap-congelado en nitrógeno líquido y se almacenan a -80 ° C indefinidamente en esta etapa.
2. Aislamiento de los núcleos de células intactas
- Descongelar las células en hielo.
- Descongelar tampón de aislamiento nuclear (NIB, Tabla 1).
- Preparar aproximadamente 5 ml de tampón NIB (Tabla 1) por cada volumen de células empaquetadas 100 l. Por cada tampón NIB 1 ml, añadir los inhibidores de proteasa y agentes estabilizantes de la siguiente manera: 1 l de 1 M de DTT, 2,5 l de AEBSF 200 mM, 2 l de 2,5 M de microcistina y 2 l de 5 M de butirato de sodio. NIB con inhibidores se conoce como la SEMILLA de ahora en adelante.
Nota: Si se estudia la fosforilación de la histona, incluir EDTA proteasa libre y cóctel inhibidor de la fosfatasa. - Eliminar quinto volumen de tampón NIB así preparado y añadir NP-40 Alternativa (Tabla 1) a una concentración final de 0,2%. El de cuatro quinto volumen restante se utiliza para el lavado.
- sedimento celular lavado en 1:10 sedimento celular a NIB sin NP-40 relación alternativa (v / v). Extraer el sobrenadante por centrifugación a 700 rcf durante 5 min.
- Lyse el cell pellet colocándolo en hielo y añadiendo pellet 01:10 célula para NIB con 0,2% NP-40 Alternativa (v / v).
- Si la extracción de muestras de tejido, homogeneizar utilizando homogeneizadores de mortero y majadero o Dounce. Las células cultivadas se pueden homogeneizar mediante pipeteo suave.
- Se incuban las células homogeneizadas en hielo durante 5 a 10 min. Las células se lisar y liberar los núcleos.
- Se centrifuga a 1.000 rcf durante de 5 - 10 min a 4 ° C. El sedimento contiene principalmente núcleos de las células, mientras que el sobrenadante contiene componentes principalmente citoplásmicos. Guarde la fracción citoplasmática si se desea.
- Lavar el sedimento de núcleos resuspendiendo suavemente en 1:10 (v / v) NIB sin NP-40 Alternativa.
Nota: Esta etapa de lavado es únicamente para eliminar los rastros de detergentes antes de la extracción de las histonas de los núcleos. - Centrifugar a 1000 rcf durante 5 min a 4 ° C y retirar el sobrenadante.
- Repita el paso 2.10 - 2.11 al menos dos veces para eliminar completamente NP-40 Alternativa. La eliminación de NP-40 Alternativa es evidente unas pipeteo suave durante la etapa de lavado ya no forma burbujas.
- Para la extracción de la histona de los tejidos:
- Enjuague el tejido fresco congelado o descongelado en helado de NIB.
- Transferir el tejido a una placa de Petri se colocaron en hielo con NIB, sólo lo suficiente para mantener el tejido húmedo.
- Dados en trozos más pequeños (<1 mm) con hoja de afeitar para aumentar la superficie de contacto para el aislamiento de núcleos.
- Transferir tejido picado a un homogeneizador enfriado previamente y lavar en NIB la pipeta hacia arriba y hacia abajo.
- Eliminar el tampón por centrifugación a 300 rcf durante 5 min.
- Añadir NIB que contiene NP-40 Alternativa a las células en un células: tampón relación de 01:10 (v / v) y homogeneizar por 5 - 10 golpes.
- Consultar la lisis celular y la homogeneización repetir según sea necesario. Un buen indicador de que las células han sido lisadas es la reducción del volumen de pellets. El sedimento debe contener sólo los núcleos.
- Centrifugar a 700 rcf durante 5 min y guardar pellets. Esta pastilla se puede extraer 1 - 2 más tiempos en 01:10 (v / v) de la punta que contiene NP-40 Alternativa; en esta etapa, las histonas se extraen de la cromatina y el sedimento se ha encogido considerablemente.
- Lavar dos veces con 2 - 3 ml de NIB sin NP40 alternativa para eliminar las trazas de detergente.
Nota: punto de parada provisional: La muestra se puede resuspende en el volumen mínimo de NIB + 5% de glicerol y se almacenó a -80 ° C.
3. Extracción y purificación de las histonas de los núcleos
Nota: Las histonas son muy ricas en restos de aminoácidos básicos, lo que les permite interactuar fuertemente con la columna vertebral de ácido fosfórico de ADN. Las histonas son proteínas entre las más básicas en el núcleo, lo que les permite ser extraídos en ácido sulfúrico enfriado con hielo (0,2 MH 2 SO 4) con un mínimo de contaminación de las proteínas no histonas, que precipitan en ácido fuerte. TCA altamente concentrado (a una concentración final de 33%) se puede usar entonces para precipitar las histonas de la sulfúricoácido. TCA se almacena como 100% en botella marrón a 4 ° C.
- Resuspender los núcleos de células en 1: 5 (v / v) en forma refrigerada 0,2 MH 2 SO 4 (Tabla 1) por pipeteo suave.
- Incubar la muestra con rotación constante o agitación suave durante 2-4 horas a 4 ° C. Típicamente, para las muestras con más de 500 l sedimento celular, una extracción de 2 horas es suficiente para extraer las histonas; de incubación más largo puede dar lugar a la extracción de otras proteínas básicas. Para precipitados nucleares más pequeñas (<200 l), 4 h de extracción proporciona un mejor rendimiento.
- Centrifugar a 3400 RCF a 4 ° C durante 5 min.
- Transferir el sobrenadante a un nuevo tubo.
- Repetir los pasos 3.3 a 3.4 para eliminar cualquier material insoluble.
- Para precipitar las histonas, añadir refrigerada 100% TCA (Tabla 1) al sobrenadante recogido (ahora histonas contienen) en la proporción de 1: 3 (v / v), con el fin de obtener una concentración de TCA final del 33%. Mezclar invirtiendo el tubo unos pocos tIME.
Nota: Las muestras se convertirán nublado con la adición de TCA, indicando la presencia de las histonas. - Incubar la mezcla en hielo durante al menos 1 hr. Para los tamaños de pellets de partida más pequeñas, se recomienda la precipitación durante la noche.
- Centrifugar a 3.400 rcf durante 5 min. La capa de histonas los lados de los tubos y también depositar en la parte inferior. Un pellet insoluble blanco también se forma en la parte inferior del tubo, que en su mayoría contiene proteínas no histonas y otras biomoléculas. Aspirar el sobrenadante mediante aspiración, con cuidado, sin raspar los lados o en el sedimento.
- Mediante el uso de una pipeta Pasteur de vidrio, enjuagar el tubo con acetona enfriada con hielo + 0,1% de HCl (Tabla 1) de manera que cubra las proteínas precipitadas revestimiento los lados y el fondo.
- Centrifugar a 3400 rcf durante 2 min y el sobrenadante aspirado, cuidadosamente sin raspar los lados o la pastilla.
- Repita los pasos 3.9 a 3.10 usando acetona al 100% enfriado con hielo.
- pellet seco con flujo de aire o con un vacuum centrifugar, o simplemente dejando el tubo abierto. La acetona se evapora rápidamente.
- Disolver las histonas con ddH2O (agua doblemente destilada) en un volumen mínimo posible disolver la capa blanca por completo. Las histonas son fácilmente solubles en agua. Para precipitados en un tubo de 1.5 ml, 100 l de ddH2O suele ser suficiente para recoger las histonas.
- Centrifugar a 3400 rcf durante 2 min y transferir el sobrenadante a un nuevo tubo.
4. Estimación de la concentración de proteína y Pureza
- Para la medición de la concentración de proteína, utilizar BCA, ensayo de proteínas de Bradford o análisis de aminoácidos (AAA). No utilice técnicas que adoptan la absorbancia a 280 nm, como las histonas son pobres en residuos de aminoácidos aromáticos.
- Verificar la pureza de las histonas extraídos por análisis de SDS-PAGE con un gel de acrilamida al 15% y tinción de Coomassie (opcional).
- Si se desean alta pureza variantes sola histonas, seguirá fraccionamiento de HPLC-UVvariantes de histonas (sección 5). Si no es así, vaya directamente a la preparación de muestras para el análisis de la histona PTM de abajo hacia arriba (sección 6).
5. Separación de histona variantes por HPLC de fase inversa (Opcional)
Nota: las variantes de histonas de alta pureza pueden obtenerse por fraccionamiento de la mezcla de la histona bruto usando HPLC de fase inversa acoplada a un detector UV. Estas histonas purificadas son útiles para estudios que requieren mayor sensibilidad y pureza. Sin embargo, para la caracterización estándar de histona PTM, este paso se puede omitir debido a que el análisis es suficientemente sensible y exhaustiva. El fraccionamiento de la histona intacta variantes idealmente requiere al menos 100 a 300 g de material de partida.
- Conectar una columna apropiada C 18 5 micras a una HPLC en función de la concentración de partida de las histonas: con aproximadamente 100 g de histonas, utilizar la columna 2,1 mm x 250 mm con una velocidad de flujo de 0,2 ml / min; con alrededor de 300 g histonas, utilizar la columna 4,6 x 250 mm conuna velocidad de flujo de 0,8 ml / min. Preparar el tampón A y B usando cristalería dedicado como sigue:
- Preparar Tampón A: 5% de acetonitrilo de grado HPLC, 0,1% de TFA en agua de calidad HPLC.
- Preparar Tampón B: 95% de acetonitrilo de grado HPLC, 0,1% de TFA en agua de calidad HPLC.
- Conectar la columna a un detector UV, y establecer la absorbancia a 210 - 220 nm.
- Se acidifica la muestra de la histona disuelto en agua con 100% de TFA para lograr una concentración final de 0,1 a 1% de TFA.
- Equilibrar la columna con 100% de tampón A durante por lo menos 15 min a la velocidad de flujo recomendada, que corresponde aproximadamente a tres volúmenes de columna. Utilizar esta señal para establecer el nivel cero de absorbancia del detector de UV.
- Preparar tubos de tamaño adecuado para recoger las fracciones de forma manual o en un colector de muestras automático.
- Inyectar la muestra en una concentración de aproximadamente 1 g / l o superior. Muestras disueltas en volúmenes más grandes podrían alterar el equilibrado de la columna duCargando anillo y conducir a la retención inferior.
- Ejecutar el gradiente, programado de la siguiente manera: de 0 a 30% de B en 1 min, de 30 a 60% de B en 90 min, y de 60 a 90% de B en 1 min.
- Recoger fracciones (ejemplo cromatograma mostrados en la Figura 2) en intervalos de 1 min utilizando un colector de fracciones automático. Recoger fracciones en tubos de tamaño apropiado para contener todo el volumen.
- Se seca muestras fraccionadas en un concentrador de vacío.
Nota: punto de parada provisional: fracciones de histona secas se pueden almacenar a temperatura ambiente durante períodos cortos (1 - 2 días) o en el congelador a -80ºC durante períodos de largo plazo.
6. La derivación química de las histonas El uso de anhídrido propiónico para el análisis de abajo hacia arriba
- (Cantidad recomendada: 50 - 100 g) Disolver muestras histonas en 40 l de 50 mM NH 4 HCO 3, pH 8,0. Si las muestras estaban en pura ddH 2 O, añadir concentrado NH 4 HCO 3 para compensar 50 mM, pH 8.0.
- Mojar una punta de pipeta P10 en la muestra a comprobar el pH con tiras indicadoras de pH sin pérdidas de muestra. NH 4 OH y ácido fórmico se pueden utilizar para ajustar el pH a 8,0.
Nota: La siguiente parte del protocolo (pasos 6.3 a 6.7) que se debe hacer en lotes de máximo de tres a cuatro muestras, a fin de mantener anhídrido propiónico reactiva. - Utilice campana de humos para las etapas posteriores donde se utiliza anhídrido propiónico. Preparar reactivo propionilación fresco mezclando anhídrido propiónico con acetonitrilo en la proporción 1: 3 (v / v). Añadir reactivo propionilación a la muestra en 1: 4 (v / v). Durante 40 histonas l, añadir 10 l de reactivo propionilación.
Nota: Es posible observar restos blanco en este paso. Sin embargo, este contiene principalmente sales y ácido propiónico, y por lo tanto necesita ser tomado ninguna acción específica. - Añadir rápidamente NH 4 OH para restablecer pH 8,0 a la solución. Nota: anhídrido propiónico reaccionar con las aminas libres de los péptidos produce propácido iónico que disminuye el pH. Por lo general, la adición de NH 4 OH a la muestra con una proporción de 1: 5 (v / v) que es apropiado para restablecer pH 8,0; por ejemplo, 8 l de NH 4 OH a 40 l de muestra.
- Mezclar inmediatamente por agitación.
- Verificar el pH con el mismo procedimiento que en el Paso 6.2.
Precaución: Cuando el pH es mayor que 10,0, el etiquetado de los otros residuos de aminoácidos con una mayor pKa es posible. - Incubar las muestras a temperatura ambiente durante 15 min.
- Repita los pasos 6.3 a 6.7, realizando estrictamente la reacción durante no más de 3 o 4 muestras por lote de reactivo propionilación.
- Las muestras secas de hasta 10 - 20 l en un concentrador de vacío. Este evapora anhídrido sin reaccionar propiónico, acetonitrilo, ácido acético y gas de amoniaco liberado de NH 4 OH. Si las muestras se sequen por completo, no se producen pérdidas significativas de la muestra.
Nota: Isopropanol se puede utilizar en lugar de acetonitrilo. Sin embargo, acetonitrilo tiene menor tensión superficial y por lo tanto másevaporación rápida. - Volver a suspender o diluir las muestras con ddH2O hasta conseguir 40 l de volumen final.
- Repita los pasos 6.2 a 6.9. Un doble ronda de propionilación histona asegura> 95% de la finalización de la reacción.
- Llene la botella anhídrido propiónico con gas argón a fin de evitar la formación de ácido acético en contacto con la humedad en la botella.
Nota: punto de parada provisional: La muestra se puede almacenar a -80 ° C reconstituida en ddH2O o secos.
7. La digestión proteolítica con tripsina
- Histonas resuspender en 50 mM NH 4 HCO 3 para lograr una concentración óptima de 1 g / l o superior. muestras más diluida conducen a disminuir la eficiencia de la tripsina.
Nota: Las histonas en este paso deben estar a pH 8,0 Si aún ácida, a continuación, añadir NH 4 HCO 3 sal a la muestra mediante el uso de la punta de la pipeta. - Añadir tripsina a las muestras de las histonas en una proporción 1:10 (peso / peso).
- Incubadoraste a 37 ° C durante 6 - 8 horas.
- Detener la digestión mediante la congelación de -80 ° C.
- Se seca la muestra a 10 - 20 l en un concentrador de vacío.
Nota: punto de parada provisional: La muestra se puede almacenar a -80 ° C.
8. propionilación de histona péptidos en N-terminales
Nota: En esta sección se describe la derivación de péptido N-terminal generado a partir de la digestión con tripsina. Tal procedimiento mejora la retención de HPLC de los péptidos más cortos (por ejemplo, amino ácido 3-8 de la histona H3), ya que el grupo propionilo aumenta la hidrofobicidad del péptido.
- Volver a suspender las muestras en 30 l de 100 mM NH 4 HCO 3.
- Repita los pasos 6.1 a 6.9.
Nota: Es normal que el secado de las muestras en vacío tarda más tiempo en este paso. - Volver a suspender o diluir las muestras con 50 - 100 l ddH 2 O + o bien TFA al 0,1% o el 0,5% de ácido acético. Nota: Se recomienda el ácido acético para los almacenamientos largos, como TFA facilitatit oxidación de metionina en el largo plazo. Por otro lado, TFA se recomienda si el escenario-vuelco (sección 9) que se realiza el mismo día, como TFA ayuda a una mejor retención cromatográfico.
Nota: punto de parada provisional: La muestra se puede almacenar a -80 ° C.
9. El desalado de la muestra con la etapa de los dedos
Nota: En esta etapa, no es la sal presente en la muestra. Las sales impiden el análisis de HPLC-MS, ya que ionizan durante electrospray, la supresión de la señal de péptidos. Las sales también se pueden formar aductos iónicos de péptidos, la reducción de la intensidad de la señal para el péptido no aducción. A medida que el péptido en aducción tendrá una masa diferente, el péptido no será identificado o cuantificado correctamente.
- Mediante el uso de una punta de pipeta P1000, perforar un disco de material de C 18 a partir de un disco de extracción en fase sólida. Empuje el minidisco de la punta P1000 mediante el uso de un capilar de sílice fundida y depositar el minidisco a la parte inferior de una punta de pipeta P100 / 200. Asegúrese de que el dISK se acuña de forma segura en la parte inferior de la punta (Figura 3).
Nota: La punta P1000 tiene un pequeño agujero en lugar de golpear el disco C18. Es apropiado para cortar el último centímetro de la punta con el fin de tener un agujero con un diámetro más grande. - Utilice dos C 18 golpes en la misma punta P100 / P200 si la desalación de más de 25 g de muestra.
- Utilice un adaptador de centrífuga para mantener la etapa de los dedos en lugar de 1,5 ml o 2 ml tubos de microcentrífuga. Utilice lenta - rotación (300 a 400 RCF); los disolventes normalmente pasan a través de la resina en menos de un minuto.
- Lavar la resina por hilado con 50 l de 100% de acetonitrilo para activar el material C 18 y eliminar posibles contaminaciones.
- Equilibrar disco por el lavado de 80 l de TFA al 0,1% por centrifugación lenta.
- Se acidifica la muestra a pH 4,0 o inferior con ácido acético. Comprobar el pH con tiras de pH para minimizar la pérdida de muestra. Cargar la muestra en el disco mediante centrifugación lenta.
- lavarmuestra por el lavado de 70 - 80 l de TFA al 0,1% por centrifugación lenta.
- Eluir la muestra por el lavado 70 l 75% de acetonitrilo y ácido acético 0,5% por centrifugación lenta. Recoger la muestra en un tubo de 1,5 ml.
- muestra seca en un concentrador de vacío.
Nota: punto de parada provisional: La muestra se puede almacenar a -80 ° C.
10. Análisis de histona péptidos
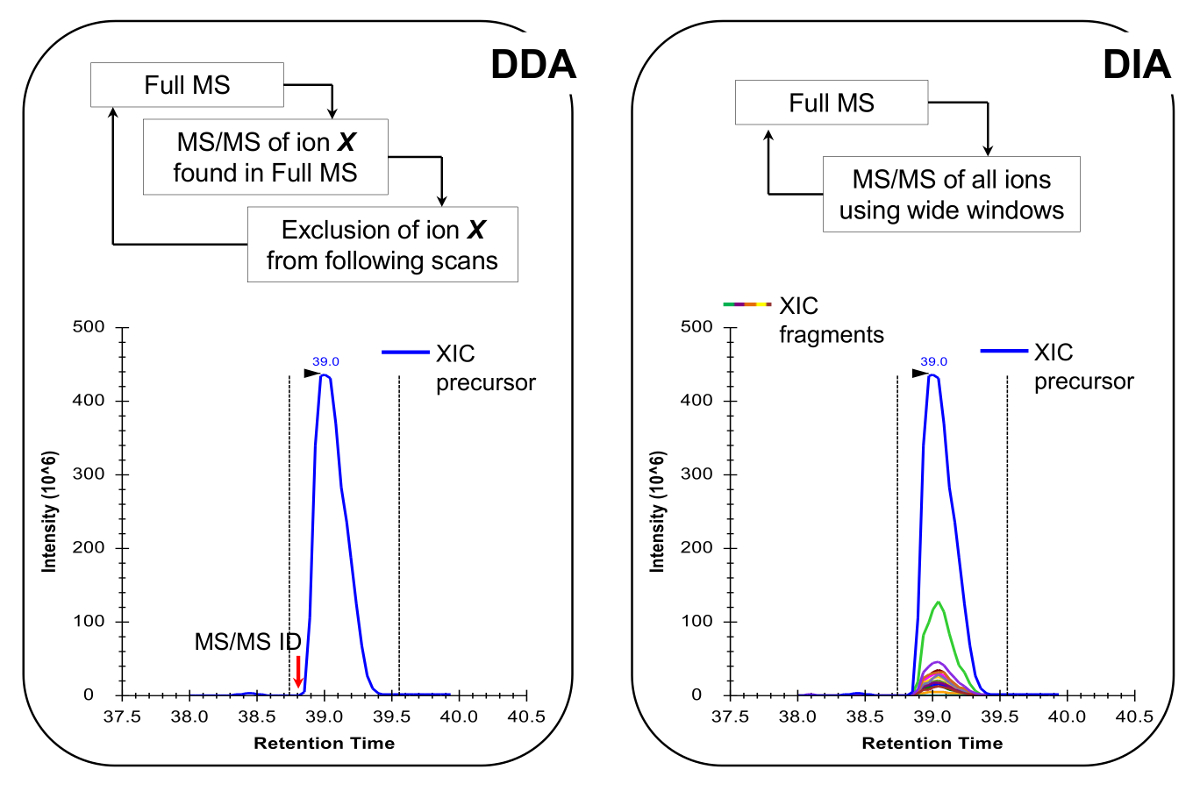
Nota: La plataforma NLC-MS debe establecerse como se hace en el análisis de péptidos tradicional. Se recomienda columna de flujo de 300 nl (75 micras Identificación columna analítica C, 18 partículas), ya que son un excelente compromiso entre la sensibilidad y la estabilidad - El uso de 200. El método de adquisición de MS puede ser o bien una combinación de la adquisición de datos dependiente (DDA) con exploraciones específicos 19 o una adquisición (DIA) de datos independiente de 20,21, ambos descritos en resultados representativos y la Figura 4.
- Preparar tampones de HPLC - A: ácido fórmico al 0,1% enagua de calidad de HPLC; B: ácido fórmico al 0,1% en acetonitrilo de calidad HPLC.
- Programar el método HPLC de la siguiente manera: de 0 a 30% de tampón B en 30 min, de 30 a 100% de B durante los siguientes 5 minutos y a isocrático 100% de B durante 8 min. Si la HPLC no está programado para el equilibrado en columna automatizada antes de la carga de la muestra, a continuación, incluyen los siguientes: gradiente 100-0% de B en 1 min y el flujo isocrático al 0% de B durante 10 min. Ajuste el caudal del análisis a 250 - 300 nl / min.
- Programar el método de adquisición MS para llevar a cabo ya sea en combinación con DDA exploraciones específicas o 19 DIA 20,21 (Figura 4). Asegúrese de que el ciclo de trabajo de MS permite una completa MS escanear cada ~ 2 segundos, con el fin de tener suficientes puntos de datos a través del pico cromatográfico, que permite la cuantificación más precisa. Nota: Con C 18 cromatografía, la anchura media de pico de línea de base es de aproximadamente 30 seg para el gradiente descrito anteriormente.
- Cargar aproximadamente 1 g de la muestra en la HPLC cCOLUMNA.
- Ejecutar el método de HPLC-MS / MS según lo programado.
Nota: En el protocolo no recomendamos detalles de columnas, instrumentos de MS o MS detalles de los parámetros, como cualquier configuración óptima que un laboratorio de proteómica individuo desarrollado será adecuado para el método. laboratorios de proteómica deben utilizar su configuración optimizada, ya que los péptidos se separan las histonas como péptidos tradicionales.
Análisis de datos 11.
- Importar los archivos de MS primas en el software para llevar a cabo la integración del área del pico. Nota: EpiProfile 22 se recomienda, ya que está optimizado para los péptidos de histonas; mediante el uso de los conocimientos tiempo de retención de elución cromatográfica se realiza la extracción de área de pico fiable de péptidos conocidos histonas. Alternativamente, Skyline 23 es otro software ideal para el propósito.
- Calcular la abundancia relativa de un péptido dado dividiendo su área de la superficie total de que el péptido en todas sus formas modificadas. Nota: En caso de Analy descubrimientoSe recomienda sis Mascot para identificar espectros de péptidos de histonas modificadas. El rendimiento de esta herramienta ha sido descrito recientemente 24. Todos los otros motores de búsqueda de base de datos para la proteómica también son utilizables, pero en nuestras pruebas que proporcionan un rendimiento inferior.
Resultados
A modo de ejemplo, se analizaron las histonas extraídas de células madre embrionarias humanas (hESCs) con y sin estimulación ácido retinoico (RA), comenzando con 200 l sedimentos celulares. La presencia de la AR en cultivo celular conduce a la diferenciación de ESC. Desde el sedimento de células, sobre 50 a 100 g de histonas se extrajeron, que es más que suficiente para llevar a cabo múltiples inyecciones LC-MS de los péptidos de histonas. Después de una derivación, la digestión y la desalinización, las muestras se cargaron en un 75 micras cm de columna x 15 C 18 (diámetro de partícula de 3 micras, tamaño de poro 300 Å) en el modo de serie con un sistema de cromatografía nano líquida de alta resolución con chips de microfluidos acoplados a un híbrido cuadrupolo lineal trampa - espectrómetro de masas Orbitrap. MS adquisición se realizó a través de DIA. En paralelo, las muestras también se analizaron con un método DDA usando un UHPLC-flujo nano acoplado a un espectrómetro de masas de trampa de iones-Orbitrap híbrido (datos no mostrados). Encada ciclo, una detección completa de MS Orbitrap se realizó con el rango de barrido de 290 a 1.400 m / z, una resolución de 60.000 (a 200 m / z) y AGC de 10 6. A continuación, el modo de adquisición dependiente de los datos se aplicó con una dinámica exclusión de 30 seg. exploraciones de MS / MS fueron seguidos de iones primarios de las más intensas. Iones con un estado de carga de un solo fueron excluidos de MS / MS. Se utilizó una ventana de aislamiento de 2 m / z. Los iones se fragmentaron usando disociación inducida por colisión (CID), con la energía de colisión de 35%. Detección de trampa de iones se utiliza con el modo de rango de exploración normal y velocidad de barrido normal con AGC de 10 4.
Se analizaron los datos en bruto MS software de adoptar para la extracción de precursores y de iones fragmento de cromatogramas, a saber Skyline 23 y 22 EpiProfile. EpiProfile ha sido optimizado para los péptidos de histonas, ya que integra la extracción inteligente área del pico debido al conocimiento previo de Peptiempo de retención marea. Por otro lado, Horizonte está optimizado para el análisis de la DIA, y por lo tanto las cifras mostradas DIA (Figuras 4 y 5A) son capturas de pantalla de este software. A partir del cromatograma de iones extraídos, el área bajo la curva se recupera, y esto se utiliza para estimar la abundancia de cada péptido. El área del pico cromatográfico se calculó para el [M + H] +, [M + 2H] 2+, y [M + 3H] 3+ iones del mismo péptido, aunque en la mayoría de los casos, el [M + 2H] 2+ era la forma predominante. Esto proporciona la abundancia en bruto de una forma modificada de un péptido dado. Con el fin de lograr la abundancia relativa de PTM, la suma de todas las diferentes formas modificadas de un péptido de histona se consideró como 100%, y el área del péptido particular, se dividió por el área total para que el péptido de la histona en todas sus formas modificadas .
péptidos histonas están presentes en una VAVariedad de formas isobáricas (Figura 5). Péptidos isobáricas, por ejemplo, K18ac y K23ac, sólo pueden ser cuantificados en el nivel de MS / MS, donde se utilizan sus iones fragmento único para determinar la relación de la especie isobáricas (Figura 5A y 5B). Esta proporción se utiliza para dividir el área del pico cromatográfico entre las dos especies. Cuando se utiliza DDA, estas formas isobáricas se incluyeron en una lista de masas específicas, debido a que estos péptidos deben ser seleccionados para la fragmentación a través de la totalidad de su elución, lo que no ocurriría en un experimento estándar DDA. La discriminación de la abundancia relativa de las especies isobáricas se lleva a cabo a continuación, mediante la supervisión del perfil de elución de los iones de fragmentos. Por otro lado, el tipo de DIA de adquisición no requiere ninguna lista de inclusión. Sin embargo, este tipo de método de adquisición no es compatible con la búsqueda de base de datos tradicional, y por lo tanto puede impedir el descubrimiento de péptidos modificados desconocidos.
Lisina acetilación (+ 42.011 Da) se discriminó de la trimethylation casi isobárica (+ 42.047 Da) mediante el uso de alta resolución de adquisición de MS (> 30.000). Por otra parte, la acetilación es más hidrofóbico que trimethylation, que conduce a la elución de péptidos acetilados más tarde de los respectivos trimethylated queridos. La forma no modificada del mismo péptido eluye incluso más tarde, debido al hecho de que la lisina se propionilado. En resumen, el orden de hidrofobicidad para un péptido con un sitio modificable es di- y trimethylated
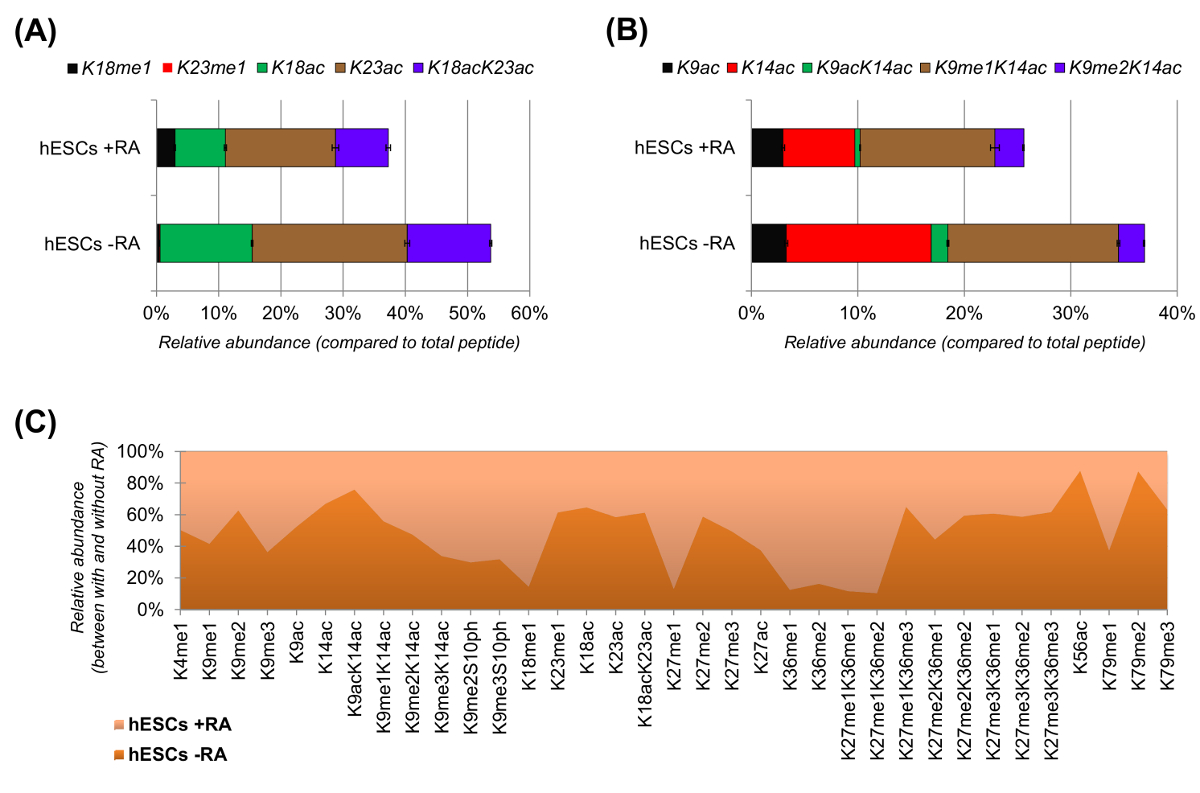
hESCs mostraron una clara reducción de los péptidos acetilados cuando son estimuladas para la diferenciación (Figura 6A y 6B). Esto no era sorprendente, ya que los resultados previos informaron mayor acetilación en los CES, en comparación con los diferenciadores 25,lo que refleja la naturaleza generalmente permisiva de la cromatina pluripotentes. Al centrarse en la histona H3, 35 diferentes formas modificadas fueron cuantificados (Figura 6C). Sin embargo, todos proteoforms histonas que pueden investigarse con este enfoque son más de 200, incluyendo todas las variantes y modificaciones de histonas de baja abundancia (datos no mostrados). Por otra parte, el análisis mostró que una alta reproducibilidad se puede obtener entre repeticiones técnica, como se evidencia por el pequeño tamaño de las barras de error (que representan ± desviación estándar). En su conjunto, esta sección se describe cómo extraer la abundancia relativa de los péptidos modificados histonas utilizando datos NLC-MS.

Figura 1:. Flujo de trabajo para MS / MS análisis de la histona de abajo hacia arriba se muestran los diez pasos para el análisis de las histonas, incluyendo una estimación del tiempo requerido para cada paso. El número de sección se da entre paréntesis como presente en el manuscrito. Sección 5, que describe el fraccionamiento de la muestra para aislar las diferentes variantes de histonas, se puede omitir a menos que exista una necesidad de un análisis altamente sensible de una variante dada. Por favor, haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Fase Inversa de gran flujo de LC para la histona variante de Fraccionamiento y de Coomassie del gel (A) cromatograma LC-UV que representa la separación de histonas intacta.. variantes de la histona H3 pueden ser discriminados unos de otros en función de su tiempo de elución. Las fracciones se pueden recoger de forma manual o mediante un colector de fracciones automático. (B) de gel de Coomassie de tres réplicas de la purificación de la histona.= "Https://www.jove.com/files/ftp_upload/54112/54112fig2large.jpg" target = "_ blank"> Haga clic aquí para ver una versión más grande de esta figura.

Figura 3:. Making of Etapa-inclinar enchufe con una punta de pipeta P1000, perforar un disco de material C 18 material de un disco de extracción en fase sólida (segundo panel). El minidisco se pegará en la punta (panel central), de modo que pueda ser empujado hacia fuera en una punta de pipeta más pequeño P100 / 200 utilizando cualquier tipo de pequeños capilares. En este ejemplo, se utilizó un diámetro externo de tubo de sílice fundida de 700 micras. El minidisco se debe empujar a la parte inferior de la punta de la pipeta P100 / 200 hasta que no pueda ir más lejos (último panel). La punta etapa está listo para el desalado de la histona, ya que tiene la capacidad suficiente para retener suficiente material de muestra para numerosas repeticiones. Específicamente, un minidisco es suficiente para 15 - 20 g de sample. Si se requiere más de la muestra, varios discos se pueden empaquetar el uno del otro. Por favor, haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4:. Representación esquemática de la ADD y DIA Métodos Al utilizar DDA, el ciclo de exploración MS se caracteriza por la selección secuencial de iones precursores de fragmentación MS / MS en función de su intensidad y estado de carga. Una vez que un ion precursor ha sido fragmentada se coloca en una lista de exclusión para evitar la selección repetitiva del mismo péptido, de modo que la MS puede "cavar" en señales menos abundantes. Este método de adquisición es la técnica de elección en la proteómica para el modo de descubrimiento. La cuantificación se logra mediante la integración de la señal de barrido completo de un ion dado junto a la MS identificados /MS espectro. En DIA, toda la gama m / z se fragmenta en cada ciclo de exploración. Este enfoque es menos adecuado para el modo de detección, pero produce un perfil cromatográfico de todos los iones, los precursores y los productos. Esto conduce a una cuantificación más confianza y la discriminación de formas isobáricas. Por favor, haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5: La cuantificación de isobáricas péptidos (A) Ejemplo de dos péptidos isobáricas comúnmente abundantes en el análisis de la histona.. El cromatograma de iones extraídos (XIC) de su masa precursora e isótopos relativos (arriba) es idéntico. Sin embargo, la XIC de los iones de productos (abajo) permite la discriminación de las dos formas isobáricas. En particular, los fragmentos de iones aparecen unicamente deben ser nosotrosed para estimar la abundancia relativa de las dos especies. (B) Representación de los iones de fragmentos únicos de los dos péptidos descritos (resaltado en rojo). (C) Lista de los péptidos comúnmente analizados en Homo sapiens que tienen al menos un equivalente isobárica. Variantes de secuencia entre los péptidos se indican las histonas en la lista. Por favor, haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 6: Los resultados representativos de las células madre embrionarias humanas con y sin tratamiento ácido retinoico (A) Cuantificación relativa del péptido de histona H3 KQLATKAAR (aa 18 - 26) en todas sus proteoforms modificados.. La abundancia relativa se estimó utilizando todos proteoforms como el 100% (el relporcentaje ative del péptido no modificado no se muestra) (B) Cuantificación relativa del péptido de histona H3 KSTGGKAPR (aa 9 -.. 17) (C) La abundancia relativa de los péptidos detectados para la histona H3 canónica con y sin tratamiento de células con ácido retinoico. La figura indica en cuál de los dos tratamientos las modificaciones dadas son más abundantes (> 50%). En general, se demuestra que la acetilación de la histona H3 disminuye en la mayoría de los residuos de lisina tras la inducción de la diferenciación celular. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
| Solución # | Composición | ||||||
| 1 | El aislamiento nuclear Buffer (NIB) stock se hace de la siguiente manera y se almacena congelado en alícuotas de 100 ml a -20 ° C; descongeladoNIB se puede almacenar a 4 ° C durante unas semanas: mM Tris 15, KCl 60 mM, NaCl 15 mM, 5 mM MgCl2, 1 mM CaCl 2, y sacarosa 250 mM. El pH del tampón se ajusta a 7,5 con HCl. | ||||||
| 2 | Inhibidores de la proteasa (añada fresca para tampones antes de su uso): 1 M ditiotreitol (DTT) en ddH2O (1,000x); AEBSF 200 mM en ddH2O (400x) | ||||||
| 3 | inhibidor de la fosfatasa (añadir fresco a los tampones antes de su uso): 2,5 M microcistina en etanol al 100% (500x) | ||||||
| 4 | inhibidor de HDAC (añadir fresco a tampones antes de su uso): 5 butirato de sodio M, hechos por valoración del ácido 5 M butírico usando NaOH a pH 7,0 (500x) | ||||||
| 5 | NP-40 Alternativa: 10% v / v en ddH 2 O | ||||||
| 6 | 0,2 MH 2 SO 4 en ddH2O | ||||||
| 7 | El ácido tricloroacético (TCA): 100% w / v en ddH2O | ||||||
| 8 | La acetona + 0,1% de ácido clorhídrico (HCl): 0,1% v / v HCl en acetona | ||||||
Tabla 1. Solutions.
Discusión
El protocolo descrito aquí está optimizado teniendo en cuenta los costos, tiempo y rendimiento. Otras preparaciones son posibles, pero tienen limitaciones, especialmente en el caso de acoplamiento con el análisis de MS. Por ejemplo, el protocolo de extracción de alto contenido de sal se puede utilizar para purificar histonas 26 en lugar de la precipitación con TCA (sección 3). protocolo de alta sal es intrínsecamente más suave, ya que no utiliza ácido fuerte. Esto preserva PTM lábiles en medio ácido y aumenta el rendimiento de las histonas extraídos, como precipitación con TCA co-precipitados de muchas otras proteínas de unión a la cromatina. Sin embargo, la extracción de alto contenido de sal conduce a muestras que contienen sal demasiado concentrada para HPLC-MS / MS. En una preparación alternativa, la digestión de la histona se puede realizar sin propionilación (sección 6-8), por ejemplo al reducir el tiempo de incubación de la tripsina y la relación enzima / sustrato 27 o el uso de ArgC como enzima de digestión 28-30. Sin embargo, se recomienda derivatización con anhídrido propiónico, como yot conduce a la generación de los péptidos más hidrófobos, que son mejor retenidos durante la cromatografía líquida.
Para la derivatización química, una variedad de anhídridos de ácidos orgánicos han sido evaluados y sus méritos ampliamente discutido 18. Sin embargo, anhídrido propiónico resultó el mejor compromiso entre la eficiencia, productos secundarios minimizados y la mejora de la hidrofobicidad del péptido. Potencialmente, anhídrido propiónico se pueden comprar en forma marcado con isótopos; Esto permite el análisis de multiplexado debido a la posibilidad de mezclar múltiples muestras y discriminar ellos en el nivel de MS en base a las diferentes masas impartidas de la etiqueta pesado. Sin embargo, este análisis conduce a un aumento de la complejidad del cromatograma LC-MS y reduce la cantidad de muestra que se puede inyectar para cada condición individual.
En este sentido, hay que destacar algunos aspectos críticos del protocolo. Lo siguiente debe ser utilizado como checklist para encontrar errores en la realización del procedimiento en caso de que se obtengan resultados negativos. En primer lugar, después de la precipitación núcleos el sedimento se debe lavar cuidadosamente con NIB sin NP-40 Alternativa (sección 2.10) hasta la eliminación total del detergente (perceptible por la falta de burbujas durante el mezclado). De no hacerlo, podría comprometer la extracción de histonas con ácidos. En segundo lugar, después de la precipitación con TCA histona (sección 3.9) lavados del precipitado con acetona es crucial. La presencia de ácido concentrado dañaría el siguiente paso si propionilación y la digestión (sección 6.1) se realizan directamente. No sería problemático en caso de fraccionamiento de la histona se lleva a cabo (sección 5). En tercer lugar, es esencial que la reacción se lleva a cabo de forma rápida propionilación (sección 06.03 a 06.07). Para ello, evitar el uso de la misma mezcla propionilación (anhídrido propiónico + acetonitrilo) durante más de 3 - 4 muestras consecutivas. Además, el pH es el aspecto más importante de la digestión con tripsina (sección 7). Si noalrededor de 8.0 (7.5 a 8.5) la digestión será ineficaz. Esto puede suceder, ya que la muestra será rico en ácido propiónico en este paso. NH4OH se puede añadir hasta que sea necesario. Además, para los investigadores familiarizados con los flujos de trabajo de la proteómica se siente normal para acidificar la muestra de interrumpir la digestión con tripsina. Esto no se debe hacer, ya que pondrá en peligro la siguiente reacción, es decir, propionilación del péptido N-terminal (sección 8.1). Por último, en el mismo número, es importante tener en cuenta para el análisis de los datos que los péptidos no modificados no son en realidad no modificado; todos los residuos de lisina libre y N-terminales serán ocupados por propionilación (56.026 Da). Por lo tanto, la realización de cromatografía iónica extracción de la masa correspondiente a la forma única secuencia peptídica conduciría a ningún resultado.
Las limitaciones del método son en su mayoría relacionados con la incapacidad de detectar PTM combinatorias, debido a las secuencias de péptidos cortos, y los sesgos en el logro de la verdadera abunbaile de una modificación, debido al hecho de que los péptidos en diferentes formas modificadas pueden ionizar con diferentes eficiencias. El primer problema puede ser resuelto mediante la combinación de esta técnica con una media hacia abajo o de arriba hacia abajo (revisado en 16). Este tipo de análisis, incluso si es técnicamente más difícil, es ideal para el estudio de las frecuencias de co-existencia de modificaciones. Además, permite una mejor discriminación de las variantes de las histonas, que no siempre pueden ser alcanzados con de abajo hacia arriba, ya que algunos péptidos tienen la misma secuencia en diferentes variantes de las histonas. El segundo problema, relacionado con la eficacia de ionización, se puede resolver utilizando una biblioteca de péptidos sintéticos 31. Este enfoque garantiza una estimación más precisa de la abundancia relativa de PTM histonas. Sin embargo, en la mayoría de los experimentos, el resultado deseado es que los cambios relativos de modificaciones dadas entre las condiciones analizadas. En este caso, dicha corrección no es necesario, debido al hecho de que todas las muestras tienen los mismos BIAs.
En conclusión, este protocolo permite el análisis de PTM histonas que se puede completar en 3 días usando nLC acoplado a MS en tándem. Las comparaciones con técnicas distintas de la MS, es decir, el uso de estrategias basadas en anticuerpos como se explica en la introducción, no son adecuados, ya que no pueden lograr aún casi este nivel de rendimiento. Además, las técnicas basadas en anticuerpos no permiten el descubrimiento de nuevas modificaciones, pero que se basan exclusivamente en confirmar y cuantificar marcas predichos. por tanto, se especula que la proteómica de abajo hacia arriba en péptidos histonas ganará popularidad en los laboratorios de proteómica debido a las ventajas intuitivas en conocer la regulación de las marcas de las histonas, que son protagonistas en la expresión génica de sintonización y por lo tanto afectar a la regulación del proteoma. Por otra parte, el protocolo descrito incluye mejoras recientes en la preparación de la muestra y el software de análisis de datos, que hacen que el análisis de la histona más triviales también para laboraconservadores que nunca tuvieron una caracterización de este tipo de péptidos hypermodified.
Divulgaciones
Los autores declaran que no tienen intereses financieros en competencia.
Agradecimientos
Este trabajo fue apoyado por la financiación de subvenciones del NIH (DP2OD007447, R01GM110174 y R01AI118891).
Materiales
| Name | Company | Catalog Number | Comments |
| Trypsin 0.25% EDTA | Invitrogen | 25200056 | For harvesting cells |
| PBS | Invitrogen | 14200075 | |
| Tris | Roche | 77-86-1 | |
| Potassium Chloride | Fisher Scientific | BP366-500 | |
| Sodium Chloride | Sigma | S9888 | |
| Magnesium Chloride hexahydrate | Sigma | M9272 | |
| Calcium Chloride, anhydrous | Sigma | C1016 | |
| Sucrose | Fisher Scientific | BP220-1 | |
| DTT | Invitrogen | 15508-013 | |
| AEBSF | EMD Millipore Corp | 101500 | |
| Microcystin | Sigma | M4194 | |
| Sodium Butyrate | Sigma | B5887 | |
| Halt Protease and Phosphatase Inhibitor Cocktail, EDTA-free (100X) | Fisher Scientific | 78445 | |
| NP-40 Alternative | CALBIOCHEM | 492016 | |
| Sulfuric Acid, ACS grade | Fisher Chemical | 7664-93-9 | |
| Trichloroacetic acid | Sigma | T6399 | |
| Acetone | Sigma | 179124 | |
| HCl | Fisher Chemical | A144-500 | |
| Bradford reagent | Biorad | 500-0006 | |
| 30% acrylamide/bis 29:1 -- 500ml | Biorad | 1610156 | |
| Coomassie | Fisher Scientific | 20278 | |
| C18 Column (5um) 2.1mm x 250mm | Grace | 218TP52 | |
| C18 Column (5um) 4.6mm x 250mm | Grace | 218TP54 | |
| HPLC grade acetonitrile | Fisher Chemical | A955-4 | |
| HPLC grade water | Fisher Scientific | W6 4 | |
| TFA | Fisher Scientific | A11650 | |
| Ammonium Bicarbonate | Sigma | A6141 | |
| ammonium hydroxide | Sigma | 338818 | |
| propionic anhydride | Sigma | 240311 | |
| Sequencing grade modified trypsin | Promega | PRV5113 | For digesting histones for MS |
| Acetic Acid | Sigma | 49199 | |
| C18 extraction disk | Empore | 2215 | |
| Formic Acid | Sigma | F0507 |
Referencias
- Waddington, C. H. Canalization of development and the inheritance of acquired characters. Nature. 150, 563-565 (1942).
- Sharma, S., Kelly, T. K., Jones, P. A. Epigenetics in cancer. Carcinogenesis. 31 (1), 27-36 (2010).
- Reik, W., Dean, W., Walter, J. Epigenetic reprogramming in mammalian development. Science. 293 (5532), 1089-1093 (2001).
- Kouzarides, T. Chromatin modifications and their function. Cell. 128 (4), 693-705 (2007).
- Tessarz, P., Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Bio. 15 (11), 703-708 (2014).
- Fischle, W., Wang, Y. M., Allis, C. D. Histone and chromatin cross-talk. Curr Opin Cell Biol. 15 (2), 172-183 (2003).
- Lee, J. S., Smith, E., Shilatifard, A. The language of histone crosstalk. Cell. 142 (5), 682-685 (2010).
- Simboeck, E., et al. A Phosphorylation Switch Regulates the Transcriptional Activation of Cell Cycle Regulator p21 by Histone Deacetylase Inhibitors. J Biol Chem. 285 (52), 41062-41073 (2010).
- Hirota, T., Lipp, J. J., Toh, B. H., Peters, J. M. Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature. 438 (7071), 1176-1180 (2005).
- Xhemalce, B., Kouzarides, T. A chromodomain switch mediated by histone H3 Lys 4 acetylation regulates heterochromatin assembly. Genes Dev. 24 (7), 647-652 (2010).
- Vermeulen, M., et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. 142 (6), 967-980 (2010).
- van Attikum, H., Gasser, S. M. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol. 19 (5), 207-217 (2009).
- Fernandez-Capetillo, O., et al. H2AX is required for chromatin remodeling and inactivation of sex chromosomes in male mouse meiosis. Dev Cell. 4 (4), 497-508 (2003).
- Santaguida, S., Musacchio, A. The life and miracles of kinetochores. Embo J. 28 (17), 2511-2531 (2009).
- Egelhofer, T. A., et al. An assessment of histone-modification antibody quality. Nat Struct Mol Biol. 18 (1), 91-93 (2011).
- Sidoli, S., Cheng, L., Jensen, O. N. Proteomics in chromatin biology and epigenetics: Elucidation of post-translational modifications of histone proteins by mass spectrometry. J Proteomics. 75 (12), 3419-3433 (2012).
- Plazas-Mayorca, M. D., et al. One-Pot Shotgun Quantitative Mass Spectrometry Characterization of Histones. J Proteome Res. 8 (11), 5367-5374 (2009).
- Sidoli, S., et al. Drawbacks in the use of unconventional hydrophobic anhydrides for histone derivatization in bottom-up proteomics PTM analysis. Proteomics. 15 (9), 1459-1469 (2015).
- Lin, S., Garcia, B. A. Examining histone posttranslational modification patterns by high-resolution mass spectrometry. Methods Enzymol. 512, 3-28 (2012).
- Sidoli, S., et al. SWATH Analysis for Characterization and Quantification of Histone Post-translational Modifications. Mol Cell Proteomics. , (2015).
- Krautkramer, K. A., Reiter, L., Denu, J. M., Dowell, J. A. Quantification of SAHA-Dependent Changes in Histone Modifications Using Data-Independent Acquisition Mass Spectrometry. J Proteome Res. , (2015).
- Yuan, Z. F., et al. EpiProfile Quantifies Histone Peptides With Modifications by Extracting Retention Time and Intensity in High-resolution Mass Spectra. Mol Cell Proteomics. 14 (6), 1696-1707 (2015).
- MacLean, B., et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 26 (7), 966-968 (2010).
- Yuan, Z. F., Lin, S., Molden, R. C., Garcia, B. A. Evaluation of proteomic search engines for the analysis of histone modifications. J Proteome Res. 13 (10), 4470-4478 (2014).
- Tan, Y., Xue, Y., Song, C., Grunstein, M. Acetylated histone H3K56 interacts with Oct4 to promote mouse embryonic stem cell pluripotency. Proc Natl Acad Sci U S A. 110 (28), 11493-11498 (2013).
- Vonholt, C., et al. Isolation and Characterization of Histones. Methods Enzymol. 170, 431-523 (1989).
- Zhang, K. L., et al. Identification of acetylation and methylation sites of histone H3 from chicken erythrocytes by high-accuracy matrix-assisted laser desorption ionization-time-of-flight, matrix-assisted laser desorption ionization-postsource decay, and nanoelectrospray ionization tandem mass spectrometry. Anal. Biochem. 306 (2), 259-269 (2002).
- Jufvas, A., Stralfors, P., Vener, A. V. Histone Variants and Their Post-Translational Modifications in Primary Human Fat Cells. Plos One. 6 (1), e15960 (2011).
- Bonaldi, T., Imhof, A., Regula, J. T. A combination of different mass spectroscopic techniques for the analysis of dynamic changes of histone modifications. Proteomics. 4 (5), 1382-1396 (2004).
- Zhao, X. L., et al. Comparative Proteomic Analysis of Histone Post-translational Modifications upon Ischemia/Reperfusion-Induced Retinal Injury. J Proteome Res. 13 (4), 2175-2186 (2014).
- Lin, S., et al. Stable-isotope-labeled histone peptide library for histone post-translational modification and variant quantification by mass spectrometry. Mol Cell Proteomics. 13 (9), 2450-2466 (2014).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados